
Abstract
Functions of the human gut virome are little understood, particularly for the hyperabundant prophages integrated in prokaryotic genomes. Here we identified 254,273 prophage sequences in 47.7% of 289,232 human gut metagenomic genomes, significantly expanding the known taxonomic and functional diversity of prophages in the human gut microbiome. Analysis of 8503 gut metagenomic samples showed the ratios of lysogens (cells harboring prophages) to non-lysogens varied widely associated with age, health condition, and geography, with the latter linked to industrialization. Notably, the alterations of the prophage-encoded genes exhibited disease-specific patterns. For inflammatory bowel diseases, the prophage-encoded futC gene, encoding α−1,2-fucosyltransferase, was less prevalent in affected patients. This enzyme was experimentally validated to direct 2-fucosyllactose (2’-FL) biosynthesis in vitro. Here we show that 2’-FL could diminish colitis in mice induced by treatment with dextran sodium sulfate. Mechanistically, 2’-FL promoted maintenance of mucosal barrier integrity, leading to intestinal IgA secretion and intraepithelial CD4⁺CD8αα⁺ T cell development mediated by the gut microbiome. Together, our findings thus link lysogeny to human age, geography, and disease, and demonstrate an immunomodulatory mechanism of prophage-encoded genes in alleviating colitis.
Similar content being viewed by others


Introduction
Viruses that infect bacteria are known as bacteriophages (hereafter phages) and make up the majority of the human gut virome. Phages generally replicate via two different strategies, the lytic and lysogenic cycles1. In the lytic cycle, phages infect bacterial cells, replicate their genomes, assemble new particles, and exit cells through lysis. Lysogeny refers to the integration of the temperate phage genomes into the bacterial genomes to form prophages. Integrated prophage genomes can remain quiescent until sensing an induction signal, such as DNA damage. Prophage genomes then excise from the host chromosomes and commence lytic replication.
The operation of these mechanisms in the gut virome has not been fully clarified. Lytic phages have been well studied both in vitro and in the gut microbiome due to their ability to kill the host cells. Multiple types of lytic phages have been assembled into phage cocktails to treat bacterial infections or diseases related to dysbiosis2,3,4. Compared to lytic phages, relatively little research has been conducted on lysogeny in the context of naturally occurring bacterial communities5.
Lysogeny may contribute to the genetic variation of lysogenic bacteria by disrupting host genes during integration or adding new phage-encoded functions6. Lysogenic conversion, whereby a bacterium acquires virulence functions from an integrated prophage, has been described in numerous bacterial pathogens7. Genes expressed by prophages can also contribute to antibiotic resistance, lyse competitive bacteria, and influence the efficacy of the immunotherapy8,9,10,11. Pilot studies have begun to catalog prophages in humans and mice12,13,14,15,16, indicating the pervasiveness of lysogeny. In a recent study, over 80% of bacterial strains derived from infant feces harbored prophages and produced phage particles in the presence of an inducing signal17. Despite its ubiquity, the functional consequences of prophages in the human gut microbiome remain understudied.
Here, we developed a pipeline to identify prophages and queried 289,232 metagenome-assembled genomes (MAGs) and 8503 publicly available gut metagenomic samples from individuals with diverse health conditions. We quantified the landscape of candidate lysogens, largely expanded known prophage diversity, and observed an altered ratio of lysogens to non-lysogens in multiple disease states. We also found disease-specific functional changes in prophage-encoded genes, notably identifying a depletion of the prophage-encoded futC genes in the gut microbiome of IBD patients. Experimental validation showed that the prophage-encoded gene could synthesize the oligosaccharide 2’-FL, which alleviates colitis by promoting intraepithelial T-cell development and intestinal IgA secretion in a microbiota-dependent way. This study provides a comprehensive view of lysogens in the human gut microbiome, highlighting the immunomodulatory roles of prophage-encoded genes and offering insights into functional mechanisms mediated by the human gut virome.
Results
Lysogens are prevalent in the human gut microbiome and encode diverse functional genes
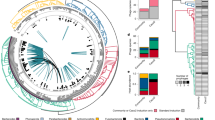
We developed a pipeline to identify candidate prophages from metageome sequencing data (Supplementary Fig. 1a, see Methods). Briefly, contigs were first assembled and subsequently grouped into MAGs through genomic binning. To maximize detection sensitivity, we integrated VirSorter2 and CheckV to identify putative phage regions within the contigs. Candidate prophages were detected by examining the presence of flanking bacterial genes adjacent to these phage regions within the same contig. To assess the accuracy of our pipeline, we induced prophages from 23 bacterial strains isolated from the gut using the DNA-damaging agent mitomycin C. The induced virus-like particles were purified, and the nucleic acids were sequenced. The induced prophage reads were then aligned to the corresponding bacterial genome (Fig. 1a and Supplementary Data 1). The induced prophage reads were mainly mapped to putative prophage sequences rather than non-phage regions (Fig. 1b). Active prophages were characterized as regions with prophage content exhibiting greater coverage compared to non-phage regions. In total, we predicted 88 prophages from these bacteria, of which 41 were identified as active prophages (Fig. 1c). Proficiency in prophage induction was correlated with the CheckV completeness score, and there was a dramatic increase in the active prophage detection ratio when the CheckV completeness score was >10% (Fig. 1c). Thus a cutoff of CheckV completeness score at 10% completeness was used for further prediction. Lysogens were defined as MAGs containing at least one prophage candidate. A total of 21 lysogens out of 23 strains were identified with default cutoffs (Fig. 1d). We further benchmarked our pipeline against other tools designed to identify prophages from metagenomic data. The results indicated that our pipeline achieved the highest precision in identifying active prophages, while performing comparably to existing tools across other metrics (Supplementary Data 2). Given that mitomycin C might not induce all prophages, we have confidence in its ability to predict lysogens reliably. Due to the challenge of determining the activity status of a prophage, we have coined the term, “prophage islands” (PIs), to describe all prophage candidates predicted by this pipeline.

a The experimental methodology for the prophages identification benchmarking (Methods) (Created with BioRender.com, https://BioRender.com/pff0n4k). b Induced virome sequencing reads coverage distribution across whole bacterial genomes and predicted prophage regions. c The relationship between virome sequencing coverage and the prophage completeness score. The left y axis presents a scatter plot depicting the correlation between the prophage completeness (x axis) and virome sequencing reads coverage (y axis, log-transformed). Deep red points signify active prophages, whereas deep blue points indicate non-active prophages. The right y axis displays a LOESS-fitted mean curve showing the correlation between the threshold of prophage completeness (x-axis) and the proportion of identified active prophages (y axis). Dashed lines indicate reference thresholds at x = 10 and y = 0.9, and the gray-shaded region shows a 95% confidence interval. d Distribution of lysogens (orange) and non-lysogens (blue) predicted by the pipeline on the 23 lab bacterial strain genomes. The circular ring on the right displays the distribution of bacteria based on the presence of active prophages. e The distribution of lysogens (orange) and non-lysogens (blue) in the UHGG database. f The distribution of PIs at the family-level and across different bacterial hosts. g Distribution of genomic distances of PIs was assessed using the Mash distance, both between and within bacterial host phyla. Mash distance 0 indicates identical genomes, and distance 1 indicates completely distinct genomes. Completely distinct genomes were excluded from this analysis. The heatmap indicates the relative number of similar PI pairs shared across different phyla. The number was corrected by the MAG numbers of the two phyla in each cell. h Relative frequency of PI shared within the same genus. The x axis shows the ratio of shared PIs within the same taxon, adjusted for the percentage of that taxon among all bacterial genomes. The median is shown as the center line, the 25th–75th percentiles define the box edges, and whiskers extend to 1.5× IQR. Results are based on 10 independent resampling iterations (effective sample sizes: n = 10, 10, 10, 6, 10, 10, 10, 10, 10, 10, 3). i Percentage of ORFs from PIs annotated by Pfam database and the most prevalent Pfam terms associated with these ORFs.
We first profiled the prophage landscape of the human gut microbiome. We applied the pipeline to 289,232 bacterial and archaeal genomes retrieved from the Unified Human Gastrointestinal Genome (UHGG) V2.0 database18, which is one of the largest available collections of the MAGs derived from the human gut microbiome. A total of 137,950 genomes containing 254,273 PIs were identified as lysogens, accounting for 47.7% of all genomes (Fig. 1e and Supplementary Data 3). The number of integrated PIs varied widely among different genomes (Supplementary Fig. 1b, c). A close association was observed between specific bacterial phenotypes and the distribution of lysogens. For example, the percentage of lysogens varied among aerobic and anaerobic bacteria, with 68.9% versus 47.3%. Frequencies also differed between Gram-positive and Gram-negative bacteria, with 45.0% versus 54.2% (Supplementary Fig. 1d, e). The lysogens were widely distributed over almost all the observed bacterial taxa. However, the percentage of lysogens varied greatly among different phyla, with Proteobacteria showing a high ratio of 62.2% (Supplementary Fig. 1f). For taxonomic profiling of human gut prophages, we selected 8306 almost complete prophages (Completeness score > 0.9) for clustering analysis. Seventy-one family-level viral operational taxonomic units (vOTUs), 436 genus-level vOTUs, and 3852 species-level vOTUs were identified. However, only 1.45% of species-level vOTUs, 12.7% genus-level vOTUs, and 46.5% family-level vOTUs were assigned to pre-existing taxonomic classifications, indicating the great diversity of human gut prophages (Supplementary Fig. 1g). The predominant families identified were Peduoviridae, comprising 17.6% of the total, and Winoviridae, accounting for 5.6% (Fig. 1f). Next, for the family-level vOTUs, we associated them with their host taxonomy and found that some prophages seem to preferentially infect certain genera of bacteria (Fig. 1f). For example, Microviridae and Autographiviridae mainly infected Phascolarctobacterium while Inoviridae only infected Vibrio.
We leveraged this dataset to quantify the host ranges of the PIs systematically. We evaluated their genomic distance values based on the taxonomic classification of their corresponding bacterial hosts, revealing lower diversities among prophages infecting bacterial hosts within the same phylum (Fig. 1g). We then characterized closely related prophages (Mash distance <0.05) shared by multiple hosts to investigate the potential for phage-mediated horizontal gene transfer across different bacterial hosts. Among 140,217 pairs of PIs shared by different bacterial genomes, 2.46% were shared across different genera (Supplementary Fig. 1h). This suggests that prophages are highly host-specific and might be horizontally transferred at the genus-level on rare occasions, which is consistent with a previous study19. The frequency of shared prophage pairs was higher within the genera Ruminococcus, Parabacteroides, and Alistipes (Fig. 1h and Supplementary Fig. 1i).
To explore the functional landscapes of the PIs in the human gut, we predicted 14,090,553 ORFs in these 254,273 PIs. All these ORFs were annotated against the Pfam database20. A total of 8,789,682 (62.4%) ORFs were annotated and categorized as specific Pfam terms (Fig. 1i). The most abundant terms involved proteins involved in phage structure, lysogenic/lytic replication cycle regulation, and DNA binding (Fig. 1i). Further functional enrichment analysis of Pfam terms with frequencies over 10,000 showed that prophages carry many cargo genes—unrelated to the phage replication cycle—that are linked to metabolism and pathogenicity (Supplementary Fig. 2a), suggesting that gut prophages may play roles in these functions. Functional annotation for the 8306 almost complete prophages using virus-tailored tools, Pharokka and PHROGs database21, revealed similar results (Supplementary Fig. 2b).
Collectively, we provided an overview of the prophage landscape by analyzing prophages in ~300,000 human gut-derived bacterial genomes. Our analysis revealed the high abundance of lysogens in the gut, uncovered novel features of prophages, identified a high diversity of previously uncharacterized prophages, and highlighted their extensive functional potential.
Lysogen frequency is linked with age, gender, and industrialization, with a core prophage-encoded cargo gene profile
We next investigated whether variations in lysogens are associated with specific human characteristics by analyzing large-scale population metagenomic datasets. We collected and analyzed 8503 human gut metagenomes from 46 datasets from 18 countries (Fig. 2a). Among them, 4312 were from healthy individuals of different ages, while others were from nine disease states, including inflammatory bowel diseases (IBD), diabetes, and others (Supplementary Data 4). Overall, 159,667 genomic bins (Completeness >50% & Contamination <10%) were obtained, of which 66,463 were lysogens (41.6%, Fig. 2b), which is comparable to the UHGG database (47.7%). In total, 24.6% of individuals had >50% of their gut bacteria as lysogens (Fig. 2b). A total of 275 samples contain only lysogens, while 191 contain only non-lysogens (Fig. 2b). The taxonomic classification of these lysogens and non-lysogens was consistent with that of the UHGG database (Fig. 2c).

a The geographic distribution, age, gender, and disease status of 8503 samples (Map data source: http://bzdt.ch.mnr.gov.cn/). See Supplementary Data 3 for detailed metadata. b The proportion of lysogens in 8503 individuals. c Taxonomic assignment of lysogens (left) and non-lysogens (right) in 8503 individuals at the phylum level. The proportion of lysogens within the bacterial community across different d age groups, e genders, f levels of industrialization, and g countries. Sample sizes (left to right): d n = 200, 493, 918, 222; e n = 802, 628; f n = 7479, 155; g n = 159, 146, 138, 18, 201, 59, 350, 861, 418, 28, 155, 81, 570, 93. h Heatmap of vOTUs (columns) across individual samples (rows). The left bar contains 10 colors representing 10 individuals, each with 10 samples from 10 different time points (rows). The heatmap cell color represents the presence (yellow) or absence (black) of the vOTU. i Principal coordinates analysis of the vOTU and functional profiles. d samples were compared using Kruskal–Wallis test with post hoc correction. e, f samples were compared using the two-sided Wilcoxon rank-sum test. d–g box plots show median, 25th–75th percentiles, and whiskers (1.5× IQR).
We then assessed the variations of lysogen frequency across different age groups using all the data from healthy individuals (n = 4312). All healthy individuals were divided into four categories, including 200 infants ( < 3 months old), 493 children (3 months old to 18 years old), 918 adults (18 years old to 65 years old), and 222 seniors (>65 years old). We found that the proportion of lysogens was highest at birth. Later, it gradually declined, reached the lowest point in adulthood, and increased slightly in old age (Fig. 2d and Supplementary Fig. 3a). A gender difference was also observed, with females showing a higher ratio of lysogens than males (Fig. 2e). For the same data, we classified the countries as highly industrialized versus unindustrialized, and found that lysogens were relatively more prevalent in the human gut in highly industrialized countries (Fig. 2f), which indicates possible influences of diet and lifestyle on lysogeny. We found that individuals in Madagascar and New Zealand had a relatively lower ratio of lysogens in their gut, while those in America, England and Spain had a higher ratio (Fig. 2g). To be noted, because detailed demographic metadata (e.g., ethnicity) were not uniformly available, we were unable to quantify demographic contributions in this analysis. We then examined the relationship between lysogen proportion and population density in each country to see if variations correlated with differing contact rates between phage predators and bacterial prey, and if lysogeny rates correlated with human population density. Our analysis revealed no significant correlation between lysogen ratios and population density (Supplementary Fig. 3b).
In the above analyses, we examined over 8500 metagenomic datasets and found that lysogen frequency was associated with human population features, particularly age, gender, and levels of industrialization. To further investigate prophage characteristics at the individual level, we re-analyzed a publicly available time-series dataset comprising metagenomic data from 10 healthy individuals sampled at 10 consecutive time points22. The PIs identified across all samples were clustered into species-level vOTUs. Substantial inter-individual variability in gut prophages was observed, with only a small number of shared PIs across individuals (Fig. 2h). The prophage taxonomic structure within each individual exhibited relative stability over time (Fig. 2h, i). Functional annotation of prophage-encoded cargo genes (genes for phage replication excluded) revealed that, despite the pronounced genomic diversity across individuals, the functional profiles of these prophages were conserved across most individuals (Fig. 2i and Supplementary Fig. 3c). Prominent functions detected included prophage genes for metabolism and bacterial virulence (Supplementary Fig. 3d).
Taken together, our analysis of global and longitudinal metagenomic datasets revealed that lysogen frequency is associated with human population features. Despite high inter-individual variability, prophage taxonomic profiles were stable within individuals, and their functional potentials were relatively conserved.
Disease-specific alterations of prophage-encoded genes and depletion of futC gene and its product in IBD patients
We next tested the association between lysogen prevalence and multiple health states by comparing the abundance of lysogens in individuals with disease to those in healthy individuals within each study (Supplementary Data 5). In total, 4505 out of the 8503 human gut metagenomes, derived from 21 studies covering 9 different disease types, were included in this analysis. The restuls revealed that the ratio of lysogens was altered in the presence of disease across a majority of disease categories (Fig. 3a). We then queried the taxonomic associations of lysogens in these studies (Supplementary Fig. 4). The proportions of lysogens were increased in Bacteroidota, Bacillota, and Pseudomonadota and decreased in Actinomycetota (Supplementary Fig. 5a). The varied proportions of lysogens in different taxa were also observed at the genus-level (Supplementary Fig. 5b). To confirm changes in lysogen ratios during disease, we assessed lysogen frequency in a Dextran sulfate sodium (DSS)-induced colitis model. The lysogen ratio increased significantly in DSS-induced colitis mouse model (Fig. 3b). We further observed temporal fluctuations in the proportion of lysogens throughout the course of the inflammation, which partially mirrored the dynamics of inflammation, suggesting a potential association between lysogen dynamics and host inflammatory status (Fig. 3c and Supplementary Fig. 5c). Thus, these results demonstrate a link between lysogen frequency and diseases.

a Ratio of lysogen proportions in disease versus healthy status across 21 public studies encompassing various disease types (Supplementary Data 4). Error bars indicate ±1 standard deviation (SD) around the mean of the disease group. The healthy group served as controls. Studies showing differences include Preterm_1 (disease vs. healthy: n = 169 vs. 99), IBD_1 (70 vs. 44), IBD_2 (60 vs. 24), Diabetes_1 (57 vs. 199), IBD_3 (126 vs. 224), IBD_4 (684 vs. 222), Diabetes_2 (169 vs. 171), Liver cirrhosis_1 (125 vs. 110), Ankylosing spondylitis_1 (91 vs. 67). b Alterations in lysogen abundance in the gut microbiome of the DSS-treated (n = 9) and control SPF mice (n = 20). The box plot shows median, 25th–75th percentiles, and whiskers (1.5× IQR). c Variations in the proportion of gut lysogens in mice during DSS treatment. Solid lines (I1, I2, and I3) represent three experimental replicates, each combining samples from two mice. d Enrichment of VFDB terms for prophage-encoded genes comparing disease to health. Each dot represents an individual study. The y-axis displays the standardized ratio of VFDB term fold enrichment scores in disease samples relative to healthy samples. The fold enrichment score was calculated by comparing the percentage of specific terms in the dataset to their percentages in the VFDB database. The colored squares covering the dots represent the number of studies with enriched (pink) or depleted (green) VFDB terms. e The differentially abundant genes under the Immune modulation term in the IBD cohort. The bar chart shows the percentage of the futC-like gene-positive individuals in disease and health. P value was calculated using two-sided chi-square testing. f Schematic representation of 2’-FL synthesis in bacteria (Created with BioRender.com, https://BioRender.com/8lb6jxc). g Genomic localization of the futC-like gene and PIs on BF9343 and the metagenome-derived contig. h Total ion chromatogram (TIC) of 2’-FL using LC-MS and the relative quantification of 2’-FL in the heterologous expression system. Empty indicates empty vectors. Data are presented as mean values ± SEM (n = 3 for all groups). i Experimental workflow for assessing the effects of 2’-FL on DSS-induced colitis. j The histological scores of colitis after 2’-FL treatment. Lower scores reflect reduced inflammation. k Representative images of colon sections stained with H&E, shown at ×40 magnification (Scale bar: 100 µm). The experiment was independently repeated twice with similar results. a, b Samples were compared using the two-sided Wilcoxon rank-sum test, and error bars represent standard deviation. j Samples were compared using ANOVA with post hoc correction.
Virulence factors (VFs) are key prophage genes that can alter bacterial fitness and affect vertebrate hosts23. We quantified prophage-encoded VF-like genes in different datasets. In total, 95,264 prophage-encoded VF-like genes from 12 groups were identified using the Virulence Factor Database (VFDB)24. We then calculated the enrichment level of these terms in different studies. We found that the VF term “Invasion” was highly enriched, and the VF term “Immunomodulation” was depleted in most of the seven IBD cohort studies (Fig. 3d). The VF term “Nutritional/metabolic factor” was under-represented in all obesity cohort studies (Fig. 3d). These results suggested that changes in the functional capacity of prophage-encoded genes are disease-specific, and they may influence pathogenesis in specific diseases.
We subsequently evaluated the differentially abundant prophage-encoded genes associated with “Immunomodulation” between healthy individuals and patients with IBD across various IBD cohorts and found that a futC-like gene was significantly depleted in IBD patients (Fig. 3e). The futC gene is homologous to the human fut2 gene. It encodes α1,2-fucosyltransferase, an enzyme essential for synthesizing 2’-fucosyllactose (2’-FL), an oligosaccharide involved in immune modulation23,25,26. However, the role of futC in the gut microbiome was not well studied. To experimentally evaluate the enzymatic activities of prophage-encoded futC-like genes, a representative sequence of a prophage-encoded futC-like gene from a Ruminoccocus MAG in the metagenomic data was selected based on clustering analysis, referred to as fpr. To further validate the enzymatic activities of the prophage-encoded futC-like genes in culturable gut-derived bacteria strains, another prophage-encoded futC-like gene, referred to as fpb, was identified in Bacteroides fragilis NCTC 9343 (Fig. 3g). Both fpr and fpb possess the glycosyltransferase family 11 domain and share highly conserved residues at the GDP-fucose binding site with the human fut2 gene (Supplementary Fig. 5d). The enzymatic activities of both genes were tested in a heterologous expression system (Supplementary Fig. 5e), and the cell lysates and supernatant were subjected to mass spectrometry analysis. The results demonstrated that both gene products possess enzymatic activity, which allows them to fucosylate lactose and produce 2’-FL (Fig. 3h).
We then tested the presence of 2’-FL in the human adult gut environment using metabolomics data collected from the iHMP IBD cohort27. Analysis revealed significantly higher levels of fecal 2’-FL in healthy individuals compared to either CD or UC patients (Supplementary Fig. 5f, g). We then administered 2’-FL to DSS-treated mice to evaluate its role in modulating colitis pathogenesis (Fig. 3i). The results demonstrated a mitigating effect on colitis in DSS-induced colitis mice, as evidenced by reduced inflammation, including significantly longer colon length, lower disease activity score, and marked histopathological improvements compared to the DSS-treated mice (Fig. 3j, k and Supplementary Fig. 5h). Consistent with these findings, 2′-FL also significantly ameliorated disease in the piroxicam-accelerated IL-10-deficient colitis (Supplementary Fig. 6). Taken together, the prophage-encoded futC-like gene, which was depleted in IBD, could synthesize 2’-FL, which could ameliorate colitis in two colitis mouse models.
Oligosaccharide 2’-FL improves mucosal immunity by promoting intraepithelial immune responses and IgA secretion
To investigate whether 2’-FL alleviates colitis by modulating the interaction between the host immune response and the microbiome, we treated SPF mice with 2’-FL for four weeks and analyzed changes in the host gut epithelial transcriptome, mucosal T/B immunity, and microbiota (Fig. 4a). RNA-seq analysis of ileal epithelial cells (IECs) showed that pathways involved in monocarboxylic acid transport and cytokine responses were enriched after treatment with 2’-FL, indicating active transport of microbial metabolites and changes in mucosal immunity in these mice (Fig. 4b, c). We then analyzed various regional immune changes in 2’-FL-treated mice, including the ileal intraepithelial region and the ileal and colonic lamina propria (LP) (Fig. 4d and Supplementary Fig. 7). We found that 2’-FL treatment did not affect regulatory T (Treg) cell or Th17 cell subsets in the gut, but we observed a significant increase in ileal CD4+CD8αα+ T cells (Fig. 4d, e), a previously described immune population with mucosal immune-enhancing functions mainly through IL-10 production28,29,30,31. In addition, we also observed an increase in ileal LP B cells and fecal IgA secretion after treatment (Fig. 4d, f, g), suggesting that 2’-FL could modulate gut mucosal immune responses via both IgA and intraepithelial T cells.

a Experimental scheme of mice experiments (Created with BioRender.com, https://BioRender.com/k7r3dmh). Mice were fed with either 2’-FL solution or regular water (Control) for 4 weeks. Profiling of the IECs transcriptome, IELs and ILPLs populations, and microbial composition was conducted. b Volcano plot of the differentially expressed genes in IECs after 2’-FL treatment. Results were shown using unadjusted P values. c Gene ontology analyses of up-regulated genes of IECs after 2’-FL treatment. d The alterations in the frequency of immune cell populations in colonic LP lymphocytes (CLPLs), ileal LP lymphocytes (ILPLs), and intraepithelial lymphocytes (IELs) across different samples relative to the mean frequency of the control (log-transformed). Representative plots and frequencies of CD4 + CD8αα+ cells (e) and CD19 + B cells (f) in the IELs population from mice treated with 2’-FL compared to control mice. g Fecal IgA levels in 2’-FL-treated mice and the control mice. Bar plot (h) and PCoA analysis (i) of gut microbiome composition for the 2’-FL-treated and control mice. j The significantly altered bacterial taxa in the gut microbiota after 2’-FL administration (ranked by effect size Coef). k Growth curves of A. muciniphila, R. gnavus, and Alistipes finegoldii (a representative species of Rikenellaceae) in different conditions. A.muciniphila strain BAA-835, Ruminococcus gnavus ATCC 29149, and Alistipes finegoldii DSM17242 were cultured in BHI (control) and BHI supplemented with 2’- FL medium (n = 4 per group per time point). Samples were compared using the two-sided Wilcoxon rank-sum test and data are presented as mean values ± SEM (***P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05). l The fecal IgA level in germ-free (GF) mice (n = 3) with monocolonization of A. muciniphila (n = 6) (Created with BioRender.com, https://BioRender.com/vazbfcp). e–g, l samples were compared using a one-tailed Welch’s t test, and data are presented as mean values ±SEM.
We next explored the possible mechanisms by which 2’-FL promotes IgA secretion and intraepithelial T cell development. Considering that 2’-FL could be consumed by the gut microbiota and affect its composition32,33, we investigated whether these immunomodulatory effects of 2’-FL were mediated through alterations in microbial composition. 16S rRNA gene sequencing revealed significant shifts in the gut microbiota of 2’-FL-treated mice (Fig. 4h, i), mainly characterized by a marked increase in the abundance of A. muciniphila, R. gnavus, and Rikenellaceae species. (Fig. 4h, j). A previous study has shown that R. gnavus could induce intraepithelial CD4+CD8αα+ T cells via conjugated linoleic acids produced by this bacterium28, indicating that 2’-FL may trigger intraepithelial T cell development via increasing R. gnavus abundance in vivo. We then sought to determine whether 2’-FL could directly promote the in vitro growth of these bacteria, and found that 2’-FL specifically promoted A. muciniphila growth while showing limited effects on other bacterial species (Fig. 4k). To determine whether the immunomodulatory effects of 2’-FL are mediated by the expansion of A. muciniphila, we conducted monocolonization experiments in gnotobiotic mice. Colonization with A. muciniphila resulted in a significant increase in intestinal IgA levels, which mirrored the effects observed with 2’-FL administration (Fig. 4l). These findings suggest that 2’-FL may enhance IgA production by directly promoting the growth of A. muciniphila.
In summary, we demonstrated that the prophage-encoded futC genes, which were depleted in IBD, could synthesize 2’-FL to ameliorate colitis via promoting both intestinal T and B cell responses in mouse models, and these processes are likely to be mediated by the microbiome modulatory role of 2’-FL.
Discussion
The dynamics of the vertebrate virome are starting to be characterized in several disease states, but the functional mechanisms are only beginning to be clarified. Prophage induction from lysogens contributes a large fraction of the human virome, and prophages encode extensive cargo genes with the potential to modulate the functions of host bacterial cells34. The influence of prophage on the gut microbiome and the consequences for host health are just starting to be studied.
Here, we developed a pipeline for identifying lysogens in metagenomic data, validated its efficacy using experimentally induced prophage sequencing data, overcoming the challenges of traditional methods that rely on culturing bacteria and inducing prophages35,36. Our pipeline integrates the prophage identification capabilities of both VirSorter2 and CheckV37,38, and incorporates an experimentally calibrated CheckV completeness score. This design enables improved performance in terms of precision compared to standalone tools such as VirSorter2, PHASTER, and geNomad39,40. However, its performance in other metrics—such as recall and F1 score—was somewhat lower than that of geNomad. Although geNomad has gained popularity as a prophage detection tool since its release, it was not incorporated into our pipeline because the manuscript was already under preparation at the time of its publication.
Recent surveys of prophages in human gut-derived bacterial genomes (n = 43,942) and environmental metagenomes (n = 1432) have highlighted their widespread distribution, remarkable diversity, and the increased prevalence of prophage-encoded antibiotic resistance genes in human-impacted habitats16,41. To our knowledge, our study represents the most comprehensive catalog of prophages in the human gut microbiome to date, based on the analysis of nearly 300,000 MAGs and over 8000 metagenomic samples. Our analysis revealed that nearly half of gut bacterial genomes contain PIs. A previous study reported a higher proportion, with over 90% of gut bacterial genomes harboring prophages16. This discrepancy can be attributed to differences in prophage identification methods and genome quality. Specifically, the previous study employed PHASTER39, which, based on our benchmarking, tends to predict a larger number of prophages compared to our pipeline (Supplementary Data 2). Moreover, their analysis primarily focused on high-abundance bacterial genomes, which are often more complete than the MAGs used in our study. Nevertheless, both studies consistently highlight the extensive diversity of prophages and lysogens within the human gut microbiome. The diversity of known prophages was greatly expanded in this study. Multiple new features of prophage biology and evolution were found. For example, we identified Microviridae as PIs in the gut microbiome, which were believed to be strictly lytic and incapable of lysogeny until metagenomic analysis on bacterial genomes42. We also identified phage families characterized by PIs with previously undocumented lysogenic life cycles, notably Winoviridae. We found a limited number of viruses in archaeal genomes, such as Thermoplasma and Methanobacterium. The detection of human gut archaeal viruses is just getting started43, and further studies are warranted to deepen our understanding in this area.
Our results revealed that this proportion of lysogenic bacteria varied widely in different settings. The percentage of lysogens was increased in the newborn and aging guts, in citizens of industrialized countries, and in most disease states, suggesting that lysogeny may be favored in an immature or compromised gut environment, which often shows reduced microbial diversity44. It has been proposed that lower bacterial diversity may lead to more frequent phage integration due to a higher incidence of phage co-infection and lower levels of bacterial cell metabolism5,45. Another recent study revealed similar findings that more prophage DNA can be identified in disease status, and proposed that disease-associated bacteria may have more prophages46. Taken together, the prevalence of lysogens or prophages is associated with distinctive gut environments. It is important to note that many observed changes in prophages may be attributed to shifts in their bacterial hosts. Additionally, inter-individual variations in prophages may also be influenced by host genetic factors, as host genetics is known to play a pivotal role in shaping the microbiome47,48. Moreover, host responses are also critical for prophage induction49. Further studies would be of great interest to explore how host genetic variation contributes to prophage dynamics.
Prophages may carry cargo genes that alter the fitness, virulence, and other characteristics of their bacterial hosts50,51. We investigated the functional potential of prophage-encoded cargo genes in the gut microbiome and found disease-specific functional signatures, suggesting that alterations in these genes may contribute to disease pathogenesis. One possible explanation is that such differences may reflect shifts in the bacterial hosts harboring these prophages. For example, the NlpC/P60 domain-containing endopeptidase—previously reported to be altered in the whole gut microbiome and associated with intestinal inflammation—was also differentially abundant among prophage-encoded genes52. Notably, we also identified prophage-encoded futC-like genes that were associated with diseases such as IBD, despite no corresponding changes observed in the bacterial genomes, indicating that phage-encoded factors may play independent roles in disease processes. We have shown biochemically that prophages can encode futC-like genes capable of synthesizing the oligosaccharide 2’-FL. The beneficial effects of 2’-FL on gut health have been documented in several studies, although the molecular mechanisms remain unclear33,53. We demonstrated in two mouse models, DSS-induced colitis and piroxicam-accelerated colitis in IL-10-deficient mice, that 2’-FL attenuates intestinal inflammation. Mechanically, our findings suggested that 2’-FL treatment can modulate mucosal immune function and intestinal IgA secretion. Firstly, 2’-FL treatment increased the levels of intraepithelial CD4⁺CD8αα⁺ T cells, which are known to improve the gut mucosal immunity28,29,30,31,54, and this effect was likely associated with the increased abundance of R. gnavus28. Additionally, our data revealed that 2’-FL can directly promote the growth of A. muciniphila to stimulate the production of gut IgA, which has been shown to play an important role in gut homeostasis55,56,57. While 2’-FL treatment in the gut promoted the outgrowth of multiple taxa, IgA induction is species-specific rather than a uniform consequence of expansion. Notably, R. gnavus, which also increased post-treatment, fails to trigger gut IgA58. Thus, from an immunomodulatory perspective, our findings may help explain why the depletion of prophage genes for 2’-FL synthesis is associated with IBD progression in patients.
In conclusion, we present an overview of lysogens in the human gut microbiome using large-scale metagenome-derived bacterial genomes and reveal a high prevalence of lysogens in the gut microbiome as well as the world's population. We observed changes in lysogen frequency across different disease states, along with the enrichment or depletion of specific prophage-encoded genes in these conditions. Our study proposes a mechanism by which prophage-encoded genes can influence host immune function through oligosaccharide synthesis. We believe that these findings provide valuable insights into the functional mechanisms of the triad of human gut virome, bacteria, and host immunity.
Methods
Animals
C57BL/6 J male mice were obtained from the Laboratory Animal Resources Center, Tsinghua University. Six to eight-week-old mice were used for experiments and were raised in specific pathogen-free conditions with a strict 12-hour light/12-hour dark shift. C57BL/6 J FOXP3-IRES-mRFP and IL-10 IRES-GFP mice were obtained from Jackson and crossed to generate the dual-reporter mice. GF C57BL/6 mice were obtained from GemPharmatech, Nanjing, China. All experiments strictly followed the Tsinghua University and Center for Excellence in Molecular Cell Science (CEMCS) animal care guidelines. Animal protocol 21-LGX1.G24-1 and S748-2108-031 were approved by Tsinghua University and CEMCS Institutional Animal Care and Use Committee, respectively.
Bacterial strains and culture conditions
A. muciniphila ATCC BAA-835, Bacteroides fragilis NCTC 9343, Ruminococcus gnavus ATCC 29149 and Alistipes finegoldii DSM17242 were used in this study. Anaerobic bacteria were grown in brain heart infusion (BHI) medium (Hopebio, HB8297-1) supplemented with 5 mg/L hemin (Sigma-Aldrich, H5533) and 2.5 mg/L vitamin K1 (Sigma-Aldrich, V3501). In addition, A. muciniphila ATCC BAA-835 was grown in supplemented BHI with 1 g/L mucin (Sangon, A002975). The bacteria were then cultured under anaerobic conditions (80% N2, 10% H2, and 10% CO2) at 37 °C in an anaerobic chamber. The 23 bacterial strains used for Lydia benchmarking are described in a previous study17.
Development of a pipeline to profile prophages in metagenome-derived bacterial genomes
To identify prophages from metagenomic sequencing data, a pipeline that integrates lysogen detection and downstream analysis was developed (https://github.com/chyghy/2025). Briefly, quality control, host read removal, and de novo assembly were performed using Sunbeam (v2.1.0)59. Contigs shorter than 1,500 nt were discarded. Next, genomic binning was conducted to obtain MAGs following the method described previously60. The high-quality reads were mapped back to contigs using Bowtie2 (v2.2.5)61, and genomic binning was performed using MetaBAT2 (v2.12.1)62. The MAG quality statistics (completeness and contamination) were evaluated by CheckM (v1.1.3)63. MAGs with completeness >50% and contamination <10% were kept for downstream analysis. CheckM was also used to perform the taxonomic assignment of these bins by assigning them to the CheckM reference tree. The assembled contigs (>10 kb) were subjected to VirSorter2 (v2.2.3) and CheckV (v0.8.1) for prophage prediction and completeness evaluation37,38. The CheckV database (Version 1.0, released on 2 February 2021) was used in this analysis. A lysogen was identified as a bin if it contained one or more contigs including both bacterial and phage regions.
In vitro prophage induction and induced VLP sequencing
Overnight cultures of isolated bacterial strains were diluted 1/100 into 10 ml medium and grown until log phase (OD600 = 0.6). Mitomycin C (Sigma-Aldrich, M4287) was then added to a final concentration of 5 μg ml−1. The OD600 values were measured, and VLPs were purified after 6 h of culture17. Specifically, the bacterial cultures were centrifuged at 4000 × g for 10 min and the supernatants were collected. Then the supernatants were filtered through a 0.22-μm-pore-size filter (SORFA). The filtrate was concentrated using a 100-kDa-molecular-mass Vivaspin® Turbo 15 centrifugal concentrator (Sartorius) to a final volume of around 500 μl. The concentrate was treated with DNase I and RNase at 37 °C for 30 min to degrade nonencapsulated nucleic acids. Then the virome libraries were made using the TruePrep Flexible DNALibrary Prep Kit for Illumina (Vazyme, TD504).
Long-read sequencing of bacterial genomes and data analysis
Bacterial DNA was isolated by MagMAX™ Viral/Pathogen Ultra Nucleic Acid Isolation Kit (ThermoFisher). Libraries were prepared using a Qitan DNALibrary prep kit (QDL-E V1.0). Sequencing was performed using a Qcell-3841 sequencing chip and Qitan DNA Sequencing Kit (QDS V1.0) on a QNome platform (QitanTech, China). Fast5 files were base-called using NiuTouGeng V3 (QitanTech, China). The raw reads were filtered using NanoPlot and NanoFilt64. Hybrid assemblies were conducted using Unicycler65 and Flye66. Pilon was used to carry out genome polishing67.
Clustering and taxonomic annotation of prophages
Prophages were clustered and assigned to taxonomic classification68. In brief, all prophages with completeness >90% were clustered into species-level vOTUs based on 95% ANI and 85% alignment fraction. We clustered prophages to genus- and family-level vOTUs using a combination of gene sharing and amino acid identity (AAI). The different levels of vOTU were assigned to specific taxonomy by clustering these prophages with 6,124 reference phage genomes from NCBI refseq database (updated on April 16, 2025) according to the workflow and criteria as above.
Functional annotation of prophage-encoded genes
Prophage sequences of each lysogen were extracted by removing its flanking host regions, which were indicated by the output of VirSorter2 or CheckV. Open reading frames (ORFs) were predicted for all prophages using Prodigal (v2.6.3)69. ORFs were annotated by mapping to Pfam database using HmmScan (HMMER 3.170; http://hmmer.org/) with E < 10−5. Pfam annotations with occurrence frequency exceeding 10,000 were then used to perform GO enrichment analysis using dcGO71. The Clusters of Orthologous Genes (COG) database was also used for functional profiling72.
The VFDB24 organized bacterial VFs into 14 categories. Here, VFs were annotated using VFDB set A, which includes representative genes associated with experimentally verified VFs. ORFs of prophages were compared against the VFDB set A using Diamond blastx (v0.9.24)73. The E value cutoff was set at 10−5 to maintain a low false-positive rate.
Calculation of prophage genomic similarity
Mash implements k-mer-based scoring and was used to compute nucleotide distance between prophage genomes74. A total of 5000 genomes were randomly selected from all the predicted prophages ten times. Pair-wise Mash distances were calculated in each subset using Mash (v1.1).
Chemically induced colitis model construction and 2’-FL treatment
All mice were randomly assigned to 4 experimental groups (n = 6 per group): control group (Control), DSS, low dose 2’-FL group (Low) and high dose 2’-FL group (High). The control mice group received regular drinking water throughout the experiment, while to establish the DSS-induced colitis model, the other three mouse groups received 3% (w/v) DSS (MP Biomedicals, SKU:0216011050) ad libitum in drinking water for 7 days and then switched to regular drinking water. To evaluate the effect of 2’-FL treatment on colitis, 200 µl phosphate-buffered saline (PBS), 200 µl PBS, 200 µl 200 mg kg−1 body weight 2’-FL (Macklin, F918027), and 200 µl 400 mg kg−1 body weight 2’-FL were administered intragastrically to the Control group, the DSS group, the Low group, and the High group from day 1 to day 7, respectively. The amount of DSS intake per mouse was recorded. There were no differences in intake between mouse strains.
Assessment of colitis symptoms
Mice were weighed every day to determine percentage weight changes. This was calculated as percent body weight change = (weight at day X / weight at day 0) × 100. The disease activity index (DAI) was calculated to evaluate the progression of colitis. It was determined by scoring weight loss (weight change × 0.2), stool consistency (0, normal; 0.5, loose stool; 1, diarrhea), and stool bleeding (0, normal brown-black; 1, yellow; 2, slight bleeding; 3, severe bleeding). Mice were sacrificed through cervical dislocation on day 12, and colon length and spleen weight were measured. The colons were embedded in paraffin and were cut into 5 μm sections. Hematoxylin-eosin staining was performed to evaluate scores of inflammation-associated histological changes, mainly by the amount of inflammatory cell infiltration, goblet cell loss, and crypt loss75.
Total microbial DNA extraction and shotgun metagenomic sequencing
Stool samples of the control and the DSS groups were collected daily and subjected to metagenomic sequencing. Approximately 200 mg of mouse stool was used for total microbial DNA extraction. Total microbial DNA was purified from each sample using the Fecal genome DNA extraction kit (TIANGEN, DP328) following the manufacturer’s instructions. A total of 50 μl total microbial DNA was obtained for each sample and stored at −20 °C. Total microbial DNA was used for the construction of the shotgun metagenomic sequencing libraries. The DNA concentration was measured using Qubit. Libraries were made using TruePrep Flexible DNALibrary Prep Kit for Illumina (Vazyme, TD504).
LC-MS-based identification and quantification of 2’-FL
The selected futC-like genes fpr and fpb were constructed with His tag on pET28a plasmid and heterologously expressed in BL21-CodonPlus (DE3)-RIPL Chemically Competent Cell (TIANGEN, EC1007). The purified proteins were verified using a Western blot using His antibody (Rabbit anti-His tag (D3I1O) antibody). The bacteria strains were cultured in LB medium, and Lactose (concentration: 1 g/L) (Macklin, HY-B212) and GDP-fucose (concentration: 1 g/L) (Solarbio, YS147449) were introduced until the OD600 reached 0.6. Cultures were then incubated for an additional 18 h. Subsequently, 10 ml of the culture was harvested and subjected to low-temperature ultrasonication (power intensity: 350 W, Ultrasonic for 2 s and the interval of intermittent ultrasound was set at 3 s, operating 50 times), followed by lyophilization of the supernatant. Quantitative analysis is performed using TSQ2404-071 instrumentation at the Metabolomics and Lipidomics Platform of Tsinghua University, with 99% HPLC (ZG-10020) pure 2’-FL (Macklin, F918026) used as the reference standard.
Isolation of mouse gut IELs and LPLs and flow cytometry analysis
2’-FL was added to the drinking water of four-week-old mice at a final concentration of 5 mg/ml for four weeks in the experimental group (2’-FL group), while the control group received no supplementation. Ileums and colons were collected from the mice of these two groups for the isolation of intraepithelial lymphocytes (IELs) and lamina propria lymphocytes (LPLs). Fat tissue was removed from the intestine, and Peyer’s patches were excised from the ileum. Intestines were cut open longitudinally and washed in cold PBS to remove feces. Then these tissues were incubated in RPMI medium containing 1 mM EDTA, 1 mM DTT, and 2% (v/v) FBS at 37 °C with shaking at 300 rpm for 20 min to dissociate epithelial cells. Suspensions were then filtered successively through 40 μm cell strainers, and the supernatant containing epithelial cells was centrifuged at 2000 rpm for 5 min. The pellet was resuspended, and the IELs were further obtained by Percoll (40%/80%) gradient centrifugation. Single-cell suspensions were subjected to flow cytometric analysis by blocking with antibodies to CD16/32 (2.4G2, 1:300 dilution) and staining with antibodies to CD45 (30-F11, 1:300 dilution), TCRβ (H57-597, 1:300 dilution), CD4 (GK1.5, 1:300 dilution), CD8α (53-6.7, 1:300 dilution), CD8β (H35-17.2, 1:300 dilution), CD19 (6D5, 1:300 dilution), CD3 (17A2,1:300 dilution), Foxp3 (FJK-16s, 1:300 dilution), Rorγt (AFKJS-9, 1:300 dilution) and with viability dye.
The intestinal tissues from the previous step (after removing the supernatant) were cut using scissors. The tissues were then digested in RPMI containing 1.5 mg/ml collagenase type II, 0.5 mg/ml dispase II, and 1.2% (v/v) FBS at 37 °C with shaking at 300 rpm for 40 min. The digest was filtered through the 40 μm cell strainer and centrifuged at 2000 rpm for 5 min. Then the LPLs were separated by Percoll (40%/80%) gradient centrifugation. Single-cell suspensions were subjected to flow cytometric analysis as described above.
16S rRNA profiling of the gut microbiota
Fecal DNA was extracted from the colonic content of the 2’-FL group and control group using a TIANamp Stool DNA kit (TIANGEN, DP328) according to the manufacturer’s instructions. To determine the bacterial taxa, the V3-V4 regions of the bacterial 16S ribosomal RNA gene were amplified by PCR using the primers CCTAYGGGRBGCASCAG and GGACTACNNGGGTATCTAAT. Raw data files were demultiplexed and quality-filtered using QIIME2 (v2024.2.0)76. Specifically, the 99_otus.fasta file was downloaded from the Greengenes database. Sequences corresponding to the V3 ~ V4 region, which match the experimental primers described before, were extracted. Based on these extracted sequences, a classifier was trained. Subsequently, this classifier was used to annotate the 16S rRNA sequencing data at different taxonomic levels. Multivariate Association with Linear Models 2 (MaAsLin2) was utilized to identify distinctive bacterial taxa. Statistical significance was assessed using unadjusted p values, with taxa having p < 0.05 reported (Supplementary Data 6).
RNA isolation of IECs and RNA-seq data analysis
Mouse ilea were used for the isolation of IECs after removing Peyer’s patches and areas of fat accumulation. The excised ileal tissues were washed in cold PBS buffer, and epithelial layers were detached by shaking at 4 °C in a chelating solution containing 5.6 mM Na2HPO4, 8 mM KH2PO4, 96.2 mM NaCl, 1.6 mM KCl, 43.4 mM sucrose, 54.9 mM d-sorbitol, 10 mM EDTA, 1 mM DTT and 2% (v/v) FBS. After 1 h of incubation, the epithelial crypt-containing supernatants were collected and centrifuged at 200 × g for 5 min to collect IECs and total RNA was extracted using Trizol. The RNA-seq library was then constructed using the Vazyme NR606 kit according to the manufacturer’s instructions. Quality control of the raw data was performed using fastp (v0.23.4)77. The clean reads were then mapped to the mm10 reference genome with HISAT2 (v2.1.0)78. Quantitative analysis was conducted using featureCounts79, followed by differential gene expression analysis with DESeq2 (v1.36.0)80. Finally, GO enrichment analysis was carried out using clusterProfiler81.
Generation of monocolonized GF mice and measurement of IgA
Male GF C57BL/6 mice (4 weeks old) were inoculated with a broth-grown A. muciniphila ATCC BAA-835 by oral gavage. Each group of mice was then housed in a sterile gnotobiotic isolator for 2 weeks. Small intestinal lumen material was collected and plated after 2 weeks of bacterial inoculation to determine colonization levels and to ensure colonization by a single bacterial strain. The fecal level of IgA in the A. muciniphila BAA-835 monocolonized GF mice was determined using the Mouse IgA ELISA Kit (Beyotime, PI471).
Bacterial growth curve
Three bacterial strains (A. muciniphila ATCC BAA-835, Ruminococcus gnavus ATCC 29149 and Alistipes finegoldii DSM17242) that were cultured overnight were diluted in fresh supplemented BHI liquid medium at a ratio of 1:100. The experimental group was supplemented with 2’-FL to achieve a final concentration of 10 mg/ml, and their OD600 values were measured separately for plotting growth curves.
Statistical analysis
Statistical tests were performed using R. Nonparametric tests were applied to compare two independent groups (Wilcoxon rank-sum test), two related groups (Wilcoxon signed-rank test), and multiple groups (Kruskal–Wallis test with Bonferroni correction). The nonparametric correlation was conducted using Spearman’s rank-order correlation (R represents Spearman’s ρ). Fisher’s exact test and Chi-square test were used to test the difference between two categorical variables. To adjust for multiple comparisons, the Benjamini–Hochberg false discovery rate (FDR) method was used. P value < 0.05 or FDR-corrected p value < 0.05 was considered significant. All acquired data were included in analyses. No statistical method was used to predetermine sample size.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
To obtain an overview of the lysogeny in the human gut microbiome, a total of 289,232 MAGs were retrieved from UHGG V2.0 database18. All the UHGG catalogs are available on the MGnify website82 (http://ftp.ebi.ac.uk/pub/databases/metagenomics/mgnify_genomes/). To further associate lysogeny with metadata, we collected metagenomic sequencing datasets from 45 studies. All the biosample information was manually collated and listed in Supplementary Data 3, which includes the following descriptions: general description, age, gender, location, and disease. The time-series metagenomic data were downloaded from the NCBI SRA database with the Project ID: PRJNA820119.
Raw sequences for shotgun metagenomic sequencing, 16S rRNA sequencing, and IECs RNA-Seq generated in this study are available in the National Center for Biotechnology Information Sequence Read Archive under BioProject ID PRJNA1190363.
Code availability
All bioinformatic scripts are available on Github (https://github.com/chyghy/2025).
References
Ptashne, M. A genetic switch (Blackwell, 1986).
Kortright, K. E., Chan, B. K., Koff, J. L. & Turner, P. E. Phage therapy: a renewed approach to combat antibiotic-resistant bacteria. Cell Host Microbe 25, 219–232 (2019).
Galtier, M. et al. Bacteriophages targeting adherent invasive Escherichia coli strains as a promising new treatment for Crohn’s disease. J. Crohn’s. Colitis 11, 840–847 (2017).
Federici, S. et al. Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell 185, 2879–2898 (2022).
Knowles, B. et al. Lytic to temperate switching of viral communities. Nature 531, 466–470 (2016).
Fortier, L.-C. & Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365 (2013).
Chen, J. & Novick, R. P. Phage-mediated intergeneric transfer of toxin genes. Science 323, 139–141 (2009).
Fluckiger, A. et al. Cross-reactivity between tumor MHC class I–restricted antigens and an enterococcal bacteriophage. Science 369, 936–942 (2020).
Modi, S. R., Lee, H. H., Spina, C. S. & Collins, J. J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 499, 219–222 (2013).
Fujimoto, K. et al. An enterococcal phage-derived enzyme suppresses graft-versus-host disease. Nature 632, 174–181 (2024).
Backman, T. et al. A phage tail–like bacteriocin suppresses competitors in metapopulations of pathogenic bacteria. Sci. 384, eado0713 (2024).
Casjens, S. Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49, 277–300 (2003).
Touchon, M., Bernheim, A. & Rocha, E. P. C. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 10, 2744–2754 (2016).
Kim, M.-S. & Bae, J.-W. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 12, 1127–1141 (2018).
López-Leal, G. et al. Mining of thousands of prokaryotic genomes reveals high abundance of prophages with a strictly narrow host range. mSystems 7, e0032622 (2022).
Pei, Z. et al. A universe of human gut-derived bacterial prophages: unveiling the hidden viral players in intestinal microecology. Gut Microbes 16, 2309684 (2024).
Liang, G. et al. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 581, 470–474 (2020).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Groussin, M. et al. Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell 184, 2053–2067.e18 (2021).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Bouras, G. et al. Pharokka: a fast scalable bacteriophage annotation tool. Bioinformatics 39, btac776 (2023).
Chen, L. et al. Short- and long-read metagenomics expand individualized structural variations in gut microbiomes. Nat. Commun. 13, 3175 (2022).
Sprenger, N. et al. FUT2-dependent breast milk oligosaccharides and allergy at 2 and 5 years of age in infants with high hereditary allergy risk. Eur. J. Nutr. 56, 1293–1301 (2017).
Liu, B., Zheng, D., Zhou, S., Chen, L. & Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917 (2022).
Engels, L. & Elling, L. WbgL: a novel bacterial α1,2-fucosyltransferase for the synthesis of 2’-fucosyllactose. Glycobiology 24, 170–178 (2014).
Seydametova, E. et al. Search for bacterial α1,2-fucosyltransferases for whole-cell biosynthesis of 2′-fucosyllactose in recombinant Escherichia coli. Microbiol. Res. 222, 35–42 (2019).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Song, X. et al. Gut microbial fatty acid isomerization modulates intraepithelial T cells. Nature 619, 837–843 (2023).
Mucida, D. et al. Transcriptional reprogramming of mature CD4+ helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat. Immunol. 14, 281–289 (2013).
Sujino, T. et al. Tissue adaptation of regulatory and intraepithelial CD4+ T cells controls gut inflammation. Science 352, 1581–1586 (2016).
Cervantes-Barragan, L. et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8αα(+) T cells. Science 357, 806–810 (2017).
Renwick, S. et al. Modulating the developing gut microbiota with 2’-fucosyllactose and pooled human milk oligosaccharides. Microbiome 13, 44 (2025).
Ryan, J. J., Monteagudo-Mera, A., Contractor, N. & Gibson, G. R. Impact of 2’-fucosyllactose on gut microbiota composition in adults with chronic gastrointestinal conditions: batch culture fermentation model and pilot clinical trial findings. Nutrients 13, 938 (2021).
Liang, G. & Bushman, F. D. The human virome: assembly, composition and host interactions. Nat. Rev. Microbiol. 19, 514–527 (2021).
Knowles, B. et al. Variability and host density independence in inductions-based estimates of environmental lysogeny. Nat. Microbiol. 2, 1–9 (2017).
Lara, E. et al. Unveiling the role and life strategies of viruses from the surface to the dark ocean. Sci. Adv. 3, e1602565 (2017).
Guo, J. et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9, 37 (2021).
Nayfach, S. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585 (2021).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
Camargo, A. P. et al. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 42, 1303–1312 (2023).
Liao, H. et al. Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments. Nat. Commun. 15, 8315 (2024).
Kirchberger, P. C. & Ochman, H. Resurrection of a global, metagenomically defined gokushovirus. Elife 9, e51599 (2020).
Li, R., Wang, Y., Hu, H., Tan, Y. & Ma, Y. Metagenomic analysis reveals unexplored diversity of archaeal virome in the human gut. Nat. Commun. 13, 7978 (2022).
Kriss, M., Hazleton, K. Z., Nusbacher, N. M., Martin, C. G. & Lozupone, C. A. Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. Curr. Opin. Microbiol. 44, 34–40 (2018).
Silveira, C. B., Luque, A. & Rohwer, F. The landscape of lysogeny across microbial community density, diversity and energetics. Environ. Microbiol. 23, 4098–4111 (2021).
Inglis, L. K., Roach, M. J. & Edwards, R. A. Prophages: an integral but understudied component of the human microbiome. Microb. Genomics 10, 001166 (2024).
Bonder, M. J. et al. The effect of host genetics on the gut microbiome. Nat. Genet. 48, 1407–1412 (2016).
Reyes, A. et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338 (2010).
Diard, M. et al. Inflammation boosts bacteriophage transfer between Salmonella spp. Science 355, 1211–1215 (2017).
Nepal, R., Houtak, G., Wormald, P.-J., Psaltis, A. J. & Vreugde, S. Prophage: a crucial catalyst in infectious disease modulation. Lancet Microbe 3, e162–e163 (2022).
Marshall, C. W., Gloag, E. S., Lim, C., Wozniak, D. J. & Cooper, V. S. Rampant prophage movement among transient competitors drives rapid adaptation during infection. Sci. Adv. 7, eabh1489 (2021).
Gao, J. et al. Gut microbial DL-endopeptidase alleviates Crohn’s disease via the NOD2 pathway. Cell Host Microbe 30, 1435–1449 (2022).
Paone, P. et al. Human milk oligosaccharide 2’-fucosyllactose protects against high-fat diet-induced obesity by changing intestinal mucus production, composition and degradation linked to changes in gut microbiota and faecal proteome profiles in mice. Gut 73, 1632–1649 (2024).
Das, G. et al. An important regulatory role for CD4+CD8 alpha alpha T cells in the intestinal epithelial layer in the prevention of inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 100, 5324–5329 (2003).
Bae, M. et al. Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature 608, 168–173 (2022).
Ansaldo, E. et al. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364, 1179–1184 (2019).
Huus, K. E., Petersen, C. & Finlay, B. B. Diversity and dynamism of IgA−microbiota interactions. Nat. Rev. Immunol. 21, 514–525 (2021).
Geva-Zatorsky, N. et al. Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943.e11 (2017).
Clarke, E. L. et al. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 7, 46 (2019).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
De Coster, W. et al. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Nayfach, S. et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 6, 960–970 (2021).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 119 (2010).
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 11, 431 (2010).
Fang, H. & Gough, J. DcGO: database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res. 41, D536–D544 (2013).
Galperin, M. Y. et al. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281 (2021).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Erben, U. et al. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 7, 4557–4576 (2014).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Mitchell, A. L. et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578 (2020).
Acknowledgements
We are grateful to members of the Liang laboratory for their help and suggestions. We thank Dr. Jintao Liu’s lab for sharing bacterial strains. We thank Dr. Jingren Zhang’s lab, particularly Dr. Haoran An and Dr. Xueting Huang, for supporting the experiment. This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0990000), the National Key R&D Program of China (2022YFA0807300, 2023YFA1800200), the Natural Science Foundation of China (92474105, 32270945), STCSM (22ZR1468700 and 22140902400) (to X.S.). It was also supported by the Natural Science Foundation of China (82341116, 32200036), the Start-up Foundation of Tsinghua University (400-53332101822), Tsinghua University Initiative Scientific Research Program, Tsinghua University Dushi Program (20241080062), SXMU-Tsinghua Collaborative Innovation Center for Frontier Medicine Foundation, and Tsinghua-Peking Center for Life Sciences (045-61000100122) (to G.L.).
Author information
Authors and Affiliations
Contributions
G.L. and X.S. supervised the project. H.G. carried out the meta-bioinformatic analysis and the colitis experiment; H.G. and Y.W. performed the biochemical experiments and the immune profiling study; Y.Z., X.J., Z.G., L.Z., Y.L. and Y.S. assisted with biochemical manipulations and bioinformatic analysis; Z.W. and J.B. carried out long-read whole genome sequencing; H.G. and G.L. wrote the manuscript; X.S., S.L., F.D.B., and J.Y. assisted in data interpretation and draft writing. All authors revised the manuscript and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zeinab Hosseinidoust, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, H., Wang, Y., Zhao, Y. et al. Human gut prophage landscape identifies a prophage-mediated fucosylation mechanism alleviating colitis. Nat Commun 16, 11541 (2025). https://doi.org/10.1038/s41467-025-66733-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66733-5
