
Abstract
Amidines are ubiquitous in medicines and natural products. However, robust synthetic access to a broad spectrum of diverse analogues remains challenging due to the intrinsic limitations of the two-electron (2e) pathway. To overcome these synthetic constraints, we seek to develop a variant that generates amidine carbon radical as a common chemical synthon based on a one-electron (1e) pathway, remodeling retro-synthetical analysis for amidine-containing compounds. Herein, we present a mild, modular, and practical photocatalytic system tailored for assembling the highly privileged amidine pharmacophore. By ingeniously combining thiourea with PPh3, an amidine carbon radical is generated. This radical species can be efficiently coupled with a wide array of electron-deficient alkenes, establishing a highly adaptable platform for synthesizing an aminodihydroquinoline framework adorned with diverse substitution patterns. The efficacy and versatility of this method are demonstrated by approximately 50 examples, showcasing its broad applicability, efficiency, selectivity, and functional group compatibility.
Similar content being viewed by others
Introduction
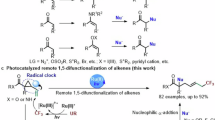
Amidines represent a crucial class of nitrogen-containing functional groups, ubiquitous in pharmaceuticals1,2,3,4 and natural products5 due to their broad bioactivity spectrum. This bioactivity arises from their ability to engage in non-covalent interactions with proteins and DNA molecular targets through hydrogen bonding, electrostatic, and cation-п interactions6,7. A prime example is Boger’s modification on vancomycin, where amidine analogues demonstrated a 500-fold increase in activity against vancomycin-resistant bacteria compared to other congeners (Fig. 1a)8,9. These findings underscore the potential amidine in crafting bioactive compounds and novel pharmaceuticals. Typically, amidines are considered as terminal functional groups rather than synthetic handles, and the most common synthetic strategies involve forming a new bond with the pre-existing skeleton10. General synthetic approaches to constructing these frameworks involve two main strategies: nucleophilic addition of primary amines to nitriles or ketenimine and Grignard reagents with carbodiimides under harsh reaction conditions11,12,13,14. Other methods encompass transformation from (thio)amide through imidoyl or thioimidate intermediates via two-step sequences15,16, and recent development using thioamide with over-stoichiometric silver promoter8. Despite these advances, several challenges persist in these transformations, including poor functional group tolerance, cumbersome chemical steps, and limited scaffold flexibility, complicating efforts to meet the growing synthetic demands17. To address these limitations, we desire to develop an innovative strategy that employs amidine radical as a universal and versatile synthon. This approach would be particularly powerful if it allowed for the facile alteration of the nitrogen unit and the alkyl skeleton, utilizing readily available starting materials. The renaissance of synthetic organic photochemistry over the past two decades has unlocked new activation modes for the classical 2e pathways by harnessing reactive yet tamable radical intermediates to facilitate the bond formations, making the formation of amidine carbon radicals possible18,19,20,21. Given the ubiquity and significance of amidine functionalities in pharmaceuticals across various therapeutic domains, developing modular and mechanistically distinct methodologies for amidine installation could expedite drug discovery processes.

a Drugs and natural products containing amidine. b Previous desulfurization with phosphine. c Our synthetic analysis for amidine carbon radical formation; d Our work based on amidine carbon radical.
In 1956, Hoffmann and coworkers carried out pioneering research, establishing the thermal/photochemical desulfurization of mercaptans using trialkylphosphites22. Based on this finding, Danishefsky23, Guo24, and Sun25 independently developed phosphine (III)-mediated cysteine-to-alanine conversions that were compatible with complex peptide and glycopeptide architectures. Subsequent breakthroughs by Jiao26, Hashmi27, and Sun28,29,30,31 revealed phosphine (III)-facilitated desulfurative radical couplings between thiols and alkenes. Most recently, Wu and coworkers reported the in-situ generation of xanthate/dithiocarbamate anions, which underwent phosphine (III)-mediated desulfurization to produce carbon radicals for bond-forming reactions32,33. The general mechanism of these transformations reveals that an alkylthiyl radical adds reversibly to phosphine (III), thereby generating a phosphoranyl radical intermediate. The subsequent β-scission is envisioned to provide a carbon radical for further transformations (Fig. 1b).
Inspired by these 1e desulfurizing strategies and the recent discovery of the formation of phosphine radical cation under photo-conditions34,35,36,37,38,39, we speculate that the phosphine radical cation generated via single electron oxidation might be well-orchestrated by thiourea in the system, which could elicit amidine carbon radical after β fragmentation, a species that remains elusive through conventional means. The ensuing amidine radical could react with various carbon double bonds to forge C–C bonds (Fig. 1c). Notably, thiourea emerges as an ideal radical precursor, owing to its modular synthesis through amines-isothiocyanates condensation. While thiourea has served as a versatile synthetic intermediate and organocatalyst40, its traditional reactivity has been primarily confined to 2e reaction mechanisms41,42. Our paradigm shift to 1e activation unlocks fundamentally distinct reactivity. Herein, we report a PPh3-mediated reductive coupling between thiourea and alkene to form aminodihydroquinolines, a privileged anticancer scaffold previously requiring multistep syntheses43. The scarcity of synthetic methods for this pharmacophore underscores the importance of our streamlined, modular approach (Fig. 1d).
Results
Reaction optimization
Initially, the generation of the amidine carbon radical was attempted using cyclohexylphenylthiourea (1) in combination with phosphine (III), and benzyl acrylate (2) was employed as a radical acceptor for the optimization of reaction conditions (Table 1 and Supplementary Table 1). The optimized reaction conditions included [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (1 mol%) as the photocatalyst, K2HPO4 (2.0 equiv.) as the base, and PPh3 (1.5 equiv.) as the S-transfer reagent in 1,2-dichloroethane (DCE) (Table 1, entry 1). Under the standard conditions, the corresponding aminodihydroquinoline (3) was obtained in 83% yield. Other photocatalysts, such as [Ru(bpy)3](PF6)2, fac–[Ir(ppy)3], 4CzIPN, and Mes-Acr-BF4, were performed inferiorly for this transformation (Table 1, entries 2 and 3). When K3PO4 was employed as the base, the yield dropped to 42% (Table 1, entry 4). Interestingly, organic bases such as TEA completely shut down the reaction, probably due to competitive reductive quenching of the excited photocatalyst through a nonproductive pathway (Table 1, entry 5). Other phosphines (III), such as diphenylmethylphospine and tricyclohexylphsophine, were less effective than triphenylphosphine (Table 1, entries 6 and 7). The solvent screening revealed that DCE was the best choice (Table 1, entry 8). Since the transformation is a net-oxidation process, oxidants such as K2S2O8, PIDA or Mn(OAc)3 were screened. However, inferior results were observed (Table 1, entry 9). It was found that the S-transfer reagent PPh3 was essential for a successful desulfurative transformation (Table 1, entry 10). Control experiments demonstrated that the reaction could not occur without the photocatalyst or light within 24 h (Table 1, entry 11).
Evaluation of substrate scopes
With the optimized reaction conditions established, we further investigated the substrate scope for both thioureas and alkenes (Fig. 2). It was found that amidine carbon radicals generated directly from thioureas could site-selectively add to the β-position of the carbonyl compounds, with no detectable branched α-position selectivity. The following carbon radical readily cyclized with the aromatic group on the thiourea to deliver aminodihydroquinoline scaffolds. Generally, aromatic substituents on thiourea bearing both electron-withdrawing groups (e.g., -F, -CN, -CF3, and -CO2Me) and electron-donating groups (e.g., -Me, -tBu, and -Ph) at the para-position could react smoothly to produce aminodihydroquinolines 3–13 in moderate to high yields (42–83%). It is noteworthy that 4-bromo (6) and 4-acyl (9) substituted thioureas tolerated the conditions well, providing an extremely important option for downstream transformations. Substituents at the meta-position gave rise to two regioisomeric products 14 in a combined yield of 34%, with a 1.3: 1 ratio. The dimethyl substituents on the meta-position and methyl substituents on the meta-position of aromatic rings had little influence on the reaction efficiency (15 and 16). Subsequently, the effect of aliphatic amine substituents on thiourea was examined. Varying the aliphatic chain length delivered the desired coupling products 17–19 in similar yields. A slightly increasing steric effect with isobutylamine, isopropylamine, 3-pentylamine, and cyclopentylamine, offered corresponding products 20–23 in 47–75% yields. Propargyl-PEG2-amine proved to be a competent substrate (24), and the terminal alkyne provided potential opportunities for biorthogonal chemistry. Leelamine, originating from rosin, also worked well for this transformation (25). Notably, highly sterically hindered amines such as benzhydrylamine, α-tertiary amine, and amantadine were tolerated in this reaction, providing corresponding products (26–29) in 35–72% yields. Notably, secondary amines, including diethylamine, 4-methylpiperidine, 3,3-difluoroazetidine, and spiroazetidine, could be coupled efficiently to furnish the corresponding aminodihydroquinolines 30–33 in 46–57% yields. Interestingly, the monosubstituted thiourea was found to deliver product 34 in 64% yield. This result is particularly noteworthy given the unsubstituted nitrogen atom presents a versatile handle for subsequent structural modifications. Due to the natural abundance and availability, various amino acids and peptides substituted thioureas were prepared for the transformation. Thioureas bearing natural amino acids, such as glycine (35), alanine (36), tyrosine (37), proline (38), and dipeptides like Ala-Gly (39) and Ala-Pro (40), were competent reaction partners and corresponding yields varied from 35 to 54%. When K3PO4 was employed as the base, the desired product 41 was obtained in high yield from diphenyl thiourea. Subsequently, many alkenes were examined. Acrylic esters derived from phenol, ethanol, and tert-butanol performed well, yielding products 42–44 in good yields. Other electron-withdrawing groups, such as nitrile and ketone, could also capture the amidine radical, giving products 45–47 in moderate yields. Different substituent patterns on alkenes were tested, revealing that 1,1-disubstituted alkenes 48–50, 1,2-disubstituted alkenes 51, and trisubstituted alkenes 52 all worked well, delivering final aminodihydroquinoline with various substituents patterns. Finally, acrylic esters derived from natural sources, such as menthol and piperonyl alcohol, provided corresponding products 53 and 54 in good yields. It is intriguing to observe that when 2-vinyl pyridine was employed as a radical acceptor, it exclusively formed a Giese-type addition product 55, albeit in moderate yield.

aReaction conditions: thiourea (0.1 mmol), 2 (0.2 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (0.001 mmol), PPh3 (0.15 mmol), K2HPO4 (0.2 mmol), DCE (2.0 mL), rt (around 35 oC), 18 W x 2 Blue LED (455 nm), 24 h. b K3PO4 was used. cWith 30 mol % K2S2O8. dReaction was performed at 80 oC with 2.0 equiv. PPh3. EWG, electron-withdrawing group.
To demonstrate the practicability of this methodology, the large-scale preparation experiment was conducted at 1 mmol, and a comparable yield was observed. Furthermore, the modularity of this transformation was demonstrated by a three-component reaction. Specifically, equimolar amounts of aryl isothiocyanate and alkylamine were mixed in the DCE for 5–10 min at room temperature. Subsequently, benzyl acrylate was added under standard conditions in one pot procedure. Remarkably, this one-pot, three-component coupling reaction proceeded efficiently, yielding products comparable to those obtained in the corresponding two-component reactions. As illustrated in Fig. 3B, a small selection of substrates was chosen to show the generality of this process, demonstrating that complexity can be rapidly generated from this method (Fig. 3).

A Large-scale synthesis. B Three-component synthesis. aReaction conditions: thiourea (0.1 mmol), 2 (0.2 mmol), DCE (2.0 mL), 5 min, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (0.001 mmol), PPh3 (0.15 mmol), K2HPO4 (0.2 mmol), rt (around 35 oC), 18 W x 2 Blue LED (455 nm), 24 h.
Mechanistic studies
To elucidate the mechanism of this photoredox transformation, several experiments were devised. The radical inhibitor TEMPO was added to the model reaction, which completely inhibited the coupling reaction, implying that a radical pathway is likely involved (Fig. 4A). Although thiols have been demonstrated to undergo facile oxidation by photocatalysts44, recent reports34,35,36,37,38,39 and our work indicate that phosphine(III) can also be readily oxidized by excited photocatalysts45. To this end, Stern-Volmer luminescence quenching (SV) analysis was performed. The results revealed that PPh3 was effective in quenching the excited state of the photocatalyst. Conversely, thiourea 1 alone did not exhibit any significant quenching effect on the excited state of the photocatalyst. Interestingly, when thiourea 1 was combined with a base, it demonstrated a modest quenching capability. Additionally, a cyclic voltammetry experiment was conducted to confirm the possibility of SET between PPh3 and photocatalyst (See SI sections 3.1 and 3.2 for the details). Furthermore, the radical mechanism of this protocol was also proven by the electron paramagnetic resonance (EPR) investigation. Spectral simulation showed good agreement with the experimental data, suggesting formation of amidine carbon radical and hydroxy radical during the transformation (Fig. 4B and SI section 3.3). Meanwhile, the isolation of amidine 55 and its dimer 56 provided strong evidence for the formation of an amidine carbon radical intermediate during the reaction. Based on these experimental findings and insights from previous studies, a SET mechanism for this desulfurization reaction is tentatively proposed in Fig. 4C. The excited photocatalyst undergoes reductive quenching by PPh3, generating a phosphorus radical cation. This species then engages in the desulfurization process via β-scission, resulting in the generation of an amidine carbon radical, which is subsequently trapped by benzyl acrylate. Due to its proximity, the carbon radical α to the ester readily undergoes cyclization with the phenyl group, furnishing the dihydroquinoline scaffold. Further oxidation then rearomatizes the intermediate to give the final product.

A Radical inhibit experiment. B Stern-Volmer and EPR studies for Ir catalyst system. C Proposed mechanism for Ir catalyst system. D Stern-Volmer studies for catalyst-free system. E Proposed mechanism for catalyst-free system. TEMPO, 2,2,6,6-tetramethyl-1-piperinedinyloxy.
Interestingly, through the course of the reaction optimization, it was observed that the desired coupling product 3 was formed in 25% yield after 144 h in the absence of photocatalyst. The reaction time could be shortened to 48 hours when a more intense light source was employed. Furthermore, the yield dropped significantly when TEMPO was added under these conditions, implying that a radical pathway is involved in the reaction mechanism even without a photocatalyst. Analysis of the UV-vis spectra revealed that no ground-state aggregation of the substrates occurred (EDA complex). For the deprotonated thiourea (DpTU), UV-vis analysis showed a bathochromic shift extending beyond 450 nm (Fig. 4Da), which is consistent with previous studies indicating that thiourea can act as a potent photocatalyst under basic conditions46. The fluorescence spectrum of DpTU showed its maximum emission wavelength at 405 nm (Fig. 4Db). Subsequently, a Stern-Volmer luminescence quenching analysis was conducted with DpTU. The results indicated that oxygen molecules were capable of efficiently quenching the photo-excited DpTU. Moreover, a saturation effect was noticed when the solution was bubbled with O2 for over 20 seconds (Fig. 4Dc). Additionally, the preformed MBH intermediate (phosphonium salt) showed weak quenching ability compared with O2 (Fig. 4Dd)33. Based on all the experimental results and literature precedents47, an alternative mechanism was proposed. In the reaction system, DpTU could reach its photo-excited state upon blue LED irradiation and generate an S-centered radical under air atmosphere. This nascent radical then couples with PPh3, followed by β-scission to yield the amidine carbon radical (Fig. 4E). While both mechanisms appear reasonable in this reaction, we suggest that the reaction performed more efficiently in the presence of Ir(III) photocatalyst since the reactions typically reach completion within 24 h with high yields under such conditions.
In vitro antifungal activities
Phytopathogenic fungi pose a significant threat to the stability and safety of agricultural production and ecosystems48,49,50,51. In this context, we have investigated the bioactivities of a series of amidine products against phytopathogenic fungi (F.graminearum), using boscalid as a positive control. As shown in Table 2, all tested compounds exhibit fair to good mycelial growth inhibition activity toward F.graminearum. Amidine compound 25, derived from leelamine, shows comparable activity to boscalid. Notably, amidine compounds 3, 5, and 51 demonstrate much more potent activities than the positive control, with inhibition rates of 72%, 65%, and 64%, respectively. The current preliminary anti-fungal activities indicate a promising prospect for further scaffold optimization based on amidine derivatives.
In conclusion, we have developed a modular and practical approach generating amidine carbon radical from thiourea under milder conditions, which reacted with various alkenes to furnish a diverse range of aminodihydroquinoline scaffolds—privileged structures in medicinal chemistry that previously required multi-step syntheses (up to four steps). Our approach streamlines access to these pharmacophores, offering new opportunities for the rapid construction of bioactive compounds and analogues. The mild reaction conditions and abroad functional group compatibility extend the application to natural products, amino acids, and peptides, making them preeminent and efficient species for potential biological and chemical applications, which is complementary to the previous methods for preparing these kinds of valued-added amidine motifs. Bioactivity screens show that the amidine products from this transformation have promising potential for growth inhibition toward F.graminearum. Encouraged by this amidine carbon radical synthon, we conduct further chemical transformations in our lab to redefine the retrosynthetic analysis of amidine-containing compounds.
Methods
General information
All reactions were performed in dry solvents under an N2 atmosphere and anhydrous conditions. DCM, THF, toluene, diethyl ether, and MeCN to be used in anhydrous reaction mixtures were dried by passage through activated alumina columns immediately prior to use. All other reagents were used as received from commercial sources. Reactions were monitored through thin-layer chromatography (TLC) on 0.25-mm silica gel plates and visualized under UV light. Flash column chromatography (FCC) was performed using Flash silica gel (60-Å pore size, 40–63 μm). NMR spectra were recorded on Bruker Avance-400 or -600 instrument, calibrated to CD(H)Cl3 as the internal reference (7.26 and 77.0 ppm for 1H and 13C NMR spectra, respectively). 1H NMR spectral data were reported in terms of chemical shift (δ, ppm), multiplicity, coupling constant (Hz), and integration. 13C NMR spectral data were reported in terms of chemical shift (δ, ppm). The following abbreviations indicated the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. High-resolution mass spectra were recorded using a SCIEX X500R LC-Q-TOF, ESI ion Source. The 18 W blue LED lamps were directly got from the supermarket. The pathogenic fungi were purchased from the Agricultural Culture Collection of China, which were preserved in agar slants at 4 °C.
General procedure for the synthesis of 3–54
The phenylthiourea derivatives (0.1 mmol), the acrylic ester derivatives (0.2 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (0.001 mmol, 1 mol%, 1.1 mg), PPh3 (1.5 equiv., 0.15 mmol, 39.3 mg), K2HPO4 (2.0 equiv., 0.2 mmol, 35.0 mg) and DCE (2 mL, 0.05 M) were added sequentially to a 4 mL clear-colored glass vial equipped with a magnetic stir bar. The reaction mixture was stirred under the irradiation of 18 W blue LEDs at room temperature. The reaction mixture was monitored by TLC until the starting materials were consumed. Then, the solution was concentrated in a vacuum and purified by column chromatography to yield the products.
In vitro antifungal activities
Each target compound was dissolved in DMSO to prepare the stock solution (10.0 g/L). The stock solution was added to the PDA medium, and the concentration of target compounds in the medium was 50.0 mg/L. Pure DMSO without the target compounds was utilized as the blank control, and boscalid was coassayed as the reference compound. Fresh dishes with a diameter of 5 mm were taken from the edge of the PDA-cultured fungi colonies and inoculated on the above three PDA media. Each treatment was tested for three replicates, and the antifungal effect was averaged. The relative inhibitory rate I (%) of all the tested compounds was calculated through the equation: I (%) = [(C − T)/(C − 5)] × 100. In this equation, I is the inhibitory rate, and C and T are the colony diameters of the blank control (mm) and treatment (mm), respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and Supplementary Information files. All data are available from the corresponding author upon request.
References
Greenhill, J. V., Pharm, B. & Lue, P. Amidines and Guanidines in Medicinal Chemistry. Prog. Med. Chem. 30, 203–326 (1993).
Soeiro, M. N. C. et al. Novel Amidines and Analogues as Promising Agents Against Intracellular Parasites: A Systematic Review. Parasitology 140, 929–951 (2013).
Wang, M. J. et al. Design, Synthesis, Mechanisms of Action, and Toxicity of Novel 20(S)-Sulfonylamidine Derivatives of Camptothecin as Potent Antitumor Agents. J. Med. Chem. 57, 6008–6018 (2014).
Zainal Abidin, A. et al. Amidine Containing Compounds: Antimicrobial Activity and Its Potential in Combating Antimicrobial Resistance. Heliyon 10, e32010 (2024).
Kumamoto, T. Amidines and Guanidines in Natural Products and Medicines. In Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts ed. Ishikawa, T. ch. 10, pp 295–313 (2009).
Zhu, Y., Nikolic, D., Van Breemen, R. B. & Silverman, R. B. Mechanism of Inactivation of Inducible Nitric Oxide Synthase by Amidines. Irreversible Enzyme Inactivation without Inactivator Modification. J. Am. Chem. Soc. 127, 858–868 (2005).
Munde, M. et al. Induced Fit Conformational Changes of a “Reversed Amidine” Heterocycle: Optimized Interactions in a DNA Minor Groove Complex. J. Am. Chem. Soc. 129, 5688–5698 (2007).
Okano, A., James, R. C., Pierce, J. G., Xie, J. & Boger, D. L. Silver(I)-Promoted Conversion of Thioamides to Amidines: Divergent Synthesis of a Key Series of Vancomycin Aglycon Residue 4 Amidines That Clarify Binding Behavior to Model Ligands. J. Am. Chem. Soc. 134, 8790–8793 (2012).
Okano, A., Isley, N. A. & Boger, D. L. Peripheral Modifications of [Ψ[CH2NH]Tpg4]Vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics. Proc. Natl Acad. Sci. Usa. 114, E5052–E5061 (2017).
Kochi, T. & Ellman, J. A. Asymmetric α-alkylation of N’-tert-Butanesulfinyl Amidines. Application to the Total Synthesis of (6R,7S)-7-Amino-7,8-Dihydro-α-Bisabolene. J. Am. Chem. Soc. 126, 15652–15653 (2004).
Bae, I., Han, H. & Chang, S. Highly Efficient One-Pot Synthesis of N-Sulfonylamidines by Cu-Catalyzed Three-Component Coupling of Sulfonyl azide, Alkyne, and Amine. J. Am. Chem. Soc. 127, 2038–2039 (2005).
Xu, H. D. et al. Copper-Catalyzed Cyclization/Aza-Claisen Rearrangement Cascade Initiated by Ketenimine Formation: An Efficient Stereocontrolled Synthesis of α-Allyl Cyclic Amidines. Angew. Chem. Int. Ed. 53, 9284–9288 (2014).
Veeranna, K. D., Das, K. K. & Baskaran, S. One-Pot Synthesis of Cyclopropane-Fused Cyclic Amidines: An Oxidative Carbanion Cyclization. Angew. Chem. Int. Ed. 56, 16197–16201 (2017).
Liu, B. et al. Direct Transformation of Terminal Alkynes into Amidines by a Silver-Catalyzed Four-Component Reaction. J. Am. Chem. Soc. 141, 1593–1598 (2019).
Xie, J. et al. Total synthesis of [Ψ[C(=S)NH]Tpg4]Vancomycin Aglycon, [Ψ[C(=NH)NH]Tpg4]Vancomycin Aglycon, and Related Key Compounds: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J. Am. Chem. Soc. 134, 1284–1297 (2012).
O’Brien, E. A., Sharma, K. K., Byerly-Duke, J., Camacho, L. A. III & VanVeller, B. A General Strategy to Install Amidine Functional Groups Along the Peptide Backbone. J. Am. Chem. Soc. 144, 22397–22402 (2022).
Aly, A. A., Brӓse, S. & Gomaa, M. A. M. Amidines: Their Synthesis, Reactivity, and Applications in Heterocyclic Synthesis. Arkivoc 2018, 85–138 (2018).
Marzo, L., Pagire, S. K., Reiser, O. & Koenig, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?. Angew. Chem. Int. Ed. 57, 10034–10072 (2018).
Crespi, S. & Fagnoni, M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 120, 9790–9833 (2020).
Huang, C. Y., Li, J. & Li, C. J. Photocatalytic C(sp3) Radical Generation via C-H, C-C, and C-X Bond Cleavage. Chem. Sci. 13, 5465–5504 (2022).
Cannalire, R. et al. Visible Light Photocatalysis in the Late-Stage Functionalization of Pharmaceutically Relevant Compounds. Chem. Soc. Rev. 50, 766–897 (2021).
Hoffmann, F. W., Ess, R. J., Simmons, T. C. & Hanzel, R. S. The Desulfurization of Mercaptans with Trialkyl Phosphites. J. Am. Chem. Soc. 78, 15652–15653 (1956).
Wan, Q. & Danishefsky, S. J. Free-Radical-Based, Specific Desulfurization of Cysteine: a Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angew. Chem. Int. Ed. 46, 9248–9252 (2007).
Gao, X. F., Du, J. J., Liu, Z. & Guo, J. Visible-Light-Induced Specific Desulfurization of Cysteinyl Peptide and Glycopeptide in Aqueous Solution. Org. Lett. 18, 1166–1169 (2016).
Shi, S., Li, R., Rao, L. & Sun, Z. A. Mild, General, and Metal-Free Method for Site-Specific Deuteration Induced by Visible Light Using D2O as the Source of Deuterium Atoms. Green. Chem. 22, 669–672 (2020).
Qin, Q., Wang, W., Zhang, C., Song, S. & Jiao, N. A Metal-Free Desulfurizing Radical Reductive C-C Coupling of Thiols and Alkenes. Chem. Commun. 55, 10583–10586 (2019).
Zhang, L. et al. Reductive C–C Coupling by Desulfurizing Gold-Catalyzed Photoreactions. ACS Catal. 9, 6118–6123 (2019).
Shi, S. et al. Three-Component Radical Homo Mannich Reaction. Nat. Commun. 12, 1006 (2021).
Liu, Y., Zhou, X., Li, R. & Sun, Z. Photocatalytic Synthesis of γ,γ-Difluoroallylic Ketones and δ,δ-Difluoroallylic Ketones via a Desulfurative/Defluorinative Alkylation ProcessThree-Component Radical Homo Mannich Reaction. Org. Lett. 26, 6424–6427 (2024).
Miao, P., Li, R., Lin, X., Raom, L. & Sun, Z. Visible-light induced metal-free cascade Wittig/hydroalkylation reactions. Green. Chem. 23, 1638–1641 (2021).
Dong, Y., Li, R., Zhou, J. & Sun, Z. Synthesis of Unsymmetrical 1,4-Dicarbonyl Compounds by Photocatalytic Oxidative Radical Additions. Org. Lett. 23, 6387–6390 (2021).
Guo, H. M. & Wu, X. Selective Deoxygenative Alkylation of Alcohols via Photocatalytic Domino Radical Fragmentations. Nat. Commun. 12, 5365 (2021).
Guo, H. M., Wang, J. J., Xiong, Y. & Wu, X. Visible-Light-Driven Multicomponent Reactions for the Versatile Synthesis of Thioamides by Radical Thiocarbamoylation. Angew. Chem. Int. Ed. 63, e202409605 (2024).
Zhang, M., Xie, J. & Zhu, C. A General Deoxygenation Approach for Synthesis of Ketones from Aromatic Carboxylic Acids and Alkenes. Nat. Commun. 9, 3517 (2018).
Zhang, M., Yuan, X., Zhu, C. & Xie, J. Deoxygenative Deuteration of Carboxylic Acids with D2O. Angew. Chem. Int. Ed. 58, 312–316 (2019).
Alvarado, J. I. M., Ertel, A. B., Stegner, A., Stache, E. E. & Doyle, A. G. Direct Use of Carboxylic Acids in the Photocatalytic Hydroacylation of Styrenes to Generate Dialkyl Ketones. Org. Lett. 21, 9940–9944 (2019).
Guo, Y., Wang, R., Song, H., Liu, Y. & Wang, Q. Visible-Light-Induced Deoxygenation/Defluorination Protocol for Synthesis of γ,γ-Difluoroallylic Ketones. Org. Lett. 22, 709–713 (2020).
Rossi-Ashton, J. A., Clarke, A. K., Unsworth, W. P. & Taylor, R. J. K. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 10, 7250–7261 (2020).
Ruzi, R., Liu, K., Zhu, C. & Xie, J. Upgrading Ketone Synthesis Direct from Carboxylic Acids and Organohalides. Nat. Commun. 11, 3312 (2020).
Parvin, T., Yadav, R. & Choudhury, L. H. Recent Applications of Thiourea-Based Organocatalysts in Asymmetric Multicomponent Reactions (AMCRs). Org. Biomol. Chem. 18, 5513–5532 (2020).
Ding, Q. & Wu, J. A Facile Route to 2,4-Dihydro-1H-Benzo[d][1,3]Thiazines via Silver-Catalyzed Tandem Dddition-Cyclization Reactions. J. Comb. Chem. 10, 541–545 (2008).
Otani, T. et al. Lewis Acid-Catalyzed or Base-Promoted Regioselective Cycloisomerization of N-Imidoyl-o-alkynylanilines for Synthesis of N-Imidoyl-(1H)-indoles and 4-Alkylidene-3,4-dihydroquinazolines. Adv. Synth. Catal. 357, 1483–1492 (2015).
Lee, E. et al. Synthesis and Anticancer Activity of Aminodihydroquinoline Analogs: Identification of Novel Proapoptatic Agents. Bioorg. Med. Chem. Lett. 23, 3976–6978 (2013).
Bao, X., Yu, W. & Wang, G. Thiols as Powerful Atom Transfer Catalyst: Opportunities in Photoredox-Mediated Reactions. Adv. Synth. Catal. 365, 2299–2309 (2023).
Zhang, K., Liu, J., Li, Y., Xu, Y. & Cai, L. Photocatalytic C(sp3)–P and C(sp2)–P Bond Formation via a Phosphorus Radical Cation. Org. Lett. 26, 9056–9061 (2024).
Dey, D. et al. Single Electron Transfer Catalysis by Diphenylthiourea under Visible Light Photoredox Conditions. Org. Chem. Front. 10, 5248–5253 (2023).
Wu, J. et al. Brønsted Acid-Catalysed Aerobic Photo-Oxygenation of Benzylic C-H bonds. Green. Chem. 25, 940–945 (2023).
Yang, Z. et al. Novel Aromatic Carboxamides from Dehydroabietylamine as Potential Fungicides: Design, Synthesis and Antifungal Evaluation. Arab. J. Chem. 15, 104330 (2022).
Wang, W. Y. et al. Design, Synthesis, Anticancer Activity and Mechanism Studies of Novel 2-Amino-4-Aryl-PyrimidineDerivatives of Ursolic Acid. N. J. Chem. 46, 2335–2350 (2022).
Yang, Z. et al. Synthesis and Antifungal/Anti-Oomycete Activity of Novel Camphor-Based Sulfonate Derivatives as Potential SDH Inhibitors. J. Mol. Struct. 1270, 133959 (2022).
Li, A. L. et al. Synthesis, Cytotoxicity and Apoptosis-Inducing Activity of Novel1H-Benzo[d]imidazole Derivatives of Dehydroabietic Acid. J. Chin. Chem. Soc. 67, 1668–1678 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22001136 to K.Z. & 22001124 to L.C.), Jiangsu Provincial Double-Innovation Doctor Program (JSSCBS20210555 to K.Z.), and the Innovation and Entrepreneurship Team of Jiangsu Province (JSSCTD202345 to L.C.), the Natural Science Foundation of Jiangsu Province (BK2024021894 to L.C.).
Author information
Authors and Affiliations
Contributions
X.Y. and J.Y. performed the experiments and analyzed the data. H.M. and G.L. performed bioactivity screen. K.Z. and L.C. designed and directed the project and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
L.C., J.Y., X.Y., and K.Z. declare the following competing interests that one Chinese patent has been registered (CN202411888030.0). G.L. and H.M. declares no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yao, X., Yu, J., Lin, G. et al. Photocatalytic generation of amidine carbon radicals for aminodihydroquinoline synthesis. Nat Commun 16, 11547 (2025). https://doi.org/10.1038/s41467-025-66701-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66701-z
