
Abstract
Stress granules (SGs) are disease-relevant dynamic ribonucleoprotein condensates formed by liquid-liquid phase separation (LLPS) of proteins and mRNAs. Understanding their regulators and developing interventions are critical for therapeutic development. O-GlcNAc transferase (OGT) has been implicated in SG regulation, but functions beyond O-GlcNAcylation remain unclear. Here we uncover that, upon induced proximity, OGT suppresses LLPS of the SG marker G3BP1 and thereby SG assembly, independent of its catalytic activity. We repurpose OGT into an SG modulator by fusing its N-catalytic and intervening domains (NI) to induced-proximity modules. This inhibitory effect arises from targeted protein immobilization that rigidifies G3BP1 under prolonged stress. This tool recognizes G3BP1’s domain organization, thus generalizes to four additional proteins featuring similar architectures, suppressing condensate formation with mobility reduction. This modular, genetically encoded strategy enables SG regulation and functional dissection by interfering material properties of critical SG proteins and illuminates the cryptic non-catalytic function of OGT.
Similar content being viewed by others


Introduction
Stress granules (SGs) are among the most extensively studied membrane‑less cytosolic ribonucleoprotein (RNP) assemblies, driven by the collective interactions of a core network of proteins and RNAs1,2,3. When cells face stressors such as oxidative damage or heat, translation is inhibited and polysomes disassemble, leading to an accumulation of uncoated mRNA1,4,5,6. These mRNAs coalesce with RNA‑binding proteins into condensates, namely SGs, via liquid–liquid phase separation (LLPS)6,7. SGs often contain proteins implicated in pathological aggregation, such as FUS, TDP‑43, and other heterogeneous nuclear RNPs (hnRNPs)5,8,9. Growing genetic evidence links aberrant SG dynamics to the pathogenesis of neurodegenerative diseases5,8,9,10,11. SGs also contribute to cytoprotective responses during viral infection12,13,14 and chemo‑/radiotherapy15. As SGs emerge as promising therapeutic targets, understanding the regulatory principles that control their dynamics is essential for developing SG‑specific interventions.
During SG assembly, G3BP1 and its paralog G3BP2 function as molecular switches for LLPS, forming multivalent interactions with various binding partners1,16. Beyond perturbing upstream signaling that triggers SG formation17,18, directly tuning the LLPS properties of core SG components is critical to shaping granule composition and dynamics19. A hierarchy of interactions—oligomerization, RNA binding, and intrinsically disordered regions (IDRs)—governs whether SGs behave like liquids or solids19,20. As exemplified by G3BP1, it contains an N-terminal NTF2-like (NTF2L) domain responsible for dimerization and a C-terminal RNA-binding domain (RBD; comprising an RRM and IDR3), both of which are required for its LLPS and SG formation. Consequently, targeting the NTF2L domain by viral peptide mimetics15 or small molecules21,22, or RNA degradation by the RNase treatment13, directly block SG formation. The long central IDR of G3BP1 also modulates SG dynamics but remains a difficult target for selective manipulation1. Many other condensate‑forming proteins share the modular architecture of G3BP1/216. However, existing regulatory approaches for G3BP1/2—primarily designed to target the NTF2L domain—are not easily transferred to these proteins, leaving few methods to control their dynamics in cells. Moreover, despite the centrality of G3BP1/2, other proteins with positive or negative cooperativity also influence SG assembly, reflecting a dynamic, balanced regulatory network1,16,19.
Post-translational modifications (PTMs) with chemical diversity add another layer of SG regulation20,23,24. Notably, O-linked N-acetylglucosamine (O-GlcNAc)—a stress-responsive monosaccharide modification installed by O-GlcNAc transferase (OGT) and removed by O-GlcNAcase (OGA)—has long been implicated in SG biology25,26,27,28. Several key factors relating to O-GlcNAc modification, including OGT, were found essential for SG assembly, leading to the discovery that O-GlcNAc modifications of components of the translational machinery promote SG assembly25. However, although robust O-GlcNAc signals were commonly found in SGs, it is difficult to observe a detectable OGT signal within SG foci. This discrepancy led us to explore other possible functions of OGT on SGs beyond maintaining O-GlcNAc homeostasis.
In this study, facilitated by a chemical-induced proximity approach, we unexpectedly uncovered a non-catalytic function of OGT in negatively regulating the material properties of key SG proteins, particularly G3BP1, both in vitro and in cells. We next screened out the non-catalytic intervening domain (Int-D) as the primary domain for the suppression of G3BP1-induced SGs under multiple stressors. Accordingly, we designed a SG-targeted suppression method using a non-catalytic OGT truncation combined with a nanobody or chemical-induced proximity, which we name as STOP. We characterized the working pattern of STOP on SG suppression and revealed that STOP substantially reduced G3BP1’s dynamics by recognizing its domain architecture. STOP is also generalizable to other SG key nodes with similar domain features and enables similar condensation suppression. Our study developed STOP as a versatile tool to reshape the material properties of phase-separated condensates of SG components, allowing for cellular activity perturbation through SG intervention. Our work also provides insightful hints for the non-catalytic function of the previously cryptic Int-D in OGT.
Results
OGT suppresses G3BP1-centric SGs independent of its transferase activity
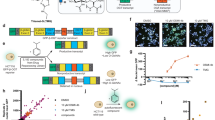
Given the reported low signal of endogenous OGT within SGs25, we wondered how SGs would be affected if we directly brought OGT into SGs using a chemical-induced proximity approach. Using the CRISPR-Cas9 technique29, we knocked in an FKBPF36V tag at the N-terminus of the OGT genomic locus in HeLa cells, allowing targeting endogenous OGT with an FKBPF36V small molecule ligand AP186730,31 (Supplementary Fig. 1). We overexpressed G3BP1 bearing a C-terminal Halotag (G3BP1-Halo) in this engineered HeLa cell line as the central node and marker protein for SG formation32, which was able to form liquid-like condensates in HeLa cells under oxidative stress induced by 500 µM sodium arsenite for 30 min (Fig. 1a). As expected, endogenous FKBPF36V-OGT was unable to enter G3BP1-Halo–centric SGs under the AP1867 treatment followed by arsenite (Fig. 1b). To achieve chemical-induced proximity between FKBPF36V-OGT and G3BP1-Halo, we resynthesized and applied a previously reported small chimeric molecule as a molecular glue—OGTAC-2—containing AP1867 and a haloalkane linking together30 (Fig. 1a and Supplementary Fig. 2a, b). Upon arsenite stress, nearly 60% of cells showed normal SG formation with colocalization of FKBPF36V-OGT and G3BP1-Halo, yet the remaining 40% of cells exhibited substantially reduced SGs, indicating that the direct recruitment of OGT to the SG marker protein, G3BP1, may suppress SG formation in HeLa cells under arsenite stress (Fig. 1c, d). However, due to the overexpression of G3BP1-Halo, endogenous OGT may not be enough to target all G3BP1-Halo proteins in most cells, leading to insufficient SG suppression. Since a previous study defined proteins affecting SG assembly by exhibiting either positive or negative cooperativity1, we speculate that OGT may be one of the factors exhibiting “negative cooperativity” for SG regulation when directly interacting with G3BP1.

a Schematic representation of the small molecule OGTAC-2–induced proximity between FKBPF36V-OGT and G3BP1-Halo under NaAsO2 stress. b–d Representative images (b, c) and quantification (d) of stress granule (SG) formation in FKBPF36V-knockin Hela cells expressing HA-tagged G3BP1-Halo with the treatments of 100 nM AP1867 (b) or OGTAC-2 (c) for 2 h and a follow-up NaAsO2 stress. Cells were fixed and stained with anti-HA (green) and anti-OGT (red) antibodies. Insets, regions of the indicated panels magnified by 10 times. Right and bottom, fluorescence intensity profiles of G3BP1-Halo and FKBPF36V-OGT along the yellow lines in the insets. HeLa cells were stressed with 500 µM NaAsO2 for 30 min unless otherwise noted. e, f Confocal imaging (e) and quantification (f) of SG formation in NaAsO2-treated HeLa cells overexpressing OGT or OGA variants with G3BP1-EGFP (green). OGT and OGA variants were stained with anti-His or anti-Myc antibodies (magenta). g Purified G3BP1-EGFP (25 μM) mixed with Cy5-labeled OGT at different concentrations (0, 1, 5, and 10 μM from left to right) in the presence of 100 ng/μL HeLa total RNA were analyzed for droplet formation at 150 mM NaCl. h Quantification of the droplet size in each group shown in (g), data are presented as mean ± s.e.m.; droplets in n = 3 fields in each group were quantified. In (d and f), data are presented as mean ± s.d. of n = 3 biologically independent sample groups with at least 30 transfected cells per group. P values were determined by unpaired two-tailed Student’s t-tests (d), Welch’s one-way ANOVA followed by Games–Howell multiple comparisons test (f), and Kruskal–Wallis test followed by Dunn’s multiple comparisons test (h). Scale bars, 20 μm (b, c, e) or 100 µm (g). Source data are provided as a Source Data file.
Since many identified components in SGs were reported to be O-GlcNAc-modfied substrates, we validated the effects of overexpressed O-GlcNAc cycling enzymes on EGFP-tagged G3BP1 (G3BP1-EGFP) in HeLa cells by monitoring SG assembly under NaAsO2 treatment. Surprisingly, both catalytically active (WT) and inactive (K852A) OGT significantly reduced the number of SG-containing HeLa cells to about 50%, showing a similar SG suppression effect to the chemical-induced G3BP1-OGT proximity. More importantly, this effect is independent of OGT’s transferase activity (Fig. 1e, f). In contrast, co-expression with OGA or its inactive variant (D174N) had little effect on SG formation, with over 95% of cells still forming SGs. Although a potential interaction between G3BP1 and OGT, rather than OGA, was observed (Supplementary Fig. 2c), we failed to detect apparent O-GlcNAc modifications on G3BP1 after either in cells or in vitro glycosylation (Supplementary Fig. 2b–d), further suggesting that the reduction in G3BP1-positive SGs is not directly linked to putative O-GlcNAc modifications on G3BP1.
These in-cell findings prompted us to investigate whether the inhibitory effect of OGT on G3BP1’s LLPS relies on the cellular environment. We performed in vitro droplet formation assays using purified G3BP1-EGFP along with increasing concentrations of purified and Cy5-labeled OGT, where no glycosylation reaction would occur. At a low concentration of 1 µM, OGT was incorporated into G3BP1-EGFP droplets with minimal changes in droplet size compared to G3BP1-EGFP alone (Fig. 1g). However, as the OGT concentration increased to 5 µM and 10 µM, the droplet size dramatically decreased (Fig. 1h). In contrast, EGFP-tagged YTHDF3 (EGFP-YTHDF3), a protein known to undergo phase separation and interact with OGT27, maintained the droplet size and OGT partitioning even at higher OGT concentrations (Supplementary Fig. 2d–f). Taken together, these results imply that OGT specifically inhibits the LLPS of G3BP1 and subsequent SG formation, both in vitro and in cells, through direct interaction independent of its enzymatic activity. This effect becomes more evident as the level of OGT increases. This observation motivates further investigation into the functional domains of OGT and the underlying mechanisms of its action.
Design of a G3BP1-targeted OGT truncation for SG suppression
Given OGT’s ability to suppress G3BP1 LLPS, we next aimed to engineer it into a G3BP1-selective tool for targeted SG regulation with minimized side effects. Previously, we have reported a set of tools for targeted protein O-GlcNAcylation and de-O-GlcNAcylation, utilizing nanobodies for intracellular protein targeting and O-GlcNAc cycling enzymes for modification33,34,35. In this study, we first employed our previously designed GFP-nanobody-fused OGT variant (nGFP-OGT4) to selectively target G3BP1-EGFP (Fig. 2a). G3BP1-EGFP coexpressed with either catalytically active or inactive nGFP-OGT4 exhibited nearly 40% reduced SG formation after NaAsO2 stress when compared with nGFP-fused truncated OGA (nGFP-OGA(Stalk)) of similar size (Fig. 2b, c). This nanobody-directed G3BP1 targeting reaffirms that the SG suppression effect is irrelevant to the enzymatic activity and size of OGT. Thus, we hypothesized that this effect may depend on the uniqueness of the OGT sequence.

a Depiction of targeting G3BP1-EGFP using GFP-nanobody (nGFP)-fused OGT variants. b, c Representative images (b) and quantification (c) of SGs in NaAsO2-treated HeLa cells coexpressing nGFP-fused OGT or OGA variants and G3BP1-EGFP. d, e Representative images (d) and quantification (e) of SGs in NaAsO2-treated HeLa cells coexpressing nGFP-fused OGT truncations with G3BP1-EGFP. f Representative images of NaAsO2-treated HeLa cells co-transfected with G3BP1-EGFP and nGFP-NI. Insets, regions of the indicated panels magnified 10 times. Right, fluorescence intensity profiles of G3BP1-EGFP (EGFP, green), nGFP-NI (HA, magenta), and the endogenous SG markers eIF4G1 and UBAP2L along the indicated line in the magnified images were plotted. eIF4G1 and UBAP2L were stained with corresponding antibodies (red). HeLa cells were stressed with 500 µM NaAsO2 for 30 min unless otherwise noted. g, h Representative images (g) and quantification (h) of HeLa cells expressing G3BP1-EGFP with or without nGFP-NI, respectively, under the treatment of sorbitol (400 mM, 1 h) or DTT (2 mM, 30 min). Data in (c, e, h) are represented as mean ± s.d. of n = 3 biologically independent samples with at least 30 co-transfected cells per group. P values were determined using Welch’s one-way ANOVA followed by Games–Howell multiple comparisons test (c, e), unpaired two-tailed Student’s t-tests (h). All nGFP-fused proteins were detected using an anti-HA antibody (magenta). Scale bars, 20 μm (b, d, f, g). Source data are provided as a Source Data file.
Based on the well-characterized structure of OGT36, we started to identify the key domains responsible for the suppression of G3BP1 phase separation and thereby repurpose OGT into a more compact tool for targeted SG regulation. The HA-tagged nGFP-OGT4 consists of an N-terminal nanobody against EGFP (nGFP), a truncated TPR domain (TPR4), the N-catalytic domain (N-cat), the intervening domain (Int-D), and the C-catalytic domain (C-cat) (Fig. 2d). Accordingly, we generated four variants of nGFP-OGT4 by deleting individual domains and compared their capability to suppress SG formation. Surprisingly, nGFP-OGT4 variants lacking either the TPR or C-cat domains were still able to suppress G3BP1-centric SGs, whereas nGFP-OGT4 variants lacking the N-cat or Int-D domains failed to suppress SG formation, with almost all cells exhibiting regular SG formation, highlighting the importance of the N-cat and Int-D domains (Fig. 2d). To further refine the tool, we fused the individual domains to nGFP for targeting G3BP1-EGFP. Notably, either N-cat or Int-D was sufficient to significantly reduce SG formation. The combination of all three fragments (nGFP, N-cat, and Int-D; termed nGFP-NI hereafter) achieved maximal suppression, reducing SG formation to nearly 90% (Fig. 2e). Given its highest efficacy and compact size, nGFP-NI was used in subsequent experiments as the tool for targeting G3BP1-EGFP in SG regulation. The high affinity between nGFP and EGFP ensured stable interaction between OGT variants and G3BP1 both before and after stress (Supplementary Fig. 3a). However, removal of nGFP from these constructs abolished their capability to suppress SG formation (Supplementary Fig. 3b–e); similarly, nGFP-NI was unable to target endogenous G3BP1 lacking EGFP as well (Supplementary Fig. 3f), both confirming that induced proximity between OGT and G3BP1 is essential for the suppression effect. Consistently, experiments in HeLa cells stably expressing nGFP-OGT4(K852A), its truncation lacking both the N-cat and Int-D domains (nGFP-∆NI), or the NI fragment alone, confirmed again that both the nGFP moiety and the NI fragment are essential for SG suppression (Supplementary Fig. 4a, b). Furthermore, we evaluated nGFP-NI in two additional cell models: U2OS cells stably expressing G3BP1-EGFP at levels lower than endogenous G3BP1, and U2OS G3BP1/2 double knockout (dKO) cells rescued with G3BP1-EGFP. In both cell lines, nGFP-NI effectively suppressed SG formation under NaAsO2 stress, demonstrating the robustness of the tool (Supplementary Fig. 4c, d).
A recent study from Taylor’s group demonstrated that G3BP1/2 have the highest centrality within the core SG network, which serve as the node for SG assembly under certain conditions, especially arsenite stress1. When applying nGFP-NI for SG suppression, we additionally examined another four known SG markers–eIF4G1, UBAP2L, G3BP2, and USP10–via immunofluorescence to explain the centrality16,37,38. We found that none of these marker proteins formed SGs independently of G3BP1 (Fig. 2f and Supplementary Fig. 5a). These results suggest that targeting G3BP1-EGFP with nGFP-NI prevented the assembly of other key SG marker proteins, reinforcing the central role of G3BP1 in SG formation under arsenite stress. However, it is important to note that G3BP1 may not exhibit the same degree of centrality under other stress conditions. To determine whether nGFP-NI is applicable to SG suppression under different stressors, we tested its effect on G3BP1-EGFP in HeLa cells subjected to osmotic stress (sorbitol) or ER stress (DTT)27. As expected, we observed a significant reduction in G3BP1 granules (Fig. 2g, h). However, unlike the arsenite treatment, other SG markers such as UBAP2L and eIF4G1 formed SGs independent of G3BP1 under sorbitol-induced stress (Supplementary Fig. 5b). These results suggest that nGFP-NI effectively suppresses G3BP1-dependent SGs and provides a strategy for distinguishing the relative importance and centrality of G3BP1 under different stress conditions. Meanwhile, we monitored other membraneless organelles during the application of nGFP-NI to G3BP1-EGFP under arsenite stress. No significant changes were observed in P-bodies or nuclear speckles (Supplementary Fig. 5c–f), underscoring the selectivity of nGFP-NI for its EGFP-tagged target protein and associated condensates.
nGFP-NI suppresses G3BP1 LLPS in an order- and concentration-dependent manner
We next investigated whether nGFP-NI acts directly on G3BP1 or indirectly disrupts SG assembly by modulating broader protein network interactions. We first examined eIF2α phosphorylation (p-eIF2α), a well-characterized trigger for translation inhibition and conventional SG formation in response to stressors18. We observed that both nGFP-NI and nGFP alone, when co-expressed with G3BP1-EGFP, resulted in similar levels of eIF2α phosphorylation in response to arsenite treatment (Supplementary Fig. 6a). Additionally, puromycin detection confirmed that translational arrest occurred as expected under both conditions13,39 (Supplementary Fig. 6b). These data suggest that early steps prior to G3BP1 involvement, such as stress recognition and translational arrest, proceeded normally, which in turn supports the notion that the inhibitory effect of nGFP-NI on SG formation does not stem from interference with the upstream SG signaling pathways, but likely from direct perturbation on G3BP1 itself. To further investigate whether nGFP-NI prevents the initiation of SG assembly or disrupts existing SGs, we employed a chemical-induced proximity approach40 with time and dose controls. We replaced the nGFP–EGFP binding pair with the FKBP–FRB pair, generating G3BP1-FRB and FKBP-NI constructs, respectively. Rapamycin acts as a molecular glue for FKBP and FRB, rapidly forming a ternary complex as its concentration increases without inducing detectable SGs (Supplementary Fig. 7a, b). When Rapamycin was applied at low concentrations prior to arsenite stress, it induced colocalization between G3BP1-FRB and FKBP-NI, while starting to inhibit SG formation once the Rapamycin concentration reached 100 nM or higher (Fig. 3a, b and Supplementary Fig. 7c). Consistent with the aforementioned in vitro droplet formation assay with an increasing amount of purified OGT (Fig. 1g, h), this inhibitory effect on SG is concentration dependent as well. In contrast, when cells were first stressed with arsenite followed by rapamycin, FKBP-NI was successfully recruited into SGs and colocalized with G3BP1-FRB, but SGs persisted (Fig. 3c, d and Supplementary Fig. 7d), indicating that the induced proximity of NI to SGs is unfortunately unable to disassemble existing SGs. These results emphasize that prior interaction between NI and G3BP1 is crucial for the suppression of arsenite-induced SGs. Meanwhile, the use of the Rapamycin-induced FKBP-FRB pair highlights the modularity of nGFP-NI, where the nGFP module can be substituted with other proteins or small molecules for versatile induced proximity.

a, b Representative images (a) and quantification (b) of SG formation in HeLa cells coexpressing FRB-fused G3BP1 (G3BP1-FRB) with FKBP-fused NI (FKBP-NI) with a pre-incubation of rapamycin (Rap) before NaAsO2 stress. c, d Representative images (c) and quantification (d) of SG formation in HeLa cells coexpressing G3BP1-FRB with FKBP-NI with NaAsO2 stress, followed by rapamycin addition. Rapamycin was added at indicated concentrations (0, 10, 100, and 1000 nM) for 10 min. Insets, regions of the indicated panels magnified 10 times. Fluorescence intensity profiles of G3BP1-FRB and FKBP-NI along the indicated line in the magnified images were plotted. HeLa cells were stressed with 500 µM NaAsO2 for 30 min unless otherwise noted. G3BP1-FRB and FKBP-NI were detected using anti-FLAG (green) and anti-HA (red) antibodies, respectively. e, f In vitro droplet formation of purified full-length G3BP1-EGFP (25 μM) at 150 mM NaCl was triggered with 100 ng/μl HeLa total RNA (e) or 10% PEG8000 (f) before (upper) or after (bottom) the incubation with Cy5-labeled nGFP (1 μM) or nGFP-NI (1 μM). Representative images (left) and size quantification of the droplets (right) in each group were shown. Data in (b, d) are presented as mean ± s.d. of n = 3 biologically independent samples with at least 30 co-transfected cells per group. Data in (e, f) are mean ± s.e.m. of all droplets identified from n = 3 randomly selected imaging frames in each group. P values were determined by Welch’s one-way ANOVA followed by Games–Howell multiple comparisons test (b, d) or two-tailed Mann–Whitney U test (e, f). Rap, Rapamycin. Scale bar, 20 µm (a, c), 100 µm (e), and 40 µm (f). Source data are provided as a Source Data file.
Furthermore, we conducted in vitro droplet formation assays of G3BP1-EGFP and nGFP-NI triggered by either total RNA or the molecular crowding reagent PEG8000 (Supplementary Fig. 7e, f). After RNA-induced LLPS, G3BP1-EGFP formed spherical droplets, even in the presence of Cy5-labeled nGFP-NI or nGFP (Fig. 3e). However, when G3BP1-EGFP was pre-mixed with nGFP-NI, it became less responsive to RNA-induced LLPS, resulting in significantly smaller droplets (Fig. 3e). Similar results were obtained when 10% PEG8000 was used to trigger G3BP1-EGFP LLPS (Fig. 3f). These results, consistent with our observations in cells, reaffirm that direct interaction between G3BP1 and nGFP-NI prior to stress is essential for inhibiting G3BP1 LLPS and SG formation. Therefore, we sought to examine the interaction between nGFP-NI and G3BP1-EGFP prior to LLPS using cross-linking mass spectrometry (XL-MS). Although a low abundance of lysine residues limited the number of cross-links obtained, we observed multiple cross-links between the NTF2L and RBD domains in the presence of nGFP alone, consistent with previous reports indicating interactions between the N- and C-termini of G3BP11 (Supplementary Fig. 8a). However, in the presence of nGFP-NI, most of these cross-links on G3BP1-EGFP disappeared, while two new cross-links between the NTF2L or RBD domains and nGFP-NI were detected (Supplementary Fig. 8b). These changes suggest that nGFP-NI may sequester G3BP1 and thereby hinder the initiation of G3BP1 LLPS.
nGFP-NI promoted G3BP1 aggregation
Next, to assess whether nGFP-NI prevents or merely delays SG assembly, we monitored SG formation over a two-hour arsenite treatment. At 0.5 h post-arsenite treatment, SGs appeared in control cells, characterized by colocalization of G3BP1-EGFP, the SG marker eIF4G1, and nGFP. In contrast, in cells expressing nGFP-NI, no apparent SGs appeared, and both G3BP1-EGFP and eIF4G1 remained diffusive (Fig. 4a). After 1 h of stress, cytoplasmic puncta appeared in nGFP-NI–expressing cells, sequestering G3BP1-EGFP, eIF4G1, and TDP-43 together (Fig. 4a and Supplementary Fig. 9a). However, while SGs in control cells retained their spherical shape after two hours of stress, the puncta in the nGFP-NI–expressing cells exhibited a more aggregated morphology (Fig. 4a). These observations prompted us to compare the fluidity of these SGs. Fluorescence recovery after photobleaching (FRAP) analyses were conducted following two hours of arsenite treatment and revealed that nGFP-NI significantly slowed the FRAP kinetics of G3BP1-EGFP compared to nGFP, indicating that the G3BP1–NI binary complex exhibited more aggregate-like material properties (Fig. 4b–e and Supplementary Fig. 9b, c). Likewise, FRAP analyses of in vitro G3BP1-EGFP droplets pre-incubated with nGFP or nGFP-NI mirrored these findings, with nGFP-NI dramatically reducing the dynamics of G3BP1-EGFP condensates (Supplementary Fig. 9d–g).

a Representative images of G3BP1-EGFP in HeLa cells coexpressing nGFP or nGFP-NI under NaAsO2 stress. Cells were treated with 500 µM NaAsO2 for the indicated time (0, 0.5, 1, 2 h). eIF4G1 was stained with its antibody (red). Insets, regions of the indicated panels magnified by 3 times (white rectangle) and 10 times (yellow rectangle). b, c FRAP analyses of G3BP1-EGFP condensates containing nGFP-mCherry (b) or nGFP-NI-mCherry (c) in HeLa cells coexpressing G3BP1-EGFP with nGFP-mCherry or nGFP-NI-mCherry with the treatment of 500 µM NaAsO2 for 2 h. Right, representative images of photo-bleached condensates at indicated time points. d, e Recovery curves (d) and immobile fraction quantification (e) of G3BP1-EGFP condensates in (b, c). f Representative images of SG formation in HeLa cells coexpressing G3BP1-EGFP with nGFP or nGFP-NI under the indicated treatments. Cells were stressed with 500 µM NaAsO2 stress for 1 h, some of which were treated with 1.5% 1,6-Hexanediol (1,6-HD) for 10 min afterwards. Insets, regions of the indicated panels magnified by 3 times. g, h Immunoblot analyses of RIPA-soluble and pellet fractions of HeLa cells transfected with indicated plasmids and treated with 500 µM NaAsO2 for different time. The total cell lysate (Total), RIPA-soluble fraction (S), and pellet fraction (P) were analyzed by immunoblotting with indicated antibodies. In (d), data are mean ± s.e.m. of n = 4 biologically independent samples. In (e), data are mean ± s.d. of n = 4 biologically independent samples. P values were determined using unpaired two-tailed Student’s t-tests (e). All nGFP-fused proteins were detected using an anti-HA antibody (magenta). Scale bar, 20 µm (a–c, f). Source data are provided as a Source Data file.
To further corroborate whether the G3BP1-EGFP and nGFP-NI complex formed solid-like aggregates, we treated cells with 1,6-Hexanediol (1,6-HD) following 1-h arsenite stress. In HeLa cells co-expressing nGFP and G3BP1-EGFP, SGs were largely disassembled by 1.5% 1,6-HD, whereas G3BP1-EGFP puncta persisted in the nGFP-NI–expressing cells, suggesting that the puncta formed by the G3BP1–NI binary complex were more solid-like, in contrast to the liquid-like SGs observed in the nGFP group (Fig. 4f). Similar findings were observed on cells when applied a 2-h recovery following 1-h arsenite exposure (Supplementary Fig. 9h). We hypothesized that the material property of nGFP-NI may cause this solidification. To this end, we performed immunoblotting to differentiate the material properties of specific proteins by analyzing RIPA-lysis-buffer and urea extracts from cells exposed to arsenite stress for 0 to 2 h (Fig. 4g, h and Supplementary Fig. 10a). Surprisingly, although nGFP-NI alone did not localize to SGs in the absence of G3BP1-EGFP, even after two hours of arsenite stress (Supplementary Fig. 3f), both nGFP-NI and nGFP-OGT4 started to appear in the RIPA-insoluble fraction following prolonged stress, with nGFP-NI exhibiting a more rapid aggregation tendency (Fig. 4g). In the presence of G3BP1-EGFP, compared to the nGFP group, the G3BP1-EGFP and nGFP-NI complex also showed increased aggregation in the RIPA-insoluble fraction over longer arsenite treatment, with inclusion of SG markers (eIF4G1 and TDP-43) (Fig. 4h and Supplementary Fig. 10b–d). These findings suggest that the aggregation propensity of nGFP-NI may sequester G3BP1-EGFP into aggregates, thereby inhibiting its LLPS. Collectively, these results indicate a possible dual effect of nGFP-NI: it suppresses the initiation of G3BP1-EGFP LLPS while promoting its solidification, leading to delayed SG formation during short arsenite exposure and the formation of solid-like, irreversible aggregates after prolonged exposure.
nGFP-NI targeted the domain architecture encoded by G3BP1
We next asked whether NI altered the material properties of G3BP1 by targeting any specific domains. G3BP1 contains an N-terminal NTF2L domain responsible for dimerization and two C-terminal RNA-binding regions (RRM and RGG), which are spanned by a long central IDRs (IDR1 and IDR2) (Fig. 5a). These domains collectively enable G3BP1 to act as a molecular switch for LLPS1. Previous studies have underscored the essential roles of G3BP1 dimerization and RNA binding in SG condensation and implied that the long central IDRs only contribute to SG dynamics rather than encoding interactions for SG assembly1,16,19. We observed a consistent inhibitory effect of nGFP-NI on the LLPS of G3BP1 lacking IDR1/2 under arsenite stress (Fig. 5b), suggesting that this inhibition does not rely on IDR1/2. An intriguing feature of the LLPS of G3BP1 is that each of its three major domains can be substituted by heterologous domains with similar properties without disrupting SG assembly1. To test whether nGFP-NI recognizes the precise sequence or a broader structural pattern of G3BP1, we replaced its NTF2L domain with glutathione S-transferase12, a protein that constitutively dimerizes, generating a chimeric protein (GST-G3BP1-EGFP). When coexpressed with nGFP in HeLa cells, GST-G3BP1-EGFP successfully reconstituted SGs under arsenite stress. However, coexpression with nGFP-NI suppressed LLPS of the chimeric protein, despite normal SG formation by endogenous G3BP1 under stress (Fig. 5c). Similarly, we replaced the RNA-binding regions of G3BP1 with tandem RRM domains from an unrelated RNA-binding protein—SRSF4—resulting in the chimeric constructs G3BP1(SRSF4 RRM1/2)-EGFP. The chimera formed SGs normally upon arsenite treatment, yet its LLPS was also blocked by nGFP-NI (Fig. 5d). These domain-swapping experiments suggest that the suppressive effect of nGFP-NI is independent of binding to any specific primary sequence within G3BP1.

a Depiction of G3BP1 domain organization. b–f Representative images (b–e) and quantification (f) of SG formation in NaAsO2-treated HeLa cells coexpressing nGFP or nGFP-NI with engineered G3BP1-EGFP variants. G3BP1(∆IDR1/2)-EGFP, G3BP1-EGFP deleting both IDR1 and IDR2 (b); GST-G3BP1-EGFP, substitution of the NTF2L domain with the GST domain (c); G3BP1(SRSF4 RRM1/2)-EGFP, substitution of G3BP1 RNA binding domain with SRSF4 RRM1/2 (d); a fully synthetic G3BP1 with GST as the dimerization domain, Ash1 as the long IDR and the tandem SRSF4 RRM1/2 (e). Insets, regions of the indicated panels magnified 10 times. Right, fluorescence intensity profiles of G3BP1-EGFP variants (EGFP, green), nGFP-NI (HA, magenta), and endogenous SG marker eIF4G1(red) along the indicated line in the magnified images were plotted. eIF4G1 was stained with its antibody (red). All nGFP-fused proteins were detected using an anti-HA antibody (magenta). HeLa cells were stressed with 500 µM NaAsO2 for 30 min (b, d, e) or 1 h (c). In (f), data are mean ± s.d. of n = 3 biologically independent samples with at least 30 transfected cells per group. P values were determined by unpaired two-tailed Student’s t-tests (f). Scale bars, 20 μm (b–e). Source data are provided as a Source Data file.
Finally, to test this hypothesis, we constructed a synthetic G3BP1 in which all three major domains were replaced by unrelated domains with analogous functions, preserving the modular architecture of the protein (Fig. 5e, f). As expected, this synthetic G3BP1 retained the ability to undergo LLPS and form SGs in the presence of nGFP. However, its arsenite-induced LLPS was still inhibited by nGFP-NI (Fig. 5e, f), further supporting the conclusion that nGFP-NI disrupts G3BP1 LLPS through recognition of a general domain architecture, independent of G3BP1’s exact amino acid sequence.
nGFP-NI is generalizable to other SG components and enables SG regulation
The success of nGFP-NI in targeting synthetic G3BP1 encouraged us to investigate whether this strategy could be generalized to suppress phase separation of other SG components, particularly those with similar modular architecture. We focused on cytosolic SG proteins that contain an oligomerization domain and multiple RBDs, interspersed with IDRs, resembling the structural features of G3BP1. These proteins typically serve as key nodes in assembling complex protein–RNA networks in SGs due to their high valency11,16. Among a panel of abundant and functionally important SG proteins, we selected CAPRIN1, PABPC1, FMR1, and FXR2 for further analysis, all of which have been reported to possess dimerization capability and multiple RBDs16,41,42 (Fig. 6a). As before, we coexpressed each protein with either nGFP or nGFP-NI in HeLa cells. In the nGFP control group, FMR1 and FXR2 formed condensates even before stress, likely due to their high intrinsic phase separation potential (Supplementary Fig. 11a, b), while CAPRIN1 and PABPC1 formed condensates only upon arsenite treatment (Fig. 6b, d). Notably, nGFP-NI suppressed the LLPS of all four proteins, resulting in a marked reduction in the percentage of SG-positive cells (Fig. 6b–f and Supplementary Fig. 11a–c). Similarly, in vitro droplet formation assays with CAPRIN1 and PABPC1 (Supplementary Fig. 11d–f), and cellular assays with FMR1 and FXR2 in U2OS G3BP1/2 dKO cells, further verified the suppressive effect of nGFP-NI (Supplementary Fig. 11g, h). These results also confirm that this effect is determined by direct interaction between the target proteins and nGFP-NI and is independent of the G3BP1-centric protein network. In contrast, SG proteins with distinct domain architectures, such as eIF4G1 and UBAP2L, were not affected by nGFP-NI (Supplementary Fig. 11i, j).

a Core scaffold proteins with high valency drive RNP network assembly in SG through multivalent interactions. b, d Representative images of SG formation in NaAsO2-treated HeLa cells coexpressing nGFP or nGFP-NI with EGFP-CAPRIN1 (b) or EGFP-PABPC1 (d). c, e Recovery curves (left) and immobile fraction (right) quantification of FRAP analyses of EGFP-CAPRIN1 condensates (c) or EGFP-PABPC1 condensates (e) in HeLa cells expressing nGFP or nGFP-NI treated with 500 µM NaAsO2 for 2 h. f Quantification results of (b, d). g, h Representative images (g) and quantification (h) of SG formation in NaAsO2-treated HeLa cells coexpressing Shuttle-tdTomato and G3BP1-EGFP with or without nGFP-NI. HeLa cells were stressed with 500 µM NaAsO2 for 30 min. i Cell viability assessment of HeLa cells transfected with indicated plasmids and G3BP1-EGFP, and treated with 500 µM NaAsO2 for 24 h was performed using flow cytometric analysis with propidium iodide (PI) staining. In (c, e), data are the mean ± s.e.m. (left) and the mean ± s.d. (right) of n = 4 biologically independent samples. In (f, i), data are mean ± s.d. of n = 3 biologically independent samples with at least 30 transfected cells per group. In (h), data are mean ± s.e.m. of n = 32, 34, 37, 50, 45, 47 (from left to right). P values were determined by unpaired two-tailed Student’s t-tests (c, e, f, i) or Welch’s one-way ANOVA followed by Games–Howell multiple comparisons test (h). All nGFP-fused proteins were detected using an anti-HA antibody (magenta). Scale bar, 20 µm (b, d, g). Source data are provided as a Source Data file.
To explore whether the mechanism of LLPS suppression was similar to that observed for G3BP1, we performed FRAP experiments on visible condensates after prolonged arsenite treatment. These analyses revealed that nGFP-NI significantly reduced the fluidity of CAPRIN1, PABPC1, FMR1, and FXR2, resulting in solid-like aggregates and delayed condensate growth (Fig. 6c, e and Supplementary Fig. 11k, l). Together, these findings demonstrate that nGFP-NI can broadly inhibit LLPS across multiple SG proteins, likely by targeting those with a multivalent modular organization similar to G3BP1 and reducing their molecular mobility. Thus, induced proximity between the target protein and the OGT fragment (NI) offers a generalizable strategy for regulating SGs by tuning the material properties of condensates.
Mounting studies revealed close associations of SGs in promoting protein aggregation, halting protein synthesis, as well as disrupting nucleocytoplasmic transport for pathogenesis of several neurodegenerative diseases17,43. Having previously characterized the impact of nGFP-NI on aggregation and translation, we next examined its effect on nucleocytoplasmic transport using a Shuttle-tdTomato reporter system44. This reporter, which contains both a nuclear localization signal (NLS) and a nuclear export signal (NES), normally shuttles from the nucleus to the cytoplasm, and becomes cytoplasmic-enriched during arsenite-induced SG formation—a hallmark of nucleocytoplasmic transport defect. As expected, overexpression of G3BP1-EGFP led to robust cytoplasmic accumulation of Shuttle-tdTomato in response to arsenite stress (Fig. 6g, h). However, when nGFP-NI targeted G3BP1-EGFP to suppress SG formation, the reporter remained largely nuclear, maintaining a nuclear-to-cytoplasmic distribution ratio comparable to unstressed conditions (Fig. 6g, h). This effect mirrors the results observed with other known SG inhibitors44, suggesting that SGs are required for sequestering nucleocytoplasmic transport factors and that disrupting SG formation can mitigate transport defects.
Additionally, since dynamic SGs have been proposed to protect cells during acute stress6, we assessed the effect of nGFP-NI on cell viability following arsenite exposure. Both HeLa and U2OS cells expressing G3BP1-EGFP and nGFP-NI exhibited the lowest survival rates compared to the controls (Fig. 6i and Supplementary Figs. 12 and 13a), indicating that impaired SG dynamics may compromise cellular stress resilience. We also applied nGFP-NI to a cytoplasmically mislocalized TDP43 mutant10, EGFP-TDP-43 NLSmut, and found that nGFP-NI’s targeting significantly reduced cell viability in both HeLa and U2OS cells, likely due to enhanced TDP43 aggregation triggered by nGFP-NI (Supplementary Fig. 13b). Beyond assessing cell viability, we used nGFP-NI to disrupt the co-condensation of G3BP1 and a disease-associated FUS mutant, FUS(R495X)45. When expressed in HeLa cells, FUS(R495X) formed SGs with G3BP1 in over 70% of cells (Supplementary Fig. 13c, d). However, co-expression with nGFP-NI abolished the formation of both FUS(R495X) and G3BP1 condensates. We propose that this failure in granule assembly results from the sequestration of G3BP1 by nGFP-NI. Taking these observations together, given the direct association between the material properties and functions of SGs, this generalizable protein-selective tool holds the potential to investigate and modulate the biological consequences of aberrant SG dynamics of a specific G3BP1-like SG protein.
Discussion
Stress granules (SGs) form when multivalent networks of proteins and RNA coalesce into liquid‑like condensates, making them an ideal model for studying intracellular phase separation. Because SG dysregulation is linked to neurodegeneration, viral infection, and treatment resistance, both defining general assembly mechanisms and creating tools to modulate SGs are of great interest15,46,47. Here, we surprisingly found that OGT—long studied for its enzymatic activity—suppresses G3BP1‑centric SGs upon induced proximity independent of O-GlcNAc installation (Fig. 1). Using induced‑proximity platforms, we showed that this negative cooperativity is mainly mediated by OGT’s intervening domain (Int‑D), a cryptic fold unique to metazoan OGTs36. We therefore repurposed OGT and generated STOP, a SG-targeted suppression method using a non-catalytic OGT truncation in nanobody- or chemical-induced proximity, by fusing the OGT N‑catalytic domain plus Int‑D (NI) to nanobody (nGFP) or FKBP modules (Figs. 2 and 3). STOP enables SGs inhibition in a stoichiometry‑ and order‑dependent manner, where pre‑stress recruitment of sufficient NI to G3BP1 is required for sufficient SG inhibition. Mechanistically, nGFP‑NI converts G3BP1 into a more solid‑like state with reduced mobility under sustained stress, thus delaying SG formation. This tool shows high selectivity toward the target protein and is versatile among several cell types.
G3BP1 is the central hub of SGs by establishing multivalent interactions to promote SG formation. Diverse modulation methods directly targeting G3BP1 have emerged recently15,21,47. Although small molecules or interfering peptides have been developed to disrupt the G3BP1 dimerization essential for LLPS initiation, these strategies are difficult to generalize to other condensate scaffolds. Since NI lacks the RNA-binding ability and is a negative factor for G3BP1 LLPS, according to the valence capping model16, high-affinity binding between nGFP-NI and G3BP1-EGFP would sequester G3BP1 and prevent the expansion of multivalent network, leading to the final SG suppression. Unlike another negative factor USP10, which disrupts G3BP1 dimerization48, nGFP-NI tunes the material properties of G3BP1 and lowers its effective valence concomitantly. This repurposing of a natural negative factor suggests a general paradigm for SG modulation, encouraging more SG positive and negative factors to be discovered and reengineered into a toolkit for precise SG modulation. Given the critical roles of SGs in several disease states, including neurodegeneration and cancer5,8,9,10,11,49,50, this tool is envisioned to discover the biological consequences of SG solidification, reveal differential G3BP1 roles in distinct types of SGs, thereby providing a potent approach in the study of condensate biology. Therefore, extending the application of STOP from current cultured cell lines to other models, such as primary or iPSC-derived neurons17, would be a critical next step to fully realize its potential.
Remarkably, STOP, represented by nGFP-NI, recognizes the domain organization of G3BP1 rather than its exact sequence (Fig. 4), enabling us to demonstrate STOP’s generality on four additional proteins that share similar modular organization (Fig. 5), which is distinct from other G3BP1 modulators generally targeting the well-structural NTF2L domain21. Proteins essential for assembling diverse condensates, such as SG, P-body, and Paraspeckle, primarily rely on stable oligomerization and RNA binding—rather than IDR-mediated weak interactions—to achieve high network connectivity for LLPS16. We speculate that targeted immobilization by STOP may impair their oligomerization and RNA-binding equilibria in the soluble phase, thereby shifting the LLPS threshold. This hypothesis could be tested by tuning STOP–target binding affinity or by comparing its effects on proteins whose LLPS is driven mainly by IDRs. It is conceivable that STOP would serve as a sequestrator for reduced connectivity and an immobilizer for reduced mobility on these proteins across diverse assemblies. Moreover, STOP is genetically encoded with modularity, which can be readily adapted to other reversible chemo- or opto-induced proximity approaches51, offering a flexible platform for tuning condensate dynamics. Knocking in short epitope tags onto endogenous proteins for use with corresponding nanobodies52, or using nanobodies or natural peptide ligands directly produced for endogenous targets53, would further enable the expansion of this method’s application to in vivo systems. As a proof-of-concept, STOP also offers a concept for modulating protein condensation through induced immobilization, complementary to other protein manipulation methods leveraging induced proximity, such as targeted protein relocalization54 and RIPTAC55.
The unexpected identification of Int-D for SG suppression prompts reconsideration of OGT’s roles in cellular regulation. Beyond the well-studied role in O-GlcNAcylation, OGT exhibits fundamental non-catalytic functions31, most of which rely on the long TPR domain to facilitate substrate recognition56. Until recently, Int-D was found to act as a nutrient and stress sensor57, and its activity in condensate modulation was not anticipated. Although we used the induced proximity approach for enhanced OGT–G3BP1 interactions, it will be intriguing to determine whether native Int-D–protein interactions exist under physiological conditions and how they might be controlled. The “adaptor protein hypothesis” posits that adaptor proteins may act like the induced proximity modules to help target OGT to selective cellular substrates for diverse functions28,58. We thus hypothesize that OGT, especially the Int-D, achieves its targeted immobilization effect on cellular condensates through the recruitment from specific adaptor proteins similar to the nanobody or rapamycin systems. Future efforts can be made on the identification of potential adaptor proteins, especially in those scenarios with abnormal SG formation.
In summary, our work introduces STOP as a modular and generalizable platform for SG modulation. By unveiling that OGT Int-D is a previously unappreciated condensate modulator, we repurposed it into a targeted protein immobilizer by adapting the induced proximity platform, and applied STOP (e.g., nGFP-NI) to six RNA-binding nodes in SGs. We conceptually validated the feasibility of tuning the material properties of cellular condensates (e.g., SGs) for condensate functional dissection and intervention. Our results provide insights into the non-catalytic functions of the poorly understood OGT Int-D as well as robust interventions for molecular understanding and therapeutic implications of LLPS-driven processes in health and disease.
Methods
Cell culture and transfection
HeLa and U2OS cell lines were cultured in Dulbecco’s Modified Eagle Medium (Gibco, C11995500BT) supplemented with 10% FBS (Gibco, 10099-141C) and 1% penicillin–streptomycin (Gibco, 15140122). HEK-293F cells were cultured in HEK293 Growell TransA (CellPlus Biotechnology, LRU2000). Cells were maintained at 37 °C and 5% CO2. TransIT-PRO (Mirus Bio, MIR 5740) and PEI (MedChemExpress, HY-K2014) were used for transient transfections. The U2OS G3BP1-EGFP stable cell line and U2OS G3BP1/2 dKO cell line were described in previous studies44,59.
Plasmids
All plasmids were constructed by cloning PCR-amplified fragments into the pcDNA3.1 vector via homologous recombination (pEASY®-Basic Seamless Cloning and Assembly Kit, TransGen Biotech, CU201), unless otherwise specified. The constructs used in this study—including OGT, OGA, catalytically inactive mutants: OGT(K852A) and OGA(D174N), nanobody fusions: nGFP-OGT4, nGFP-OGT4(K852A) and nGFP-OGA(Stalk), the NLS-tdTomato-NES reporter and EGFP-YTHDF3—were adopted or reproduced from our previous studies27,33,34,44. Variants of OGT (N-Cat, Int-D, NI, and K852A) were generated with a C-terminal 6 × His tag. The nGFP-OGT4 series (including truncation mutants ΔTPR, ΔN-Cat, ΔInt-D, and ΔC-Cat) were constructed with a C-terminal HA tag based on the nGFP-OGT4 backbone. SG regulators (eIF4G1, UBAP2L, CAPRIN1, FXR2, FMR1, PABPC1, and RNA-binding domain (SRSF4 RRM1/2) were amplified from their cDNAs and cloned into pcDNA3.1 with an N-terminal FLAG tag, pcDNA3.1_G3BP1-EGFP-FLAG was a gift from Prof. Peng R. Chen (Peking University). Synthetic modules (FRB, FKBP, HaloTag, GST, Ash1) were assembled into pcDNA3.1_G3BP1-EGFP or nGFP-NI constructs. Constructs for protein expression in HeLa cells were validated in Supplementary Fig. 14a. Bacterial expression plasmids (G3BP1-EGFP, EGFP-YTHDF3, EGFP-CAPRIN1, EGFP-PABPC1, OGT, OGT(K852A), and nGFP-NI) were generated using the pET28a vector containing an N-terminal 6 × His tag. Lentiviral expression plasmids (nGFP-OGT4, nGFP-ΔNI, and NI) were generated using the pLenti vector containing an N-terminal HA tag. All constructs were verified by Sanger sequencing (Tsingke Biotechnology), and summarized in Supplementary Table 1.
Antibodies and reagents
Commercial antibodies were used under the following conditions: anti-His (Cell Signaling Technology [CST] #12698; 1:400 for immunofluorescence [IF], 1:1000 for immunoblotting [IB]), anti-Myc (CST #2276; 1:1000 IF/IB), anti-OGT (CST #24083; 1:400 IF, 1:1000 IB), anti-HA (Proteintech #66006-2-Ig, 1:400 IF; CST #3724, 1:800 IF, 1:1000 IB), anti-eIF4G (CST #2469; 1:300 IF, 1:1000 IB), anti-UBAP2L (CST #40199; 1:300 IF), anti-G3BP1 (Abcam #ab56574; 1:300 IF, 1:1000 IB), anti-G3BP2 (Abcam #ab86135; 1:300 IF), anti-USP10 (Abcam #ab109219; 1:300 IF), anti-FLAG (CST #14793, 1:800 IF, 1:1000 IB; sigma #F3165, 1:1000 IB), anti-TDP-43 (Proteintech #10782-2-AP; 1:300 IF, 1:1000 IB), anti-peIF2α (CST #9721; 1:1000 IB), anti-eIF2α (CST #5324; 1:1000 IB), anti-puromycin (Millipore #MABE343, 1:1000 IB), anti-O-GlcNAc (RL2) (Abcam #ab2739; 1:1000 IB), anti-ACTIN (Proteintech #66009-1-Ig; 1:10,000 IB), anti-DCP1A (Proteintech #22373-1-AP; 1:400 IF), and anti-SC35 (Abcam #ab11826; 1:300 IF). Secondary antibodies included Alexa Fluor 647/568 conjugates (Thermo #A-21244/#A-11004; 1:1000 IF), IRDye 800CW/680CW (LI-COR #926-32211/#926-68071/#926-32210; 1:10,000 IB), and HRP conjugates: anti-Mouse IgG (HUABIO #HA1006; 1:10,000 IB), anti-Rabbit IgG (HUABIO #HA1001; 1:10,000 IB).
Protein expression and purification
G3BP1-EGFP, EGFP-YTHDF3, EGFP-CAPRIN1, EGFP-PABPC1, and OGT were purified according to previously reported methods1,27,41,60. Briefly, pET28a-based plasmids were transformed into E. coli BL21(DE3) cells. Single colonies were grown in LB medium with kanamycin (50 μg/mL) at 37 °C to OD600 0.6–0.8, then induced with 0.5 mM IPTG at 25 °C for 16 h (G3BP1-EGFP and EGFP-YTHDF3) or 16 °C for 16 h (EGFP-CAPRIN1, EGFP-PABPC1, OGT, OGT(K852A), and nGFP-NI). For nGFP production, HEK293F cells were transfected with corresponding plasmids using PEI and cultured for 96 h.
Bacterial cells were harvested by centrifugation (4000 × g, 20 min, 4 °C), resuspended in lysis buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 1× protease inhibitors), and lysed by sonication (150 W, 3 s on/5 s off, ice-cooled, 30 min total). Lysates were clarified by centrifugation (12,000 × g, 30 min, 4 °C). For G3BP1-EGFP, EGFP-YTHDF3, EGFP-CAPRIN1, EGFP-PABPC1, OGT, and OGT(K852A), supernatants were loaded onto Ni-NTA columns pre-equilibrated with lysis buffer, washed with buffer containing 80 mM imidazole, and eluted with 200 mM imidazole. Eluted proteins were concentrated using 10 K/30 K Amicon spin columns (Merck-Millipore, UFC901096/ UFC903096) (4000 × g, 30 min, 4 °C) and buffer-exchanged into storage buffers (G3BP1-EGFP and EGFP-CAPRIN1: 20 mM Tris-HCl pH 7.5, 400 mM NaCl, 0.5 mM DTT, 10% glycerol; EGFP-YTHDF3: 20 mM HEPES pH 7.4, 300 mM KCl, 6 mM MgCl2, 0.02% NP-40, 20% glycerol; EGFP-PABPC1: 5 mM Tris pH 7.4, 250 mM NaCl, 0.25 mM TCEP, 10% glycerol; OGT and OGT(K852A): 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 2 mM DTT, 20% glycerol).
For nGFP-NI, after initial centrifugation and lysis, the insoluble pellet was resuspended in PBS-Urea buffer (PBS, 8 M urea, pH 7.4), recentrifuged (12,000 × g, 20 min, 4 °C), and the supernatant loaded onto a Ni-NTA column. The column was washed with PBS-Urea buffer containing 50 mM imidazole and eluted with 500 mM imidazole. The eluate was further purified by a Superdex™ 200 Increase 10/300 GL column (Cytiva, cat. 28990944). The column was equilibrated with storage buffer (PBS, 300 mM NaCl, 10% glycerol, pH 7.4) and fractions containing nGFP-NI were collected.
HEK293F cell supernatants (for nGFP) were collected by centrifugation (10,000 × g, 10 min, 4 °C) and purified using Ni-NTA chromatography as described for bacterial proteins. Purified nGFP was buffer-exchanged into PBS with 10% glycerol (pH 7.4). Protein concentrations were quantified via NanoDrop One spectrophotometer (Thermo) and stored at −80 °C. The Coomassie-stained gel image of the above purified proteins is shown in Supplementary Fig. 14b.
Immunofluorescence and microscopy
Cells were seeded into 4-well confocal dishes (JingAn Biological, J40204) and exposed to indicated stress conditions. Post-treatment, cells were fixed with 4% paraformaldehyde (Leagene, DF0134) for 15 min, washed three times with PBS, and blocked for 1 h at room temperature in PBS containing 5% BSA and 0.3% Triton X-100. Cells were then incubated sequentially with primary and secondary antibodies (diluted in 1% BSA/0.3% Triton X-100/PBS) for 1 h each at room temperature, with three PBS washes between steps. Nuclei were counterstained with DAPI (Thermo, R37606) followed by three PBS washes. Imaging was performed using a Dragonfly spinning-disk confocal microscope (Andor Technology, CR-DFLY-202-2540).
Immunoblotting
Cells were washed with PBS and lysed on ice for 30 min in RIPA buffer (Applygen, C1053-500) containing 1× EDTA-free protease inhibitor cocktail (MCE, HY-K0010). Lysates were clarified by centrifugation (12,000 × g, 20 min, 4 °C), and supernatants were mixed with 1× SDS buffer, followed by denaturation at 95 °C for 5 min. Proteins were separated on 4–12% Bis-Tris gradient gels (Genscript, M00654) and transferred to nitrocellulose membranes (Millipore, HATF00010). Membranes were blocked with 5% non-fat milk/TBST for 1 h at room temperature, then sequentially incubated with primary and secondary antibodies for 1 h each at room temperature, with three 1 × TBST washes between steps. Signals were visualized using an Azure Imager C600 (Azure Biosystems).
Immunoprecipitation
HeLa cells were cultured to >90% confluency in 6-well plates, harvested, and washed with PBS. Cells were lysed in RIPA buffer supplemented with 1× EDTA-free protease inhibitor cocktail, incubated on ice for 20 min, and clarified by centrifugation (12,000 × g, 10 min, 4 °C). Supernatants were incubated with anti-FLAG (Selleck, B26101; Sigma, M8823) or anti-HA affinity beads (Selleck, B26201) at 4 °C overnight under gentle rotation. Beads were washed thrice with 500 µL PBST, and immunoprecipitated complexes were eluted by boiling in 1× SDS buffer (95 °C, 5 min). Eluted proteins were resolved via SDS-PAGE, transferred to nitrocellulose membranes, and analyzed by immunoblotting.
Liquid-liquid phase separation
G3BP1-EGFP, nGFP, nGFP-NI, and OGT were buffer-exchanged into LLPS buffer (150 mM NaCl, 50 mM HEPES pH 7.5) using 10/30 kDa MWCO filters (Merck-Millipore, UFC5010/5030), while EGFP-YTHDF3 was buffer-exchanged into PBS via the same method. OGT, nGFP, and nGFP-NI were Cy5-labeled via 4 h room-temperature incubation. G3BP1-EGFP phase separation was induced by adding HeLa total RNA (Yeasen, 19231ES) or 10% PEG8000 (Coolaber, CP8241). Samples were mixed, loaded into a custom imaging chamber (glass coverslip adhered to a slide with double-sided tape, ~10 µL volume), and allowed to phase-separate for 3 min. Droplets were imaged using a Dragonfly spinning-disk confocal microscope (Andor Technology, CR-DFLY-202-2540) with a 20× or 63× objective, and images were analyzed in ImarisViewer (v10.2.0) and ImageJ.
Fluorescence recovery after photobleaching (FRAP)
For FRAP assays, in vitro phase-separated droplets and cellular condensates were analyzed as follows: In vitro samples were prepared as described previously, while cellular experiments involved seeding cells in 4-well confocal dishes, transfecting with target plasmids, and applying stress treatments 24 h post-transfection. Live-cell imaging was performed under physiological conditions (37 °C, 5% CO2) using a ZEISS LSM 900 microscope equipped with a live-cell system. Target condensates were photobleached with a 488 nm laser (80% intensity), and fluorescence recovery was monitored at defined intervals. Raw intensity data were normalized in ImageJ, with pre-bleach signals set to 100% and recovery expressed as relative percentages of initial intensity to calculate dynamic exchange rates.
In vitro glycosylation assay
Reaction mixtures (10 μM target protein, 1 μM OGT, 1 mM UDP-GlcNAc [Millipore, U4375]) were prepared in reaction buffer (25 mM Tris pH 7.5, 50 mM NaCl, 0.5 mM EDTA, 2 mM DTT, 10% glycerol) and incubated at 30 °C for 4 h with 200 rpm agitation. Post-reaction, samples were denatured in 1× SDS buffer (95 °C, 5 min), resolved by SDS-PAGE, and subjected to immunoblotting detected by indicated antibodies.
Separation of RIPA lysis buffer-soluble and insoluble fraction
According to a previously reported method61 with minor modifications, stress granule isolation was performed at 4 °C. Briefly, cells were washed once with PBS and processed in parallel: (1) For total lysates, cells were lysed in Urea buffer (8 M urea, 2% SDS, 50 mM Tris-HCl pH 7.4), mixed with 1× SDS buffer. (2) For fractionation, cells were lysed in RIPA buffer (with 1× EDTA-free protease inhibitor cocktail) on ice for 30 min, sequentially centrifuged (4000 × g, 5 min; 18,000 × g, 20 min) to collect the soluble fraction (S), which was denatured as above. The insoluble pellet was washed with RIPA buffer (18,000 × g, 5 min), resolubilized in Urea buffer, and denatured to generate the pellet fraction (P). All samples were resolved by SDS-PAGE for immunoblotting.
Puromycin incorporation assay
The puromycin incorporation assay was modified from Kedersha et al.62. HeLa cells were treated with or without 500 μM NaAsO2 for 30 min and treated with 10 µg/mL puromycin (Gibco, A1138-03) for 15 min prior to lysis. Whole-cell lysates were analyzed by SDS-PAGE followed by Coomassie staining or Western Blot using an anti-puromycin antibody.
Cell viability
Cell viability was assessed 24 h post-transfection by inducing stress with 500 μM NaAsO2 for an additional 24 h. Cells were harvested (500 × g, 3 min), washed twice with PBS, and resuspended in 500 μl PBS. Propidium iodide (PI) (Biosharp, BL708A) was added (1 μg/mL final) and incubated in the dark at room temperature for 20 min to label dead/damaged cells. GFP fluorescence (transfection marker) and PI emission (excitation: 488 nm; emission: 617 nm) were simultaneously analyzed via flow cytometry (Thermo, Attune NxT). Live (GFP⁺/PI−) and dead (GFP⁺/PI⁺) cells were gated based on fluorescence intensity, with data processed using FlowJo.
MTT assay
Cell viability was assessed via the rapid MTT assay after seeding cells in 96-well plates. Culture medium was replaced with 100 µL fresh medium, followed by 10 µL of 12 mM MTT stock (MedChemExpress, HY-15924) per well; medium-only wells served as negative controls. Following 37 °C incubation for 2–5 h, medium was aspirated, and 50 µL DMSO was added to dissolve formazan crystals. Plates were further incubated at 37 °C for 10 min, thoroughly mixed, and absorbance measured at 540 nm.
Cross-linking of recombinant proteins and MS analysis
In a 100 μL reaction volume, 6 μM G3BP1-EGFP was incubated with 2 μM nGFP or nGFP-NI and 100 μM BS3 (Thermo, 21595) in reaction buffer (400 mM NaCl, 50 mM HEPES, pH 7.5) at room temperature for 30 min, then quenched with 50 mM Tris (pH 7.5) for 15 min. Proteins were precipitated with methanol/chloroform, centrifuged at 15,000 × g for 10 min, and the pellets were washed twice with methanol before resuspension in 6 M urea. After adding 10 mM TCEP (Sigma, C4706) and 20 mM iodoacetamide (Sigma, I6125) at 37 °C for 30 min, 1× TEAB (Sigma, T7408) was added to dilute urea to 1 M, followed by trypsin (Promega, V5111) for enzymatic digestion at 37 °C with rotation for 16 h. Resulting peptides were dried by vacuum centrifugation, fractionated using Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo, 84868), and finally resuspended in 0.1% formic acid (FA) for MS analysis. XL-MS analysis was performed once.
Tandem MS
LC−MS/MS analysis was performed using an UltiMateTM 3000 RSLCnano System (Thermo) coupled to an Orbitrap Exploris 480 equipped with a nanospray flex ion source (Thermo). Mobile phases A and B were water with 0.1% (v/v) FA and 80% acetonitrile with 0.1% (v/v) FA. Peptides were separated using a linear gradient from 10% to 28% B within 45 min, followed by an increase to 32% B within 6 min and further to 99% B within 2 min and re-equilibration. The MS1 spectra (350–1500 m/z) were obtained at a resolution of 60,000, an AGC target of 3 × 106 and a maximum injection time of 50 ms. HCD fragmentation was performed on the most abundant precursors exhibiting a charge state from 2 to 6 of an AGC target of 1 × 105 by isolating them in the quadrupole mode at 1.6 m/z and with fragmentation energy of 30%.
MS data analysis
Crosslinked peptides were identified using pLink 2 software (v.2.3.9) with the following search parameters: precursor mass tolerance 10 ppm, fragment mass tolerance 20 ppm, peptide length per chain 6–60 amino acids, peptide mass per chain 600–6000 Da, variable modification of Cys (+57.02146), trypsin as the enzyme, and up to 3 missed cleavages per chain. For crosslinked samples, searches were performed against FASTA files containing sequence information of nGFP, nGFP-NI, and G3BP1-EGFP.
Statistics and reproducibility
Colocalization analysis was performed using the Fiji (version 2.9.0). GraphPad Prism 9 was used for statistical analysis. Data are reported as mean ± s.d. or mean ± s.e.m. Normality (Shapiro–Wilk test) and homogeneity of variances (Brown–Forsythe test) were checked before choosing the statistical test. Two-group comparisons used unpaired two-tailed Student’s t-test (parametric) or Mann–Whitney U test (non-parametric), while >2 groups were analysed by Welch’s one-way ANOVA with Games–Howell post-hoc (parametric) or Kruskal–Wallis with Dunn’s multiple-comparison test (non-parametric). All P values were obtained from GraphPad Prism 9. P < 0.05 was considered to be statistically significant. For extremely significant results (e.g., P < 1 × 10−15), the software restricts output to 15 decimal places; in these cases, we report the smallest displayable value to ensure transparency and reproducibility. The sample size (n) represents biological replicates per experiment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Unless otherwise stated, all data supporting the results of this study can be found in the article, supplementary, and source data files. Source Data are provided with this paper. The XL-MS data have been deposited to the ProteomeXchange Consortium through the iProX63,64 repository with the dataset identifier PXD068817 [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD068817]. Source data are provided with this paper.
References
Yang, P. et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345 (2020).
Markmiller, S. et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604 (2018).
Yao, Z. et al. The divergent effects of G3BP orthologs on human stress granule assembly imply a centric role for the core protein interaction network. Cell Rep. 43, 114617 (2024).
Gwon, Y. et al. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 372, eabf6548 (2021).
Lu, S. et al. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol. 24, 1378–1393 (2022).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017).
Bracha, D., Walls, M. T. & Brangwynne, C. P. Probing and engineering liquid-phase organelles. Nat. Biotechnol. 37, 1435–1445 (2019).
Szewczyk, B. et al. FUS ALS neurons activate major stress pathways and reduce translation as an early protective mechanism against neurodegeneration. Cell Rep. 42, 112025 (2023).
Fang, M. Y. et al. Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103, 802–819 (2019).
Sun, H. et al. Polystyrene nanoparticles trigger aberrant condensation of TDP-43 and amyotrophic lateral sclerosis-like symptoms. Nat. Nanotechnol. 19, 1354–1365 (2024).
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Zhang, X. et al. Molecular mechanisms of stress-induced reactivation in mumps virus condensates. Cell 186, 1877–1894 (2023).
Manjunath, L. et al. APOBEC3B drives PKR-mediated translation shutdown and protects stress granules in response to viral infection. Nat. Commun. 14, 820 (2023).
Yang, Z. et al. Interaction between host G3BP and viral nucleocapsid protein regulates SARS-CoV-2 replication and pathogenicity. Cell Rep. 43, 113965 (2024).
Freibaum, B. D. et al. Identification of small molecule inhibitors of G3BP-driven stress granule formation. J. Cell Biol. 223, e202308083 (2024).
Sanders, D. W. et al. Competing protein-RNA interaction networks control multiphase intracellular organization. Cell 181, 306–324 (2020).
Cui, Q., Liu, Z. & Bai, G. Friend or foe: the role of stress granule in neurodegenerative disease. Neuron 112, 2464–2485 (2024).
Wolozin, B. & Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 20, 649–666 (2019).
Guillen-Boixet, J. et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 181, 346–361 (2020).
Millar, S. R. et al. A new phase of networking: the molecular composition and regulatory dynamics of mammalian stress granules. Chem. Rev. 123, 9036–9064 (2023).
Ciancone, A. M. et al. Global discovery of covalent modulators of ribonucleoprotein granules. J. Am. Chem. Soc. 145, 11056–11066 (2023).
Dong, T. et al. G3BP1/2-targeting PROTAC disrupts stress granules dependent ATF4 migracytosis as cancer therapy. J. Am. Chem. Soc. 147, 446–461 (2025).
Duan, Y. et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 29, 233–247 (2019).
Li, J. et al. Post-translational modifications in liquid-liquid phase separation: a comprehensive review. Mol. Biomed. 3, 13 (2022).
Ohn, T., Kedersha, N., Hickman, T., Tisdale, S. & Anderson, P. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 10, 1224–1231 (2008).
Zhang, X., Shu, X. E. & Qian, S. B. O-GlcNAc modification of eIF4GI acts as a translational switch in heat shock response. Nat. Chem. Biol. 14, 909–916 (2018).
Chen, Y. et al. O-GlcNAcylation determines the translational regulation and phase separation of YTHDF proteins. Nat. Cell Biol. 25, 1676–1690 (2023).
Yang, X. & Qian, K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465 (2017).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Ma, B. et al. Targeted protein O-GlcNAcylation using bifunctional small molecules. J. Am. Chem. Soc. 146, 9779–9789 (2024).
Levine, Z. G. et al. Mammalian cell proliferation requires noncatalytic functions of O-GlcNAc transferase. Proc. Natl. Acad. Sci. USA 118, e2016778118 (2021).
Los, G. V. et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 (2008).
Ge, Y. et al. Target protein deglycosylation in living cells by a nanobody-fused split O-GlcNAcase. Nat. Chem. Biol. 17, 593–600 (2021).
Ramirez, D. H. et al. Engineering a proximity-directed O-GlcNAc transferase for selective protein O-GlcNAcylation in cells. ACS Chem. Biol. 15, 1059–1066 (2020).
Ge, Y. & Woo, C. M. Writing and erasing O-GlcNAc from target proteins in cells. Biochem. Soc. Trans. 49, 2891–2901 (2021).
Lazarus, M. B., Nam, Y., Jiang, J., Sliz, P. & Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469, 564–567 (2011).
Asano-Inami, E. et al. The association of UBAP2L and G3BP1 mediated by small nucleolar RNA is essential for stress granule formation. Commun. Biol. 6, 415 (2023).
Wang, T. et al. Intracellular energy controls dynamics of stress-induced ribonucleoprotein granules. Nat. Commun. 13, 5584 (2022).
Wu, Z. et al. FAM69C functions as a kinase for eIF2alpha and promotes stress granule assembly. EMBO Rep. 24, e55641 (2023).
Hernández-Candia, C. N., Pearce, S. & Tucker, C. L. A modular tool to query and inducibly disrupt biomolecular condensates. Nat. Commun. 12, 1809 (2021).
Song, D. et al. Yin and yang regulation of stress granules by Caprin-1. Proc. Natl. Acad. Sci. USA 119, e2207975119 (2022).
Kozlov, G. et al. Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc. Natl. Acad. Sci. USA 98, 4409–4413 (2001).
Zhang, K. et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 (2015).
Zhang, K. et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173, 958–971 (2018).
Dormann, D. et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857 (2010).
Shao, W. et al. Tracking stress granule dynamics in live cells and in vivo with a small molecule. Anal. Chem. 93, 16297–16301 (2021).
Uechi, H. et al. Small-molecule dissolution of stress granules by redox modulation benefits ALS models. Nat. Chem. Biol. 21, 1577–1588 (2025).
Kristensen, O. Crystal structure of the G3BP2 NTF2-like domain in complex with a canonical FGDF motif peptide. Biochem. Biophys. Res. Commun. 467, 53–57 (2015).
Gao, X. et al. Stress granule: a promising target for cancer treatment. Br. J. Pharmacol. 176, 4421–4433 (2019).
Grabocka, E. & Bar-Sagi, D. Mutant KRAS enhances tumor cell fitness by upregulating stress granules. Cell 167, 1803–1813 (2016).
Stanton, B. Z., Chory, E. J. & Crabtree, G. R. Chemically induced proximity in biology and medicine. Science 359, eaao5902 (2018).
Xu, J. et al. Protein visualization and manipulation in Drosophila through the use of epitope tags recognized by nanobodies. eLife 11, e74326 (2022).
Zhang, Y. et al. Probing condensate microenvironments with a micropeptide killswitch. Nature 643, 1107–1116 (2025).
Ng, C. S. C., Liu, A., Cui, B. & Banik, S. M. Targeted protein relocalization via protein transport coupling. Nature 633, 941–951 (2024).
Raina, K. et al. Regulated induced proximity targeting chimeras-RIPTACs-A heterobifunctional small molecule strategy for cancer selective therapies. Cell Chem. Biol. 31, 1490–1502 (2024).
Potter, S. C. et al. Dissecting OGT’s TPR domain to identify determinants of cellular function. Proc. Natl. Acad. Sci. USA 121, e2401729121 (2024).
Blankenship, C. M. et al. Motif-dependent binding on the intervening domain regulates O-GlcNAc transferase. Nat. Chem. Biol. 19, 1423–1431 (2023).
Griffin, M. E. et al. Functional glycoproteomics by integrated network assembly and partitioning. Preprint at https://www.biorxiv.org/content/10.1101/2023.06.13.541482v1.full (2023).
Figley, M. D., Bieri, G., Kolaitis, R. M., Taylor, J. P. & Gitler, A. D. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 34, 8083–8097 (2014).
de Rozieres, C. M. & Joseph, S. Influenza A virus NS1 protein binds as a dimer to RNA-free PABP1 but not to the PABP1.Poly(A) RNA complex. Biochemistry 59, 4439–4448 (2020).
Gao, J. et al. Poly(ADP-ribose) promotes toxicity of C9ORF72 arginine-rich dipeptide repeat proteins. Sci. Transl. Med. 14, eabq3215 (2022).
Kedersha, N. et al. G3BP–Caprin1–USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 212, 845–860 (2016).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2022).
Acknowledgements
This study is supported by the National Key R&D Program of China (2024YFC3407200 to Y.G.), the National Natural Science Foundation of China (92478103 and 22277080 to Y.G.), Guangdong Basic and Applied Basic Research Foundation (2023B1515020109 to K.Z.), the Major Program of Shenzhen Bay Laboratory (S241101001 to Y.G. and S241101003 to K.Z.), the Startup Fund from Shenzhen Bay Laboratory (21230102 to Y.G.). We thank the Biomedical Research Core Facilities of Shenzhen Bay Laboratory for the technical support.
Author information
Authors and Affiliations
Contributions
Y.G. conceived the project. Y.G., K.Z., and N.W. designed the experiments. N.W. performed most experiments and analyzed the data. F.H. performed the experiments relating to validations on G3BP1 material properties. S.L. generated the CRISPR-knockin cell line and H.W. synthesized OGTAC-2. S.M. and W.F. helped the protein purification experiments and provided valuable suggestions. N.W., Y.G., and K.Z. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Michael Boyce, Dhwanit Dave, Cong Liu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, N., Hou, F., Ma, S. et al. Targeted stress granule regulation by engineering a non-catalytic O-GlcNAc transferase. Nat Commun 17, 163 (2026). https://doi.org/10.1038/s41467-025-66689-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66689-6
