
Abstract
Single-cell technologies have transformed our understanding of cellular heterogeneity through multimodal data acquisition. However, robust cell alignment remains a major challenge for data integration and harmonization, including batch correction, label transfer, and multi-omics integration. Many existing methods constrain alignment based on rigid feature-wise distance metrics, limiting their ability to capture accurate cell correspondence across diverse cell populations and conditions. We introduce scGALA, a graph-based learning framework that redefines cell alignment by combining graph attention networks with a score-driven, task-independent optimization strategy. scGALA constructs enriched graphs of cell-cell relationships by integrating gene expression profiles with auxiliary information, such as spatial coordinates, and iteratively refines alignment via self-supervised graph link prediction, where a deep neural network is trained to identify and reinforce high-confidence correspondences across datasets. In extensive benchmarks, scGALA identifies over 25 percent more high-confidence alignments without compromising accuracy. By improving the core step of cell alignment, scGALA serves as a versatile enhancer for a wide range of single-cell data integration tasks.
Similar content being viewed by others

Introduction
Single-cell technologies have revolutionized our understanding of cellular heterogeneity and function, enabling the generation of rich, high-dimensional datasets across diverse molecular modalities1,2,3. From transcriptomics and epigenomics to proteomics and spatial measurements, these techniques allow researchers to probe cell states and interactions at unprecedented resolution4,5,6. However, this technological leap introduces a fundamental challenge: how to accurately align and integrate cells across datasets generated from different conditions, batches, or modalities, while preserving biological relevance7,8,9. Cell alignment serves as a foundation for numerous downstream tasks, including batch effect correction10,11, label transfer12,13, multi-omics integration14,15, and spatial alignment16,17, making it a critical step in the single-cell analysis pipeline.
Despite considerable progress, current cell alignment methods face substantial limitations. Linear approaches such as Canonical Correlation Analysis (CCA)18, implemented in Seurat’s anchoring strategy19, aim to uncover shared correlation structures across datasets. These methods are computationally efficient and effective when the datasets have similar linear structure, but they often fail to capture the complex, non-linear biological relationships that underlie cell states20,21. In contrast, non-linear methods, including those based on Mutual Nearest Neighbors (MNN)22, are better suited for detecting local correspondences in expression space. However, these methods still struggle when aligning cells across modalities or conditions with substantial technical variation, where geometric proximity is distorted by noise or batch effects23. Furthermore, both linear and non-linear methods generally rely on expression-derived features and fail to incorporate auxiliary information such as spatial coordinates or known biological relationships—critical cues that are especially important in spatial transcriptomics5,24,25. As a result, these methods can yield suboptimal alignments that overlook biologically meaningful correspondences.
Recent advances in graph learning offer new opportunities to model complex relationships in biological systems26,27,28,29,30,31. In particular, graph-based methods have demonstrated strong performance in capturing non-linear and long-range dependencies32,33,34, and link prediction techniques have proven effective in inferring missing or hidden connections in networked data35,36,37. Motivated by these developments, we introduce scGALA (\(\underline{{{{\bf{s}}}}}{{{\rm{ingle}}}}\)-\(\underline{{{{\bf{c}}}}}{{{\rm{ell}}}}\,\underline{{{{\bf{G}}}}}{{{\rm{raph}}}}\,\underline{{{{\bf{A}}}}}{{{\rm{ttention}}}}\,\underline{{{{\bf{L}}}}}{{{\rm{ink}}}}\)-prediction based \(\underline{{{{\bf{A}}}}}{{{\rm{lignment}}}}\)), a computational framework that reconceptualizes the alignment problem as a graph-based link prediction task. scGALA builds comprehensive graphs to model cell-cell relationships across datasets, integrating both gene expression and auxiliary information. It then applies a multi-scale Graph Attention Network (GAT)38,39 to predict high-confidence links between corresponding cells. A key feature of scGALA is its iterative score-based optimization strategy40,41, which refines alignments by prioritizing matches with high predicted link probabilities. This allows the method to progressively improve alignment accuracy while capturing both local and global graph structure. Importantly, scGALA’s architecture allows the integration of diverse auxiliary features, such as spatial coordinates or known cell-cell relationships42,43, which extends its applicability across a range of single-cell data integration tasks.
We benchmarked scGALA across diverse tasks to demonstrate its comprehensive capabilities. As an all-in-one integration pipeline, scGALA consistently outperformed existing methods in batch correction, label transfer, multi-omics integration, and spatial alignment, achieving up to 67.8% improvement in clustering accuracy metrics. As a universal booster, scGALA enhanced the performance of ten state-of-the-art integration tools, including Seurat19, INSCT44, and scCross14, by improving their underlying cell alignment. In spatial transcriptomics applications, scGALA effectively imputed missing gene expression while maintaining biological fidelity, expanding gene coverage three-fold while preserving spatial organization information (GraphST-based spatial clustering ARI of 0.51 for scGALA imputed data versus 0.47 for limited gene coverage data). For mosaic integration, scGALA bridged separate dual-omics datasets (RNA+ATAC and RNA+ADT) into virtual tri-omics data, identifying 25% more high-confidence alignments than conventional approaches. In cross-modality imputation, scGALA accurately generated RNA profiles from ATAC-seq measurements with high biological fidelity (Pearson correlation = 0.93 in terms of marker genes), preserving cell type-specific markers, pathway enrichment, and intercellular communication patterns. Across all tasks and datasets, scGALA consistently demonstrated superior alignment accuracy and biological coherence compared to existing methods.
Results
Overview of scGALA
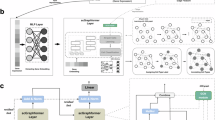
scGALA is a graph-based learning framework that enables accurate and scalable cell alignment across single-cell datasets by formulating alignment as a masked link prediction problem over cell-cell graphs (Fig. 1a). For each dataset pair, intra-dataset graphs are built using K-nearest neighbors (KNN) from molecular profiles, and inter-dataset edges are initialized via mutual nearest neighbors (MNN). These graphs are input into a Graph Attention Network (GAT), trained using a self-supervised masked link prediction strategy in which a random subset of edges is hidden during each epoch and the model is optimized to reconstruct them. This encourages the discovery of cross-dataset correspondences beyond those observed during training. The predicted alignments are further refined through iterative, score-based optimization and merged with initial MNN matches to produce a robust and high-confidence cell mapping, a strategy that differentiates scGALA from prior alignment frameworks that rely solely on mutual nearest-neighbor heuristics.

a Cell Alignment Backbone Module via Graph Link Prediction. The foundational alignment framework operates on each pair of datasets by constructing cell-cell graphs and learning alignments via masked link prediction. Intra-dataset graphs are built using K-nearest neighbors (KNN) based on molecular profiles, optionally incorporating spatial coordinates, while inter-dataset edges are initialized using mutual nearest neighbors (MNN). A Graph Attention Network (GAT) is trained on these graphs using a self-supervised strategy in which a subset of edges is randomly masked during training and the model is optimized to reconstruct them, encouraging the discovery of cross-dataset correspondences. The predicted links are iteratively refined via score-based optimization and merged with the MNN-based priors to yield a high-confidence alignment backbone that enables subsequent integration analyses. b All-in-One Data Integration Pipeline. Building upon the enhanced cell alignment backbone from panel (a), scGALA supports core tasks for single-cell data integration and harmonization, including: (1) batch correction, by removing technical variation guided by predicted alignments while preserving biological heterogeneity; (2) label transfer, by propagating annotations through alignment-based matching; (3) multi-omics integration, by aligning and constructing unified representations across modalities; and (4) spatial alignment, by incorporating spatial coordinates to enable spatial-aware alignment across tissue slices. c Advanced Multimodal Functionalities. Leveraging the improved cell alignment methodology independently, scGALA enables distinct advanced multi-omics functionalities not widely supported by existing methods, including: (1) mosaic integration, where datasets with partially overlapping modalities (e.g., RNA+ATAC and RNA+protein) are jointly integrated to reconstruct unified tri-modal profiles; (2) cross-modality multi-omics imputation and generation, where unmeasured modalities (e.g., RNA for ATAC cells) are inferred using alignment-derived mappings to support full-transcriptome analysis; and (3) spatial transcriptomics enhancement, where scRNA-seq data is aligned with spatial datasets to impute missing gene expression (such as in Xenium), increasing resolution and enabling downstream analyses such as spatial domain identification and spatial marker discovery.
Built upon this enhanced alignment backbone, scGALA can serve in two complementary roles: as a “universal booster” for existing alignment-based methods and as a “standalone framework” for advanced applications. As a universal booster, scGALA replaces the intermediate alignment module in existing pipelines with its enhanced alignments, directly improving core single-cell integration tasks (Fig. 1b). These include batch correction by mitigating technical variation while preserving biological heterogeneity; label transfer by mapping annotations from reference to query datasets; multi-omics integration by aligning modalities into a unified representation; and spatial alignment by incorporating spatial coordinates for context-aware mapping across platforms. Since many widely used integration tools already perform these tasks, our focus is not to replicate existing modules but to demonstrate that scGALA can universally enhance them, providing a drop-in improvement across diverse pipelines and establishing scGALA as a broadly generalizable backbone for integration. In addition to these foundational capabilities, scGALA can also independently perform a set of distinct advanced multi-omics functionalities (Fig. 1c) that are not widely enabled by existing methods. These include mosaic integration, where datasets with partially overlapping modalities (e.g., RNA+ATAC and RNA+ADT) are jointly aligned to construct unified tri-modal profiles; cross-modality multi-omics imputation, which infers unmeasured profiles (e.g., RNA from ATAC) to enable transcriptome-scale analyses in unimodal datasets; and spatial transcriptomics enhancement, in which aligned reference scRNA-seq data are used to impute missing gene expression in spatial datasets, thereby increasing resolution and supporting downstream tasks such as spatial domain identification and spatial marker discovery.
scGALA enables all-in-one single-cell data integration and harmonization across multiple tasks
To comprehensively evaluate scGALA’s core capabilities as an integrated analysis pipeline, we conducted the experiments in this section on the Rodent Research-3 dataset45 from the NASA Open Science Data Repository. This dataset contains matched single-cell RNA sequencing and ATAC sequencing data of 21178 cells, generated using the 10x Multiome protocol on one hemisphere of each mouse brain, and spatially resolved transcriptomics data of 29770 spots, acquired using the 10x Genomics Visium Gene Expression protocol on the other hemisphere. This multimodal and spatially structured dataset provides an ideal benchmark for testing scGALA across a broad range of integration challenges.
We focused on four key tasks—batch correction, label transfer, multi-omics integration, and spatial alignment—which collectively represent the most common and essential challenges in single-cell data integration and harmonization. These tasks span across modalities, batches, and spatial axes, and are routinely encountered in multi-condition or large-cohort studies. A unified solution that can address all four tasks consistently is crucial for enabling scalable and biologically coherent single-cell analysis. Through systematic evaluation including both visual and quantitative comparisons, we demonstrate that scGALA’s graph-based cell alignment framework enhances the performance of existing methods across all four tasks.
Our analysis reveals that scGALA significantly improves Seurat’s batch correction capabilities while preserving biological signals. The UMAPs provide an intuitive illustration of better separation of cell types with minimized batch effects when using scGALA-enhanced Seurat. As shown in the red circle of Fig. 2a, the UMAP of the Seurat-corrected data demonstrates a mixture of other cell types within the cluster dominated by cell type Neurogenesis I, while in the scGALA-enhanced results, the corresponding cluster shows less cell type mixing. This is further illustrated by the clearer isolation of Neuroendocrine Activity cells in UMAP regions highlighted by the purple circle. This improvement is quantitatively supported by increased biological conservation metrics, with the Adjusted Rand Index (ARI)46 improving by an average of 14.7% (P = 6.07 × 10−5) and the Normalized Mutual Information (NMI)47 increasing by 7.7% (P = 1.41 × 10−5) compared to standard Seurat (Fig. 2b). Furthermore, as shown in Supplementary Table S1, 15 out of a total of 18 clusters demonstrate improved batch-specific Average Silhouette Width (ASW)48 scores (P = 6.90 × 10−5) in scGALA-enhanced results. The increased ASW/Batch scores and Graph Connectivity scores49 further confirm the enhanced integration quality while maintaining cell type-specific features.

a UMAP visualizations comparing batch correction results from Seurat (baseline) and scGALA, colored by batch (left) and cell type (right). Circled regions highlight improved cell type separation achieved by scGALA. b Normalized integration metrics (range 0-1) evaluating biological conservation (e.g., cell type purity) and batch effect removal (higher values indicate better performance). c UMAPs showing label transfer results (Spearman correlation vs. ground truth) using Seurat and scGALA, colored by transferred cell type labels. d Label transfer performance metrics including accuracy and Cohen’s kappa, with 95% confidence intervals. e Multi-omics integration of scRNA-seq and scATAC-seq using scCross (baseline) and scGALA, colored by modality domain (left) and integrated cell type labels (right). f Multi-omics integration metrics: box plots showing biological conservation and domain harmonization metrics. Line plot of FOSCTTM scores (lower values indicate better performance) as a function of increasing cell numbers. g Spatial alignment results comparing spatial coordinate plots of STAligner (baseline) and scGALA, colored by annotated spatial region labels. h Spatial integration metrics evaluating biological conservation, modality mixing, and alignment accuracy across slices. Each score in panels b, d, f, and h is derived from N = 10 bootstrapping replicates using different random seeds (technical replicates). Boxes indicate the interquartile range (IQR, 25th to 75th percentile), with the line inside each box representing the median. Whiskers extend to the most extreme data points within 1.5 times the IQR from the quartiles. P values calculated using one-sided Student’s t-test: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001; ns, not significant. Exact P values are provided in the Source Data. Source data are provided as a Source Data file.
In label transfer applications, scGALA enhances cell type annotation accuracy between reference and query datasets. The UMAP visualization shown in Fig. 2c reveals improved classification precision across cell types, particularly for populations that are prone to misidentification, such as Neurogenesis I and Neurovasculature, which yielded a 38.5% improvement in Spearman correlation. This enhancement is characterized in Fig. 2d by wider 95% confidence interval bounds for prediction accuracy (AccuracyLower: P = 6.79 × 10−12; and AccuracyUpper: P = 2.75 × 10−13), and significantly higher overall accuracy (P = 4.16 × 10−10) and Cohen’s kappa (P = 1.90 × 10−11) statistics, which show average increases of 52.8% and 66.8% respectively. These results demonstrate scGALA’s ability to establish more accurate cell type correspondences across datasets, facilitating reliable label harmonization and cross-dataset integration.
For multi-omics integration, scGALA enhances scCross’s ability14 to integrate scRNA-seq and scATAC-seq profiles into a unified representation. The integrated analysis shows improved coordination between transcriptomic and chromatin accessibility signals, as evidenced by more coherent clustering patterns in the joint embedding space visualized in Fig. 2e. The FOSCTTM50 analysis shown in the right-side panel of Fig. 2f demonstrates consistently superior preservation of biological relationships between modalities across varying sample sizes (P = 1.34 × 10−5). From all the metrics shown in the left-side panel of Fig. 2f, although the domain label-based ASW scores do not show considerable improvements, the biological conservation metrics, including ARI (P = 4.48 × 10−5), NMI (P = 8.80 × 10−6), and ASW (P = 4.09 × 10−13), all show significant increases, calculated using a one-sided Student’s t-test51. These findings indicate the advantage of scGALA in producing more biologically meaningful alignments, which provide stronger cell type-specific signals for downstream applications. This enhancement may also support more reliable inferences of regulatory relationships between the chromatin state and gene expression, demonstrating scGALA’s ability to unify transcriptomic and chromatin accessibility signals within a coherent framework.
In spatial alignment applications, scGALA significantly improves STAligner’s performance52 by leveraging both molecular profiles and spatial information through its enhanced cell alignment framework. The experiment showcases spatial alignment between two adjacent slices (Sample 158 A1 and Sample 158 B1), comparing results from the standard STAligner and the scGALA-enhanced version. As shown in the spatial alignment plots in Fig. 2g with visualization of alignments on retrosplenial neurons in Sample 158 A1, the enhanced algorithm achieves more precise matching of the corresponding tissue regions across consecutive slices, with more alignments deemed correct based on region annotations. Quantitative assessment shows a statistically significant increase in spatial alignment accuracy52 (Fig. 2h), with improved preservation of both spatial relationships and molecular profiles, as demonstrated by higher NMI (19.5% average improvement, P = 1.71 × 10−10) and slice-specific ASW scores (16.7% average improvement, P = 3.20 × 10−14), based on the intermediate result of integrated representations across slices and ground truth region labels. In the calculation of spatial alignment accuracy, alignments are considered correct when aligned cells have matched region labels. This improvement enables a more accurate reconstruction of three-dimensional tissue organization from sequential sections and facilitates the integrated analysis of spatially resolved transcriptomics data, underscoring scGALA’s capacity to extend beyond molecular modalities to robust spatial harmonization.
scGALA boosts a broad spectrum of methods by enhancing cell alignment
Beyond establishing scGALA as a standalone integration framework, we evaluated its applicability as a general enhancement module for established computational methods in single-cell data analysis. Since scGALA’s core enhancement function lies in improving the fundamental cell alignment step that underlies many integration methods, we first validated the quality of scGALA’s alignment capabilities by directly comparing its cell-cell correspondence identification against dedicated alignment methods.
We benchmarked scGALA’s ability to identify cell-cell relationships (i.e., cell alignments) against state-of-the-art methods designed for this task, including random walk MNN (rwMNN) from the iMAP framework53 and BATMAN54. Alignment experiments were conducted between a CITE-seq dataset55,56 of 161,764 bone marrow mononuclear cells and a PBMC Multiome dataset from 10x Genomics57 containing 10,412 cells from healthy donors. As shown in Supplementary Fig. S1, scGALA consistently outperforms both methods, achieving an average AUROC of 0.767 compared to 0.690 for rwMNN (P = 8.55 × 10−31) and 0.663 for BATMAN (P = 2.84 × 10−31). Notably, this improvement in alignment accuracy is obtained while maintaining comparable quality, as indicated by similar Spearman correlation distributions between aligned pairs (scGALA: 0.496; BATMAN: 0.502; rwMNN: 0.485; Supplementary Fig. S1c). Confusion matrices further confirm that scGALA produces alignments with higher cell-type specificity (Supplementary Fig. S1d). These results demonstrate that scGALA achieves both greater coverage and equal or superior quality compared to existing alignment methods, which is the foundation of scGALA’s enhancement ability.
Since scGALA improves established methods through enhancing their intermediate cell alignment modules, we next performed a direct comparison between the alignments generated by scGALA and the initial anchors identified by Seurat to demonstrate the enhancement mechanism. This experiment is conducted on the same datasets as in Supplementary Fig. S1: the CITE-seq dataset56 and the PBMC Multiome dataset57. As shown in Supplementary Fig. S2, scGALA effectively enriches the initial Seurat anchors with around 145% more alignments identified exclusively by scGALA (Supplementary Fig. S2ab). Importantly, the quality of these newly identified alignments is comparable to the original Seurat anchors, as shown by their similar Spearman correlation distributions for gene expression between aligned cell pairs (average correlation: Seurat 0.492, scGALA Unique 0.472) (Supplementary Fig. S2a). To ensure a fair comparison of accuracy, we selected a subset from scGALA results using the same scoring method as Seurat and matched in number to the Seurat anchors. This matched set achieved a slightly higher AUROC of 0.814 compared to Seurat’s 0.808, confirming scGALA’s ability to identify high-quality cell-cell correspondences (Supplementary Fig. S2c). The confusion matrices further visualize this, showing that scGALA maintains high cell-type specificity while vastly increasing the number of alignments (Supplementary Fig. S2d).
This fundamental improvement in the core cell alignment step directly translates to enhanced performance in downstream applications. Building on the demonstration of its core capabilities, we integrated scGALA’s graph learning and score-based optimization approach into several widely used pipelines featuring both classic and recent top-performing tools. Crucially, we focused on methods whose core algorithms rely on an intermediate cell-cell alignment module, as these are the methods that scGALA is designed to enhance. Using benchmark datasets that capture representative challenges in batch correction, label transfer, multi-omics integration, and spatial alignment, we assessed performance changes in both visual and quantitative outcomes. Across all tested settings, scGALA improved the integration quality of the baseline methods, supporting its utility as a versatile alignment enhancer that can be incorporated into diverse analytical workflows.
For batch correction, we tested scGALA with Seurat (unsupervised), INSCT (in both supervised and unsupervised modes)44, scDML (unsupervised)11 Scanorama (unsupervised)10, iMAP (unsupervised)53, and STACAS (semi-supervised)20 on the Mouse Brain ATAC (Gene) dataset of 11270 cells from the scIB benchmark suite49. Due to space constraints, the quantitative metrics of Scanorama, iMAP, and STACAS are provided in Supplementary Fig. S3b. UMAP visualizations showed that scGALA-enhanced INSCT (supervised) yielded more coherent cell type clusters with reduced batch effects, particularly in the resolution of closely related subtypes such as Inhibitory and Excitatory Neurons (Fig. 3a; and Supplementary Fig. S3a, S4). These qualitative improvements were supported by quantitative metrics: scGALA-enhanced methods achieved an average increase of 29.7% in ARI (e.g., 48.6% average improvement for INSCT Unsupervised, P = 9.03 × 10−5) and 17.0% in NMI (e.g., 8.8% average improvement for Scanorama, P = 3.66 × 10−6) compared to their original implementations (Fig. 3b; and Supplementary Fig. S3b). Improvements were also observed in batch effect reduction, with average gains of 7.5% in Graph Connectivity (e.g., 37.6% average improvement for INSCT Unsupervised, P = 3.32 × 10−12) and 6.7% in batch-based ASW (e.g., 23.4% average improvement for scDML, P = 9.19 × 10−12). Even in cases where batch-specific metrics decreased slightly (e.g., Graph Connectivity in scDML, ASW in Seurat), the average decline was minimal (1.9%), indicating that scGALA introduces negligible trade-offs. Notably, the largest performance boosts were observed in methods with moderate baseline performance, suggesting that scGALA’s learned alignments can compensate for method-specific limitations.

a UMAPs comparing batch correction by INSCT (supervised baseline) and scGALA-enhanced INSCT (replacing alignment module), colored by batch (left) and cell type (right). b Normalized metrics (0-1) quantifying biological conservation (e.g., cell type purity) and batch effect removal. Higher values indicate better performance. c Label transfer accuracy (Spearman correlation vs. ground truth) comparing Seurat (baseline) and scGALA-enhanced Seurat, with UMAPs colored by transferred cell type labels. d Label transfer performance metrics: accuracy, Cohen’s kappa, and 95% confidence intervals (box plots). e Multi-omics integration (RNA + ATAC) comparing Seurat (baseline) and scGALA-enhanced Seurat, colored by modality domain (left) and joint cell type labels (right). f Multi-omics integration metrics: biological conservation and domain harmonization (box plots), and FOSCTTM (lower is better; line plot) across varying cell numbers. g Spatial alignment of tissue slices: spatial coordinate plots (X/Y/Z tissue positions) comparing STAligner (baseline) and scGALA-enhanced STAligner, colored by spatial region labels. h Spatial integration metrics: biological conservation, modality mixing, and alignment accuracy (box plots). Each score in panels b, d, f, and h is derived from N = 10 bootstrapping replicates using different random seeds (technical replicates). Boxes indicate the interquartile range (IQR, 25th to 75th percentile), with the line inside each box representing the median. Whiskers extend to the most extreme data points within 1.5 times the IQR from the quartiles. P values calculated using one-sided Student’s t-test: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001; ns, not significant. Exact P values are provided in the Source Data. Source data are provided as a Source Data file.
To evaluate label transfer, we assessed scGALA’s effect on Seurat, scGCN58, Conos13, and Monet59 using a 4-patient breast cancer dataset60 of 10689 cells, derived from Chromium Flex (snRNA-seq) FFPE samples61. The dataset was split unevenly (30% reference, 70% query), and synthetic batch effects were introduced to the query data with batch and noise strength both set to 0.3. When scGALA was applied, all tested methods showed marked improvements in transferring accurate cell type labels. For example, in Seurat, the Spearman correlation between transferred and true labels improved by 60.0% (Fig. 3c; and Supplementary Fig. S3c), with visible benefits for closely related cell types such as Natural T-regulatory Cells and Cytotoxic T Cells. More visual results are shown in Supplementary Fig. S5. Across all methods, scGALA led to an average increase of 6.1% in overall classification accuracy (e.g., 14.7% average improvement for Seurat, P = 6.01 × 10−6) and 19.2% in Cohen’s kappa (e.g., 36.2% average improvement for Conos, P = 2.66 × 10−15), with consistently higher 95% confidence intervals represented with the maximum value (AccuracyUpper, e.g., 4.0% average improvement for scGCN, P = 2.28 × 10−14) and minimum value (AccuracyLower, e.g., 4.0% average improvement for Monet, P = 2.68 × 10−11) in the interval (Fig. 3d; Supplementary Fig. S3d). These gains demonstrate that scGALA enhances the reliability of label transfer under strong batch and technical noise, enabling more robust cell type inference across experiments.
For multi-omics integration, we used the Mouse Cortex SNARE-seq dataset62 containing paired scRNA-seq and scATAC-seq data of 9190 cells. scGALA was integrated with scCross, GCN-SC63, Seurat, and Conos13. The resulting embeddings showed a more distinct clustering of cell types and improved agreement between modalities in the joint latent space (Fig. 3e; and Supplementary Fig. S3e, S6). These visual improvements were reflected in quantitative metrics, where the scGALA-enhanced methods achieved average increases of 19.3% in ARI (e.g., 32.5% average improvement for GCN-SC, P = 1.66 × 10−5) and 13.6% in NMI (e.g., 20.0% average improvement for Conos, P = 1.12 × 10−12). Furthermore, FOSCTTM curves confirmed an improved cross-modality correspondence, with scGALA-enhanced pipelines consistently producing closer embeddings (e.g., 12.4% average improvement for scCross, P = 7.41 × 10−4) for true matching cells (Fig. 3f; and Supplementary Fig. S3f). These results suggest that scGALA strengthens the integrative capacity of multi-omics methods, enabling more accurate reconstructions of regulatory landscapes and joint cellular states.
To assess spatial alignment, we combined scGALA with STAligner, INSCT, Seurat, and STADIA64 to align tissue sections from human dorsolateral prefrontal cortex samples profiled with 10x Visium65. The dataset included four slices (A-D) from three individuals (I-III), consisting of sequencing data on 47681 spots in total. We evaluated alignment between slices A and B of Sample I (Fig. 3g; and Supplementary Fig. S3g, S7). scGALA-enhanced methods produced better spatial alignment with improved preservation of anatomical boundaries and tissue structure. Quantitatively, scGALA yielded average improvements of 19.2% in spatial alignment accuracy52 (e.g., 9.3% average improvement for STADIA, P = 4.37 × 10−4) and 11.5% in NMI (e.g., 8.4% average improvement for STAligner, P = 1.21 × 10−4) based on integrated representations and spatial region annotations (Fig. 3h; and Supplementary Fig. S3h). Even in methods that do not explicitly model spatial information, such as Seurat, scGALA’s spatially informed alignments improved downstream spatial coherence (33.4% average improvement of spatial alignment accuracy, P = 2.54 × 10−10), demonstrating the added value of incorporating graph-based spatial relationships.
scGALA advances mosaic multi-omics integration
Contemporary multi-omics technologies typically generate paired measurements that capture two modalities from the same cell66,67, such as RNA+ATAC or RNA+protein abundance. While these “dual-omics” approaches have advanced our understanding of cellular states, they remain limited in their scope, capturing only partial views of the complex molecular landscape within cells68,69. A significant challenge in the field is mosaic integration70,71, where separate dual-omics datasets are integrated into comprehensive “tri-omics” or higher-order multimodal representations that provide more complete cellular characterization72,73. scGALA addresses this challenge by leveraging its enhanced cell alignment framework to integrate multiple dual-omics datasets through shared modalities, establishing a bridge for comprehensive mosaic data integration while preserving biological relevance across all measured features.
As illustrated in Fig. 4a, scGALA employs its enhanced cell alignment ability to integrate distinct dual-omics datasets that share a common modality, in this case, the RNA component from both RNA+ATAC and RNA+ADT measurements. The workflow begins by establishing cell alignments between datasets based on their shared RNA profiles, creating initial alignments through MNN. These initial alignments are then significantly enhanced through scGALA’s graph attention network, which leverages both local and global connectivity patterns to refine existing matches and discover additional biologically relevant alignments that conventional methods miss. By identifying these expanded cell alignments, scGALA effectively creates a bridge between modalities that were not simultaneously measured, enabling the construction of a virtual tri-omics dataset (RNA+ATAC+ADT) that provides a more comprehensive molecular characterization without requiring new experimental techniques. This integration capability represents a potential advancement, as it allows researchers to leverage existing datasets to gain insights that would otherwise require more complex and costly experimental designs74,75.

a Schematic workflow of scGALA integrating two dual-omics datasets (e.g., RNA+ATAC and RNA+ADT) into a unified tri-omics dataset via refined cell alignment. b Cell type-specific alignment precision: Confusion matrices comparing MNN (baseline), scGALA-enhanced (MNN + scGALA-exclusive), and scGALA-exclusive alignments. Diagonal enrichment highlights scGALA's improved accuracy in tri-omics integration. c Spearman’s correlation distribution of aligned cell pairs from MNN (baseline) versus scGALA-exclusive matches. Comparable distributions confirm scGALA maintains alignment quality while expanding coverage. d Composition of scGALA alignments (doughnut chart): scGALA identifies previously undiscovered cell pairs (exclusive to scGALA) while retaining high-confidence MNN matches, enabling comprehensive multi-omics unification. e ROC curves quantifying alignment accuracy for cell type matching, with AUROC values demonstrating scGALA's superior performance in cross-modality integration. f UMAP visualization of cross-dataset alignment, with connecting lines indicating alignments from MNN (baseline), scGALA-enhanced (MNN + scGALA-exclusive), and scGALA-exclusive alignments (blue: correct cell type matches; pink: mismatches). Source data are provided as a Source Data file.
We conducted the integration process between a CITE-seq dataset55 of 161764 bone marrow mononuclear cells56 and a PBMC Multiome dataset published by 10x Genomics57 of 10412 cells from healthy donors. To showcase the integration performance in aligning multiple datasets, we sampled subsets, identified by ‘PN’ in ‘orig.ident’ annotation that denotes different patients, from the CITE-seq dataset and integrated them with the Multiome dataset respectively. To evaluate the biological fidelity of these integrations, we examined the cell type-specific alignment precision achieved by scGALA compared to standard MNN approaches. The confusion matrices displayed in Fig. 4b, Supplementary Fig. S8a, S9a and S10a reveal a visible improvement in alignment accuracy when using scGALA’s enhanced framework. The quantitative comparison of alignment accuracy is provided in Fig. 4e with AUROC value and ROC curve, which will be discussed later. While standard MNN establishes some correct alignments between cells of the same type, the matrices show considerable off-diagonal elements indicating mismatched cell types. In contrast, scGALA’s confusion matrices exhibit stronger diagonal enrichment, which is further proven by the confusion matrices of scGALA Unique alignments, where the CD4 T cell subsets (CD4 Naive, CD4 TCM and CD4 TEM) are visibly benefited with more accurate alignments. The unique alignments result in average precisions of 0.723 (P7) and 0.737 (P8), which are considerably higher than the precisions of MNN alignments: 0.581 (P7) and 0.562 (P8). This pattern demonstrates that scGALA not only expands alignment coverage but does so while maintaining, and often improving, biological coherence. The improved alignment precision directly translates to more reliable integration of measurements across modalities, ensuring that molecular features from different omics layers are correctly associated with their corresponding cellular identities. This biological fidelity is essential for downstream analyses that seek to understand the regulatory relationships between different molecular features. To further validate the alignment accuracy through solid ground truth, we conducted experiments on the tri-omics NEAT-seq dataset76 that performed simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression of 11300 cells. We duplicated the gene expression data and added varying strength of Gaussian noise and dropout effect to mimic the technical variation between real experiments. The simulated and the original gene expression data form a pair of datasets to use as alignment input. Thus, the mosaic integration performance can be evaluated according to a solid one-to-one correspondence ground truth. The recall and AUROC results shown in the Supplementary Fig. S11 indicate consistently better performance of the scGALA alignments.
A critical consideration in expanding alignment coverage is whether the additional matches identified by scGALA maintain comparable quality to those found by MNN. Our analysis in Fig. 4c, Supplementary Fig. S8b, S9b and S10b demonstrates that scGALA Unique alignments, those not identified by MNN, maintain similar correlation distributions to the alignments shared with MNN. The comprehensive breakdown in Fig. 4d, Supplementary Fig. S8c, S9c and S10c further illustrates that scGALA discovers alignments that MNN fails to find while preserving quality, with approximately 20% of all alignments being exclusive to scGALA’s enhanced approach. This expansion in alignment coverage, without considerable sacrifice in quality, enables more comprehensive integration of features across modalities and reduces the sparsity issues that often plague multi-omics analyses77,78. The comparable quality of the additional matches identified by scGALA is also demonstrated in the experiments conducted on the MPAL CITE-seq and DAb-seq datasets of 72131 cells79, as shown in Supplementary Fig. S12.
To quantitatively validate the accuracy of scGALA’s cross-dataset alignments, we performed receiver operating characteristic (ROC) analysis based on cell type annotations. As shown in Fig. 4e, scGALA Unique alignments achieves higher area under the ROC curve (AUROC) values (0.774) compared to MNN (0.715). This enhanced alignment accuracy indicates more reliable feature integration across modalities. Further visual validation of alignment accuracy is provided in Fig. 4f, Supplementary Fig. S8d, S9d and S10d, where three-dimensional UMAP embedding of the integrated data reveals clear correspondence between matching cells across datasets. Cells connected by scGALA alignment links demonstrate appropriate cell type correspondence, with minimal instances of cross-type misalignments. These results demonstrate that scGALA reliably advances mosaic multi-omics integration by expanding alignment coverage while maintaining quality.
scGALA empowers cross-modality imputation and generation
A persistent challenge in single-cell multi-omics analysis stems from the reality that many datasets contain measurements from only one modality when comprehensive characterization would ideally require multiple80,81. This limitation often demands costly and technically challenging paired experiments to obtain complementary data types from the same cells or tissues74,75. To address this constraint, we implemented a GAT-based model within scGALA’s graph-based cell alignment framework to enable accurate cross-modality imputation, specifically focusing on generating RNA expression profiles from chromatin accessibility data in current experiments. This capability extends scGALA beyond an integration tool to a comprehensive multi-omics analysis platform that can significantly expand the utility of existing single-modality datasets without requiring additional experimental measurements.
Using the same PBMC Multiome dataset as in the last section, we generated RNA expression profiles from chromatin accessibility data and compared them with snRNA data from the same dataset. Our evaluation of scGALA’s cross-modality generation capabilities revealed considerable performance in preserving cell type-specific gene expression patterns. As demonstrated in Fig. 5a, the generated RNA profiles from ATAC-seq input data showed strong correlation of the mean expression levels with ground truth RNA-seq measurements across diverse cell types. Although deep learning stochasticity may increase the fraction of cells with low-level marker gene expression, the mean expression levels remain well preserved (Pearson correlation = 0.93, P < 1 × 10−300). This preservation of cell type-specific transcriptional signals is critical for downstream analyses that rely on marker gene expression, as it enables accurate cell type classification from generated data. The consistency across both abundant (CD14 Mono and CD4 Naive) and rare (cDC and pDC) cell populations indicates that scGALA effectively captures the regulatory relationship between chromatin accessibility and gene expression, regardless of cellular prevalence in the dataset.

a Cross-modal RNA generation accuracy: Dot plots compare scGALA-generated RNA (from input ATAC data) to ground truth RNA, quantified by Pearson’s correlation of cell type-specific signature gene expression. b Cell type clustering fidelity: UMAPs of scGALA-generated RNA (left) and ground truth RNA (right), colored by cell type labels. Adjusted Rand Index (ARI) quantifies clustering concordance with ground truth. c Functional relevance of generated RNA: Gene Ontology Biological Process (GOBP) enrichment analysis of signature genes. Heatmaps compare (\(-{\log }_{10}(\,{\mbox{FDR}})\)) values for scGALA-generated vs. ground truth RNA, with Pearson’s correlation confirming conserved biological processes. d Cell-cell interaction preservation: Heatmap of CellChat-derived interaction strengths (Secreted Signaling and Cell-Cell Contact) for scGALA-generated RNA vs. ground truth. Pearson’s correlation validates scGALA's ability to retain multi-omics signaling dynamics. P-values are obtained from two-sided Pearson correlation tests. Source data are provided as a Source Data file.
Beyond preserving individual gene expression patterns, scGALA-generated RNA profiles maintained the global structure of cell types and their relationships. The UMAP visualizations in Fig. 5b demonstrate remarkable similarity between clustering patterns of generated and ground truth RNA data, with comparable ARI values of 0.68 and 0.67 based on ground truth annotations. This is particularly evident in the accurate clustering of closely related cell types like CD4 T cell subsets. The high fidelity in global structure preservation demonstrates that scGALA not only generates accurate gene expression values but also preserves the complex multidimensional relationships between cells, enabling reliable cell type identification using standard RNA-seq analysis workflows even when only ATAC-seq data is available for input.
The biological relevance of scGALA-generated data extends beyond preserving cellular identities to maintaining functional signatures that reflect underlying biological processes. Gene Ontology Biological Process (GOBP) enrichment analysis82 revealed strong correlations between GOBP terms inferred from generated and ground truth RNA profiles (Fig. 5c). The correlation of GOBP term enrichment scores reached 0.87 across all major cell types (P = 9.82 × 10−17), with particularly high fidelity in terms related to immune reactions, such as “neutrophil activation involved in immune response” and “antigen receptor-mediated signaling pathway”. This preservation of biological pathway signatures demonstrates that scGALA captures not just expression levels of individual genes but the coordinated regulatory programs that define cellular function. This functional fidelity enables researchers to perform pathway-level analyses and draw biological insights from generated data with confidence that the results accurately reflect cellular processes in the original tissue.
Furthermore, scGALA-generated RNA profiles preserved complex intercellular communication patterns that depend on accurate expression levels of multiple interacting genes. Analysis of cell-cell interaction networks derived from generated versus ground truth RNA data revealed strong correlations in CellChat-derived interaction strengths based on cell secreted signaling and cell-cell contact interaction predictions (Pearson correlation coefficient = 0.94, P = 5.18 × 10−164), as visualized in Fig. 5d. The preservation of these interaction patterns was particularly evident in Plasmacytoid dendritic cells (pDCs) related interactions. This level of fidelity in capturing interaction networks demonstrates that scGALA-generated data can reliably support sophisticated multicellular analyses that depend on accurate representation of the complex molecular machinery governing cell-cell communication. Such capability is especially valuable for tissue-level studies where understanding cellular cooperation and signaling is essential for deciphering pathological mechanisms or developmental processes83.
scGALA improves the gene coverage of spatially resolved transcriptomics data
Spatially resolved transcriptomics technologies like Xenium have revolutionized our understanding of tissue architecture and cellular organization by preserving spatial context during transcriptomic profiling84,85. While these methods offer high-resolution spatial data, they typically capture only a few hundred genes compared to the thousands measured by conventional single-cell RNA sequencing approaches, limiting comprehensive spatial analysis of gene expression programs86,87. This technological constraint restricts the ability to investigate complex biological processes and regulatory networks within their native spatial contexts88. To address this limitation, we leveraged scGALA’s sophisticated alignment capabilities to develop a gene imputation approach that enhances gene coverage in spatial transcriptomics data by integrating information from reference RNA sequencing datasets. We used the Rodent Research-3 dataset45 as described in the integration pipeline evaluation section. This dataset includes single-nucleus RNA sequencing and spatial transcriptomics data from opposite hemispheres of mouse brains, showcasing significant biological similarity between the two distinct sequencing profiles. Based on this dataset, we systematically evaluated scGALA’s ability to generate biologically accurate expanded gene profiles while preserving spatial information. This approach enables researchers to maximize the utility of spatially resolved transcriptomics data without requiring additional experimental procedures or reagents, thereby extracting more comprehensive biological insights from existing datasets.
To rigorously assess imputation accuracy, we designed a controlled evaluation framework using spatial transcriptomics data from the CF7 samples identified with ‘bio origin’ annotation. Our simulation mimics the low-coverage constraints common in emerging spatial profiling technologies. For example, Xenium (10x Genomics) typically captures 250–800 genes per experiment89,90,91, Molecular Cartography (Resolve Biosciences) detects approximately 100 genes92,93, and MERFISH (Vizgen) panels typically target 250–800 genes depending on configurations94,95,96. Starting with a ground truth dataset containing 14,630 genes, we simulated sparse input data by selecting only 500 highly variable genes (HVGs) and then imputed additional 1,050 genes based on the HVGs from reference snRNA datasets using scGALA’s reference-guided approach. Additional experiments featuring varying numbers of HVGs sampled are shown in Supplementary Figs. S13, S14, S15, and S16 to assess the robustness of imputation to gene selection. This experimental design allowed us to directly compare the imputed expression profiles with the actual measured values, providing a quantitative assessment of imputation fidelity.
The UMAP visualizations shown in Fig. 6a of cell embeddings derived from the ground truth, simulated sparse data, scGALA-imputed data, and imputed-only data revealed that scGALA successfully recovered the underlying cell type structure. Quantitative evaluation using the Adjusted Rand Index (ARI) demonstrated concordance between cell clusters identified in the imputed data and those in the ground truth dataset. Even imputed-only genes resulted in a 0.49 ARI, which is quite close to the ground truth with a 0.52 ARI. This high degree of clustering agreement indicates that scGALA’s imputation process effectively preserves biologically meaningful cell type-specific expression patterns while expanding gene coverage.

a Imputation accuracy across datasets: UMAPs compare ground truth (14,630 genes), simulated sparse data (500 high-variance genes), scGALA-imputed data (1,545 genes; using alignment-guided graph reconstruction), and imputed-only genes (1,050 genes). Adjusted Rand Index (ARI) quantifies clustering concordance with ground truth cell types. b Functional fidelity of imputed data: Gene Ontology Biological Process (GOBP) enrichment analysis comparing scGALA-imputed dataset and simulated sparse dataset showing (\(-{\log }_{10}(\,{\mbox{FDR}})\)) values across cell types. The number before term name indicates cell types. Non-significant terms are labeled as “ns''. P-values calculated using one-sided Student’s (t)-test. c Cell type-specific marker gene recovery: Dot plots show Pearson’s correlation between imputed and ground truth expression for key marker genes, validating scGALA's precision in preserving biological signals. P-value is obtained from two-sided Pearson correlation test. d Spatial domain preservation: Spatial clustering using GraphST on ground truth and scGALA-imputed data (retaining original spatial coordinates). ARI quantifies agreement between scGALA-imputed spatial domains and ground truth annotations (clustering resolution optimized for ground truth labels). Source data are provided as a Source Data file.
To validate that imputed genes reflect genuine biological signals rather than computational artifacts, we examined cell type-specific marker gene recovery (Fig. 6c). Pearson correlation analysis between imputed and ground truth expression values for established marker genes yielded a Pearson correlation coefficient of 0.96 (P = 4.86 × 10−312). Key neuron-specific markers such as Slc17a6 (excitatory neuronal marker97), which is related to the transmission of nerve impulses and Pou3f1 (neural development marker98) which participates in the neural fate commitment, showed expression patterns closely mirroring their actual measured values, both in magnitude and cell type specificity.
Comprehensive GOBP enrichment analysis (Fig. 6b) demonstrated that scGALA-imputed data not only preserved existing functional signatures but enhanced pathway detection compared to using only the measured genes. The imputed dataset recovered four additional GOBP terms present in the ground truth but undetectable in the sparse data. For detected pathways like myelination99, the imputed data showed 14.3% stronger enrichment significance compared to using simulated sparse data (P = 0.036), indicating improved sensitivity for biological pathway analysis.
Crucially, scGALA preserved the spatial organization inherent in the original measurements. Spatial domain segmentation using GraphST100 (Fig. 6d) revealed that domains identified from scGALA-imputed data showed better correspondence with ground truth annotations (ARI = 0.51) compared to using only simulated sparse data (ARI = 0.47). The imputed-only genes maintained comparable spatial structure (ARI = 0.46), confirming that scGALA effectively reconstructs spatially-aware gene expression patterns even when the imputed genes are never spatially measured.
To consolidate the applicability of scGALA to spatial technologies with limited gene throughput, we applied scGALA to a real-world Xenium human breast cancer dataset101, which has a limited panel of 541 genes and is accompanied by ground truth spatial domain annotations.
First, we performed imputation directly on the original Xenium data to impute from 541 genes to 2268 genes. As shown in Supplementary Fig. S17a, the imputed-genes-only dataset (1895 genes) yields similarly defined cell clusters in the UMAP embedding compared to the original data. Although the comparison of UMAP is not directly quantified due to the lack of cell type annotations, when we performed spatial domain identification using GraphST, the imputed data resulted in a significantly higher ARI of 0.52 compared to the original data (ARI = 0.41) (Supplementary Fig. S17b). This demonstrates that the imputed genes add meaningful biological information that enhances the identification of spatial structures.
Second, to solve the issue of no ground truth to compare with, which prevents us from quantitative assessing the biological fidelity of the imputed genes, we conducted a controlled experiment similar to our original analysis in Fig. 6. We created a simulated sparse dataset by sampling 300 highly variable genes from the original 541-gene panel. We then used scGALA to impute the remaining genes. This allowed us to compare the imputed expression of marker genes to their actual measured values in the original data. As shown in the spatial plots in Supplementary Fig. S17c, the imputed data recovered the spatial organization with high fidelity (ARI = 0.49), comparable to the simulated sparse input (ARI = 0.44). Furthermore, the dot plot in Supplementary Fig. S17d shows a high Pearson correlation of 0.96 (P = 7.35 × 10−126) between the scGALA imputed data and ground truth data in the expression levels of key cell type marker genes, confirming that scGALA imputation accurately preserves biological signals.
Discussion
The growing diversity and complexity of single-cell datasets—ranging across modalities, platforms, and biological contexts—have introduced a persistent challenge in computational biology: how to robustly align and integrate heterogeneous data while preserving biological fidelity. Traditional approaches for cell alignment have largely relied on assumptions of proximity in low-dimensional expression space, often using linear projections or local neighborhood matching. These assumptions, however, can break down in the presence of substantial modality gaps, complex non-linear cell relationships, or batch-specific distortions. To overcome these limitations, we developed scGALA, a graph-based deep learning framework that reconceptualizes cell alignment as a graph link prediction problem. By integrating molecular features and auxiliary information into a unified graph attention network, scGALA enables the inference of robust, context-aware cell correspondences. This shift from geometric to relational reasoning provides a more principled and flexible foundation for aligning single-cell data across varying experimental and technological conditions.
scGALA’s core contribution lies in its universal enhancement of single-cell integration, both as a plug-in module and as a standalone tool that enables new analytical capabilities. At the heart of scGALA is a graph link prediction mechanism that significantly improves alignment accuracy, identifying approximately 25% more valid cell correspondences compared to state-of-the-art methods, without sacrificing biological coherence. Since cell alignment is a critical component underlying most single-cell analysis workflows, enhancing its accuracy and robustness has far-reaching consequences. We demonstrate that simply replacing conventional alignment modules with scGALA improves the performance of existing tools across diverse integration tasks such as batch correction, label transfer, multi-omics integration, and spatial alignment. In batch correction scenarios, scGALA’s ability to form coherent cross-batch correspondences leads to more effective mitigation of technical variation while preserving cellular heterogeneity, as evidenced by improved clustering and batch-mixing metrics. For label transfer tasks, scGALA leverages its graph structure to propagate annotations with higher accuracy, especially in challenging cases involving subtle cell type differences. In the context of multi-omics integration, scGALA successfully bridges modalities like RNA and ATAC, capturing nontrivial biological relationships that improve the coherence of joint embeddings. When applied to spatial transcriptomics, scGALA incorporates spatial coordinates as graph features, achieving more accurate registration of corresponding cells across tissue sections and conditions.
Beyond improving existing workflows, scGALA delivers advanced multi-omics functionalities and enables three distinct applications that address pressing limitations in current single-cell analysis. First, it introduces a mosaic integration strategy that bridges disjoint doublet datasets, such as RNA-ATAC and RNA-ADT, to computationally construct a tri-omics dataset (RNA-ATAC-ADT), thus bypassing the need for specialized triple-modal sequencing protocols while maintaining coherence across modalities. This approach maximizes the utility of available data resources while providing deeper insight into the molecular complexity of cellular systems than what would be possible from any single dataset alone. Second, scGALA demonstrates the capacity to impute one modality from another, most notably predicting transcriptomic profiles from chromatin accessibility data with high accuracy. This cross-modality generation extends from gene-level expression to pathway-level activity and even cell-cell communication profiles. By enabling researchers to computationally derive one data type from another with high biological fidelity, scGALA extends the utility of existing single-modality datasets and provides opportunities for integrative analysis across previously incompatible datasets. This capability is particularly valuable for archived samples, such as Formalin-Fixed Paraffin-Embedded (FFPE) tissue and biological specimens in biobanks, where multimodal profiling may not be feasible. Third, scGALA addresses the sparsity inherent in current spatial transcriptomics platforms by imputing gene expression beyond the limited gene panels typically measured. Using both spatial proximity and transcriptional similarity, scGALA reconstructs spatial transcriptomics profiles with high fidelity (Spearman correlation > 0.85 to ground truth), thus improving the interpretability of spatial data without requiring additional experiments. Notably, scGALA is designed to be modular and easily integrated into existing analysis pipelines, including Seurat and Conos, requiring minimal modification and thereby facilitating widespread adoption. The lightweight design (476K parameters) and plug-and-play modularity of scGALA support its use as a drop-in replacement for conventional cell alignment modules in existing workflows, enabling scalable application to large single-cell and spatial transcriptomics datasets.
Despite scGALA’s demonstrated effectiveness, a key limitation lies in its reliance on initial anchors derived from methods such as mutual nearest neighbors (MNN) or canonical correlation analysis (CCA). These anchors serve as the foundation for constructing the initial graph structure, and their quality can directly influence the accuracy of downstream cell alignment. In scenarios involving highly divergent datasets, modality gaps, or rare cell populations, the initial anchor set may be sparse or error-prone, potentially leading to suboptimal link predictions and reduced integration performance. The experiment results shown in Supplementary Fig. S18, where we imposed a range of dropout effects and added varying ratios of random errors into the initial MNN alignments, demonstrated that scGALA is robust to the quality of initial alignments. A promising direction to fully address this limitation is to move from hard and deterministic anchors to a probabilistic graph construction framework that assigns confidence scores to each potential alignment link. These confidence scores, derived from model uncertainty or learned similarity metrics, would enable scGALA to weight cell pairs according to their estimated reliability. Such a strategy would not only reduce the sensitivity to poor initial matches but also allow downstream analyses to explicitly incorporate alignment uncertainty. This refinement would make the model more robust in challenging settings and provide end-users with greater interpretability and control, particularly for integrating data across highly heterogeneous biological systems.
By providing a unified, graph-based solution to the challenges of single-cell data alignment and integration, scGALA enables researchers to maximize the value of increasingly complex and multimodal datasets. Its ability to enhance both foundational tasks and emerging applications positions it as a versatile component in the modern single-cell analysis toolkit. Importantly, scGALA empowers the research community to extract deeper biological insights from limited or incomplete data, facilitates the reuse of archival datasets, and reduces reliance on costly experimental procedures. As the field of single-cell genomics continues to evolve with ever-increasing scale and multimodality of datasets, tools like scGALA that support generalizable, scalable, and biologically grounded integration will be essential for advancing discoveries across developmental biology, immunology, cancer research, and regenerative medicine.
Methods
Data preprocessing
The input data for scGALA consists of expression profiles (e.g., gene count tables) and auxiliary information, such as spatial coordinates, when available. For gene count table preprocessing, we implement a comprehensive quality control and normalization pipeline through SCANPY’s established preprocessing functions102. Initial quality assessment begins with computing three fundamental cell-specific metrics: total count depth per cell, number of unique genes detected per cell, and percentage of counts derived from mitochondrial genes103. Cells are filtered using thresholds determined through the distinct separation between high-quality and low-quality cells in the violin plot of the quality metric distributions. For published datasets that are already preprocessed, we skip the quality control step and retain the cells and genes as provided in the preprocessed data.
Following quality control, we perform library size normalization, scaling each cell’s expression to 10,000 counts, and then apply log-transformation with a pseudocount of 1 to stabilize variance and approximate log-normal expression patterns104. To reduce computational complexity while preserving biological signals, we select the top 2000 highly variable genes (HVGs) using Scanpy with the batch key provided to identify HVGs within each batch separately before merging105. For cell alignment between scATAC datasets, which is required in cross-modality generation, we first generate gene activity scores from the scATAC-seq data to create a compatible representation with RNA expression. Using Signac’s GeneActivity function, we quantify scATAC-seq counts in the 2 kb-upstream region and the gene body of each gene to estimate transcriptional activity106. This estimation presents biological signals from the original scATAC data in a less noisy and computationally demanding way. These gene activity scores then undergo the same preprocessing steps as RNA data, including normalization, log-transformation, and HVG selection. For alignment tasks, the expression data is scaled after edge construction to facilitate deep learning model training. However, for specialized applications such as multi-omics generation and spatial gene imputation, the inputs remain unscaled to better preserve biological signals, and genes can be manually selected based on experimental requirements.
Graph construction
The graph construction process integrates multiple data types into a comprehensive cell-cell relationship network. In this network, the nodes represent the cells in each dataset and the edges indicate that linked cells are biologically similar or related. For the k-th pair of datasets, with expression profiles \({{{{\bf{X}}}}}_{1}^{k}\) and \({{{{\bf{X}}}}}_{2}^{k}\), from the total Np pairs of input datasets, we combine them into a unified node feature matrix \({{{{\bf{X}}}}}^{k}=\left[\begin{array}{c}{{{{\bf{X}}}}}_{1}^{k}\\ {{{{\bf{X}}}}}_{2}^{k}\end{array}\right]\in {{\mathbb{R}}}^{n\times d}\). Similarly, when spatial coordinates are available, we combine \({{{{\bf{S}}}}}_{1}^{k}\) and \({{{{\bf{S}}}}}_{2}^{k}\) into \({{{{\bf{S}}}}}^{k}=\left[\begin{array}{c}{{{{\bf{S}}}}}_{1}^{k}\\ {{{{\bf{S}}}}}_{2}^{k}\end{array}\right]\in {{\mathbb{R}}}^{n\times 2}\). The advantages of leveraging auxiliary spatial information are demonstrated in Supplementary Fig. S19a. The graph’s edge structure is constructed through multiple interconnected steps. First, we establish intra-dataset connections using KNN107 based on expression profiles, with KKNN defaulting to 20 as is commonly adopted in the methods used in our evaluations. This generates adjacency matrices \({{{{\bf{A}}}}}_{1,X,{{{\rm{KNN}}}}}^{k}\) and \({{{{\bf{A}}}}}_{2,X,{{{\rm{KNN}}}}}^{k}\) for datasets \({{{{\bf{X}}}}}_{1}^{k}\) and \({{{{\bf{X}}}}}_{2}^{k}\) respectively. When spatial coordinates are available, additional intra-dataset edges \({{{{\bf{A}}}}}_{1,S,{{{\rm{KNN}}}}}^{k}\) and \({{{{\bf{A}}}}}_{2,S,{{{\rm{KNN}}}}}^{k}\) are constructed using KNN on spatial distances.
For inter-dataset connections, we employ MNN22 to identify corresponding cells between datasets with KMNN defaulting to 20, as is mostly adopted in existing methods shown in evaluations that utilize MNN as their cell alignment modules. To ensure robust distance calculations, we first apply cosine normalization108 to the input data. The MNN process constructs an adjacency matrix \({{{{\bf{A}}}}}_{X,{{{\rm{MNN}}}}}^{k}\) by identifying pairs of cells from the k-th dataset pair that are mutually recognized as nearest neighbors in each other’s datasets. Specifically, for a cell in dataset \({{{{\bf{X}}}}}_{1}^{k}\), we find its nearest neighbors in dataset \({{{{\bf{X}}}}}_{2}^{k}\), and vice versa. A cell pair is included in the adjacency matrix if each cell is among the nearest neighbors of the other. These pairs form the basis for inter-dataset edges, with matrix entries set to 1 for MNN pairs and 0 otherwise.
The complete edge set’s construction demonstrates the flexibility in incorporating different types of cell alignments. While MNN serves as our default approach for establishing inter-dataset connections (AX,MNN), the framework readily accommodates alternative anchoring methods. For instance, when enhancing Seurat’s integration pipeline, we utilize CCA anchors18 to construct the inter-dataset edges AX,CCA, as these better align with Seurat’s underlying methodology. This adaptability in edge construction enables scGALA to leverage method-specific strengths while maintaining its graph-based enhancement capabilities.
The unified adjacency matrix combines all edge types through:
where \({{{{\bf{A}}}}}_{X,{{{\rm{anchor}}}}}^{k}\) represents the inter-dataset edges constructed by any suitable anchoring method (e.g., MNN or CCA anchors) between the k-th dataset pair from the total Np pairs of input datasets. \({\bigvee }_{i=1}^{2}\) represents the binary union process that combines the edges from different edge types into one adjacency matrix. \({\mathbb{I}}(\parallel {{{\bf{S}}}}{\parallel }_{{{{\rm{F}}}}} > 0)\) denotes an indicator function that evaluates to 1 when the Frobenius norm of the matrix S is positive, indicating that spatial information is available. This generalized formulation results in a graph \({{{\mathcal{G}}}}=({{{\bf{X}}}},{{{\bf{A}}}})\), where \({{{\bf{X}}}}={\left[\begin{array}{c}{{{{\bf{X}}}}}^{1},{{{{\bf{X}}}}}^{2},\cdots,{{{{\bf{X}}}}}^{{N}_{p}}\end{array}\right]}^{T}\in {{\mathbb{R}}}^{n\times d}\) represents the node features comprising combined expression profiles, and A ∈ {0, 1}n×n denotes the adjacency matrix constructed from various edge types.
This flexible graph construction approach enables scGALA to not only capture both local and global cell relationships but also smoothly integrate with the anchoring strategies of existing integration methods. The resulting graph structure provides a robust foundation for subsequent link prediction and cell alignment tasks, effectively leveraging both transcriptomic and spatial information when available, while maintaining compatibility with method-specific cell correspondence identification approaches.
scGALA architecture
The scGALA framework consists of two complementary components: a multi-scale GAT for link prediction and a score-based search algorithm for alignment refinement. We adopted the GAT model instead of GCN for its ability to obtain more alignments with comparable alignment quality, as shown in Supplementary Fig. S19b. The GAT component follows a Variational Graph Autoencoder (VGAE)109 structure to learn and predict cell-cell relationships across datasets, while the score-based search component iteratively optimizes these predictions to establish robust cell alignments.
The GAT architecture implements a sophisticated attention mechanism across Nh parallel heads, where each head h ∈ {1, 2, . . . , Nh} independently processes the input graph at a different scale, producing latent representations of varying dimensionality dh. This multi-scale design enables simultaneous capture of both fine-grained and coarse-grained cell-cell relationships39. Through a shared trainable attention mechanism, each head computes attention coefficients between connected nodes and aggregates neighboring features accordingly to form a comprehensive representation of each node:
where \({\Theta }_{h}\in {{\mathbb{R}}}^{{d}_{h}\times d}\) represents a learnable linear transformation specific to head h that projects the input features into a lower-dimensional space (d), and \({{{{\bf{a}}}}}_{h}\in {{\mathbb{R}}}^{2{d}_{h}}\) is the learnable attention vector that determines how to combine the features of connected nodes, and ∥ denotes vector concatenation. The LeakyReLU activation function, with a negative slope of 0.2, introduces non-linearity while preventing vanishing gradients for negative inputs110. The denominator performs softmax normalization over all neighbors \({{{\mathcal{N}}}}(i)\) of node i, ensuring that the attention coefficients sum to 1 for each node.
The resulting feature vector \({{{{\bf{w}}}}}_{i}^{(h)}\in {{\mathbb{R}}}^{{d}_{h}}\) encodes both the structural and feature-based information from the node’s local neighborhood. These attended features then undergo further transformation through head-specific Multi-Layer Perceptrons (MLPs)111 to produce the parameters of a variational distribution. The final latent representation Z is then constructed by transforming concatenated samples from Nh independent Gaussian distributions, each parameterized by its respective head’s output, into target latent dimension \({{{\bf{Z}}}}\in {{\mathbb{R}}}^{n\times {d}_{z}}\):
where \({\parallel}^{{\rm{N}}}_{{{\rm{h}}}=1}\) denotes the concatenation of the samples from Nh independent Gaussian distributions.
During training, the model reconstructs both the graph structure and node features using separate decoders. The edge reconstruction probability matrix \({{{{\bf{A}}}}}^{{\prime} }\in {[0,1]}^{n\times n}\) is obtained by computing the inner products of latent representations, followed by a sigmoid activation. Meanwhile, the node features are reconstructed using a single Linear layer:
Here, σ(⋅) is the sigmoid function that maps the inner products to values between 0 and 1, representing the probability of an edge existing between nodes. We use a simple Linear layer as the feature decoder to encourage the GAT encoder to better capture the input expression profiles.
The training process employs a comprehensive loss function that balances multiple objectives to achieve effective cell alignment while maintaining biological meaningfulness. The total loss comprises four main components:
The first term, \({{{{\mathcal{L}}}}}_{{{{\rm{recons}}}},X}\), represents the reconstruction loss for cell expression profiles, which serves as an auxiliary task to ensure the model learns meaningful cellular representations. The likelihood \({p}_{{\varphi }_{X}}({{{{\bf{X}}}}}_{i}| {{{\bf{Z}}}})\) is modeled as: \({p}_{{\varphi }_{X}}({{{{\bf{X}}}}}_{i}| {{{\bf{Z}}}})={\prod }_{i=1}^{{N}_{h}}\, {{{\mathcal{N}}}}({{{{\bf{x}}}}}_{i}| {{{{\boldsymbol{\mu }}}}}_{i},{{{\bf{I}}}})\), where μi = MLPdecoder(zi) is the reconstructed feature vector for cell i. For graphs with available spatial information, we replace the standard reconstruction loss with triplet loss112, represented as the second term \({{{{\mathcal{L}}}}}_{{{{\rm{triplet}}}}}\), which explicitly preserves spatial relationships. Given a set of spatial edges \({{{{\mathcal{E}}}}}_{S}=\{(i,j)\vert {({\bigvee }_{k=1}^{2}{{{{\bf{A}}}}}_{k,S,{{{\rm{KNN}}}}})}_{ij}=1\}\), for each edge \((i,j)\in {{{{\mathcal{E}}}}}_{S}\) (spatially close cell pair i,j), we randomly sample a negative cell k that is not spatially adjacent to cell i, which forms \({{{\mathcal{T}}}}=\{(i,j,k)| {{{{\bf{z}}}}}_{j}\in {{{{\mathcal{N}}}}}_{S}(i),{{{{\bf{z}}}}}_{k}\notin {{{{\mathcal{N}}}}}_{S}(i)\}\). The margin parameter α is set to 1, the default in PyTorch. This loss encourages the model to embed spatially adjacent cells closer together in the latent space while pushing non-adjacent cells apart, effectively preserving the local spatial structure in the learned representations.
The third term models the edge reconstruction probability \({p}_{{\varphi }_{A}}({{{{\bf{A}}}}}_{i}| {{{\bf{Z}}}})\) through inner products in the latent space: \({p}_{{\varphi }_{A}}({{{{\bf{A}}}}}_{i}| {{{\bf{Z}}}})={\prod }_{j=1}^{N}\mathop{\prod }_{k=1,k\ne j}^{N}p({{{{\bf{A}}}}}_{jk}| {{{{\bf{z}}}}}_{j},{{{{\bf{z}}}}}_{k})\). The KL divergence term \({{{{\mathcal{L}}}}}_{{{{\rm{KL}}}}}\) regularizes the latent space by encouraging the approximate posterior qθ(Z∣Xi, Ai) to match a standard normal prior p(Z). To enhance model robustness and prevent overfitting, the edges of input graph are randomly masked at the beginning of each training step. This self-supervised learning strategy forces the model to learn generalizable patterns rather than memorizing specific edge configurations and enables the model to predict unseen edges, facilitating the ability of link prediction. We selected a default masking ratio of 30%, as it provides a sufficient number of edges for both robust training and effective validation. To formally assess the impact of this hyperparameter, we conducted a sensitivity analysis by varying the masking ratio from 20% to 50%. The results demonstrate that scGALA’s performance is highly robust to this parameter (Supplementary Fig. S20). The Receiver Operating Characteristic (ROC) curves for each ratio are nearly overlapping, with Area Under the ROC Curve (AUROC) values showing minimal variation (from 0.755 to 0.767) (Supplementary Fig. S20a) with 30% yielding a marginally higher average AUROC of 0.767. This stability is further supported by the cell type-specific confusion matrices, which show consistently accurate alignments regardless of the masking ratio (Supplementary Fig. S20b).
In our framework, we apply a uniform random masking strategy to both intra- and inter-dataset edges. The rationale for also masking intra-dataset edges is to encourage the model to learn robust representations of cell identity and neighborhood structure, which improves the quality of cell embeddings and, in turn, enhances the model’s ability to predict correct inter-dataset alignments. To validate this uniform strategy, we performed an ablation study comparing it against two alternatives: masking only inter-dataset edges ("Inter-Only”) and masking only intra-dataset edges ("Intra-Only”) (Supplementary Fig. S21). The results show that our default Uniform masking (AUROC: 0.767) and the Inter-Only strategy (AUROC: 0.764) perform comparably and are both significantly superior to the Intra-Only strategy (AUROC: 0.735) (Supplementary Fig. S21ab). The slight advantage of the Uniform approach suggests that learning to reconstruct the intra-dataset graph provides valuable information that refines cell embeddings. This is achieved without sacrificing the quality of the newly identified alignments, as all three strategies produced similar Spearman correlation distributions (Supplementary Fig. S21c). These results support our choice of a uniform masking strategy as the default.
Then, the hyperparameters λ and β control the balance between reconstruction accuracy and latent space regularity. The model is trained using the Adam optimizer, with an initial learning rate of 1 × 10−3 and a cosine annealing schedule. Training continues until convergence, defined as no improvement in validation loss for 10 consecutive epochs.
The score-based search algorithm constitutes the second major component of scGALA, designed to refine the initial cell-cell relationships predicted by the GAT model. It preserves better biological signals than traditional threshold-based alignments, which determines the alignments through threshold-based filtering on the edge probabilities generated by graph model, as shown in Supplementary Fig. S19c. Building upon the classical Gale-Shapley algorithm for stable matching problems113, our approach introduces flexible matching capabilities that allow cells to form multiple alignment pairs while preventing over-alignment through a carefully designed quadratic penalty term. This modification enables the discovery of biologically meaningful many-to-many cell relationships while maintaining control over the total number of alignments per cell.
The optimization objective is formulated as a constrained maximization problem over the binary alignment matrix \({{{\bf{P}}}}\in {\{0,1\}}^{{n}_{1}\times {n}_{2}}\):
where Pij represents the binary alignment decision between cell i and cell j from different datasets, \({{{{\bf{A}}}}}_{ij}^{{\prime} }\) denotes the alignment probability predicted by the GAT model, and λ controls the strength of the over-alignment penalty. The first term encourages alignments between cells with high predicted correspondence, while the sparsity regularization term prevents excessive many-to-many matches through progressive penalization of additional alignments.
The greedy algorithm implements an iterative optimization strategy that alternates between datasets to ensure balanced consideration of both populations. For each iteration, the process consists of two phases: forward alignment from dataset \({{{{\bf{X}}}}}_{1}^{k}\) to dataset \({{{{\bf{X}}}}}_{2}^{k}\), and reverse alignment from dataset \({{{{\bf{X}}}}}_{2}^{k}\) to dataset \({{{{\bf{X}}}}}_{1}^{k}\). Starting from zero, it greedily adds or replaces existing alignments to refine the result. A visual overview of the preprocessing workflow is provided in Supplementary Fig. S22. During each phase, cells are processed based on their maximum alignment probabilities in \({{{{\bf{A}}}}}^{{\prime} }\). For cell i considering alignment with cell j, the marginal alignment score is computed as:
where \({\sum }_{j=1}^{{n}_{2}}{{{{\bf{P}}}}}_{i,j}\) and \({\sum }_{i=1}^{{n}_{1}}{{{{\bf{P}}}}}_{i,j}\) represent the current number of alignments for cells i and j respectively. An alignment is accepted if \({\Delta }_{ij}^{{{{\rm{add}}}}} > 0\). When \({\Delta }_{ij}^{{{{\rm{add}}}}}\le 0\), the algorithm attempts to improve the alignment by replacing an existing lower-confidence match. For a candidate replacement of cell l’s alignment with j, the replacement score is calculated as:
The replacement is executed if \({\Delta }_{ij,lj}^{{{{\rm{replace}}}}} > 0\), ensuring that each modification improves the overall objective score. This process continues alternating between datasets until either no changes occur for three consecutive iterations or a maximum number of iterations is reached. This limit is set to n1 ⋅ 0.02 and is capped between 50 and 100 iterations.
The quadratic form of the penalty term is specifically designed to impose progressively stronger constraints as the number of alignments increases. This non-linear penalty effectively prevents over-alignment by making each additional alignment increasingly costly, particularly for cells that already have multiple matches. The parameter λ is empirically set to balance between alignment flexibility and specificity, with larger values promoting more conservative alignment patterns.
After the score-based optimization converges, the final alignment matrix \(\hat{{{{\bf{A}}}}}\) is constructed by combining the optimized alignment plan P with the initial inter-dataset edges: \(\hat{{{{\bf{A}}}}}={{{\bf{P}}}}\vee {{{{\bf{A}}}}}_{X,{{{\rm{anchor}}}}}\). This choice to combine the initial high-confidence anchors with scGALA’s predictions was validated through an ablation study (Supplementary Fig. S23). We compared the performance of using: (1) only the initial anchors, (2) only the alignments uniquely predicted by scGALA, and (3) the combined set used in our final model. The results demonstrated that the combined approach yielded the most accurate and robust alignments. For instance, when enhancing Seurat, the combined strategy achieved an average label transfer accuracy of 0.8263, a significant improvement over using either initial anchors alone (0.7201) or only the newly predicted links (0.7866) (Supplementary Fig. S23f). Furthermore, the alignments discovered exclusively by scGALA constitute around 21% of the final pairings and exhibit a quality comparable to the initial anchors, as shown by their similar Spearman correlation distributions of gene expression (Supplementary Fig. S23a–e). This confirms that scGALA enriches the alignment map with high-quality cell pairs that are previously undiscovered, and the combined strategy produces the most comprehensive results. This integration ensures that high-confidence anchor pairs are preserved while allowing for additional biologically meaningful alignments discovered through the score-based optimization process. The resulting alignment matrix provides a comprehensive cell-cell correspondence map that captures both strong one-to-one matches and carefully controlled many-to-many relationships, enabling flexible yet reliable cell alignment across datasets.
All-in-one integration and harmonization pipeline
Having established the core graph-based alignment framework, we next describe how scGALA implements fundamental single-cell data integration tasks. The scGALA framework enhances existing single-cell integration methods by replacing their core cell alignment modules while preserving their established workflows. This enhancement strategy capitalizes on the robust graph-based cell alignments generated through our GAT architecture and score-based optimization, smoothly integrating these improved alignments into various downstream applications. By maintaining the fundamental structure of each method while upgrading their alignment capabilities, scGALA achieves superior performance across multiple integration tasks without requiring significant modifications to established pipelines.
For each integration method, the enhancement process primarily involves substituting the original cell correspondence identification mechanism with scGALA’s alignment output \(\hat{{{{\bf{A}}}}}\). In Seurat’s batch correction pipeline, we modify the CCAIntegration module by replacing the CCA-based anchors with scGALA-derived alignments, maintaining the subsequent correction steps that leverage these anchors for batch effect removal. Similarly, for label transfer applications, Seurat’s FindTransferAnchors function is adapted to utilize scGALA’s alignments instead of the default PCA-based anchor identification. The scCross multi-omics integration pipeline is enhanced by incorporating scGALA-generated alignments into its MNN prior generation step, while STAligner’s spatial alignment capabilities are improved by updating its MNN dictionary construction with scGALA’s refined cell alignments.
To demonstrate the versatility and effectiveness of this enhancement strategy, we developed a comprehensive evaluation framework utilizing the Rodent Research-3 dataset45 from the NASA Open Science Data Repository. This spatial multi-omics dataset combines single-cell RNA sequencing (21,178 cells), ATAC sequencing (21,178 cells), and spatially-resolved transcriptomics data (29,770 spots), all generated from mouse brain hemispheres using 10x Multiome and Visium protocols respectively. This unique dataset enables the assessment of all four primary integration applications—batch correction, label transfer, multi-omics integration, and spatial alignment—within a single cohesive experimental context. The evaluation framework implements rigorous quantitative metrics tailored to each application’s specific objectives while maintaining consistency in the assessment methodology across different integration tasks. Statistical significance evaluated using one-sided Student’s t-test is adopted to determine whether scGALA is capable of producing significant improvements51. As the evaluation processes of all-in-one pipeline and universal booster are the same, the details of the specific evaluation framework for each task will be elaborated on in the next section.
scGALA as universal booster
To demonstrate the broad applicability of scGALA’s enhancement capabilities as a universal booster for existing methods, we extended our evaluation to encompass multiple established methods within each application category, testing their performance on commonly used benchmark datasets specifically designed for each task. For batch correction, we evaluated Seurat (unsupervised), INSCT (supervised and unsupervised modes)44, scDML (unsupervised)11, Scanorama (unsupervised)10, iMAP (unsupervised)53, and STACAS (semi-supervised)20 using the Mouse Brain ATAC (Gene) dataset of 11,270 cells from the scIB benchmark suite49. The scGALA enhancement process involved replacing their respective cell alignment module with scGALA and adapting the output formats to fit their original workflows: modifying CCA-based anchor identification for Seurat, updating MNN dictionary construction for INSCT variants and Scanorama, adapting MNN pair calculations for scDML and iMAP. The detailed code modifications can be accessed in our public code repository.
For batch correction performance assessment, we employ a dual-metric approach that evaluates both biological conservation and technical variation removal. Biological conservation is quantified through three complementary metrics: Adjusted Rand Index (ARI)46, which measures the similarity between clustering results, and Normalized Mutual Information (NMI)47, which quantifies the shared information between two clustering results with values between 0 and 1, for cluster alignment accuracy based on the Leiden graph-clustering method114, and Average Silhouette Width (ASW)48, which evaluates how well-separated clusters are by measuring the average similarity of points within their assigned clusters compared to other clusters, for assessing the preservation of biological structure. The effectiveness of batch effect removal is measured using graph connectivity analysis49, which assesses integration quality by measuring the connections between cells from different batches in a nearest-neighbor graph, and batch-specific ASW score, which measures how well batches are mixed in each cell type and therefore affected by both batch mixing and cell type isolation in each cluster. Specifically, for cells i with batch label \({{{\mathcal{B}}}}\) of a cell type Cj, the batch-specific ASW score of that cell type is:
where \(a(i;{{{\mathcal{B}}}})\) is the average distance between cell k and all other cells in the same batch, \(b(k;{{{\mathcal{B}}}})\) is the minimum (over all the other batches) of the average distance between k and cells in that batch. All metrics in the Batch Correction evaluation are obtained using the scIB benchmark suite49 and scaled between 0 and 1, with 0 indicating suboptimal performance and 1 indicating optimal performance. The UMAP visualizations are plotted using Scanpy102 with the Leiden algorithm114 as the cell clustering backbone.
Label transfer evaluation expanded to include Seurat, scGCN58, Conos13, and Monet59, using a 4-patient breast cancer dataset60 of 10,689 cells derived from Chromium Flex (snRNA-seq) FFPE samples61. The scGALA enhancement process involved replacing inter-dataset graph generation components with scGALA for scGCN and Conos, modifying reference-query anchors for Seurat, and replacing MNN alignments for Monet.
To rigorously evaluate label transfer performance under realistic conditions, we implemented two key data preprocessing strategies: uneven cell type splitting and batch effect simulation. For the splitting procedure, we developed an adaptive sampling approach that generates reference and query datasets with intentionally imbalanced cell type distributions while maintaining a specified overall split ratio. This is achieved by assigning random split ratios (uniformly distributed between 0.1 and 0.9) to each cell type independently, followed by a global adjustment step that ensures the target overall ratio is preserved through selective redistribution of cells between sets. For batch effect simulation, we applied a two-component batch effect model to the query dataset. The model combines multiplicative gene-specific effects, drawn from a normal distribution centered at 1 with controlled variance, with additive random noise. The strength of both components can be adjusted through parameters controlling the variance of the multiplicative effects and the magnitude of the random noise, respectively. For a gene expression matrix X, the batch effect simulation can be expressed as: Xbatch = X ⊙ (1 + αϵb) + βϵn, where ⊙ represents element-wise multiplication, \({\epsilon }_{b} \sim {{{\mathcal{N}}}}(0,1)\) is the gene-specific batch effect, \({\epsilon }_{n} \sim {{{\mathcal{N}}}}(0,1)\) is random technical noise, and α, β are strength parameters for batch effect and noise respectively. The resulting modified expression profiles maintain biological signals while exhibiting realistic technical variations, creating challenging but representative conditions for evaluating label transfer methods. This preprocessing framework enables systematic assessment of method robustness under conditions where cell type representations are unbalanced and technical variations exist between reference and query datasets. The combination of uneven splitting and simulated batch effects provides a standardized yet challenging evaluation scenario that closely mirrors real-world integration challenges.
Then, the label transfer accuracy is evaluated through a multi-faceted statistical framework that combines overall accuracy measurements with detailed confidence analysis. Beyond simple accuracy calculations, we adopt Cohen’s Kappa coefficient, which measures inter-rater reliability while accounting for the possibility of agreement occurring by chance, and compute 95% confidence intervals, providing robust statistical bounds for the reliability of label transfer results. This approach enables precise quantification of improvement in annotation transfer capabilities while accounting for statistical uncertainty. All metrics are obtained using confusionMatrix function from the R package caret115, directly comparing ground truth labels and transferred labels.
In multi-omics integration assessment, we used the Mouse Cortex SNARE-seq dataset62 containing paired scRNA-seq and scATAC-seq data of 9190 cells, accessible through GEO accession GSE126074. We incorporated GCN-SC63 with modifications to its MNN pair calculation with scGALA enhanced alignments. With scCross, Seurat, and Conos enhanced by scGALA as stated previously, we also evaluated their integration ability tailored for multi-omics data.
Multi-omics integration evaluation employs an extended metric suite that builds upon the biological conservation measures used in batch correction assessment. In addition to ARI, NMI, and ASW calculations, we introduced domain-specific graph connectivity and ASW analysis using scIB by replacing the batch labels with domain labels in batch-specific graph connectivity and ASW analysis. Moreover, we implemented the Fraction of Samples Closer Than True Match (FOSCTTM) methodology50, which evaluates integration quality by measuring the proportion of cells from one modality that are closer to a cell’s true match in another modality, with lower values indicating better integration. The FOSCTTM curve analysis provides particular insight into the quality of cell matching over varying sample size.
Spatial alignment evaluation examined STAligner, INSCT, Seurat, and STADIA64 to align tissue sections from human dorsolateral prefrontal cortex samples profiled with 10x Visium65, accessed through the spatialLIBD Bioconductor package116. The dataset included four slices (A-D) from three individuals (I-III), totaling 47,681 spots. We evaluated alignment between slices A and B of Sample I. The scGALA enhancement process involved updating MNN dictionary construction for STAligner, augmenting the MNN adjacent matrix construction for STADIA, and the same modifications as described for INSCT and Seurat above.
Spatial alignment assessment introduces unique metrics that account for both molecular and spatial aspects of integration quality. While retaining the core biological conservation metrics, we implement specialized spatial integration measures including slice-specific graph connectivity and ASW analysis using scIB by replacing the batch labels with slice labels in batch-specific graph connectivity and ASW analysis. For cross-slice alignment accuracy, we employed the Spatial Match function from scSLAT117, which uses embedding to match cells from different slices according to cosine similarity and then calculate the accuracy based on matching cell type labels. This comprehensive evaluation framework enables quantification of both transcriptional and spatial correspondence accuracy, providing a complete picture of integration performance in the spatial context.
This comprehensive evaluation framework demonstrates scGALA’s capability to enhance various integration methods across diverse experimental contexts. By maintaining consistent evaluation metrics while testing multiple methods on commonly used benchmark datasets, we establish the broad applicability and robust performance improvements achieved through scGALA enhancement. The framework provides strong evidence for scGALA’s utility as a universal booster for single-cell integration methods, capable of improving performance across diverse applications while maintaining the core functionality and unique advantages of each original method.
Advanced multi-omics functionalities
Beyond fundamental integration tasks, scGALA implements advanced multi-omics functionalities that leverage scGALA’s unique graph-based alignment architecture to solve problems that remain challenging or impossible with conventional approaches. These advanced capabilities extend beyond traditional integration tasks to enable unseen biological insights from complex and heterogeneous datasets spanning multiple modalities and technologies. These functionalities include mosaic integration and cross-modality imputation, which enhance data alignment and infer unmeasured profiles, and spatial transcriptomics enhancement that improves resolution by imputing missing gene expression in spatial datasets.
Mosaic Integration and Cross-modality Imputation: Single-cell multimodal analysis presents unique challenges in data integration, requiring sophisticated approaches to align and analyze data across different molecular modalities. The scGALA framework addresses these challenges through two distinct but complementary capabilities built upon the graph-based framework established in previous sections while introducing specialized components for multimodal analysis: mosaic integration for combining datasets with overlapping modalities, and cross-modal generation for imputing missing modalities.
For mosaic integration, scGALA focuses on unifying dual-omics datasets (e.g., paired scRNA+scATAC and scRNA+scADT measurements) that share a common modality into a comprehensive tri-omics dataset by leveraging shared modalities as alignment bridges. The process begins with careful preprocessing of each modality following the established protocol. The shared RNA modality serves as the primary basis for alignment. The graph construction for mosaic integration follows the basic framework described earlier. Given dual-omics dataset pairs with shared RNA modality, we construct the input graph data as \({{{{\mathcal{G}}}}}_{{{{\rm{Multi}}}}}=({{{\bf{X}}}},{{{\bf{A}}}})\), where we use the RNA expression profiles to construct the intra-dataset and inter-dataset edges. The edge structure incorporates both intra-dataset relationships through KNN and inter-dataset connections through the chosen anchoring method (e.g., MNN or CCA). The input graph then undergoes the GAT-based link prediction and score-based search optimization as previously described. The resulting enhanced alignments from scGALA enable the construction of a unified tri-omics dataset by establishing robust cell-cell alignments that preserve the biological relationships across all three modalities.
For mosaic integration evaluation, we used the CITE-seq dataset55 of 161,764 bone marrow mononuclear cells56 (accessible through GEO accession GSE128639) and the PBMC Multiome dataset of 10,412 cells published by 10x Genomics57. To showcase integration performance across multiple datasets, we sampled subsets (identified by ‘PN’ in ‘orig.ident’ annotation denoting different patients) from the CITE-seq dataset and integrated them with the Multiome dataset respectively.
The integration process begins by establishing cell alignments between datasets based on their shared RNA profiles using MNN as initial anchors, followed by scGALA’s graph attention network enhancement. To evaluate biological fidelity, we examined cell type-specific alignment precision through confusion matrices comparing three alignment strategies: MNN baseline (ARNA,MNN), scGALA-enhanced (\(\hat{{{{\bf{A}}}}}\)), and scGALA-exclusive (P, exclusive alignments identified by scGALA). Quality control was assessed through Spearman’s correlation distribution analysis of aligned cell pairs with SciPy118 and composition analysis via doughnut chart visualization. Alignment accuracy was validated using ROC analysis with scikit-learn119 based on cell type annotations, with AUROC values quantifying cell type matching performance. Visual validation was provided through three-dimensional UMAP embedding using SLAT117, with alignment links indicating correspondence quality.
To validate alignment accuracy with solid ground truth, we conducted experiments on the NEAT-seq dataset76 (accessible through GEO accession GSE178707) containing simultaneous profiling of proteins, chromatin accessibility, and gene expression in 11,300 cells. We duplicated gene expression data and added varying Gaussian noise and dropout effects to simulate technical variation, enabling evaluation against one-to-one correspondence ground truth. Additional validation with the same experiment process was performed using MPAL CITE-seq and DAb-seq datasets79 of 72,131 cells (GEO accession GSE232074).
The cross-modal generation capability of scGALA extends its graph-based framework to address a critical challenge in single-cell multi-omics analysis: generating RNA expression profiles for cells where only ATAC-seq measurements are available. This functionality enables comprehensive molecular characterization of cells by leveraging information from a reference multi-omics dataset (containing paired scRNA-seq and scATAC-seq measurements) to predict RNA profiles for query cells with only ATAC-seq data. The approach implements a two-stage framework that first establishes robust cell-cell alignments through graph-based alignment, then utilizes these alignments to guide RNA profile generation through a specialized Graph Attention Network architecture.
In the Graph Data Generation stage, we begin by preprocessing both the reference multi-omics dataset \({{{{\bf{D}}}}}_{{{{\rm{ref}}}}}=\{{{{{\bf{X}}}}}_{{{{\rm{RNA}}}}}^{{{{\rm{ref}}}}},{{{{\bf{X}}}}}_{{{{\rm{ATAC}}}}}^{{{{\rm{ref}}}}}\}\) and the query ATAC dataset \({{{{\bf{X}}}}}_{{{{\rm{ATAC}}}}}^{{{{\rm{query}}}}}\) following our established protocol. A critical step involves generating gene activity scores from both ATAC datasets to create a representation compatible with RNA expression patterns. Using the Signac package’s GeneActivity function, we compute activity scores \({{{{\bf{X}}}}}_{{{{\rm{GAS}}}}}^{{{{\rm{ref}}}}}\) and \({{{{\bf{X}}}}}_{{{{\rm{GAS}}}}}^{{{{\rm{query}}}}}\) by quantifying ATAC-seq counts in the 2kb-upstream region and gene body of each gene. These activity scores undergo the same preprocessing steps as RNA data, including library size normalization to 10,000 counts, log-transformation with a pseudocount of 1, and selection of highly variable genes. Subsequently, the gene activity scores from both datasets are merged into a unified feature matrix: \({{{{\bf{X}}}}}_{{{{\rm{GAS}}}}}=\left[\begin{array}{c}{{{{\bf{X}}}}}_{{{{\rm{GAS}}}}}^{{{{\rm{ref}}}}}\\ {{{{\bf{X}}}}}_{{{{\rm{GAS}}}}}^{{{{\rm{query}}}}}\end{array}\right]\). This combined representation serves as input to scGALA’s cell alignment framework, which identifies corresponding cells between the reference and query datasets. The resulting alignment matrix \({\hat{{{{\bf{A}}}}}}_{{{{\rm{ATAC}}}}}\) captures both one-to-one and carefully controlled many-to-many relationships between cells based on their chromatin accessibility patterns.
Using these alignments, we construct a new graph data structure specifically designed for cross-modal generation as \({{{{\mathcal{G}}}}}_{{{{\rm{gen}}}}}=({{{{\bf{X}}}}}_{{{{\rm{gen}}}}},{{{{\bf{A}}}}}_{{{{\rm{gen}}}}})\), where \({{{{\bf{X}}}}}_{{{{\rm{gen}}}}}=\left[\begin{array}{c}{{{{\bf{X}}}}}_{{{{\rm{RNA}}}}}^{{{{\rm{ref}}}}}\\ {{{{\bf{X}}}}}_{{{{\rm{GAS}}}}}^{{{{\rm{query}}}}}\end{array}\right]\) is the node feature matrix in \({{\mathbb{R}}}^{n\times d}\) and \({{{{\bf{A}}}}}_{{{{\rm{gen}}}}}=\left({\bigvee }_{i=1}^{2}{{{{\bf{A}}}}}_{i,{{{\rm{KNN}}}}}\right)\vee {\hat{{{{\bf{A}}}}}}_{{{{\rm{ATAC}}}}}\) represents the adjacency matrix. Here, Ai,KNN represents intra-dataset edges constructed using K-nearest neighbors within each dataset, and \({\hat{{{{\bf{A}}}}}}_{{{{\rm{ATAC}}}}}\) provides the inter-dataset connections derived from scGALA’s alignment process. The node features combine reference RNA expression and query gene activity scores, and the adjacency matrix incorporates both intra-dataset and inter-dataset edges.
The GAT-based Cross-modal Generation stage implements a specialized architecture optimized for translating chromatin accessibility patterns into gene expression profiles. The model consists of three GAT layers (N = 3) with increasing complexity:
with each GAT layer, as mentioned in scGALA Architecture section, implementing multi-head attention that encodes both the structural and feature-based information from the node’s local neighborhood to facilitate effective information propagation across the graph.
The model is trained to minimize a weighted mean squared error loss:
where SNN(i, j) is the shared neighbor overlap between anchor i in the reference dataset and anchor j in the query dataset, computed based on the kscore nearest neighbors. Q0.01(SNN) and Q0.90(SNN) are the 0.01 and 0.90 quantiles of the raw SNN scores, respectively. The weight for each anchor pair is capped between 0 and 1, and we use the default kscore value as in Seurat.
The training process employs the Adam optimizer with an initial learning rate of 1 × 10−3 and implements a step-based learning rate scheduler (step size = 500, gamma = 0.5) which decays the learning rate of each parameter group by gamma every step size epochs for stable convergence. Early stopping monitors the training loss with a patience of 10 epochs to prevent overfitting. During inference, the trained model processes the query cells’ gene activity scores through the graph attention layers to generate corresponding RNA expression profiles.
For cross-modality imputation evaluation, we used the PBMC Multiome dataset57 to generate RNA expression profiles from chromatin accessibility data. The framework implements a two-stage approach as introduced above: first establishing robust cell-cell alignments through graph-based alignment, then utilizing a specialized GAT architecture for RNA profile generation.
The evaluation framework for cross-modal generation encompasses multiple complementary metrics designed to assess both technical accuracy and biological relevance of the generated profiles. Cross-modal generation accuracy is quantified through Pearson’s correlation analysis of expression patterns of cell type-specific signature genes obtained by Scanpy102 between generated and ground truth RNA profiles. Cell type clustering fidelity is evaluated using the Adjusted Rand Index (ARI) to compare clustering results between generated and ground truth data in UMAP space constructed with Scanpy. The functional relevance of generated profiles is assessed through Gene Ontology Biological Process (GOBP) enrichment analysis of signature genes82 using pathway enrichment function from OmicVerse120, with the correlation of \(-{\log }_{10}(\,{\mbox{FDR}})\) values between generated and ground truth RNA confirming the preservation of biological processes. Finally, cell-cell interaction dynamics are validated using CellChat analysis121, comparing interaction strengths for both Secreted Signaling and Cell-Cell Contact pathways between generated and ground truth profiles. This comprehensive evaluation framework ensures that the generated RNA profiles not only accurately reflect gene expression patterns but also preserve essential biological characteristics and cellular interaction dynamics.
Spatial Transcriptomics Enhancement: Spatially resolved transcriptomics technologies, while revolutionary, often provides limited gene coverage compared to traditional single-cell RNA sequencing methods. For instance, current spatial technologies like Xenium typically sequence only 300-500 genes61,89,90, creating a significant gap in transcriptome-wide analysis capabilities. To address this limitation, we developed a specialized module within scGALA that leverages reference scRNA-seq data to impute genome-wide expression profiles for spatial transcriptomics data. This imputation framework integrates our graph-based cell alignment approach with an efficient implementation of ClusterGCN122, enabling comprehensive transcriptome reconstruction while maintaining computational feasibility for large-scale spatial datasets.
The imputation process follows a two-stage framework that first establishes robust cell alignments between spatial and reference data, then leverages these relationships for feature expansion. In the initial graph data generation stage, we first perform scGALA-enhanced cell alignment between reference scRNA data XRef and the spatial transcriptomics data XST. The node features of the input graph consist of common genes between spatial and reference datasets: \({{{{\bf{X}}}}}_{{{{\rm{Align}}}}}=\left[\begin{array}{c}{{{{\bf{X}}}}}_{{{{\rm{Ref}}}}}[:{n}_{s}]\\ {{{{\bf{X}}}}}_{{{{\rm{ST}}}}}[:{n}_{s}]\end{array}\right]\), where ns denotes the number of genes measured in the spatial data that also exist in the reference scRNA data. This graph undergoes scGALA’s cell alignment framework, which identifies corresponding cells between the reference scRNA data and spatial transcriptomics data. The resulting alignments \({\hat{{{{\bf{A}}}}}}_{{{{\rm{ST}}}}}\) are utilized as the inter-dataset edges for the new graph data constructed specifically for imputation:
where node features Ximp now encompass the full gene set, with reference scRNA nodes containing complete transcriptome profiles and spatial nodes containing measured genes padded with zeros. For reference data nodes r ∈ XRef, features are reordered as xr = [xr,common; xr,unique], where xr,common represents genes shared with spatial data. For spatial nodes s ∈ XST, features are structured as xs = [xs,common; 0], where zero padding matches the dimension of unique reference genes. This structured arrangement enables direct computation on the common feature space while maintaining the complete gene set for imputation. The edge set Aimp incorporates both KNN-based intra-dataset connections and inter-dataset edges derived from the scGALA cell alignment results, forming a comprehensive graph that enables information flow from reference to spatial nodes during imputation.
The core imputation model employs ClusterGCN122, which processes dense subgraphs identified through graph clustering to reduce memory requirements while maintaining accuracy:
here D is the corresponding degree matrix and W(l) is the learnable weight matrix. ClusterGCN operates by sampling blocks of nodes that form dense subgraphs and restricts neighborhood search within these subgraphs, significantly improving computational efficiency.
The network consists of two ClusterGCN layers with the following ReLU activation functions and Dropout function with a probability of 50%, processing the graph data in manageable chunks while preserving the essential relationship information for accurate imputation. The training process employs an adaptive optimization strategy with the Adam optimizer (initial learning rate = 1 × 10−3) and a StepLR scheduler (step size = 500, gamma = 0.5) to ensure stable convergence. We implement an early stopping mechanism with increased patience (30 epochs) and minimum delta (1e-4) compared to our standard cell alignment training, accounting for the more gradual convergence patterns observed in imputation tasks.
The loss function combines mean squared error (MSE) terms for both RNA and spatial domains:
where \({{{{\mathcal{V}}}}}_{R}\) and \({{{{\mathcal{V}}}}}_{S}\) represent the sets of RNA and spatial nodes respectively, and \(\hat{{{{\bf{x}}}}}\) represents the reconstructed expression values. This formulation ensures that the model learns to accurately reconstruct the full transcriptome for RNA nodes while maintaining fidelity to the measured genes in spatial nodes.
After the training convergence, imputation is performed through a forward pass of the spatial nodes through the trained network. The output provides genome-wide expression profiles for each spatial location, with the measured genes serving as an internal validation metric for imputation accuracy. This approach effectively leverages the rich information available in scRNA-seq reference data while preserving the crucial spatial context of the original measurements, enabling comprehensive transcriptome-wide analysis of spatial gene expression patterns.
To evaluate imputation quality, we implemented a multi-faceted validation framework examining both global and local accuracy metrics using the Rodent Research-3 dataset that contains both spatial transcriptomics and reference snRNA-seq data. Starting with ground truth data (14,630 genes), we simulated sparse input by selecting 500 HVGs, then imputed additional 1,050 genes using scGALA’s reference-guided approach. Additional experiments with varying HVG numbers (300, 350, 400, 450) assessed the robustness to gene selection.
We compared clustering results and transcriptome structure preservation through UMAP visualization (Scanpy) and Adjusted Rand Index (ARI) calculations (scIB) across different data representations: ground truth, simulated sparse data (sampled high-variance genes), scGALA-imputed data (using alignment-guided graph reconstruction), and imputed-only genes. Biological relevance was assessed through GOBP enrichment analysis using pathway enrichment function from OmicVerse120 comparing cell type-specific differential expression patterns between ground truth and imputed data, with GOBP term significance correlations (− Log10(FDR)) quantifying functional preservation. Additionally, we validated spatial domain preservation using the GraphST algorithm and examined cell type-specific marker gene expression patterns through dot plot visualization with Pearson correlation assessments.
Furthermore, we applied scGALA to a Xenium human breast cancer dataset101 (541 genes, accessible through SubcellularSpatialData R/Bioconductor package with dataset ID: EH8567) to demonstrate practical applicability on real spatial transcriptomics data with limited gene panels. Controlled experiments involved sampling 300 HVGs from the original panel to enable quantitative comparison against measured values, confirming high biological fidelity with Pearson correlation and improved spatial domain identification as introduced above.
This comprehensive evaluation framework demonstrated that our imputation approach effectively leverages the rich information available in scRNA-seq reference data while preserving both crucial spatial context and cell type-specific expression patterns, enabling reliable transcriptome-wide analysis of spatial gene expression.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets used in the evaluation of scGALA are all publicly available. The processed data of Rodent Research-3 dataset45 is accessible via NASA Open Science Data Repository under study ID OSD-352. The preprocessed data of Mouse Brain ATAC (Gene) benchmark dataset from scIB49 is available as preprocessed AnnData objects on Figshare. The processed data of breast cancer dataset60 is available at cellxgene database123. The processed data of Mouse Cortex dataset profiled with SNARE-seq62 is available at the Gene Expression Omnibus (GEO) database under accession number GSE126074. The processed data of 10x Genomics Visium ST data from human dorsolateral prefrontal cortex tissues65 is available from the Bioconductor package spatialLIBD116. The processed expression matrices of CITE-seq dataset on bone marrow mononuclear cells56 is available through GEO database under accession number GSE128639. The PBMC Multiome dataset published by 10x Genomics57 can be accessed in 10x Genomics open database. The MPAL CITE-seq and DAb-seq datasets79 can be accessed through GEO database under accession number GSE232074. The NEAT-seq dataset76 can be accessed through GEO database under accession number GSE178707. The Xenium human breast cancer dataset101 is accessible through the SubcellularSpatialData R/Bioconductor data package101 with identifier EH8567. The example datasets associated with this study are available on FigShare at https://doi.org/10.6084/m9.figshare.28728617124. Source data are provided with this paper.
Code availability
The code used to develop the model, perform the analyses, and generate results in this study is publicly available and has been deposited in GitHub at https://github.com/mcgilldinglab/scGALA, under the MIT license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://doi.org/10.5281/zenodo.17409944125.
References
Cheow, L. F. et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat. Methods 13, 833–836 (2016).
Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017).
Schier, A. F. Single-cell biology: beyond the sum of its parts. Nat. Methods 17, 17–20 (2020).
Giladi, A. et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 38, 629–637 (2020).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Gut, G., Herrmann, M. D. & Pelkmans, L. Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018).
Fouché, A. & Zinovyev, A. Omics data integration in computational biology viewed through the prism of machine learning paradigms. Front. Bioinforma. 3, 1191961 (2023).
Chen, S. et al. Integration of spatial and single-cell data across modalities with weakly linked features. Nat. Biotechnol. 42, 1096–1106 (2024).
Muller, E., Shiryan, I. & Borenstein, E. Multi-omic integration of microbiome data for identifying disease-associated modules. Nat. Commun. 15, 2621 (2024).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Yu, X., Xu, X., Zhang, J. & Li, X. Batch alignment of single-cell transcriptomics data using deep metric learning. Nat. Commun. 14, 960 (2023).
Du, Z.-H. et al. scpml: pathway-based multi-view learning for cell type annotation from single-cell RNA-seq data. Commun. Biol. 6, 1268 (2023).
Barkas, N. et al. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat. Methods 16, 695–698 (2019).
Yang, X., Mann, K. K., Wu, H. & Ding, J. sccross: a deep generative model for unifying single-cell multi-omics with seamless integration, cross-modal generation, and in silico exploration. Genome Biol. 25, 198 (2024).
Wani, S. A. & Quadri, S. Evaluation of computational methods for single cell multi-omics integration. Procedia Computer Sci. 218, 2744–2754 (2023).
Biancalani, T. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021).
Zeira, R., Land, M., Strzalkowski, A. & Raphael, B. J. Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022).
Weenink, D. et al. Canonical correlation analysis. vol. 25 of Proceedings of the Institute of Phonetic Sciences of the University of Amsterdam, 81–99 (University of Amsterdam Amsterdam, 2003).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Andreatta, M. et al. Semi-supervised integration of single-cell transcriptomics data. Nat. Commun. 15, 872 (2024).
Manicka, S., Johnson, K., Levin, M. & Murrugarra, D. The nonlinearity of regulation in biological networks. NPJ Syst. Biol. Appl. 9, 10 (2023).
Haghverdi, L., Lun, A. T., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Zhu, B. et al. Robust single-cell matching and multimodal analysis using shared and distinct features. Nat. Methods 20, 304–315 (2023).
Zhu, J. et al. Mapping cellular interactions from spatially resolved transcriptomics data. Nat. Methods 21, 1830–1842 (2024).
Wan, X. et al. Integrating spatial and single-cell transcriptomics data using deep generative models with spatialscope. Nat. Commun. 14, 7848 (2023).
Gao, Z. et al. Hierarchical graph learning for protein–protein interaction. Nat. Commun. 14, 1093 (2023).
Jang, Y. J. et al. Accurate prediction of protein function using statistics-informed graph networks. Nat. Commun. 15, 6601 (2024).
Yi, H.-C., You, Z.-H., Huang, D.-S. & Kwoh, C. K. Graph representation learning in bioinformatics: trends, methods and applications. Brief. Bioinforma. 23, bbab340 (2022).
Hewett, M. et al. Pharmgkb: the pharmacogenetics knowledge base. Nucleic Acids Res. 30, 163–165 (2002).
Pavlopoulos, G. A. et al. Bipartite graphs in systems biology and medicine: a survey of methods and applications. GigaScience 7, giy014 (2018).
Wang, J. et al. scgnn is a novel graph neural network framework for single-cell RNA-seq analyses. Nat. Commun. 12, 1882 (2021).
Ben-Haim, T. & Raviv, T. R. Graph neural network for cell tracking in microscopy videos. European Conference on Computer Vision, 610–626 (Springer, 2022).
Tang, Z. et al. Sigra: single-cell spatial elucidation through an image-augmented graph transformer. Nat. Commun. 14, 5618 (2023).
Gravina, A., Lovisotto, G., Gallicchio, C., Bacciu, D. & Grohnfeldt, C. Long range propagation on continuous-time dynamic graphs. Forty-first International Conference on Machine Learning (2024).
Ghasemian, A., Hosseinmardi, H., Galstyan, A., Airoldi, E. M. & Clauset, A. Stacking models for nearly optimal link prediction in complex networks. Proc. Natl Acad. Sci. 117, 23393–23400 (2020).
Zhang, M. & Chen, Y. Link prediction based on graph neural networks. Advances in Neural Information Processing Systems 31 (2018).
Parisi, F., Caldarelli, G. & Squartini, T. Entropy-based approach to missing-links prediction. Appl. Netw. Sci. 3, 1–15 (2018).
Veličković, P. et al. Graph attention networks. International Conference on Learning Representations (2018).
Guo, Z., Wang, F., Yao, K., Liang, J. & Wang, Z. Multi-scale variational graph autoencoder for link prediction. Proceedings of the Fifteenth ACM International Conference on Web Search and Data Mining, 334–342 (Association for Computing Machinery, 2022).
Irving, R. W. & Manlove, D. F. The stable roommates problem with ties. J. Algorithms 43, 85–105 (2002).
Duckworth, W., Manlove, D. F. & Zito, M. On the approximability of the maximum induced matching problem. J. Discret. Algorithms 3, 79–91 (2005).
Ektefaie, Y., Dasoulas, G., Noori, A., Farhat, M. & Zitnik, M. Multimodal learning with graphs. Nat. Mach. Intell. 5, 340–350 (2023).
Chami, I., Abu-El-Haija, S., Perozzi, B., Ré, C. & Murphy, K. Machine learning on graphs: A model and comprehensive taxonomy. J. Mach. Learn. Res. 23, 1–64 (2022).
Simon, L. M., Wang, Y.-Y. & Zhao, Z. Integration of millions of transcriptomes using batch-aware triplet neural networks. Nat. Mach. Intell. 3, 705–715 (2021).
Masarapu, Y. et al. Spatially resolved multiomics on the neuronal effects induced by spaceflight in mice. Nat. Commun. 15, 4778 (2024).
Hubert, L. & Arabie, P. Comparing partitions. J. Classif. 2, 193–218 (1985).
Strehl, A. & Ghosh, J. Cluster ensembles—a knowledge reuse framework for combining multiple partitions. J. Mach. Learn. Res. 3, 583–617 (2002).
Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Demetci, P., Santorella, R., Sandstede, B., Noble, W. S. & Singh, R. Scot: single-cell multi-omics alignment with optimal transport. J. Computational Biol. 29, 3–18 (2022).
Mishra, P., Singh, U., Pandey, C. M., Mishra, P. & Pandey, G. Application of Student’s t-test, analysis of variance, and covariance. Ann. Card. Anaesth. 22, 407–411 (2019).
Zhou, X., Dong, K. & Zhang, S. Integrating spatial transcriptomics data across different conditions, technologies and developmental stages. Nat. Computational Sci. 3, 894–906 (2023).
Wang, D. et al. iMAP: Integration of multiple single-cell datasets by adversarial paired transfer networks. Genome Biol. 22, 63 (2021).
Mandric, I., Hill, B. L., Freund, M. K., Thompson, M. & Halperin, E. BATMAN: Fast and accurate integration of single-cell RNA-seq datasets via minimum-weight matching. iScience 23, 101185(2020).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
10x Genomics. 10x genomics PBMC from a healthy donor - granulocytes removed through cell sorting (10k). Single Cell Multiome ATAC + Gene Exp. Dataset by Cell Ranger ARC 1.0.0 (2020).
Song, Q., Su, J. & Zhang, W. scgcn is a graph convolutional networks algorithm for knowledge transfer in single cell omics. Nat. Commun. 12, 3826 (2021).
Wagner, F. Monet: An open-source Python package for analyzing and integrating scRNA-Seq data using PCA-based latent spaces. Preprint at bioRxiv: https://doi.org/10.1101/2020.06.08.140673 (2020).
Dong, Y. et al. Transcriptome analysis of archived tumor tissues by Visium, Geomx DSP, and Chromium methods reveals inter- and intra-patient heterogeneity. bioRxiv https://doi.org/10.1101/2024.11.01.621259 (2024).
Janesick, A. et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 14, 8353 (2023).
Chen, S., Lake, B. B. & Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 37, 1452–1457 (2019).
Gao, H. et al. A universal framework for single-cell multi-omics data integration with graph convolutional networks. Brief. Bioinforma. 24, bbad081 (2023).
Li, Y. & Zhang, S. Statistical batch-aware embedded integration, dimension reduction, and alignment for spatial transcriptomics. Bioinformatics 40, btae611 (2024).
Maynard, K. R. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 24, 425–436 (2021).
Baysoy, A., Bai, Z., Satija, R. & Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 24, 695–713 (2023).
Lee, J., Hyeon, D. Y. & Hwang, D. Single-cell multiomics: technologies and data analysis methods. Exp. Mol. Med. 52, 1428–1442 (2020).
Canzler, S. et al. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 94, 371–388 (2020).
Hayes, C. N., Nakahara, H., Ono, A., Tsuge, M. & Oka, S. From omics to multi-omics: A review of advantages and tradeoffs. Genes 15, 1551 (2024).
He, Z. et al. Mosaic integration and knowledge transfer of single-cell multimodal data with MIDAS. Nat. Biotechnol. 42, 1594–1605 (2024).
Ghazanfar, S., Guibentif, C. & Marioni, J. C. Stabilized mosaic single-cell data integration using unshared features. Nat. Biotechnol. 42, 284–292 (2024).
Mohr, A. E., Ortega-Santos, C. P., Whisner, C. M., Klein-Seetharaman, J. & Jasbi, P. Navigating challenges and opportunities in multi-omics integration for personalized healthcare. Biomedicines 12, 1496 (2024).
Ivanisevic, T. & Sewduth, R. N. Multi-omics integration for the design of novel therapies and the identification of novel biomarkers. Proteomes 11, 34 (2023).
Kashima, Y. et al. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 52, 1419–1427 (2020).
Hu, Y. et al. Single cell multi-omics technology: methodology and application. Front. Cell Developmental Biol. 6, 28 (2018).
Chen, A. F. et al. Neat-seq: simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression in single cells. Nat. Methods 19, 547–553 (2022).
Miao, Z., Humphreys, B. D., McMahon, A. P. & Kim, J. Multi-omics integration in the age of million single-cell data. Nat. Rev. Nephrol. 17, 710–724 (2021).
Feldner-Busztin, D. et al. Dealing with dimensionality: the application of machine learning to multi-omics data. Bioinformatics 39, btad021 (2023).
Peretz, C. A. et al. Multiomic single cell sequencing identifies stemlike nature of mixed phenotype acute leukemia. Nat. Commun. 15, 8191 (2024).
Vandereyken, K., Sifrim, A., Thienpont, B. & Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 24, 494–515 (2023).
Ogbeide, S., Giannese, F., Mincarelli, L. & Macaulay, I. C. Into the multiverse: advances in single-cell multiomic profiling. Trends Genet. 38, 831–843 (2022).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Jha, N. K. et al. Molecular mechanisms of developmental pathways in neurological disorders: a pharmacological and therapeutic review. Open Biol. 12, 210289 (2022).
Cooper, R. A., Thomas, E., Sozanska, A. M., Pescia, C. & Royston, D. J. Spatial transcriptomic approaches for characterising the bone marrow landscape: pitfalls and potential. Leukemia 1–5 (2024).
Takano, Y. et al. Spatially resolved gene expression profiling of tumor microenvironment reveals key steps of lung adenocarcinoma development. Nat. Commun. 15, 1–17 (2024).
Choe, K., Pak, U., Pang, Y., Hao, W. & Yang, X. Advances and challenges in spatial transcriptomics for developmental biology. Biomolecules 13, 156 (2023).
Lee, J., Yoo, M. & Choi, J. Recent advances in spatially resolved transcriptomics: challenges and opportunities. BMB Rep. 55, 113 (2022).
Chen, J., Wang, Y. & Ko, J. Single-cell and spatially resolved omics: advances and limitations. J. Pharm. Anal. 13, 833 (2023).
Liu, Q. et al. Single-cell, single-nucleus and xenium-based spatial transcriptomics analyses reveal inflammatory activation and altered cell interactions in the hippocampus in mice with temporal lobe epilepsy. Biomark. Res. 12, 103 (2024).
Umek, N. et al. In situ spatial transcriptomic analysis of human skeletal muscle using the Xenium platform. Cell and Tissue Research 1–12 (2025).
Marco Salas, S. et al. Optimizing xenium in situ data utility by quality assessment and best-practice analysis workflows. Nature Methods 1–11 (2025).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Hu, S. et al. Single-cell spatial transcriptomics reveals a dynamic control of metabolic zonation and liver regeneration by endothelial cell Wnt2 and Wnt9b. Cell Reports Medicine 3 (2022).
Liu, J. et al. Concordance of merfish spatial transcriptomics with bulk and single-cell RNA sequencing. Life Science Alliance 6 (2023).
Zhang, M. et al. Spatially resolved cell atlas of the mouse primary motor cortex by Merfish. Nature 598, 137–143 (2021).
Choi, J. et al. Spatial organization of the mouse retina at single cell resolution by merfish. Nat. Commun. 14, 4929 (2023).
Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018).
Pandey, S., Shekhar, K., Regev, A. & Schier, A. F. Comprehensive identification and spatial mapping of habenular neuronal types using single-cell RNA-seq. Curr. Biol. 28, 1052–1065 (2018).
Nave, K.-A. & Werner, H. B. Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev.Biol. 30, 503–533 (2014).
Long, Y. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with graphs. Nat. Commun. 14, 1155 (2023).
Bhuva, D. D. et al. Library size confounds biology in spatial transcriptomics data. Genome Biol. 25, 99 (2024).
Wolf, F. A., Angerer, P. & Theis, F. J. Scanpy: large-scale single-cell gene expression data analysis. Genome Biol. 19, 1–5 (2018).
McCarthy, D. J., Campbell, K. R., Lun, A. T. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Cole, M. B. et al. Performance assessment and selection of normalization procedures for single-cell RNA-seq. Cell Syst. 8, 315–328 (2019).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. & Satija, R. Single-cell chromatin state analysis with Signac. Nature Methods (2021).
Cover, T. & Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 13, 21–27 (1967).
Luo, C. et al. Cosine normalization: Using cosine similarity instead of dot product in neural networks. International conference on artificial neural networks, 382–391 (Springer, 2018).
Kipf, T. N. & Welling, M. Variational graph auto-encoders. NIPS Workshop on Bayesian Deep Learning (2016).
Xu, J., Li, Z., Du, B., Zhang, M. & Liu, J. Reluplex made more practical: Leaky ReLU. IEEE Symposium on Computers and Communications (ISCC), 1–7 (IEEE, 2020).
Murtagh, F. Multilayer perceptrons for classification and regression. Neurocomputing 2, 183–197 (1991).
Balntas, V., Riba, E., Ponsa, D. & Mikolajczyk, K. Learning local feature descriptors with triplets and shallow convolutional neural networks. vol.1 of Procedings of the British Machine Vision Conference, 3 (British Machine Vision Association, 2016).
Teo, C.-P., Sethuraman, J. & Tan, W.-P. Gale-shapley stable marriage problem revisited: Strategic issues and applications. Manag. Sci. 47, 1252–1267 (2001).
Traag, V. A., Waltman, L. & Van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 1–12 (2019).
Kuhn, M. Building predictive models in r using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Pardo, B. et al. spatiallibd: an r/bioconductor package to visualize spatially-resolved transcriptomics data. BMC Genomics 23, 434 (2022).
Xia, C.-R., Cao, Z.-J., Tu, X.-M. & Gao, G. Spatial-linked alignment tool (slat) for aligning heterogenous slices. Nat. Commun. 14, 7236 (2023).
Virtanen, P. et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 17, 261–272 (2020).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Zeng, Z. et al. Omicverse: a framework for bridging and deepening insights across bulk and single-cell sequencing. Nat. Commun. 15, 5983 (2024).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Chiang, W.-L. et al. Cluster-gcn: An efficient algorithm for training deep and large graph convolutional networks. Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining, 257–266 (2019).
Megill, C. et al. Cellxgene: a performant, scalable exploration platform for high dimensional sparse matrices. bioRxiv https://doi.org/10.1101/2021.04.05.438318 (2021).
Jiang, G. et al. scgala advances graph link prediction–based cell alignment for comprehensive data integration and harmonization https://doi.org/10.6084/m9.figshare.28728617 (2025).
Jiang, G. et al. scgala advances graph link prediction–based cell alignment for comprehensive data integration and harmonization https://doi.org/10.5281/zenodo.17409944 (2025).
Acknowledgements
This work is supported by grants from the Canadian Institutes of Health Research (CIHR) [PJT-180505 to J.D]; the Funds de recherche du Québec - Santé (FRQS) [295298 to J.D., 295299 to J.D., 366764 to J.D.]; the Natural Sciences and Engineering Research Council of Canada (NSERC) [RGPIN2022-04399 to J.D.]; and the Meakins-Christie Chair in Respiratory Research [to J.D.]. This research was enabled in part by support provided by Calcul Québec (calculquebec.ca) and the Digital Research Alliance of Canada (alliancecan.ca). The illustrations of scRNA-seq and scADT-seq present in Fig. 1b, 1c, and 4a are adapted from Servier Medical Art (https://smart.servier.com), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Author information
Authors and Affiliations
Contributions
J.D. guided the study, initiated the cooperation project that led to this work and planned the experiments. G.J. contributed to the design and implementation of the methodology and conducted the experiments. J.D. and G.J. participated in data collection and the analysis of computational experiments. I.C. proposed the research question and participated in the result analysis. All authors (J.D., G.J., K.S., G.F., D.W., I.C., H.W.) contributed to writing the manuscript. Each author has read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yuxuan Hu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, G., Song, K., Fonseca, G.J. et al. scGALA advances graph link prediction-based cell alignment for comprehensive data integration and harmonization. Nat Commun 16, 11656 (2025). https://doi.org/10.1038/s41467-025-66644-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66644-5
