
Abstract
Chemical glycosylation facilitates the scalable synthesis of structurally well-defined carbohydrates for functional studies and therapeutic development, with progress being driven by donors and strategies. Herein, we present an efficient and versatile O-glycosylation method utilizing newly designed, bench-stable and readily accessible oxazolidinone-based glycosyl carbamates as donors. This reaction is catalyzed by 2-pentafluorophenyl pyridinium salts under mild conditions and demonstrates elegant performance compared to conventional promoters. This robust protocol facilitates orthogonal, iterative and latent-active glycosylations for streamlined synthesis of oligosaccharides. Mechanistic studies, including NMR, deuteration and kinetic isotope effect experiments, establish that the pyridinium catalyst initiates glycosylation by first binding glycosyl acceptors. It then activates carbamate donors via CO₂ release to generate the oxocarbenium ion, which is identified as the rate-determining step of the glycosylation process.
Similar content being viewed by others
Introduction
Carbohydrates are essential for diverse biological processes1, yet their structural complexity renders isolation from natural sources challenging. Chemical synthesis provides an efficient, scalable approach2,3, facilitating therapeutic development and mechanistic studies. Recent advances, including automated chemical synthesis4,5, pre-activation methods6, orthogonal glycosylation7, latent-active strategies8, and iterative approaches9, have streamlined glycan assembly by minimizing intermediate purification. However, their efficacy relies on glycosyl donors and glycosylation protocols, underscoring the need for innovative donor design and synthetic methodologies.
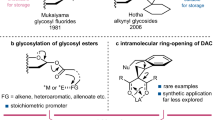
An ideal glycosyl donor requires an optimal stability-reactivity balance, facile accessibility, and inert leaving groups to ensure selective glycosylation. Classical donors like glycosyl halides10 and acetimidates11,12 remain fundamental, but advancements include sulfur13,14, phosphorus15,16, epoxy17,18, ether19,20, and particularly ester-based21,22,23,24,25,26,27 alternatives. Enhanced reactivity has been achieved through isomerization, cyclization, precipitation, and strain-release strategies (Fig. 1a). However, practical implementation is hindered by the lack of cost-effective, mild synthetic protocols with stable precursors and the non-recyclable nature of leaving groups, which generate inactive waste, adversely impacting atomic economy.

a Strategies for improving donor reactivity; b Development of oxazolidinone-based glycosyl donors for glycosylation with newly designed catalyst.
Gas-release activation represents a promising strategy for driving chemical reactions28, however, its application in carbohydrate chemistry remains underdeveloped14. Glycosyl carbamates29,30,31,32,33,34 are a unique donor class of donors that can be synthesized under mild conditions, and activated by CO2 release to drive glycosylation. Since Ley’s seminal report29 on imidazole-based donors, this class has been used for O-glycoside synthesis. However, their inherent instability necessitates fresh preparation and reliance on stoichiometric ZnBr₂ for promotion. Notably, imidazole exchange side reactions compromise glycosylation efficiency35. Subsequent developments included Kunz’s alkene-functionalized donor30, which, although activated by N-iodosuccinimide, undergoes cyclization instead of gas release due to the poor leaving ability of the allylamine group. Kiessling’s sulfonyl carbamate donor31 improved stability and reactivity but still required stoichiometric TMSOTf for high stereoselectivity. Redlich’s trichloroacetamide carbamate32 demonstrated enhanced reactivity, while its practical application was hampered by decomposition during purification33. Notably, both sulfonamide and trichloroacetamide leaving groups compete in glycosylation36,37, potentially disrupting reaction pathways. Despite these advances, carbamate donors still rely on conventional promoters, compromising their orthogonality with other synthetic strategies. Additionally, the stereoselective catalytic glycosylation employing glycosyl carbamates via gas-releasing remains in its infancy.
To improve the utility of glycosyl carbamates, we developed oxazolidinone-based glycosyl carbamates as donors, leveraging their commercial availability, good stability, and suppressed nucleophilicity38 that minimizes side reactions. We established an efficient O-glycosylation system combining these carbamates with a 2-pentafluorophenyl pyridinium catalyst (Fig. 1b), offering distinctive advantages: (1) good donor stability and straightforward synthesis; (2) broad compatibility; (3) activation under mild conditions via a unique catalytic mechanism; (4) recyclable oxazolidinone leaving groups improving atomic economy; (5) orthogonal activation enabling seamless integration with existing methods.
Results
Development of oxazolidinone-based glycosyl carbamate
The oxazolidinone-based glycosyl carbamates were synthesized by reacting hemiacetal S1 with acyl chloride S2 (prepared in one step from oxazolidinone with triphosgene39) in the presence of DIPEA. With this method, diverse donors (1a-1k) encompassing various sugar types and protecting groups were successfully prepared (Fig. 2). Notably, the donor exhibited good stability, maintaining integrity for >3 months at room temperature (Supplementary Fig S27).

Isolated yields. Bn = benzyl; Ac = acetyl; Me = methyl; TBDPS = tert-butylphenylsilyl Reaction condition: S1 (3.3 mmol), S2 (3.3 mmol), DIPEA (2.0 equiv.) RT, 4 h. aS1 (6.2 g, 12.6 mmol) was used.
Reaction development
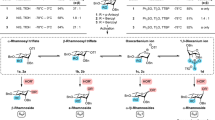
The reactivity of these newly designed donors in glycosylation was investigated using donor 1a and 4-fluorophenol 2a as model substrates under varying conditions (Table 1). Initial screening of common promoters (TMSOTf, TfOH, TsOH, BF3·Et2O, SnCl4) revealed minimal reactivity (entry 1) or uncontrolled selectivity (entries 2–5). Phosphoric acid A marginally improved selectivity but suffered from low yield (entry 6). Pyridinium catalysts40,41 were then explored; while B failed to initiate the reaction (entry 7), introducing a phenyl group at the ortho position did not enhance reactivity (entry 8). Substitution with a strongly electron-withdrawing pentafluorophenyl group significantly improved performance, yielding the product 3a in 71% yield with excellent β-selectivity (entry 9). However, a bromide counterion reduced both yield and selectivity (entry 10). Solvent screening showed that high-polarity solvents inhibited the reaction (entry 11). Pleasingly, increasing the stoichiometry of 1a to 1.5 equiv, and using only 5 mol% catalyst D improved the yield to 96% with recovering 93% of the oxazolidinone (entry 12, Supplementary Fig S12). Surprisingly, when 1a-α was employed as the donor, the reaction proceeded very slowly, even with a catalyst loading of 20% (entry 13). This observation suggests the 2-OAc group in the β-anomer may play a crucial role in facilitating the departure of anomeric leaving group42. Further studies on alternative donor 1h confirmed that neighboring group participation was essential for high selectivity and reactivity (entry 14). Notably, the disarmed peracetylated donor 1i proved completely unreactive (entry15). The failure of glycosylation with 1l32 underscores the good reactivity profile of our donor (entry 16). Furthermore, control experiments confirmed the indispensable role of the catalyst (entry 17).
Substrate scope investigation
β-O-Aryl glycosides are valuable scaffolds in medicinal chemistry43. However, the selective synthesis of these compounds remains challenging because the reaction is under thermodynamic control, favoring the formation of C-aryl glycosides and leading to undesired rearrangement44. Using optimized conditions, we evaluated various phenolic nucleophiles with glycosyl carbamate donors (Fig. 3). Remarkably, the reactions proceeded with excellent stereocontrol, affording exclusively the 1,2-trans O-aryl glycosides irrespective of the phenol electronic properties (3a-3c). Pleasingly, this method showed no scale-up effect. The reaction performed on a 1 mmol scale proceeded well, affording excellent yield and stereoselectivity (3c). Notably, β-naphthol, which typically favors C-glycosylation44, exclusively formed the desired O-glycoside 3d. The method also accommodated pharmaceutically relevant scaffolds including 2-hydroxycoumarin (3e), estrone (3f), and 2-hydroxylanthraquinone (3g). The scope was further extended to D-xylose, D-ribose, and D-mannose (3h-3j). While ribose-derived donors reacted efficiently under standard conditions, the xylosylation and mannosylation proceeded sluggishly with catalyst D. Notably, switching to triflate analog F significantly enhanced the reaction, affording the α-mannoside (3j) in good yield. Intriguingly, the α-configured ribosyl donor also yielded exclusively the β-product (3i), underscoring neighboring group participation. Furthermore, this protocol was successfully applied to the Me-protected derivative, demonstrating the generality of the method (3k).

Reaction condition: 1a (0.075 mmol), 2a (0.05 mmol), CH2Cl2 (1 mL), D (5 mol%), RT, 12 h, under N2; aD (20 mol%), 40 °C; bF (20 mol%), 40 °C; cF (20 mol%), 50 °C, d1a (0.15 mmol), F (20 mol%), 40 °C; e36 h, no catalyst; fF (40 mol%). Isolated yields. Bn benzyl, Ac acetyl, Ph phenyl.
The glycosylation method also demonstrated good generality across diverse alcohol acceptors (Fig. 3), delivering glycosides (3l-3v) in high yields (85-99%) with excellent stereoselectivity. A wide range of carbohydrate acceptors reacted smoothly with donor 1a. This versatility facilitated the preparation of β-(1 → 6)-, (1 → 4)-, (1 → 3)-, and (1 → 2)-linked disaccharides (3l-3s) efficiently regardless of variations in protecting groups or hydroxyl position.
Aliphatic alcohols, ranging from small-chain substrates (methanol, 3t; isopropanol, 3u) to long-chain analogs (dodecanol, 3v), were seamlessly converted. Specifically, selective glycosylation occurred preferentially at the aliphatic OH over phenolic OH in 4-hydroxyphenethyl alcohol (3w). The method was further validated by nearly quantitative glycosylation of the bioactive natural product podophyllotoxin (3x), a precursor to anticancer agents etoposide and teniposide45. The protocol demonstrated broad versatility, successfully converting multiple pyranosyl carbamate donors into their corresponding glycosides (3y-3ac) in 75–99% yields.
Remarkable scope and stereoselectivity were also demonstrated in the glycosylation of carboxylic acids46 using this protocol. Both aromatic (benzoic acid derivatives, 3ad-3ag) and pharmacologically relevant aliphatic molecules (indomethacin 3ah, tripterine 3ai, naproxen 3aj, aspirin 3ak), formed exclusiveβ-glycosyl esters. The protocol also accommodated amino acid derivative (acetyl-L-phenylalanine, 3al) and various sugar configurations (3am-3ap). Control experiments confirmed that the pyridinium catalyst was indispensable, with no product formation occurring in its absence. The method further enabled efficient synthesis of aminooxy glycosides (3aq-3as) from N-hydroxysuccinimide, N-hydroxyphthalimide and oxime, providing streamlined access to biologically important aminooxy glycosides47. Intriguingly, phosphoric acid acceptors (diphenyl/dibenzyl hydrogen phosphates) reacted smoothly with donor 1a to afford phosphate-linked glycosides48 (3at-3au). It is noteworthy that the uncatalyzed reations exhibited significantly slower kinetics, underscoring the critical role of the catalyst in this transformation (3at).
Application in oligosaccharide synthesis
Our oxazolidinone-based glycosyl carbamates enable oligosaccharide assembly through unique reactivity profiles distinct from established conditions. As shown in Fig. 4a, donor 1a exhibited negligible reactivity under NIS/TMSOTf 24 conditions and remained inactive under gold-catalyzed conditions22. Among comparative donors (5, 6, 7, 8), only 7 participated in our pyridinium-catalyzed system. This excellent orthogonality allows for selective, stepwise oligosaccharide construction through strategic donor selection. Initially, we established orthogonal glycosylation between our donor 1a and Yu’s donor 9, successully obtaining trisaccharide 10 through a one-pot, two-step reaction (Fig. 4b, Reaction 1). Furthermore, a one-pot sequence involving glycosylation of donor 1k and Xiao’s donor 11, followed by selective TBDPS deprotection and subsequent coupling with Schmidt’s donor 13, afforded tetrasaccharide 14 (Fig. 4b, Reaction 2). Then, we rapidly constructed tetrasaccharide 15 through three consecutive pyridinium-catalyzed glycosylation steps employing an iterative strategy (Fig. 4c). Interestingly, latent-active strategy glycosylation proved successful (Fig. 4d). Donor 1a reacted selectively with the hydroxy group at C6 position of 16, leaving the anomeric hydroxy group untouched. Subsequent installation of carbamate moiety afforded 18, which served as a competent donor for futher glycosylation with 2 l to produce trisaccharide 19. Notably, the C2 acetyl group served as a useful handle for controlled assembly of oligosaccharide 20 through selective deprotection-glycosylation sequences (Fig. 4e).

a Orthogonality investigation; b Orthogonal strategy for synthesis of tetrasaccharide; c Iterative strategy for synthesis of tetrasaccharide; d Latent-active strategy for synthesis of trisaccharide; e Sequential glycosylation. Isolated yields. ABz 2-(hexyn-1-yl)benzoyl, PVB 2-(1-phenylvinyl)benzoyl, ⭘ 2,3,4-tri-O-benzoyl-6-O-(tert-butyldiphenylsilyl)-D-glucosyl, Bn benzyl, Ac acetyl, Bz benzoyl, TBDPS tert-butyldiphenylsilyl, Bu butyl.
Mechanistic studies
To elucidate the reaction mechanism, we conducted a series of experiments. Initial attempt employing deuterated catalyst d-F, even with a stoichiometric amount, revealed no deuterium transfer to the oxazolidinone (Fig. 5a), ruling out the direct donor activation. We therefore proposed that the catalyst might initially bind the glycosyl acceptor to form an intermediate. In the NMR titration of catalyst F with phenol 2a, an upfield proton shift of the catalyst and disappearance of the phenolic OH signal (Fig. 5b) indicated hydrogen bonding between the phenol -OH and catalyst -NH, in line with a previous report40 and supporting initial catalyst-acceptor interaction. Subsequent kinetic studies using MeOD and MeOH demonstrated a primary kinetic isotope effect (KIE) of only 1.3, suggesting O-H bond cleavage is not the rate-determining step (Fig. 5c)49. Additionally, the 1,2-trans product configuration implied oxocarbenium involvement50. Parallel experiments with deuterated donor d-1j vs 1j yielded a secondary KIE of 1.18 (Fig. 5d), supporting sp3 to sp2 rehybridization51. These findings collectively suggest that the oxocarbenium generation as the rate-determining step. Based on experimental evidence and literature precedents50, a plausible mechanism is proposed in Fig. 5e. The reaction starts with the formation of Int 1; which subsequently activates the glycosyl donor 1, leading to the generation of the oxocarbenium (Int 2), with concomitant release of CO2 and oxazolidinone 21. In this process, the hydroxyl proton of Int 1 is proposed to interact with the carbonyl groups of the glycosyl donor 1 through hydrogen bonding, while the neighboring participation effect of the 2-OAc group facilitates the departure of the anomeric leaving group, thereby promoting the efficient formation of the oxocarbenium Int 2. Concurrently, Int 1 is converted into Int 3. Finally, Int 2 is intercepted either by the glycosyl acceptor 2 (path 1) or Int 3 (path 2) to afford the product 3.

a Byproduct analysis with deuterated catalyst (NMR yields); b NMR titration for catalyst and phenol; c Kinetic isotope effect analysis for glycosyl acceptors (MeOH and MeOD); d Competition experiment; e Proposed mechanism. Bn benzyl, Ac acetyl, Me methyl.
Discussion
In conclusion, we have developed an efficient atom-economical glycosylation method for O-glycosides synthesis using stable glycosyl carbamates and cost-effective pyridinium salts as catalysts, with the CO2 release and oxazolidinone recovery. This protocol exhibits broad applicability, accommodating diverse glycosyl donors and acceptors while enabling orthogonal, iterative, and latent–active strategies for oligosaccharide construction. Mechanistic studies suggest that the pyridinium catalyst initially interacts with the glycosyl acceptor to generate a reactive intermediate, promoting carbamate activation. Kinetic isotope effect analysis implicates oxocarbenium formation as the likely rate-determining step.
Methods
General procedure for pyridinium-catalyzed O-glycosylation
To an oven-dried vial was added glycosyl carbamate 1 (0.075 mmol, 1.5 equiv.), glycosyl acceptor 2 (0.05 mmol, 1.0 equiv.), catalyst D (5 mol%) and anhydrous CH2Cl2 (1 mL) under nitrogen atmosphere. The solution was stirred at room temperature for 12 h. The resulting mixture was concentrated and the residue was purified by silica gel column chromatography to afford the product 3.
Procedure for gram-scale reaction
In a glove box filled with nitrogen, to an oven-dried 25 mL tube equipped with a stirring bar were added 1a (0.9078 g, 1.5 mmol, 1.5 equiv.), acceptor 2c (124.2 mg, 1.0 mmol, 1.0 equiv.), D (66.0 mg, 20 mol%), and anhydrous CH₂Cl₂ (7 mL, 0.14 M). The reaction mixture was stirred at 40 °C for 12 h and then purified by column chromatography on silica gel with petroleum ether/ethyl acetate (5:1) as eluent to afford 3c as a white solid (580.2 mg, 97% yield).
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information Files. Additional data are available from the corresponding author upon request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers 2433567 (1a). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Bertozzi, C. R. & Kiessling, L. L. Chemical glycobiology. Science 291, 2357–2364 (2001).
Boltje, T. J., Buskas, T. & Boons, G. J. Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat. Chem. 1, 611–622 (2009).
Nielsen, M. M. & Pedersen, C. M. Catalytic glycosylations in oligosaccharides synthesis. Chem. Rev. 118, 8285–8358 (2018).
Yao, W. et al. Automated solution-phase multiplicative synthesis of complex glycans up to a 1080-mer. Nat. Synth. 1, 854–863 (2022).
Plante, O. J., Palmacci, E. R. & Seeberger, P. H. Automated Solid-phase synthesis of oligosaccharides. Science 291, 1523–1527 (2001).
Yao, W. & Ye, X. Donor preactivation-based glycan assembly: from manual to automated synthesis. Acc. Chem. Res. 57, 1577–1594 (2024).
He, H. et al. An orthogonal and reactivity-based one-pot glycosylation strategy for both glycan and nucleoside synthesis: access to TMG-chitotriomycin, lipochitoolifosaccharides and capuramycin. Chem. Sci. 12, 5143–5151 (2021).
Das, A. & Jayaraman, N. Aglycon reactivity as a guiding principle in latent-active approach to chemical glycosylations. Carbohydr. Res. 508, 108404 (2021).
Liu, K. et al. Iterative synthesis of 2-deoxyoligosaccharides enabled by stereoselective visible-light-promoted glycosylation. Angew. Chem. Int. Ed. 61, e202204923 (2022).
Singh, Y., Geringer, S. A. & Demchenko, A. V. Synthesis and glycosidation of anomeric halides: evolution from early studies to modern methods of the 21st century. Chem. Rev. 122, 11701–11758 (2022).
Schmidt, R. R. New methods for the synthesis of glycosides and oligosaccharides—are there alternatives to the Koenigs-Knorr method? [New synthetic method (56)]. Angew. Chem. Int. Ed. 25, 212–235 (1986).
Yu, B. & Sun, J. Glycosylation with glycosyl N-phenyltrifluoroacetimidates (PTFAI) and a perspective of the future development of new glycosylation methods. Chem. Commun. 46, 4668–4679 (2010).
Lian, G., Zhang, X. & Yu, B. Thioglycosides in carbohydrate research. Carbohydr. Res 2015, 13–22 (2015). 403.
Shang, W. & Niu, D. Radical pathway glycosylation empowered by bench-stable glycosyl donors. Acc. Chem. Res. 56, 2473–2488 (2023).
Plante, O. J., Palmacci, E. R., Andrade, R. B. & Seeberger, P. H. Oligosaccharide synthesis with glycosyl phosphate and dithiophosphate triesters as glycosylating agents. J. Am. Chem. Soc. 123, 9545–9554 (2001).
Li, Q., Levi, S. M., Wagen, C. C., Wendlandt, A. E. & Jacobsen, E. N. Site-selective, stereocontrolled glycosylation of minimally protected sugars. Nature 608, 74–79 (2022).
Danishefsky, S. & Chow, K. Stereospecific Vorbrueggen-like reactions of 1,2-anhydro sugars. an alternative route to the synthesis of nucelosides. J. Org. Chem. 55, 4211–4214 (1990).
Tanaka, M. et al. Diastereoselective desymmetric 1,2-cis-glycosylation of meso-diols via chirality transfer from a glycosyl donor. Nat. Commun. 11, 2431 (2020).
Hu, Z., Tang, Y. & Yu, B. Glycosylation with 3,5-dimethyl-4-(2’-phenylethynylphenyl)phenyl (EPP) glycosides via a dearomative activation mechanism. J. Am. Chem. Soc. 141, 4806–4810 (2019).
Zhang, J. et al. Photosensitizer-free visible-light-promoted glycosylation enabled by 2-glycosyloxy tropone donors. Nat. Commun. 14, 8025 (2023).
Lopez, L. C. & Fraser-Reid, B. n-pentenyl esters versus n-pentenyl glycosides. synthesis and reactivity in glycosidation reactions. J. Chem. Soc., Chem. Commun. 1991, 159–161 (1991).
Yu, B. Gold(I)-catalyzed glycosylation with glycosyl o-alkynylbenzoates as donors. Acc. Chem. Res. 51, 507–516 (2018).
Mishra, B., Neralkar, M. & Hotha, S. Stable alkynyl glycosyl carbonates: catalytic anomeric activation and synthesis of a tridecasaccharide reminiscent of mycobacterium tuberculosis cell wall lipoarabinomannan. Angew. Chem. Int. Ed. 55, 7786–7791 (2016).
Li, P. et al. Glycosyl ortho-(1-phenylvinyl)benzoates versatile glycosyl donors for highly efficient synthesis of both o-glycosides and nucleosides. Nat. Commun. 11, 405 (2020).
Ma, X. et al. Traceless” directing group enables catalytic SN2 glycosylation toward 1,2-cis-glycopyranosides. J. Am. Chem. Soc. 143, 11908–11913 (2021).
Wang, H. et al. Isoquinoline-1-carbohylate as a traceless leaving group for chelation-assisted glycosylation under mild and neutral reaction conditions. Angew. Chem. Int. Ed. 56, 15698–15702 (2017).
Ding, H., Lyu, J., Zhang, X., Xiao, X. & Liu, X. Efficient and versatile formation of glycosidic bonds via catalytic strain-release glycosylation with glycosyl ortho-2,2-dimethoxycarbonylcyclopropylbenzoate donors. Nat. Commun. 14, 4010 (2023).
Hui, C., Wang, S. & Xu, C. Dinitrogen extrusion of diazene in organic synthesis. Chin. Chem. Lett. 33, 3695–3700 (2022).
Ford, M. J. & Ley, S. V. A simple, one-pot, glycosidation procedure via (1-imidazolylcarbonyl) glycosides and zinc bromide. Synlett 1990, 255–256 (1990).
Kunz, H. & Zimmer, J. Glycoside synthesis via electrophile-induced activation of N-allyl carbamates. Tetrahedron Lett. 34, 2907–2910 (1993).
Hinklin, R. J. & Kiessling, L. L. Glycosyl sulfonylcarbamates: new glycosyl donors with tunable reactivity. J. Am. Chem. Soc. 123, 3379–3380 (2001).
Knoben, H., Schlüter, U. & Redlich, H. Synthesis of N-unsubstituted, mono- and disubstituted carbohydrate-1-o-carbamates and their behaviour in glycoside syntheses. Carbohydr. Res. 339, 2821–2833 (2004).
Shirahata, T. et al. Sequential one-pot glycosylation with glycosyl N-trichloroacetylcarbamate and trichloroacetate including dehydrative approach using 1-hydroxy sugars. Tetrahedron 67, 6482–6496 (2011).
Gurung, P. B., Thapa, P., Hettiarachchi, I. L. & Zhu, J. Cationic gold(I)-catalyzed glycosylation with glycosyl N-1,1-dimethylpropargyl carbamate donors. Org. Biomol. Chem. 20, 7006–7010 (2022).
Chandrashekar, C., Okamoto, R., Izumi, M. & Kajihara, Y. Chemical modification of the N termini of unprotected peptides for semisynthesis of modified proteins by utilizing a hydrophilic protecting group. Chem. Eur. J. 25, 10197–10203 (2019).
van de Vrande, K. N. A., Filippov, D. V. & Codée, J. D. C. Formation of glycosyl trichloroacetamides from trichloroacetimidate donors occurs through an intermolecular aglycon transfer reaction. Org. Lett. 25, 6128–6132 (2023).
Nielsen, M. M., Mała, P., Baldursson, E. & Pedersen, C. M. Self-promoted and stereospecific formation of N-glycosides. Chem. Sci. 10, 5299–5307 (2019).
Kur’yanov, V. O. et al. Synthesis of heteroaromatic N-β-glycosides of N-acetylglucosamine under the conditions of phase transfer catalysis: I. Glucosaminides of 2-oxobenzazoles. Russ. J. Bioorg. Chem. 32, 552–557 (2006).
Ganiu, M. O., Nepal, B., Van Houten, J. P. & Kartika, R. A decade review of triphosgene and its applications in organic reactions. Tetrahedron 76, 131553 (2020).
Jiao, Q. et al. Anion-bridged dual hydrogen bond enabled concerted addition of phenol to glycal. Adv. Sci. 11, 2308513 (2024).
Ghorai, J., Almounajed, L., Noori, S. & Nguyen, H. M. Cooperative catalysis in stereoselective O- and N-glycosylations with glycosyl trichloroacetimidates mediated by singly protonated phenanthrolinium salt and trichloroacetamide. J. Am. Chem. Soc. 146, 34413–34426 (2024).
Premathilake, H. D., Mydock, L. K. & Demchenko, A. V. Superarming commun glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. J. Org. Chem. 75, 1095–1100 (2019).
Dimakos, V. & Taylor, M. S. Recent advances in the direct O-arylation of carbohydrates. Org. Biomol. Chem. 19, 514–524 (2021).
Li, Y., Wei, G. & Yu, B. Aryl C-glycosylation of phenols with glycosyl trifluoroacetimidates. Carbohydr. Res. 341, 2717–2722 (2006).
Clark, P. I. & Slevin, M. L. The clinical pharmacology of etoposide and teniposide. Clin.-Pharmacokinet. 12, 223–252 (1987).
Liu, Z., Liu, D., Zhu, D. & Yu, B. Stereoselective synthesis of β-glycosyl esters via 1-hydroxybenzotriazole mediated acylation of glycosyl hemiacetals. Org. Lett. 25, 5372–5377 (2023).
Thadke, S. A., Neralkar, M. & Hotha, S. Facile synthesis of aminooxy glycosides by gold (III)-catalyzed glycosidation. Carbohydr. Res. 430, 16–23 (2016).
Zhang, X. et al. Stereoselective gold (I)-catalyzed approach to the synthesis of complex α-glycosyl phosphosaccharides. Nat. Commun. 13, 421 (2022).
Westheimer, F. H. The magnitude of the primary kinetic isotope effect for compounds of hydrogen and deuterium. Chem. Rev. 61, 265–273 (1961).
Crich, D. Mechanism of a chemical glycosylation reaction. Acc. Chem. Res. 43, 1144–1153 (2010).
Kurtz, K. A. & Fitpatrick, P. F. pH and secondary kinetic isotope effects on the reaction of D-amino acid oxidase with nitroalkane anions: evidence for direct attack on the flavin by carbanions. J. Am. Chem. Soc. 119, 1155–1156 (1997).
Acknowledgements
We appreciate the National Natural Science Foundation of China (22201041 (C.X.), 22208055 (S.W.)), the Fuzhou University (511041 (C.X.)), the Fujian Provincial Natural Science Foundation of China (2020J011002 (W.C.), 2025J01783 (T.L.)), and the Joint Funds for the Innovation of Science and Technology, Fujian Province (2023Y9140 (W.C.)) for financial support. We greatly thank Dr. Sebastian Hui for his valuable assistance in language editing.
Author information
Authors and Affiliations
Contributions
X.Q. conducted the majority of the experimental work. L.K., Q.J., W.Z., W.L., J.L. helped with expansion of substrate scope. W.C., T.L. and S.W. discussed the project. C.X. conceived the idea and supervised the project. C.X. and X.Q. prepared this manuscript. All authors contributed to data analysis and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rima Thakur and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, X., Ke, L., Jiao, Q. et al. A universal O-glycosylation platform enabled by pyridinium catalysis using gas-releasing oxazolidinone-based carbamates donors. Nat Commun 17, 141 (2026). https://doi.org/10.1038/s41467-025-66857-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66857-8
