
Abstract
Human milk oligosaccharides (HMOs) have highly diverse and branched structures that present a significant challenge for chemical synthesis. Here we show that masking the amino group in glucosamine with a p-nitrobenzyloxycarbonyl (pNZ) protecting group enhances coupling and deprotection efficiency during automated glycan assembly (AGA) of homogeneous HMOs, including the lacto-N-tetraose (LNT), lacto-N-neotetraose (LNnT), lacto-N-fucopentaose (LNFP), lacto-N-difuco-hexaose (LNDFH), and lacto-N-neohexaose (LNnH) series. Deprotection strategies are developed to achieve excellent purity of linear and branched HMO structures. The end-to-end tractability of pNZ-protected oligosaccharides underscores the robustness of this approach, while three orthogonal deprotection pathways offer synthetic versatility for HMO compounds.
Similar content being viewed by others


Introduction
Breast milk is the primary source of nutrition for infants during their early development, supporting a wide range of physiological functions1,2,3. Human milk oligosaccharides (HMOs) are the third most abundant component of breast milk and are highly diverse in structure and function4,5. Comprehensive LC–MS surveys and databases have reported more than 200 HMOs, from which over 150 distinct structures were elucidated across both neutral and sialylated HMOs6,7,8,9. However, understanding the functional roles of HMOs remains limited due to difficulties in obtaining these complex glycans in pure form and sufficient quantities. Since isolating large amounts of pure HMOs from natural sources is challenging, synthetic approaches are essential to explore the biological potential of HMOs10,11.
Various strategies have been developed to prepare HMOs, including chemical synthesis12,13,14, enzymatic synthesis15, chemo-enzymatic synthesis16,17,18, microbial fermentation19, and mammalian cell culture20. The backbone of HMOs, consisting of a lactose unit at the reducing end, extended by N-acetylglucosamine (GlcNAc) and galactose (Gal), is often decorated with fucose (Fuc) and/or N-acetylneuraminic acid (Neu5Ac). Notably, GlcNAc contains a C-2 amino group. The choice of protecting group at this position is crucial, as it can significantly impact glycosylation reactivity21. Protecting groups that can be removed under mild and selective conditions offer a unique access point for fluorescent labeling22, bio-orthogonal chemistry23, and modulation of pharmacological properties of HMOs.
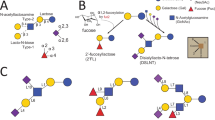
Automated glycan assembly (AGA) accelerates the synthetic process of a wide variety of complex oligosaccharides24,25,26,27,28,29. In solution-phase oligosaccharide synthesis, amine groups are commonly masked using trichloroethylchloroformate (Troc) and phthaloyl (Phth). These participating protecting groups promote the formation of 1,2-trans glycosides and suppress the formation of 1,2-oxazoline byproducts compared to natural acetyl (Ac) group. However, the Troc group is base-labile, making it incompatible with AGA protocols where concentrated basic solutions are introduced periodically. In contrast, Phth is base-stable and thus compatible with basic steps, but its removal typically relies on hydrazinolysis or related nucleophiles, which can demand large excess of reagents and, depending on substrate, elevated temperature or prolonged reaction times30,31. These conditions can compromise sensitive glycosidic bonds and other temporary protecting groups, making them not applicable to further modifications or global deprotection following AGA (Fig. 1a). Trichloroacetyl (TCA) and carboxybenzyl (Cbz) groups were previously explored to overcome the challenges (Fig. 1b)32,33. The greater stability of TCA and Cbz groups often comes at a cost: multiple glycosylation cycles and the need to convert to a more reactive phosphate leaving group to overcome otherwise sluggish couplings. Simultaneous cleavage of multiple TCA and Cbz protecting groups often requires extended hydrogenolysis and can suffer from selectivity issues, particularly when Cbz is also present on linkers, leading to parallel unmasking of multiple amines and complicating selective N-acetylation. Prior experiments reported incomplete TCA reduction, fucose cleavage, and solubility problems33,34.

a Common groups for solution-phase synthesis; (b) Common groups for AGA; (c) This work: pNZ as N-protecting group in AGA. PG: protecting group, LG: leaving group (thioether or glycosyl phosphate).
Here, we aimed to develop reliable syntheses of a collection of diverse neutral HMOs using AGA on a commercial Glyconeer® synthesizer as well as home-built instruments. A key aspect of this development was the introduction of the p-nitrobenzyloxycarbonyl (pNZ)35,36,37, a carbamate-type protecting group. Initially, pNZ was employed in peptide chemistry for the protection of amino side chains, particularly to mask the amine functionalities of several amino acids. In these solution-phase studies36,38, pNZ-protected donors exhibit lower reactivity compared to other amino-protected donors, necessitating higher temperatures and longer reaction times. However, in solid phase synthesis, this moderate reactivity helps suppress donor decomposition and side reactions, thereby improving synthetic efficiency24,39,40. pNZ-protected glucosamine building blocks displayed excellent stability and yielded exclusively 1,2-trans glycosides. In addition, these shelf-stable thioglycosides can be used directly without the need to convert them into glycosyl phosphates (Fig. 1c). The preparation of these building blocks was straightforward and efficient. In this work, pNZ was key to the assembly of branched and linear HMO structures through AGA. We also demonstrated that pNZ is a very versatile and orthogonal protecting group, which can be selectively removed in three different ways.
Results and discussion
Preparation of N-pNZ protected building blocks
To accommodate the diverse native linkages found in HMOs, we designed four N-pNZ-protected glucosamine building blocks optimized for AGA. These building blocks feature a permanent benzyl ether (Bn) protecting group as well as two orthogonal temporary protecting groups: fluorenylmethoxycarbonyl (Fmoc) and levulinoyl (Lev). The glucosamine set includes 4-O-Fmoc (3), 4-O-Lev (4), 3-O-Fmoc (6), and 3-O-Lev-4-O-Fmoc (8) (Fig. 2). In brief: thioglycoside 1 containing a C-2 pNZ group was synthesized from D-glucosamine in four steps36. Next, one-pot silylation and benzylation were employed: Addition of 1,1,1,3,3,3-hexamethyldisilazane (HMDS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) in anhydrous acetonitrile (MeCN) afforded per-O-silylation of 141. Subsequently, 4,6-O-benzylidenation and 3-O-benzylation by addition of benzaldehyde (PhCHO), triethylsiliane (Et3SiH) and trifluoromethanesulfonic acid (TfOH)42, followed by regioselective ring opening of the 4,6-O-benzylidene group with trifluoroacetic acid (TFA) and triethylsilane (Et3SiH) gave intermediate 2. 4-O-Fmoc glucosamine donor 3 and 4-O-Lev glucosamine donor 4 were subsequently prepared from 2.

a 1 HMDS, TMSOTf, MeCN, r.t. 2) PhCHO, Et3SiH, TfOH, CH2Cl2, 0 °C to r.t.; b TFA, TFAA, Et3SiH, CH2Cl2, 0 °C to r.t.; c FmocCl, pyridine, CH2Cl2, r.t.; d EDC, DMAP, LevOH, CH2Cl2, 0 °C to r.t.; e PhCH(OMe)2, CSA, MeCN/THF r.t.; f PhBCl2, Et3SiH, CH2Cl2, −78 °C.
3-O-Fmoc glucosamine donor 6 was prepared from 1 by 4,6-O-benzylidenation with benzaldehyde dimethyl acetal (PhCH(OMe)2) and a catalytic amount of camphor sulfonic acid (CSA) in MeCN/THF, followed by introduction of Fmoc group at the C-3 position. The intermediate was subjected to reductive opening of the 4,6-O-benzylidene ring using a combination of dichlorophenylborane (BPhCl2) and TMSOTf, yielding intermediate 5. Since Fmoc is a base-labile protecting group, acidic benzylation was carried out to furnish 641.
3-O-Lev-4-O-Fmoc glucosamine donor 8, bearing two temporary protecting groups at the C-3 and C-4 positions, was subsequently prepared. The Lev group was introduced at C-3 following 4,6-O-benzylidene protection. Selective reductive ring opening and introducing Fmoc at C-4 afforded building block 8 in excellent yield.
All synthetic intermediates were readily isolated by simply washing under hot ethanol (ca. 60 °C) as excess reagents and by products were completely soluble, whereas the desired products precipitated and were filtered from the crude solution before a final flash column chromatography.
AGA using N-pNZ protected glucosamine building blocks
The diLacNAc tetramer, Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ 10, served as target molecule for optimizing AGA conditions. Generally, glycosylation efficiency and stereoselective outcomes are influenced by many factors, including concentration, temperature, protecting groups, and solvent40,43,44,45,46,47,48. For AGA, the reaction vessel was loaded with a set amount of modified polystyrene resin carrying a photolabile linker. To prepare the resin for the first glycosylation, an acidic wash was followed by glycosylation on solid support. The instrument delivered a solution of building block and activator solution, under an inert atmosphere and precise temperature. Trace amounts of unreacted hydroxyl groups were capped via acidic acetylation, followed by orthogonal removal of a temporary protecting group at the designated elongation site. The cycle was programmed to repeat fully autonomously until the desired glycan structure was built49.
We recently identified optimal initiation and coupling temperatures for 3-O-Fmoc galactoside 940,50. The initiation temperature (T₁) was set at −40 °C for five minutes, followed by coupling temperature (T₂) at −20 °C for 20 min. These conditions were kept unchanged, while efforts focused on optimizing the conditions for 4-O-Fmoc glucosamine donor 3. After 30 min incubation at −20 °C, most of 3 remained unactivated. Gratifyingly, at −10 °C this glycosyl donor was fully activated (Fig. 3a, b and Supplementary Fig. 1). Following photocleavage and normal-phase HPLC purification, tetrasaccharide 10 was isolated in 27% overall yield. The analytical HPLC of the crude sample revealed traces of deletion sequences. A further increase in the glycosylation temperature to 0 °C resulted in accelerated decomposition of 3 and significantly reduced the yield of the target compound 10. Thus, the coupling temperature for 3 was set at −10 °C for 30 min.

a Glycosylation at −20 °C; b Glycosylation at −10 °C; c 1% TMSOTf as acidic wash solution; d 0.1% TMSOTf as acidic wash solution; e 2/1 ratio of CH2Cl2/Dioxane; f 4/1 ratio of CH2Cl2/Dioxane.
To address the traces of deletion sequences observed, we examined the concentrations of TfOH and TMSOTf, as their acidity can critically affect glycosidic bond activation and stability. For TfOH, an optimal threshold concentration of 0.15 equivalents per glycosyl donor was observed. The effect of TMSOTf concentration in the acidic wash module was also examined (Fig. 3c, d). Under concentration of 1% v/v TMSOTf, signs of glycosidic bond cleavage was observed. Reducing TMSOTf concentration tenfold to 0.1% v/v completely suppressed cleavage, raised isolated yield to 37% as observed by crude HPLC analysis. Next, the solvent ratio and an extra basic wash with 10% pyridine solution were examined, but these had only minor impact on the glycosylation efficiency (Fig. 3e, f and Supplementary Fig. 2).

Automated synthesis of protected oligo-LacNAc and HMOs. Overall yields indicated for each product.
Automated synthesis of pNZ-protected oligo-LacNAc & HMOs (Fig. 4)
With optimized coupling conditions in hand, the substrate scope of HMOs was explored. The synthesis of HMOs fragments containing LacNAc type-2 linkage (Galβ1-4GlcNAcβ1-3) yielded tetramer 10 (37%), hexamer 18 (26%), and octamer 19 (20%) using 3-O-Fmoc galactoside 9 and 4-O-Fmoc glucosamine donor 3.
Oligo-LacNAc and HMOs differ in the first monosaccharide at the reducing end: while oligo-LacNAc begins with glucosamine, HMOs contain glucose. 4-O-Fmoc glucoside 11, from our building block repertoire32, was coupled at an initiation temperature (T1) of -20 °C for five minutes, followed by a coupling temperature (T2) at 0 °C for 20 min. Removal of Fmoc protecting group exposed a C-4 hydroxyl group for glycosylation using building blocks 9 and 3 to furnish linear tetrasaccharide 20 (36%) and hexasaccharide 21 (28%).
Oligosaccharides containing the LacNAc type-1 epitope (Galβ1-3GlcNAc), are abundant in HMOs and associated with a variety of developmental and pathological conditions51. To construct the linear Galβ1-3GlcNAc 22, 3-Fmoc glucosamine donor 6 was coupled at 0 °C for 40 min to overcome its lower reactivity to give tetrasaccharide 22 in 32% yield.
Many neutral HMOs do contain fucose as a crucial epitope33. The Fucα1-3GlcNAc branch was introduced into the linear LacNAc type-2 tetramer, yielding pentasaccharide 23. A selective deprotection strategy was implemented to install both Galβ1-4GlcNAc and Fucα1-3GlcNAc linkages. First, the Lev-protecting group on the terminal glucosamine was selectively removed using a solution of hydrazine acetate, exposing a hydroxyl group for the attachment of per-benzyl fucoside 13. Thereafter, the 4-O-Fmoc group of glucosamine was removed with piperidine, revealing a second hydroxyl group for glycosylation with 3-O-Fmoc galactoside 9. This stepwise strategy furnished pentasaccharide 23 in 29% isolated yield. The same strategy for deprotection of Fmoc and Lev groups was applied in the synthesis of LacNAc type-1 pentasaccharide 24, to introduce the Fucα1-4GlcNAc branch. In order to prevent complications during Fmoc deprotection, the 2,3-O-di-benzoyl galactoside 14 was introduced as the terminal unit. Building block 14 required a higher activation temperature and the coupling was performed with T1 at -20 °C for five minutes, followed by T2 at 0 °C for 20 min to yield 21% of pentasaccharide 24.
For symmetrically branched HMOs, the 6-O-Lev-3-O-Fmoc galactoside 15 provided the appropriate branching points to construct both the GlcNAcβ1-6Gal and GlcNAcβ1-3Gal linkages. However, the presence of an additional electron-withdrawing group on building block 15 necessitated a higher activation temperature when compared to 3-O-Fmoc galactoside 950. During the synthesis of hexasaccharide 25a, the 6-O-Lev and 3-O-Fmoc groups on 15 were sequentially removed, exposing two hydroxyl groups. Bis-glycosylation using 4-Fmoc glucosamine donor 3 was performed at both the 6-OH and 3-OH positions of the galactose unit. A subsequent bis-glycosylation step with 3-O-Fmoc galactoside 9 produced branched pentasaccharide 25a in 29% isolated yield. For comparison, the N-pNZ-protected glucosamine donor 3 was replaced by N-TCA-protected 17a and N-Cbz 17b glucosamine donors to obtain hexasaccharides 25b and 25c, respectively. The pNZ-protected donor afforded a higher isolated yield (29%) over multiple glycosylation cycles, compared to the TCA-protected (16%) and the Cbz-protected donor (6%). In the case of the TCA-protected donor, the absence of prominent deletion sequences suggested that glycosidic linkages may undergo acid-catalyzed hydrolysis during the synthesis52. In contrast, the use of the Cbz-protected donor resulted in the deletion of sequences due to poorer glycosylation efficiency (Supplementary Fig. 3). Another symmetrically branched HMO containing the Fucα1-3GlcNAc motif was examined. The efficiency of the fucosylation and the final galactosylation was slightly reduced, likely due to steric effect. By increasing the equivalents of building blocks 9 and 13 from 6.5 to 10, target octasaccharide 26 was obtained in 19% isolated yield. After coupling, the hydrolyzed building blocks were recovered by rerouting the reaction mixture to the fraction collector.
To synthesize unsymmetrically branched decasaccharide HMO 27, we introduced 4-O-Lev glucosamine donor 4 to facilitate a more complex synthetic pathway. This building block was activated similar to 4-O-Fmoc glucosamine donor 3, as different ester groups did not markedly impact the activation temperature50. The architecture of decasaccharide 27 featured two distinct branching points on two identical galactose terminals. Initially, the 6-O-Lev group on the first galactose was removed to expose the C-6 hydroxyl group that was then glycosylated with 4-O-Lev glucosamine donor 4. Next, the 4-O-Lev on this glucosamine was removed, and the resulting hydroxyl group was used to react with 6-O-Lev-3-O-Fmoc galactoside 15. The resulting tetrasaccharide contained two Fmoc and one Lev groups that were promptly cleaved to reveal three hydroxyl groups. A tris-glycosylation step with 4-O-Fmoc glucosamine donor 3 was followed by another tris-glycosylation with galactose building block 9, completing the synthesis of decasaccharide 27 in 16% isolated yield. The automation sequence for the unsymmetrically branched heptasaccharide 28 combined elements of the sequences used to synthesize glycans 23 and 27. The 6-O-Lev on galactose was removed, followed by glycosylation with 4-O-Lev glucosamine donor 4 and 2,3-O-di-benzoyl galactoside 14. Subsequently, the 3-O-Fmoc on galactose 15 was removed to expose the hydroxyl group. The Lewis-X epitope, Galβ1-4(Fucα1-3)GlcNAc, was introduced using the protocol applied for pentasaccharide 23, yielding 17% of heptasaccharide 28. Hexasaccharide 29 contains two fucose branches at glucose and glucosamine. Initially, 3-O-Lev, 4-O-Fmoc glucoside 12 was introduced to construct the Fucα1-3Glc and Galβ1-4Glc linkage. The Fmoc group of galactose was removed to expose the C-3 hydroxyl group that was then glycosylated with 3-O-Lev-4-O-Fmoc glucosamine donor 8. However, due to steric hindrance, the reactivity of the C-3 hydroxyl group in galactose diminished, necessitating an increase in coupling time from 30 to 45 min. The Lewis-A epitope Galβ1-3(Fucα1-4)GlcNAc was added using the automation protocol employed for the synthesis of pentasaccharide 24 to yield 17% of hexasaccharide 29.
α1,2-Fucosylated glycans represent a distinctive subset of HMOs, characterized by the presence of α1,2-linked fucose residue, also commonly associated with the H-antigen5. These structures are predominantly found in the milk of so-called secretor mothers, whose active FUT2 gene enables the enzymatic addition of fucose in an α1,2 linkage to precursor glycans. Owing to their structural uniqueness and biological relevance, α1,2-fucosylated HMOs are believed to play critical roles in shaping the infant gut microbiota and providing protection against pathogenic infections53. 2-Fmoc galactoside 16 was used to synthesize pentasaccharide 30 (21%) and hexasaccharide 31 (15%) yield.
Modification of protected oligo-LacNAc and HMOs
The pNZ group was removed by reduction of the nitro group in a two-step protocol. pNZ was first reduced to the p-aminobenzyloxycarbonyl derivative that underwent a spontaneous 1,6-electron pair shift, afforded quinonimine methide and carbamic acid. The transient carbamic acid subsequently decomposed to release the free amine35. The transformation was accelerated in the presence of an acid catalyst35. Three reducing conditions including the commonly used Tin(II) chloride (SnCl2)35, Zinc–copper couple (Zn/Cu)54, and the recently reported tetrahydroxydiboron (B2(OH)4)/4,4’-bipyridine55 were examined. All conditions successfully reduced the pNZ group to the free amine with moderate to high yield (70–89%). The boronic acid method was chosen for pNZ deprotection, owing to its overall efficiency, mildness, and the avoidance of toxic heavy metals. In the second step, N-acetylation was conducted to further modify the amino group of glucosamine residues in oligo-LacNAc and HMO structures, resulting in the formation of N-acetylglucosamine within these structures. Then, a solution of sodium methoxide in MeOH was used to remove all ester-protecting groups, followed by hydrogenation with Pd/C catalyst to remove all remaining protecting groups, including benzyl ethers and benzyloxycarbonyl (Cbz), to afford the desired HMOs (Fig. 5a).

a HMOs deprotection conditions; b Collection of neutral linear, symmetrical- and unsymmetrical-branched LacNAc-based oligosaccharides and HMOs.
Following reverse-phase HPLC purification, oligo-LacNAc derivatives 32-34 were isolated in overall yields ranging from 43% to 55% (Fig. 5b). The linear HMOs LNnT 35 and pLNnH 36 were synthesized in 55% and 52% yield, respectively. The tetrameric HMO LNT 37, incorporating a Galβ1-3GlcNAc linkage, was obtained in 57% yield. Minor deletion sequences lacking fucose residues were detected after the global deprotection step, although strongly diminished when compared to syntheses involving building blocks protected with N-TCA groups33. Linear mono-fucosylated HMOs, including LNFP III 38 and LNFP II 39, were produced in yields comparable to their non-fucosylated counterparts (ca. 50%). Branched structures, including symmetrical LNnH 40, DFLNnH 41, and unsymmetrically branched iLND 42, FLNnH II 43 were synthesized efficiently by routine adaptation of the automation sequence. Finally, di-fucosylated HMOs, including LNDFH II 44, bearing Fucα1-3 and Fucα1-4 epitopes, along with LNFP I 45 and LNDFH I 46, featuring Fucα1-2 motifs, were successfully obtained. The reducing ends of the oligo-LacNAc and HMOs were covalently linked to a five-carbon aliphatic spacer bearing a terminal amine released from the photocleavable solid support for immediate conjugation on glass surfaces for glycan array fabrication or to various biomolecules of interest, such as carrier proteins for vaccine development or adjuvants. The pNZ protecting group can be selectively removed, providing non-acetylated amino glycans, for example chitooligosaccharides, that serve as a versatile handle for further transformations including bioconjugation or incorporation into natural products32.
In conclusion, the p-nitrobenzyloxycarbonyl group was examined as a protecting group for the C-2 amine in glucosamine during automated glycan assembly. Glucosamine building blocks carrying pNZ protection enabled the efficient automated synthesis of human milk oligosaccharides and oligo-LacNAc structures. The new building blocks were prepared conveniently using a simple purification method. Investigations into temperatures, concentrations, and automation sequences led to a robust routine protocol for these challenging and manually labor-intensive target molecules. The high end-to-end efficiency of pNZ protection facilitated the synthesis of linear, branched, structurally diverse, and complex HMO variants using the Glyconeer® automated synthesizer. Moreover, three methods for the selective removal of the pNZ group were applied to enable product customization.
Methods
General
The synthesis and characterization of monomer building blocks (3, 4, 6 and 8), along with the synthetic protocols and HPLC chromatograms for AGA of protected-oligosaccharides (10 and 18–31) and deprotected-oligosaccharides (32–46), are provided in Supplementary Information. 1H, 13C and two-dimensional (2D) NMR spectra of the compounds described in the article are available in Supplementary Information.
AGA
The automated synthesizer executes a series of commands that are combined into modules to achieve specific transformations (see Supplementary Information)
Acidic wash module
Once the temperature of the reaction vessel has adjusted to the desired temperature of −20 °C by the cooling device, 1 mL of the Acidic Wash Solution is delivered to the reaction vessel. After three minutes, the solution is drained. Finally, the resin is washed with 3 mL CH2Cl2 (bubbling = 15 s) and drained.
Glycosylation using thioglycosides module
Upon draining the CH2Cl2 in the reaction vessel, 1 mL of Building Block Solution containing the appropriate building block is delivered from the building block storing component to the reaction vessel through. After the temperature reaches the desired temperature (T1), Activator Solution (1 mL) is delivered to the reaction vessel from the respective activator storing component to the reaction vessel. The glycosylation mixture is incubated for the selected duration (t1) at the desired T1, then the reaction temperature is linearly ramped to T2. Once T2 is reached, it is maintained and the reaction mixture is incubated for an additional time (t2). Once the incubation time is finished, the reaction mixture is drained and the resin is washed with CH2Cl2 (1 × 2 mL for 15 s), then dioxane (1 × 2 mL for 15 s), and finally CH2Cl2 (2 × 2 mL for 15 s).
Pyridine wash module
The resin is washed with DMF (2 × 3 mL for 15 s). Then Pre-capping Solution (2 mL) is delivered into the reaction vessel and incubated for 3 min. The resin is then washed with CH2Cl2 (3 × 2 mL for 15 s).
Capping module
The resin is washed with DMF (2 × 3 mL for 15 s). Then Pre-capping Solution (2 mL) is delivered into the reaction vessel and incubated for 3 min. The resin is then washed with CH2Cl2 (3 × 2 mL for 15 s). Upon washing, Capping Solution (4 mL) is delivered and the temperature is adjusted and maintained 25 °C. The resin and the reagents are incubated for 20 min. The solution is then drained from the reactor vessel and the resin is washed with CH2Cl2 (3 × 3 mL for 15 s).
Fmoc deprotection module
The resin is first washed with DMF (3 × 3 mL for 15 s), and then Fmoc Deprotection Solution (2 mL) is delivered to the reaction vessel. After 5 min the reaction solution is drained and the resin is washed with DMF (3 × 3 mL for 15 s) and CH2Cl2 (3 × 3 mL for 15 s). After this module the resin is ready for the next glycosylation cycle.
Lev deprotection module
The resin was washed with DMF (3 × 30 s) and DCM (1.3 mL) added to the reaction vessel. Lev Deprotection Solution (0.8 mL) was added to the reaction vessel, and the temperature was adjusted to 25 °C. After 30 min, the reaction solution was drained and the entire cycle was repeated twice more. After Lev deprotection was completed, the resin was washed with DMF, THF and DCM.
Cleavage and normal phase purification
Resin cleavage
After automated synthesis, the resin was removed from the reaction vessel, suspended in CH2Cl2 (20 mL), and photocleaved in a continuous-flow photoreactor. A Vapourtec E-Series easy-MedChem, equipped with a UV-150 Photochemical reactor having a UV-150 Medium-Pressure Mercury Lamp (arc length 27.9 cm, 450 W) surrounded by a long-pass UV filter (Pyrex, 50% transmittance at 305 nm) was used. A Pump 11 Elite Series (Harvard Apparatus syringe pump at a flow rate of 1.0 mL/min was used to pump the mixture through a FEP tubing (i.d. 3.0 inch, volume: 12 mL) at 20 °C. The reactor was washed with 20 mL CH2Cl2 at a flow rate of 2.0 mL/min. The output solution was filtered to remove the resin and the solvent was evaporated in vacuo.
Analytical NP-HPLC
The crude product was dissolved in 4 mL of ethyl acetate (EtOAc) and analyzed using analytical HPLC (Agilent 1200 Series system). A YMC-Diol-300-NP column (150 mm × 4.600 mm I.D.) was used with a flow rate of 1.00 mL/min.
Preparative NP-HPLC
The crude product was dissolved in a 1:2 mixture of hexane and EtOAc and conducted on an Agilent 1200 Series system. A YMC-Diol-300-NP column (150 mm × 20 mm I.D.) was used with a flow rate of 15.00 mL/min.
General deprotection procedure
Protected glycan, B2(OH)4 (22 mg), and 4,4’-bipyridine (0.1 mg) were all added to a sample vial. Then, DMF (3 mL) was added to a reaction mixture. Over 10 min, the reaction mixture changed color from transparent to purple and subsequently to yellow. After 15 min, 1 M of HCl in dioxane (50 μL) was added, and the reaction mixture was stirred at 40 °C for overnight. After the reaction, the reaction mixture was diluted with water and ethyl acetate. The organic layer was collected and washed with brine and evaporated in vacuo. Then, the crude compound was dissolved in Ac2O/pyridine (4 mL, 1:1) and stirred for 3 h. Upon the completion of acetylation, the solvent was evaporated in vacuo. The acetylated crude compound was dissolved in THF (5 mL) and sodium methoxide (0.5 M solution in MeOH, 0.5 mL) was added. The mixture was stirred at room temperature for two hours. Amberlite IR-120 (H+ form) was then added to quench. After neutralization, the reaction mixture was filtered and the solvent was removed in vacuo. The crude compound was used for hydrogenolysis without further purification. The crude compound from methanolysis was dissolved in a mixture of EtOAc:tBuOH:H2O (2:1:1, 4 mL) with AcOH (0.1 mL). Pd/C (10%) was added, and the solution was purged with N2 and H2 for ten minutes. The suspension was stirred under H2 balloon for overnight. The insoluble material was removed by a CHROMAFIL ®Xtra, RC 0.45 syringe filter. The solid was washed once with MeOH and several times with water. The filtrate was collected and concentrated in vacuo.
Reverse-phase purification
Analytical RP-HPLC
The crude product was dissolved in 3 mL of H2O and analyzed using analytical HPLC (Agilent 1200 Series system). A Hypercarb column (150 mm × 4.6 mm I.D.) was used with a flow rate of 0.7 mL/min.
Preparative NP-HPLC
The crude product was dissolved in H2O and conducted on an Agilent 1200 Series system. A Hypercarb column (150 mm × 10 mm I.D.) was used with a flow rate of 3.0 mL/min.
Data availability
The raw data used to generate the figures and tables in this manuscript are available from the corresponding author on request.
References
Dinleyici, M., Barbieur, J., Dinleyici, E. C. & Vandenplas, Y. Functional effects of human milk oligosaccharides (HMOs). Gut Microbes 15, 2186115 (2023).
Bode, L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22, 1147–1162 (2012).
Bode, L. Human milk oligosaccharides: prebiotics and beyond. Nutr. Rev. 67, 183–191 (2009).
Gan, J., Cao, C., Stahl, B., Zhao, X. & Yan, J. Advances and challenges for obtaining human milk oligosaccharides: extraction from natural sources and synthesis by intentional design. Trends Food Sci. Technol. 141, 104203 (2023).
McDonald, A. G., Mariethoz, J., Davey, G. P. & Lisacek, F. In silico analysis of the human milk oligosaccharide glycome reveals key enzymes of their biosynthesis. Sci. Rep. 12, 10846 (2022).
Tseng, H.-W. et al. Controllable enzymatic synthesis of natural asymmetric human milk oligosaccharides. JACS Au 4, 4496–4506 (2024).
Ninonuevo, M. R. et al. A strategy for annotating the human milk glycome. J. Agric. Food Chem. 54, 7471–7480 (2006).
Wu, S., Tao, N., German, J. B., Grimm, R. & Lebrilla, C. B. Development of an annotated library of neutral human milk oligosaccharides. J. Proteome Res. 9, 4138–4151 (2010).
Wu, S., Grimm, R., German, J. B. & Lebrilla, C. B. Annotation and structural analysis of sialylated human milk oligosaccharides. J. Proteome Res. 10, 856–868 (2011).
Xu, L. L. & Townsend, S. D. Synthesis as an expanding resource in human milk science. J. Am. Chem. Soc. 143, 11277–11290 (2021).
Bode, L. et al. Overcoming the limited availability of human milk oligosaccharides: challenges and opportunities for research and application. Nutr. Rev. 74, 635–644 (2016).
Bandara, M. D., Stine, K. J. & Demchenko, A. V. Chemical synthesis of human milk oligosaccharides: lacto-N-hexaose Galβ1→3GlcNAcβ1→3 [Galβ1→4GlcNAcβ1→6] Galβ1→4Glc. J. Org. Chem. 84, 16192–16198 (2019).
Bandara, M. D., Stine, K. J. & Demchenko, A. V. Chemical synthesis of human milk oligosaccharides: lacto-N-neohexaose (Galβ1 → 4GlcNAcβ1→)2 3,6Galβ1 → 4Glc. Org. Biomol. Chem. 18, 1747–1753 (2020).
Singh, Y. et al. Chemical synthesis of human milk oligosaccharides: para-Lacto-N-hexaose and para-Lacto-N-neohexaose. Chem. - Eur. J. 29, e202302288 (2023).
Zeuner, B. & Meyer, A. S. Enzymatic transfucosylation for synthesis of human milk oligosaccharides. Carbohydr. Res. 493, 108029 (2020).
Prudden, A. R. et al. Synthesis of asymmetrical multiantennary human milk oligosaccharides. Proc. Natl Acad. Sci. 114, 6954–6959 (2017).
Li, T. et al. An automated platform for the enzyme-mediated assembly of complex oligosaccharides. Nat. Chem. 11, 229–236 (2019).
Ooi, K.-E., Zhang, X.-W., Kuo, C.-Y., Liu, Y.-J. & Yu, C.-C. Chemoenzymatic synthesis of asymmetrically branched human milk oligosaccharide lacto-N-hexaose. Front. Chem. 10, 905105 (2022).
Palur, D. S. K., Pressley, S. R. & Atsumi, S. Microbial production of human milk oligosaccharides. Molecules 28, 1491 (2023).
McManaman, J. L. & Neville, M. C. Mammary physiology and milk secretion. Adv. Drug Deliv. Rev. 55, 629–641 (2003).
Tsutsui, M. et al. Efficient synthesis of antigenic trisaccharides containing N-acetylglucosamine: protection of NHAc as NAc2. Eur. J. Org. Chem. 2020, 1802–1810 (2020).
Gomez, A. M. & Lopez, J. C. Bringing color to sugars: the chemical assembly of carbohydrates to BODIPY dyes. Chem. Rec. 21, 3112–3130 (2021).
Agrahari, A. K. et al. Cu(I)-catalyzed click chemistry in glycoscience and their diverse applications. Chem. Rev. 121, 7638–7956 (2021).
Crawford, C. J. et al. Automated synthesis of algal fucoidan oligosaccharides. J. Am. Chem. Soc. 146, 18320–18330 (2024).
Crawford, C. J. & Seeberger, P. H. Advances in glycoside and oligosaccharide synthesis. Chem. Soc. Rev. 52, 7773–7801 (2023).
Niggemeyer, G. B., Danglad-Flores, J. A. & Seeberger, P. H. Automated synthesis of C1-functionalized oligosaccharides. J. Am. Chem. Soc. 147, 1649–1655 (2025).
Ganesh, N. V., Fujikawa, K., Tan, Y. H., Stine, K. J. & Demchenko, A. V. HPLC-assisted automated oligosaccharide synthesis. Org. Lett. 14, 3036–3039 (2012).
Tang, S.-L. & Pohl, N. L. B. Automated solution-phase synthesis of β-1,4-mannuronate and β-1,4-mannan. Org. Lett. 17, 2642–2645 (2015).
Yao, W. et al. Automated solution-phase multiplicative synthesis of complex glycans up to a 1,080-mer. Nat. Synth. 1, 854–863 (2022).
Enugala, R., Carvalho, L. C. R., Dias Pires, M. J. & Marques, M. M. B. Stereoselective glycosylation of glucosamine: the role of the N-Protecting Group. Chem. Asian J. 7, 2482–2501 (2012).
Debenham, J., Rodebaugh, R. & Fraser-Reid, B. Recent advances in N-Protection for amino sugar synthesis. Liebigs Ann. 1997, 791–802 (1997).
Tyrikos-Ergas, T. et al. Systematic structural characterization of chitooligosaccharides enabled by automated glycan assembly. Chem. Eur. J. 27, 2321–2325 (2021).
Guberman, M., Bräutigam, M. & Seeberger, P. H. Automated glycan assembly of Lewis type I and II oligosaccharide antigens. Chem. Sci. 10, 5634–5640 (2019).
Tyrikos-Ergas, T., Sletten, E. T., Huang, J.-Y., Seeberger, P. H. & Delbianco, M. On resin synthesis of sulfated oligosaccharides. Chem. Sci. 13, 2115–2120 (2022).
Isidro-Llobet, A., Guasch-Camell, J., Álvarez, M. & Albericio, F. p-Nitrobenzyloxycarbonyl (pNZ) as a temporary Nα-protecting group in orthogonal solid-phase peptide synthesis—avoiding diketopiperazine and aspartimide formation. Eur. J. Org. Chem. 2005, 3031–3039 (2005).
Grann Hansen, S. & Skrydstrup, T. Studies directed to the synthesis of oligochitosans—preparation of building blocks and their evaluation in glycosylation studies. Eur. J. Org. Chem. 2007, 3392–3401 (2007).
Isidro-Llobet, A., Álvarez, M. & Albericio, F. Semipermanent p-nitrobenzyloxycarbonyl (pNZ) protection of Orn and Lys side chains: prevention of undesired α-Fmoc removal and application to the synthesis of cyclic peptides. Tetrahedron Lett. 46, 7733–7736 (2005).
Boullanger, P., Jouineau, M., Bouammali, B., Lafont, D. & Descotes, G. The use of N-alkoxycarbonyl derivatives of 2-amino-2-deoxy-d-glucose as donors in glycosylation reactions. Carbohydr. Res. 202, 151–164 (1990).
Lahmann, M. & Oscarson, S. Investigation of the reactivity difference between thioglycoside donors with variant aglycon parts. Can. J. Chem. 80, 889–893 (2002).
Tuck, O. T., Sletten, E. T., Danglad-Flores, J. & Seeberger, P. H. Towards a systematic understanding of the Influence of temperature on glycosylation reactions. Angew. Chem. Int. Ed. 61, e202115433 (2022).
Joseph, A. A. et al. TMSOTf-catalyzed silylation: streamlined regioselective one-pot protection and acetylation of carbohydrates. Eur. J. Org. Chem. 2012, 744–753 (2012).
Despras, G., Urban, D., Vauzeilles, B. & Beau, J.-M. One-pot synthesis of d-glucosamine and chitobiosyl building blocks catalyzed by triflic acid on molecular sieves. Chem. Commun. 50, 1067–1069 (2014).
Chatterjee, S., Moon, S., Hentschel, F., Gilmore, K. & Seeberger, P. H. An Empirical understanding of the glycosylation reaction. J. Am. Chem. Soc. 140, 11942–11953 (2018).
Chang, C. W. et al. Automated quantification of hydroxyl reactivities: prediction of glycosylation reactions. Angew. Chem. Int. Ed. 60, 12413–12423 (2021).
Moon, S., Chatterjee, S., Seeberger, P. H. & Gilmore, K. Predicting glycosylation stereoselectivity using machine learning. Chem. Sci. 12, 2931–2939 (2021).
Trinderup, H. H., Andersen, S. M., Heuckendorff, M. & Jensen, H. H. How do various reaction parameters influence anomeric selectivity in chemical glycosylation with thioglycosides and NIS/TfOH activation? Eur. J. Org. Chem. 2021, 3251–3259 (2021).
Andreana, P. R. & Crich, D. Guidelines for O-glycoside formation from first principles. ACS Cent. Sci. 7, 1454–1462 (2021).
Volbeda, A. G., van der Marel, G. A. & Codée, J. D. C. Protecting Groups (Wiley, 2019).
Lin, M.-H. et al. Enabling technologies in carbohydrate chemistry: automated glycan assembly, flow chemistry and data science. ChemBioChem 24, e202200607 (2023).
Lin, M.-H., Kuo, Y.-T., Danglad-Flores, J., Sletten, E. T. & Seeberger, P. H. Parametric analysis of donor activation for glycosylation reactions. Chem. Eur. J. 30, e202400479 (2024).
Fischöder, T., Laaf, D., Dey, C. & Elling, L. Enzymatic synthesis of N-acetyllactosamine (LacNAc) type 1 oligomers and characterization as multivalent galectin ligands. Molecules 22, 1320 (2017).
Hahm, H. S. et al. Automated glycan Assembly of complex oligosaccharides related to blood group determinants. J. Org. Chem. 81, 5866–5877 (2016).
Bode, L. & Jantscher-Krenn, E. Structure-function relationships of human milk oligosaccharides. Adv. Nutr. 3, 383S–391S (2012).
Franz, R. & Neumann, H.-G. The reduction of aromatic nitro compounds with Zn/Cu. A new synthesis of N-acetoxy-N-acetyl-arylamines. Carcinogenesis 7, 183–184 (1986).
Jang, M., Lim, T., Park, B. Y. & Han, M. S. Metal-free, rapid, and highly chemoselective reduction of aromatic nitro compounds at room temperature. J. Org. Chem. 87, 910–919 (2022).
Acknowledgements
We gratefully acknowledge the Max-Planck Society for its generous financial support. Y.-T.K. and K.L.M.H. acknowledge financial support from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 956758.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
K.L.M.H. and P.H.S. conceived and supervised the experiments and contributed to manuscript preparation. M.-H.L. and Y.-T.K. performed the synthetic work, conducted data analysis, and prepared the initial draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.-H.L. and Y.-T.K. declare no competing interests. K.L.M.H. and P.H.S. declare a competing interest in GlycoUniverse GmbH & Co. KGaA, the company that commercializes the Glyconeer® automated synthesizer and glycan products, including all building blocks used in this study.
Peer review
Peer review information
Nature Communications thanks Somnath Yadav and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, MH., Kuo, YT., Le Mai Hoang, K. et al. p-nitrobenzyloxycarbonyl protective group as key to automated glycan assembly of neutral human milk oligosaccharides. Nat Commun 16, 10941 (2025). https://doi.org/10.1038/s41467-025-66557-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66557-3
