
Abstract
The electrochemical synthesis of solid compounds is a critical emerging area in electrocatalysis; however, a major challenge is still remaining on severe catalysts scaling driven by strong solid-solid interactions, giving rise to rapid electrode deactivation. In this study, for the example of simultaneous synthesis of Mg(OH)2 and H2 from natural seawater electrolysis, we demonstrate that Pt catalyst with surface-coordinated halogens (F, Cl, Br, I) can alleviate this scaling effect (i.e., solidophobicity) by like-charge repulsion and thus regulating the local environment. Specifically, Pt-I coordination results in supersolidophobicity, achieving a successful extraction of Mg(OH)2 ( > 99% purity) while stably producing H2 (under 100 mA cm-2 for over 5000 hours). A combination of experimental and theoretical studies reveals that, due to the like charge repulsion between I- and in situ generated OH-, the Pt-I catalyst regulates the surface pH gradient which increases the distance from the electrode surface to the Mg(OH)2 nucleus (>4 μm) and facilitates homogeneous nucleation. Additionally, the scalability of the Pt-I catalyst, along with the techno-economic analysis and life cycle assessment of the natural seawater electrolysis technology are systematically demonstrated.
Similar content being viewed by others

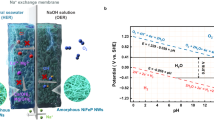

Introduction
The production of green hydrogen fuel through water electrolysis is crucial for the advancement of renewable energy systems1,2,3,4. According to the World Energy Council, global hydrogen demand is projected to reach at least 148 million tons per year by 20505,6. However, compared to gray hydrogen, which is produced via methane reforming or coal gasification, the cost of green hydrogen production remains significantly higher (~US$2 and ~US$5 per kg H2 for gray and green hydrogen, respectively)7. In addition to the cost reduction of renewables, coupling water electrolysis with other value-increment reactions is considered an effective strategy to offset energy input costs8,9,10. The general approach involves replacing the oxygen evolution reaction (OER) with alternative anodic reactions that exhibit lower thermodynamic equilibrium potential and/or reduced kinetic overpotential11,12,13, such as electrooxidation of biomass molecules14, plastic wastes15, and organic compounds16. These alternatives have been extensively explored, achieving significant progress17,18,19. Another strategy is to capitalize on the surface alkaline environment generated in situ during the hydrogen evolution reaction (HER) at the cathode, which facilitates the extraction of valuable metal resources from the electrolyte by forming easily separable hydroxide precipitates. For instance, the electrolysis of natural seawater enriched with Mg2+ (> 0.79 g/L) enables the simultaneous production of H2 and high-value Mg(OH)2 at the cathode20.
Although the aforementioned process elevates the benefits of seawater electrolysis, the severe adhesion of precipitates to the electrode surface, which leads to rapid electrode deactivation, limits its sustainability21,22,23. According to classical nucleation theory, heterogeneous nucleation on the electrode surface is thermodynamically more favorable than homogeneous nucleation due to the significantly lower nucleation energy barrier24,25. Therefore, from a kinetic perspective, it is essential to regulate the local reaction environment at the cathode during the hydrogen evolution reaction (HER) to favor homogeneous nucleation of Mg(OH)2 in the electrolyte. However, the lack of in-depth studies on the local reaction environment significantly complicates solidophobic catalyst design26. In principle, the catalyst should simultaneously achieve high HER activity under near-neutral environment and exhibit solidophobic surface properties.
In this study, we demonstrate that the coordinated halogens (F, Cl, Br, I) on Pt catalysts are critical for regulating the local reaction environment while maintaining high HER activity. In experiments, the Pt-I catalyst loaded on a Ti substrate (Pt-I/Ti) exhibits the best solidophobic behavior (i.e., supersolidophobicity) and maintains stable operation at 100 mA cm−2 in natural seawater electrolyte for over 5000 h. In contrast, the unmodified Pt/Ti electrode is rapidly covered by Mg(OH)2, leading to a drastic degradation in current density (100 mA cm−2, < 1 h, Fig. 1a). The stability of Pt-I/Ti is attributed to its ability to regulate the local surface reaction environment. This regulation arises from the like-charge repulsion between the surface halogen ligands and OH− generated during the HER process, which drives OH− away from the electrode and allows it to react with Mg2+ in the bulk seawater (Fig. 1b). In situ experiments and theoretical calculations indicate that surface-coordinated halogens remain stable throughout the HER process, with the peak OH− concentration occurring ~8 μm away from the surface, much farther than on an unmodified electrode (~3 μm). The practical applicability of this supersolidophobic electrode is demonstrated by assembling it in a scaled electrolyzer (electrode area: 100 cm2), which exhibits stable performance (10 A, > 1000 h) for the simultaneous production of green H2 and Mg(OH)2 (5.54 g h−1, > 99% purity). The double-product strategy contributes to reducing the H2 production cost and environmental impact in seawater electrolysis.

a Conventional Pt/Ti cathode which will be gradually covered by Mg(OH)2 precipitates. b Halogen-modified Pt-X/Ti (X: F, Cl, Br, I) cathode which can be stably operated due to the like-charge repulsion towards OH-.
Results and discussion
Preparation and characterization of the Pt-X/Ti (X: F, Cl, Br, I) electrocatalyst
The synthesis of Pt-X/Ti involves first electrodeposition of H2PtCl6 on Ti substrate to form Pt-Cl/Ti electrode, which is subsequently utilized to fabricate Pt-F/Ti, Pt-Br/Ti and Pt-I/Ti electrodes by exchanging the coordinated halogens, as schemed in Fig. 1a and Fig. S1 (details can be seen in Methods). The contrastive sample of Pt/Ti without coordinated halogens was also prepared, characterized and evaluated. The scanning electron microscopy (SEM), selected area electron diffraction (SAED) and X-ray diffraction (XRD) results of five samples exhibited negligible morphological differences of Pt and the same predominant exposed crystallographic plane (1,1,1) (Fig. S2-6), consistent with the samples obtained via electrodeposition methods reported previously27.
The energy dispersive spectroscopy (EDS) results of Pt-X/Ti from transmission electron microscopy (TEM) are shown in Fig. 2b–e, where the distributions of halogen elements align well with those of Pt. The results from ion chromatography and Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) indicate that the four samples exhibit similar halogen/platinum content ratios (Fig. S7). Time of Flight Secondary Ion Mass Spectrometry (TOF-SIMS) was further employed to characterize the interaction between halogens and Pt nanoparticles28,29, as shown in Fig. 2f-i and Fig. S8. The 3D TOF-SIMS results demonstrated that PtX2- (X: F, Cl, Br, I) were primarily existed on the surface of four samples, while the concentrations gradually decreased with the increase of depth, directly proving that halogens coordinated with Pt were predominantly located at the surface. In contrast, negligible halogens can be detected on the surface of Pt nanoparticles for the Pt/Ti electrode without halogen-modification (Fig. S9).

a Schematic illustration of the synthesis process of Pt-X/Ti. EDS mapping and 3D TOF-SIMS images of (b, f) Pt-F/Ti, (c, g) Pt-Cl/Ti, (d, h) Pt-Br/Ti, and (e, i) Pt-I/Ti.
Electrocatalytic performance in natural seawater
The HER activities of Pt/Ti, Pt-X/Ti (X: F, Cl, Br, I) and Pt foil were firstly evaluated in a standard three-electrode system with natural seawater (from Ningbo Bay after sediment removal, Fig. S10, 11, and Supplementary Table 1) as the electrolyte. As shown in Fig. 3a, the Pt-X/Ti required small overpotentials (445, 428, 450 and 465 mV for Pt-F/Ti, Pt-Cl/Ti, Pt-Br/Ti and Pt-I/Ti, respectively) for achieving industrial current density (~100 mA cm−2), better than those of Pt/Ti electrode and Pt foil (472 and 550 mV, respectively). The polarization curves without iR correction are presented in Fig. S13a. Moreover, the Tafel slope of Pt-X/Ti (163–196 mV dec−1) and the charge transfer resistance (3.4–9.5 Ω) further confirmed the advanced HER kinetics (Fig. S12b and 13, Supplementary Table 2)30,31,32. The enhanced HER activity can be explained by the enhanced first-electron-affinity due to surface coordinated halogens33. Density functional theory (DFT) calculations of the HER process in a neutral environment, as shown in Fig. S14, 15, reveal that the coordinated halogens weaken the adsorption energy of H* at adjacent Pt sites, thereby accelerating the Heyrovsky step and promoting the HER process34. The chlorine weakens the adsorption energy the most while iodine shows the weakest effect, consistent with the difference in HER activities.

a Polarization curves with iR correction of Pt/Ti, Pt-X/Ti and Pt foil in natural seawater (pH=7.71 ± 0.02, the solution resistance: 5 ± 0.5 Ω). The scan rate was 5 mV s−1. b Short-term durability test of Pt/Ti and Pt-X/Ti in natural seawater electrolysis under 100 mA cm−2. c Five representative polarization curves with iR correction of Pt-I/Ti in different areas of a scaled electrode (electrode size: 100 cm2, the solution resistance: 5 ± 0.5 Ω). d Long-term durability tests of Pt-I/Ti electrode under 100 mA cm−2 (the solution resistance: 7.7 ± 0.3 Ω). e Micro-XRF mapping of scaled Pt-I/Ti electrode after stability tests. f High-resolution XPS spectra of I 3 d for fresh and used Pt-I/Ti. g In situ electrochemical Raman spectroscopy of Pt-I/Ti from OCP to −1.0 V vs. RHE. Source data for Fig. 3 are provided as a Source Data file.
Compared with HER activity, stability issue requires much more attention due to the severe scaling effect on electrode surface, resulting in fast electrode deactivation21,22,35,36. Thus, in order to screen the stabilities of different cathodes, the commercial boron-doped diamond (BDD) and 0.5 M Na2SO4 were adopted as anode and anolyte to ensure the stability of the anodic side, and the experiments were conducted in a three-electrode system equipped with a cation exchange membrane (Fig. S16, 17). Under the current density of 100 mA cm−2, the applied bias of Pt/Ti electrode without halogen-modification quickly increased ~100 mV within 30 min (Fig. 3b), which was majorly due to the decrease of working area caused by the coverage of white flocculent scale (Mg(OH)2, Fig. S18-20). In sharp contrast, all four Pt-X/Ti electrodes exhibited much enhanced stabilities under the same conditions. Specifically, Pt-F/Ti, Pt-Cl/Ti, and Pt-Br/Ti electrodes displayed potential increases of ~33 mV, ~29 mV, and ~17 mV, respectively, while Pt-I/Ti electrode maintained stable performance with negligible potential increase, demonstrating its supersolidophobic property. It is worth noting that, through comprehensive evaluation, the Pt-I/Ti electrode ranks among the first class for HER in natural seawater environment (Fig. S21, 22)37,38,39,40. Meanwhile, optical images and XRD patterns of the four electrodes before and after the reaction further supported the difference in stabilities (Fig. S23, 24).
Benefiting from the simplicity of synthesis process, the Pt-I/Ti electrode can be easily scaled up to 100 cm2 in the laboratory, as shown in the inset of Fig. 3c and Fig. S25. The large-area consistency was demonstrated by selecting and evaluating, five different regions of the 100 cm2 electrode in terms of the HER activity and stability in natural seawater (Fig. 3c and Fig. S26). Moreover, as shown in Fig. 3e and Fig. S27-30, Micro X-ray Fluorescence (Micro-XRF) and SEM results revealed that most regions exhibited relatively identical elements distribution and morphology, further proving the scaling-up feasibility of this Pt-I/Ti electrode. More importantly, for practical application, the life time of the electrode is required at least thousands of hours (> 1000 h)41,42. Therefore, in this case, we have extended the HER testing of Pt-I/Ti electrode in natural seawater over 5000 h and observed stable operation with a negligible increase in applied bias (< 10 mV) (Fig. 3d and Fig. S31), which demonstrates competitive stability among the natural seawater cathodes reported previously (Supplementary Table 3)40,43,44,45,46,47.
The stability of Pt-I/Ti electrode heavily lies on the firmness of Pt-I coordination, which has been demonstrated in several ex situ and in situ characterizations. The Micro-XRF pattern of Pt-I/Ti electrode after 1000 h test demonstrated Pt and I elements maintain a uniform distribution. (Fig. 3e and Fig. S32, 33). As shown in Fig. 3f, the high-resolution X-ray photoelectron spectroscopy (XPS) spectra of I 3 d for fresh and used Pt-I/Ti exhibited nearly identical peaks48,49, confirming the stable presence of coordinated iodine on Pt surface. The presence of coordinated halogens (F, Cl, Br) were also confirmed by the observation of ν(Pt-X, X: F, Cl, Br) peaks in Far-Infrared and XPS results (Fig. S34-36). In addition, in situ Raman spectroscopy revealed that, with the increase of applied bias from OCP to −1.0 V vs. RHE, the ν(Pt-I) remained detectable and unchanged at ~147 cm−150,51, while the δ(Pt-OH) at 1063 cm−1 did not appear at all33,52, as shown in Fig. 3g (experimental set-up is shown in the Fig. S37). This phenomenon demonstrates that the coordinated iodine is stable and cannot be replaced by the generated OH- during the HER process, and other halogens (F, Cl, Br) also show the same behavior (Fig. S37). The observable ν(S-O) peak at 980 cm−1 is attributed to the background signal from the electrolyte (Experimental details and Fig. S38).
Mechanistic investigation
Indeed, the synthesis of hydroxides on the electrode surface (i.e., Mg(OH)2 in this case) is closely related to the local reaction environment (i.e., pH) near the cathode during the HER process26,53. To further investigate the mechanism on how halogen coordination regulates the local reaction environment, a two-dimensional (2D) COMSOL Multiphysics model was firstly constructed to map out the pH distribution near the cathode surface (Fig. S39). It is found that the Pt/Ti electrode without halogen-modification exhibited a maximum pH value (~10.2) at ~3 μm from the electrode surface while the pH peak (~10.3) for the Pt-I/Ti electrode shifted to ~8 μm under a current density of 100 mA cm-2, as shown in Fig. 4a and Fig. S40. To verify this simulation, we developed a micro-region pH measurement system capable of in situ surface pH monitoring, and incorporated a stepper motor to measure the pH variation with distance (experimental set-up is shown in the Fig. S41). As shown in Fig. 4b and Fig. S42, the local pH of Pt/Ti electrode surface rapidly increased, achieving the minimum pH required for Mg(OH)2 precipitation (pH ≈ 9.6) at ~2 μm and complete precipitation (pH ≈ 11.1) at ~4 μm. In sharp contrast, for the Pt-I/Ti electrode, the pH reached 9.6 at ~4 μm and exceeded 11.1 at ~8 μm. These theoretical and experimental results both revealed that the surface diffusion of OH- was accelerated on the Pt-I/Ti electrodes, which were confirmed by other current densities (Fig. S43-45). It is worth noting that, for both electrodes, the maximum local pH during the HER process was lower than the minimum pH (~13.0) for starting the precipitation of Ca2+, giving rise to the potential in Ca-Mg separation from seawater. Further, open circuit potential (OCP) decay transients was used to verify interfacial pH swings during electrocatalysis54, and the results (Fig. 4c and Fig. S46, 47) demonstrated that the Pt-I/Ti electrode exhibited stronger resistance to pH increases which also indicated the accelerated OH- diffusion on electrode surface. It should be noted that the different OH- diffusion properties are consistent with *OH binding strength on Pt-X/Ti, as observed in the CO stripping experiments55,56, where Pt-I/Ti electrode exhibited the strongest repulsive capacity (Fig. S48).

a COMSOL simulation of the pH distribution near the Pt/Ti and Pt-I/Ti electrodes under 100 mA cm−2. b Micro-region pH measurements as a function of distance during the HER process of Pt/Ti and Pt-I/Ti electrodes at 100 mA cm−2. Measurements were taken at least three times and the average pH values are presented with the standard deviation as error bars. c Interfacial pH swing calculated for Pt/Ti and Pt-I/Ti electrodes from the post-electrolysis open circuit potential decay transient. d, e Snapshots of classical MD simulations of electrolyte systems with or without iodine above the Pt exposure surface. f Number of OH- versus distance above Pt and Pt-I exposure surface from classical MD simulation results. g, h Schematic illustrations of Mg(OH)2 formation on Pt and Pt-I surfaces. Source data for Fig. 4 are provided as a Source Data file.
Molecular dynamics (MD) simulations were used to validate the ion (e.g., Cl- or OH-) exclusion ability of the electrode surface57,58. The Pt and Pt-I slabs covered with solvent layers were constructed as model systems to explore the distribution of OH− close to the electrode surface (Fig. 4d, e, and Fig. S49). It is observed that the pure Pt surface would be surrounded by OH−, while the OH− ions on the Pt-I surface were repelled away from the surface. The number of OH− within 30 Å of the surface was halved (Fig. 4f), and the diffusion coefficients of OH- increased from 8.64 × 10−5 to 9.49 × 10−5 cm2 s−1 (Fig. S50).
Combined with the experimental and theoretical results, the HER process on electrode surfaces with or without halogen-modification in natural seawater can be illustrated in Figs. 4g, h. For the pure Pt surface, the diffusion of OH- generated by hydrogen evolution is dependent on the electrode repulsion and concentration gradient. Nevertheless, for the Pt-X surface, besides the above forces, like-charge repulsion from the halogen layer led to the faster OH- diffusion and expanded low-pH region. Thus, the combination of Mg2+ and OH- occurred closer to the pure Pt surface, which facilitated the heterogeneous nucleation process (i.e., Mg(OH)2 scaling). In contrast, for the Pt-X surface, as the position for Mg2+ precipitation was much farther from the electrode, homogeneous nucleation of Mg(OH)2 in the electrolyte was preferred (i.e., supersolidophobicity).
Performance and techno-economic analysis of scale electrolyzer
Compared with individual Mg2+ extraction and hydrogen production from seawater, this study achieves simultaneous production of H2 and high-purity Mg(OH)2 from seawater in virtue of the stable and supersolidophobic cathode (Fig. 5a). Next, the practical applicability of the supersolidophobic electrode was demonstrated in a large-scale direct seawater electrolyzer (electrode area: 100 cm2), assembled by a cation exchange membrane, an IrO2-Ti anode (Fig. S51-53), and a scaled Pt-I/Ti cathode, as schemed in Fig. 5b and Fig. S54-55. Natural seawater and 0.5 M Na2SO4 were used as the catholyte and anolyte, respectively. Fig. 5c demonstrated that the lab-made electrolyzer could be stably operated with a cell potential of ~2.75 V under 10 A for over 1000 h. During this process, the white Mg(OH)2 product was continuously produced at a rate of 5.54 g h−1, with a purity exceeding 99% and a particle size of ~35 μm (Fig. S56-61 and Supplementary Table 4). The catholyte pH was maintained at ~9.5 by flowing the natural seawater, resulting in a OH− availability of ~53.1% (Fig. S62). In contrast, for the traditional chemical precipitation method, the OH− availability was only ~16.7% when the solution pH was adjusted to ~9.6. With the pH increased further to ~11.0 by adding NaOH dropwise, the OH- availability rose to ~36.1%, although the purity of Mg(OH)2 ~ 91.4% (Fig. 5d, Fig. S63, 64, and Supplementary Table 5 and details can be seen in the Supplementary Discussion 1). The effective utilization of local pH indicates much enhanced OH− availability, which also represents a notable advantage of this value-increment natural seawater electrolysis strategy.

a Schematic diagram of three strategies including extraction of Mg(OH)2 from seawater, H2 production via seawater electrolysis and extraction of Mg(OH)2 coupled with H2 production from seawater. b Internal construction of the 100 cm2 flow-type electrolyzer. c Stability tests of the natural seawater electrolyzer under 100 mA cm−2 at 25 °C, with the inset showing the Mg(OH)2 obtained during this process. d OH− availability of chemical precipitation and seawater electrolysis to synthesis Mg(OH)2. e Brief division of natural seawater electrolysis total costs and benefits. f Life Cycle Assessment (LCA) of the seawater electrolysis-based magnesium extraction and hydrogen production process. Source data for Fig. 5 are provided as a Source Data file.
The TEA results were shown in Fig. 5e and Fig. S65, which summarized the preliminary and rough estimation of expenses associated with the direct conversion of natural seawater into valuable H2 and Mg(OH)2 (details can be seen in the Supplementary Discussion 2). The co-produced Mg(OH)2 could balance the hydrogen production cost, bringing the hydrogen production cost down to US$0.61 per kg of H2, which was below the DOE 2025 and 2030 targets (US$1.40 and US$1.00 per kg of H2, respectively)59,60,61(Fig. S65). As shown in Fig. S66, 67, variations in each economic parameter from −10% to +10% of their original values produce linear trends with specific slopes. Compared to the original value, electricity cost shows a relatively smaller effect on hydrogen production costs. However, the slopes of the curves related to the prices of magnesium hydroxide and sodium hydroxide are steeper, indicating that any change in these parameters could have a significant impact on hydrogen production costs. We also conducted a Life Cycle Assessment (LCA) to quantify its environmental impact compared to traditional hydrogen production processes (Fig. 5f, and Supplementary Table 6 and 7). In this assessment, the system expansion approach was adopted as the allocation method, assuming that the co-produced byproducts could equivalently replace the same quantities of conventionally produced materials in the market, thereby generating positive environmental benefits62. As shown in Fig. 5f, the process produces 8 t of O2 and 1.45 t of Mg(OH)2 along with 1 t of H2. Compared with average H2 production, the seawater electrolysis-based magnesium extraction and hydrogen production process reduces abiotic depletion potential (ADP) and global warming potential (GWP), while exhibiting higher values only in the ADP-element and ozone layer depletion potential (ODP) categories. The results indicate that this double-product strategy can reduce the overall environmental impact.
In summary, this study developed a series of halogen-modified Pt-X (X: F, Cl, Br, I) catalysts aiming at meeting the strict requirement (both high HER activity and solidophobic surface property) of cathode in natural seawater electrolysis. The supersolidophobic Pt-I/Ti cathode demonstrated stable performance for over 5000 h under 100 mA cm−2 in natural seawater electrolysis, while also enabling the co-electrosynthesis of high-purity Mg(OH)2 through the utilization of localized alkaline environment. Detailed characterizations, alongside COMSOL and classical MD simulations, revealed that the local reaction environment regulation and supersolidophobicity arise from the iodine modification on the electrode surface, inducing like-charge repulsion between I− and in situ generated OH- during the HER process and promoting homogeneous nucleation of solid product. Considering the high economic value of double-product advantage, the proposed seawater electrolysis strategy would reduce the sell price and environmental impact of green hydrogen. Furthermore, we can foresee that this strategy in constructing supersolidophobic electrode will be of significant important for future electro-synthesis of solid compounds.
Methods
Chemicals
Chloroplatinic (IV) acid (H2PtCl6, Aladdin Industrial, ≥99.95%), potassium nitrate (KNO3, China National Pharmaceutical Group Corporation, ≥99.0%), sodium fluoride (NaF, Macklin reagent, 99.99%), sodium bromide (NaBr, Macklin reagent, 99.9%), sodium iodide (NaI, Macklin reagent, 99.9%), sodium sulfate (Na2SO4, Macklin reagent, ≥99.0%), sulfuric acid (H2SO4, China National Pharmaceutical Group Corporation, ≥96.0%), sodium hydroxide (NaOH, Aladdin Industrial, 97%), sodium phosphate dibasic (Na2HPO4, Aladdin Industrial, 99.9%), sodium dihydrogen phosphate (NaH2PO4, Aladdin Industrial, 99.9%), 5,5-Dimethyl-1-pyrroline N-oxide (DMPO, Aladdin Industrial, 97%). Ti mesh (99.5%, 100 mesh) and IrO2-Ti felt (thickness is 0.25 mm, loading amount: 2 mg cm−2) were purchased from Suzhou sinero technology Co., LTD. Carbon fiber paper (TGP-H-060, 0.19 mm) was purchased from TORAY Industries. Boron doped diamond (BDD) was purchased from Suzhou shure technology Co., LTD. Cation exchange membrane (Nafion 117, thickness is 183 μm) was purchased from Suzhou sinero technology Co., LTD. All deionized water was prepared with OmniaPure ultra-pure water system (resistivity of 18.2 MΩ). All chemical reagents were used as received without further purification.
Preparation of Ti mesh matrix electrode
First, the Ti mesh was cut into pieces with the size of 1 cm × 2 cm. Second, these pieces were sequentially degreased through sonication in acetone, ethanol, and deionized water for 10 min each. Subsequently, they were cleaned with 2 M H2SO4 in an ultrasound bath for 10 min to remove the surface impurity layer. Finally, the Ti mesh was washed with deionized water until attaining a neutral pH and dried at 60 °C for 3 h.
Preparation of Pt-Cl/Ti electrode
The Pt-Cl/Ti electrode was synthesized through electrochemical deposition strategy in the three-electrode system, wherein the Ti mesh, graphite paper and saturated calomel electrode (SCE) electrode served as the working electrode, counter electrode, and reference electrode, respectively. The electrodeposition solution contained 0.0015 M H2PtCl6 and 0.1 M KNO3. The Pt-Cl/Ti was prepared using the i-t method at a potential of −0.3 V for 15 min, followed by washed with deionized water and dried at 60 °C for 3 h. Pt loading amount: Pt-Cl/Ti (0.254 mg cm-2).
Preparation of Pt-X/Ti (X: F, Br, I) electrodes by coordination exchange
The Cl ligand can be removed under the irradiation of visible light in water. CEL-HXUV300E from China Education Au-light with a spectral region from 320 nm to 780 nm was used as the light source with an output power density of 3.75 mW cm−2. After 30 min of irradiation, the electrode was immersed in 2 M NaX (X: F, Br, I) at 40 °C for 4 h under magnetic stirring. Finally, the Pt-X/Ti (X: F, Br, I) electrode was washed with deionized water and dried at 60 °C for 3 h. Pt loading amount: Pt-F/Ti (0.253 mg cm−2), Pt-Br/Ti (0.248 mg cm−2) and Pt-I/Ti (0.247 mg cm−2).
Preparation of Pt/Ti electrode
The Pt/Ti electrode was synthesized by calcining the Pt-Cl/Ti electrode in a tube furnace with a 5% H2/Ar flow under 200 °C for 4 h. Pt loading amount: Pt/Ti (0.248 mg cm−2).
Preparation of Pt-X/CFP (X: F, Cl, Br, I) and Pt/CFP electrodes
The synthesis method for the Pt-X/CFP (X: F, Cl, Br, I) and Pt/CFP electrodes is similar to that for the Pt-X/Ti (X: F, Cl, Br, I) and Pt/Ti electrodes, with the only difference being the substitution of the substrate with carbon fiber paper.
Preparation of scaled Pt-I/Ti electrode
The synthesis procedure of scaled Pt-I/Ti electrode is similar to that of the 1 cm × 2 cm Pt-I/Ti electrode, with the only difference being the substitution of the Ti matrix (10 cm × 10 cm). The electrode was then electrodeposited using a laboratory-made electrodepositing cell for 20 min, followed by washing with deionized water and drying at 60 °C for 3 h.
Seawater Pretreatment
Seawater pretreatment only sediment removal by filtration system (filter paper diameter = 150 mm).
Material characterizations
Scanning electron microscopy (SEM) images were taken on Hitachi S4800 working at 20 keV. The aberration-corrected HAADF-STEM measurements were taken on a JEM-ARM200F instrument at 200 keV. The crystal structures of the samples were examined by X-ray diffraction (XRD, Bruker D8) with Cu Kα radiation (λ = 0.154178 nm). The surface structures of Pt-X/Ti (X: F, Cl, Br, I) electrodes were analyzed by PHI nano TOF II TOF-SIMS (ULVAC-PHI, Japan). X-ray photoelectron spectroscopy (XPS) measurements were performed by using a Thermo ESCALAB 250 high-performance electron spectrometer using monochromatized Al Ka (hν = 1486.6 eV) as the excitation source. Raman measurements were conducted by using a RENISHAW inVia Raman spectrometer. Far-Infrared spectra were obtained on a Bruker Vertex 70 V. The content of cations in seawater was measured by Agilent ICP 720 inductively coupled plasma optical emission spectrometer (ICP-OES). Micro X-ray Fluorescence (Micro-XRF) images were taken on a Bruker M4 TORNADO. The product particle size was measured by Malvern Zetasizer Nano ZS90. Faradaic efficiencies were tested by using gas chromatography (GC, Agilent 7890B) method30. The electron paramagnetic resonance (EPR) spectra were collected on a Bruker Elexsys 500 spectrometer at 25 °C. Micro-region pH measurements were performed by using a commercial glass-type pH microelectrode with a tip diameter of 500 μm (Unisense, pH-500c), together with an internal reference.
Electrochemical measurements
Except for the electrolyzer stabilization process, all of the electrochemical measurements were measured on electrochemical workstation (ChenHua CH instrument 760E) with a three-electrode configuration at 25 °C. Except in stability tests, the fabricated samples, graphite paper and saturated calomel electrode (SCE, saturated KCl) served as the working electrode, counter electrode, and reference electrode, respectively. In a stability test, the fabricated samples, commercial boron-doped diamond (BDD, 3 cm × 4 cm) and saturated calomel electrode (SCE, saturated KCl) acted as the reference electrode, counter electrode, and reference electrode. The as-fabricated electrodes were clamped by pure Ti holder (100%) to make electrical contact. The electrolyte (100 mL natural seawater, pH = 7.71 ± 0.02 and 100 mL 0.5 M Na2SO4, pH = 7.0 ± 0.02) was purged with pure N2 (for HER) or O2 (for OER) for at least 30 min. The polarization curves of HER were documented at a rate of 5 mV s−1. LSV is measured independently three times.
The reference electrodes were calibrated in an electrolyte saturated with high-purity hydrogen, using two platinum foils (1 × 1 cm2) as the working and counter electrodes, respectively. Cyclic voltammetry (CV) was performed at a scan rate of 5 mV s−1. The thermodynamic potential of the hydrogen electrode reaction was determined as the average of the two potentials at which the current crossed zero during the CV scans, and this value was used to convert the measured potentials to the reversible hydrogen electrode (RHE). All potentials of measured curves were corrected with iR (100%)63 compensation and then converted to RHE according to the following two equations:
The service life of the electrodes in the electrolyzer was tested using a direct current (DC) power supply (MAISHENG, MP1560D) with a constant current density in a two-electrode mode, and the cell potential was recorded every hour, commercial IrO2-Ti (10 cm × 10 cm) acted as the counter electrode. Electrochemical impedance spectroscopy (EIS) curves were obtained by a frequency range from 1000 kHz to 0.1 Hz under different potentials.
All the solutions used as electrolytes are prepared freshly and used immediately, and are stored in a dark place.
MEA measurement
The MEA setup with the geometric area of 100 cm2 was designed and manufactured by the lab and equipped with a designed flow channel. The as prepared electrodes (Pt-I/Ti, 10 cm × 10 cm) and commercial IrO2-Ti felt (10 cm × 10 cm) electrode were selected as HER and OER electrodes, respectively. The cation exchange membrane was Nafion 117 (Dupont, USA). The membranes were pretreated by boiling sequentially in 3% H2O2 solution for 1 h and 0.5 M H2SO4 for 3 h, followed by rinsing in deionized water. The stability was carried out on DC power supply. The anolyte (0.5 M Na2SO4, 20 L) and catholyte (natural seawater, 20 L) were replaced every 6 h to maintain a constant pH, with the cathode effluent pH consistently stabilized at ~9.5.
Electrochemical in situ Raman measurements
Electrochemical in situ Raman spectroscopy was carried out using a RENISHAW inVia Raman spectrometer equipped with a Leica TCS SP8 CARS microscope and a Spectra-Physics 532 nm Ar laser. All in situ Raman measurements were performed with a custom-made spectro-electrochemical cell. The electrochemical activities were measured in a three-electrode configuration at 25 °C. The as prepared electrodes, Pt wire and Ag/AgCl (saturated KCl) electrode served as the working electrode, counter electrode, and reference electrode, respectively. The electrolytes were 0.5 M Na2SO4. Noted that, to prevent side reactions, a sulfate solution is used. The laser beams were focused on the sample through the quartz window to collect the Raman spectrum. The chronoamperometry method was employed to applied different potentials to the electrode with 100 mV intervals in the range of 0 V to −1.0 V vs. RHE. The acquisition time for each spectrum was 10 s with 3 sweeps from 100.92 to 1811.54 cm−1. The optical images of custom-built electrochemical cells for in situ Raman measurements is provided in Supplementary Fig. 37.
CO stripping measurements
The CO stripping experiment was performed in a three-electrode H-cell configuration. The system employed a graphite rod as the counter electrode, a saturated calomel electrode (SCE, saturated KCl) as the reference electrode, and the as-prepared electrode as the working electrode. Initially, 10% CO/Ar gas was introduced into a CO-saturated 0.5 M H2SO4 electrolyte, allowing CO adsorption onto the working electrode at a constant potential of 0.1 V (vs. RHE) for 30 min. Subsequently, the electrode was transferred to an Ar-saturated 0.5 M H2SO4 electrolyte for CV analysis. Prior to measurement, the solution was purged with Ar gas, and CO stripping CV curves were recorded at a scan rate of 20 mV s−1.
Micro-region pH measurements
To test the pH on the electrode surface, a laboratory-made micro-region pH system equipped with a microscope (ZOOM-650, Shenzhen Micro Vision Technology Co., Ltd.) and a stepper motor was used to measure pH variation with distance (Marzhauser Wetzlar MM33 with tilting device, step size: 1 μm). The electrolyte was 0.5 M Na2SO4, and the as prepared electrodes, Pt foil, and saturated calomel electrode (SCE, saturated KCl) served as the working electrode, counter electrode, and reference electrode, respectively. Initially, the glass-type pH microelectrode tip (Unisense, pH-500c) was positioned ~1 μm from the electrode surface using the stepper motor. A constant current was then applied, and the pH electrode was controlled to move away from the electrode surface in 1 μm increments. At each test point, the pH electrode was held for 1 s before moving to the next point. Each set of measurements was repeated three times to minimize error. The optical image of custom-built system for micro-region pH measurements is provided in Supplementary Fig. 41.
Open circuit potential (OCP) decay transients measurements
The open circuit potential (OCP) decay transients can be used to verify interfacial pH swings during electrocatalysis54. The electrolyte was 100 mL 0.5 M NaPi (Phosphate buffer, pH=6.8 ± 0.02), and the as prepared electrodes, Pt foil, and saturated calomel electrode (SCE, saturated KCl) served as the working electrode, counter electrode, and reference electrode, respectively. Multi-step chronopotentiometry (CP) was employed to galvanostatically polarize the working electrode at current densities of 10, 25, 50 and 100 mA cm−2 for 2 min, with a data collection interval of 0.25 s, allowing a steady-state potential to be reached. Subsequently, a second CP step at the same current density, but with a higher data collection interval of 0.0002 s, was performed for 1 second. A third CP step at 0 mA cm−2 was used to collect the open circuit potential decay transient with a data collection interval of 0.0002 s for 1 s. Finally, a fourth CP step at 0 mA cm−2, with a lower data collection interval of 0.25 s, was conducted to capture the remainder of the open circuit potential decay. The data collection intervals were carefully selected to ensure high time resolution during the termination of polarization, while avoiding the data buffering capacities of the potentiostat.
Theoretical computation details
All density functional theory (DFT) calculations were carried out using the Vienna Ab initio Simulation Package (VASP) with the projector augmented-wave (PAW) method64. The exchange-correlation interactions were described using the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional. A plane-wave energy cutoff of 450 eV was employed, and partial occupancies of the Kohn-Sham orbitals were treated using the Gaussian smearing method with a width of 0.2 eV. The self-consistent calculations apply a convergence energy threshold of 10−5 eV, and the force convergency was set to 0.05 eV Å−1. The p(3 × 3) Pt (111) surface was built with 4 layers, where tow of the layers at the bottom was fixed in all the calculations. The coordinated halogen atoms (F, Cl, Br, I) was adsorbed on the top site of Pt, and the relevant affected site for HER was considered closed to the adsorbed halogen atoms. The k-point of 3 × 3 × 1 was used. The reaction free energy of H+ involved steps was calculated following the computational hydrogen electrode (CHE) model65. Free energy corrections were considered at the temperature of 298 K, following:
Here, ΔE, ΔGZPE, ΔGU, and ΔS represent the DFT-calculated energy change, the zero-point energy correction, the internal energy correction, and the entropy correction, respectively66.
All Molecular dynamic (MD) simulations were applied to calculation the number distribution and diffusion coefficients of OH− in seawater at pristine Pt (111) and I− adsorbed Pt (111) surfaces under an external electric field of 0.2 V Å−1 along z direction. The solid–liquid interface calculations were conducted with COMPASS III force field using the Forcite module in Materials Studio (MS) 2020. The dimensional lengths of the Pt substrate are 24.89 Å × 23.95 Å in xoy plane, which is composed of four layer Pt atoms. The solution contains 24 OH−, 36 Na+, 2 Mg2+, 16 Cl− and 1200 H2O molecules in a rectangular box with length scales of 24.89 Å × 23.95 Å × 61.86 Å. After geometry optimization, the aqueous solution was placed on pristine Pt (111) and I− adsorbed Pt (111) surfaces. All MD calculations were performed under the NVT ensemble (T = 298.0 K) with a time step of 1 fs and a total simulation time of 400 ps, during which simulation trajectories were recorded every 4000 steps. The running time was long enough for system energy and temperature reaching stable. The temperature was controlling by a Nose–Hoover thermostat. The Ewald scheme and atom-based cutoff method (i.e., a radius of 12.5 Å) were applied to treat electrostatic and van der Waals (vdW) interactions, respectively. The number profiles of OH− along perpendicular direction were analyzed. The solid–liquid interfaces snapshots were displayed. The diffusion coefficients of OH− can be estimated from the slope of the mean square displacement (MSD) curves by the Einstein relationship.
The atomic coordinates of the optimized crystal structures can be found in Supplementary Data 1.
LCA analysis
An LCA was conducted using the SimaPro software following the international standard ISO 14040. The CML 2001 method was selected to evaluate the environmental impacts of the studied system, and background data were obtained from the Ecoinvent 3.11 database. The production of 1 t of hydrogen was defined as the functional unit, and the system boundary was set on a cradle-to-gate basis, covering the production processes of hydrogen and magnesium hydroxide in the seawater electrolysis-based magnesium extraction and hydrogen production process. A life-cycle inventory was established to clearly distinguish the system inputs and outputs. The main inputs include seawater, sodium hydroxide, catalyst materials, and electricity, while the outputs consist of hydrogen, oxygen, and magnesium hydroxide. A system expansion approach was applied for the attributional LCA, assuming that the co-produced byproducts could substitute conventionally produced materials in the market and thereby generate environmental benefits. Based on the CML 2001 method, the characterization analysis quantified the specific contributions of each environmental impact category (AP, GWP, FAETP, MAETP, TETP, ADP, EP, HTP, ADP element, ODP, and POCP) to represent the overall life-cycle environmental impacts.
Data availability
Source data are provided with this paper.
References
Turner, J. A. Sustainable hydrogen production. Science 305, 972–974 (2004).
Dresselhaus, M. S. & Thomas, I. L. Alternative energy technologies. Nature 414, 332–337 (2001).
Glenk, G. & Reichelstein, S. Economics of converting renewable power to hydrogen. Nat. Energy 4, 216–222 (2019).
Logan, B. E., Shi, L. & Rossi, R. Enabling the use of seawater for hydrogen gas production in water electrolyzers. Joule 5, 760–762 (2021).
Electric power research institute (EPRI), PwC, working paper: hydrogen demand and cost dynamics, https://www.worldenergy.org/assets/downloads/Working_Paper_-_Hydrogen_Demand_And_Cost_Dynamics_-_September_2021.pdf?v=1634916141 (2021).
Wappler, M. et al. Building the green hydrogen market – current state and outlook on green hydrogen demand and electrolyzer manufacturing. Int. J. Hydrog. Energy 47, 33551–33570 (2022).
Majumdar, A., Deutch, J. M., Prasher, R. S. & Griffin, T. P. A framework for a hydrogen economy. Joule 5, 1905–1908 (2021).
Wi, T.-U. et al. Upgrading carbon monoxide to bioplastics via integrated electrochemical reduction and biosynthesis. Nat. Synth. 3, 1392–1403 (2024).
Sendeku, M. G. et al. Deciphering in-situ surface reconstruction in two-dimensional CdPS3 nanosheets for efficient biomass hydrogenation. Nat. Commun. 15, 5174 (2024).
Elgazzar, A. & Wang, H. Beyond molecular transformations in electrochemical porous solid electrolyte reactors. Nat. Chem. Eng. 2, 3–7 (2025).
Wang, T. et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 5, 66–73 (2022).
Liu, T. et al. Redox-mediated decoupled seawater direct splitting for H2 production. Nat. Commun. 15, 8874 (2024).
Yan, D., Mebrahtu, C., Wang, S. & Palkovits, R. Innovative electrochemical strategies for hydrogen production: from electricity input to electricity output. Angew. Chem. Int. Ed. 62, e202214333 (2023).
Li, Z. et al. Electrocatalytic synthesis of adipic acid coupled with H2 production enhanced by a ligand modification strategy. Nat. Commun. 13, 5009 (2022).
Zhou, B. et al. Membrane-free electrocatalytic co-conversions of pbs waste plastics and maleic acid into high-purity succinic acid solid. Angew. Chem. Int. Ed. 63, e202411502 (2024).
Liu, C., Chen, F., Zhao, B.-H., Wu, Y. & Zhang, B. Electrochemical hydrogenation and oxidation of organic species involving water. Nat. Rev. Chem. 8, 277–293 (2024).
Li, J. & Duan, H. Recent progress in energy-saving hydrogen production by coupling with value-added anodic reactions. Chem. 10, 3008–3039 (2024).
Lin, J. et al. Tandem microplastic degradation and hydrogen production by hierarchical carbon nitride-supported single-atom iron catalysts. Nat. Commun. 15, 8769 (2024).
Cha, H. G. & Choi, K.-S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 7, 328–333 (2015).
Yi, L. et al. Solidophobic surface for electrochemical extraction of high-valued Mg(OH)2 coupled with H2 production from seawater. Nano Lett. 24, 5920–5928 (2024).
Lu, X. et al. A sea-change: Manganese doped nickel/nickel oxide electrocatalysts for hydrogen generation from seawater. Energy Environ. Sci. 11, 1898–1910 (2018).
Tong, W. et al. Electrolysis of low-grade and saline surface water. Nat. Energy 5, 367–377 (2020).
Kirk, D. W. & Ledas, A. E. Precipitate formation during sea water electrolysis. Int. J. Hydrog. Energy 7, 925–932 (1982).
Erdemir, D., Lee, A. Y. & Myerson, A. S. Nucleation of crystals from solution: classical and two-step models. Acc. Chem. Res. 42, 621–629 (2009).
Meng, J. & Wang, S. Advanced antiscaling interfacial materials toward highly efficient heat energy transfer. Adv. Funct. Mater. 30, 1904796 (2020).
Chen, C. et al. Local reaction environment in electrocatalysis. Chem. Soc. Rev. 53, 2022–2055 (2024).
Li, Y. et al. Under-water superaerophobic pine-shaped pt nanoarray electrode for ultrahigh-performance hydrogen evolution. Adv. Funct. Mater. 25, 1737–1744 (2015).
Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl. Acad. Sci. 116, 6624–6629 (2019).
Fan, R. et al. Ultrastable electrocatalytic seawater splitting at ampere-level current density. Nat. Sustain. 7, 158–167 (2024).
Xu, W. et al. Scalable fabrication of Cu2S@NiS@Ni/NiMo hybrid cathode for high-performance seawater electrolysis. Adv. Funct. Mater. 33, 2302263 (2023).
Lin, D. & Lasia, A. Electrochemical impedance study of the kinetics of hydrogen evolution at a rough palladium electrode in acidic solution. J. Electroanal. Chem. 785, 190–195 (2017).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 334, 1256–1260 (2011).
Zhang, T. et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 13, 6875 (2022).
Zhou, Z. et al. Electrocatalytic hydrogen evolution under neutral pH conditions: current understandings, recent advances, and future prospects. Energy Environ. Sci. 13, 3185–3206 (2020).
Jin, H. et al. Emerging materials and technologies for electrocatalytic seawater splitting. Sci. Adv. 9, eadi7755 (2023).
Kang, W. et al. Scaling-free cathodes: enabling electrochemical extraction of high-purity nano-CaCO3 and -Mg(OH)2 in seawater. Environ. Sci. Technol. 58, 14034–14041 (2024).
Lv, Q. et al. Featherlike NiCoP holey nanoarrys for efficient and stable seawater splitting. ACS Appl. Energy Mater. 2, 3910–3917 (2019).
Yu, L. et al. Hydrogen generation from seawater electrolysis over a sandwich-like NiCoN|NixP|NiCoN microsheet array catalyst. ACS Energy Lett. 5, 2681–2689 (2020).
Ma, Y. Y. et al. Highly efficient hydrogen evolution from seawater by a low-cost and stable CoMoP@C electrocatalyst superior to Pt/C. Energy Environ. Sci. 10, 788–798 (2017).
Bao, D. et al. Dynamic creation of a local acid-like environment for hydrogen evolution reaction in natural seawater. J. Am. Chem. Soc. 146, 34711–34719 (2024).
Chen, H. et al. Stable seawater electrolysis over 10,000 h via chemical fixation of sulfate on NiFeBa-LDH. Adv. Mater. 36, 2411302 (2024).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673–678 (2022).
Liang, J. et al. Electroreduction of alkaline/natural seawater: self-cleaning Pt/carbon cathode and on-site co-synthesis of H2 and Mg hydroxide nanoflakes. Chem 10, 3067–3087 (2024).
Liang, J. et al. Efficient bubble/precipitate traffic enables stable seawater reduction electrocatalysis at industrial-level current densities. Nat. Commun. 15, 2950 (2024).
Zhang, X.-L. et al. In situ ammonium formation mediates efficient hydrogen production from natural seawater splitting. Nat. Commun. 15, 9462 (2024).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Yang, C. et al. Electron-donating Cu atoms induced high proton supply and anti-poisoning ruthenium clusters for superior direct seawater hydrogen production. Adv. Funct. Mater. 34, 2404061 (2024).
Li, X. et al. Enhanced redox kinetics and duration of aqueous I2/I− conversion chemistry by MXene confinement. Adv. Mater. 33, 2006897 (2021).
Zhou, P. et al. Single-atom Pt-I3 sites on all-inorganic Cs2SnI6 perovskite for efficient photocatalytic hydrogen production. Nat. Commun. 12, 4412 (2021).
Cooney, R. P., Hendra, P. J. & Fleischmann, M. Raman spectra from adsorbed iodine species on an unroughened platinum electrode surface. J. Raman Spectrosc. 6, 264–266 (1977).
Barańska, H., Kuduk-Jaworska, J. & Cacciari, S. Raman study of neutral platinum–imidazole complexes*. J. Raman Spectrosc. 28, 1–7 (1997).
Dong, J.-C. et al. In situ Raman spectroscopic evidence for oxygen reduction reaction intermediates at platinum single-crystal surfaces. Nat. Energy 4, 60–67 (2019).
Zhu, Y. et al. An anti-scaling strategy for electrochemical wastewater treatment: leveraging tip-enhanced electric fields. Environ. Sci. Technol. 58, 13145–13156 (2024).
Sauvé, E. R. et al. Open circuit potential decay transients quantify interfacial pH swings during high current density hydrogen electrocatalysis. Joule 8, 728–745 (2024).
Wang, Y. et al. Pt–Ru catalyzed hydrogen oxidation in alkaline media: oxophilic effect or electronic effect? Energy Environ. Sci. 8, 177–181 (2015).
Duan, Y. et al. Bimetallic nickel-molybdenum/tungsten nanoalloys for high-efficiency hydrogen oxidation catalysis in alkaline electrolytes. Nat. Commun. 11, 4789 (2020).
Berendsen, H. J., Grigera, J.-R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. -Us 91, 6269–6271 (1987).
Liu, W. et al. Ferricyanide armed anodes enable stable water oxidation in saturated saline water at 2 A/cm2. Angew. Chem. Int. Ed. 135, e202309882 (2023).
Hydrogen and Fuel Cell Technologies Office Multi-Year Program Plan, https://www.energy.gov/eere/fuelcells/hydrogen-and-fuel-cell-technologies-office-multi-year-program-plan (2024).
Glenk, G., Holler, P. & Reichelstein, S. Advances in power-to-gas technologies: cost and conversion efficiency. Energy Environ. Sci. 16, 6058–6070 (2023).
Boettcher, S. W. Introduction to green hydrogen. Chem. Rev. 124, 13095–13098 (2024).
Zhang, C. et al. Concentrated formic acid from CO2 electrolysis for directly driving fuel cell. Angew. Chem. 136, e202317628 (2024).
Heenan, A. R., Hamonnet, J. & Marshall, A. T. Why Careful iR compensation and reporting of electrode potentials are critical for the CO2 reduction reaction. ACS Energy Lett. 7, 2357–2361 (2022).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Guo, C. et al. Computational design of spinel oxides through coverage-dependent screening on the reaction phase diagram. ACS Catal. 12, 6781–6793 (2022).
Acknowledgements
This work was supported by the National Key Research and Development Project (Grant No. 2023YFB4005100 [Z.Y.L.]), the Pioneer R&D Program of Zhejiang Province-China (Grant No. 2025C01161 [Z.Y.L.]), the Ningbo Yongjiang Talent Introduction Programme (Grant No. 2021A-036-B [Z.Y.L.]), the Ningbo S&T Innovation 2025 Major Special Program (Grant No. 2022Z205 [Z.Y.L.]) and National Natural Science Foundation of China (Grant No. 52201285 [X.C.], Grant No. 22405283 [W.Y.Z.], Grant No. 22379154 [Z.Y.L.], Grant No. 22509212 [S.X.Z.] and Grant No. 52274297 [X.L.T]).
Author information
Authors and Affiliations
Contributions
Z.Y.L. and X.L.T. supervised the project. Z.Y.L. and L.Y. designed the experiments and analyzed the results. L.Y. and C.H.C. carried out most of the experiments. Y.J.W. designed and manufactured the electrolyzers. X.L.T., W.W.X. and X.C. discussed and gave suggestions on the design of the experiments. W.Y.Z. took part in the electrochemical measurements and discussions. S.X.Z., H.C.C., J.C.Z. and J.B.W. helped some experiments. W.B.G. performed the COMSOL simulation. T.Y.Q. and X.L.T. performed the LCA analysis. Z.Y.L. and L.Y. prepared the manuscript. All the authors discussed the results and assisted during the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Menghao Yang who co-reviewed with Mohamed Ait Tamerd; Min-Rui Gao, Vitaly ORDOMSKY and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yi, L., Chen, C., Wen, Y. et al. Supersolidophobic Pt catalyst for long-term natural seawater electrolysis with hydrogen production and magnesium extraction. Nat Commun 16, 11493 (2025). https://doi.org/10.1038/s41467-025-66473-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66473-6
