
Abstract
The interplay among host genetic architecture, gut microbiota, metabolites and bone metabolism remains poorly understood. This study aims to comprehensively investigate the shared genetic factors, causal relationships, and the involvement of blood metabolites between gut microbiota and bone mass, based on datasets from European populations. Here, we estimate the polygenic SNP heritability for 1104 gut microbiota taxa, only 96 (8.7%) showed significant polygenic heritability. We identify 14 distinct gut microbiota taxa with pleiotropic effects on estimated heel bone mineral density (eBMD), 11 of them are found to have a causal association with eBMD. Following sensitivity and validation analyses, we find that the gut microbiota taxa family Bifidobacteriaceae; genus Bifidobacterium and species Bifidobacterium adolescentis exert causal effects leading to decreased eBMD. Mediation analyses indicate that the impact of these taxa on eBMD may be driven by their influence on circulating stearidonate (18:4n-3) levels via n-3 pathway, with mediating proportion from 77.53% to 87.24%. In conclusion, the genetically informed negative association between bifidobacterial taxa and bone mass may reflect the host–microbe interactions at LCT/MCM6 locus and represent an adaptive microbial response to lactose intolerance, thus, it could potentially be mitigated through supplementation with bifidobacterial probiotics and n-3 polyunsaturated fatty acids.
Similar content being viewed by others

Introduction
Osteoporosis is a bone disease characterized by decreased bone mineral density (BMD) and the occurrence of bone microstructural damage. The evaluation of this condition is primarily measured using dual energy X-ray absorptiometry scans (DXA), with osteoporotic fractures serving as the main clinical outcome1. In the United States, over 2 million osteoporosis-related fractures occurred in 2005, resulting in a treatment cost of $17 billion2. BMD is a highly heritable trait with estimated heritability ranging from 50 to 85% in family and twin studies3,4. Over the past decade, genome-wide association studies (GWAS) have identified hundreds of genetic loci associated with BMD, osteoporosis, and osteoporotic fractures5.
The host gut microbiota (GM), a complex community of microbes inhabiting the gastrointestinal tract, has recently been recognised for its potential role in bone regulation6. Yan et al. demonstrated that colonisation of the GM in mice promotes skeletal growth and remodelling by stimulating the hormone insulin-like growth factor 1 (IGF-1), as compared to germ-free mice7. Li et al. also found that GM could influence bone loss associated with sex steroid deficiency in mice8. Certain microbiota taxa, such as Bifidobacterium longum, were shown to affect mineral absorption, including calcium, magnesium, and phosphate9, and may also play a crucial role in synthesising vitamins B and K10, thereby potentially affecting bone development11,12. Furthermore, Studies have shown that GM may influence bone development by producing short-chain fatty acids (such as butyrate)13, and affecting peptide like glucagon-like peptide 1 which are related to bone metabolism14.
Previous studies have explored the relationship between GM and BMD using the mendelian randomisation (MR) approaches15,16. However, these studies primarily examined bacterial taxa at a broader taxonomic level, such as order or family, rather than at a more specific level like genus. The summary datasets were derived from the TwinsUK GWAS for GM17, in which the sample size was relatively small. The reliability of genetic associations depends heavily on the selection of instrumental variables18,19. Fortunately, large-scale GWAS datasets that could offer robust SNP-trait associations were publicly available recently. Qin et al. identified single nucleotide polymorphisms (SNPs) associated with gut taxonomies within a Finnish cohort (FINRISK) in 5959 participants20. The Dutch Microbiome Project (DMP) with 7738 participants confirmed these associations near the LCT and ABO genes21. A separate investigation in five German cohorts (German) involving nearly 9000 individuals also revealed a significant effect of host blood type genes on GM composition22. Finally, the MiBioGen consortium undertook a comprehensive analysis that integrated genome-wide genotyping and 16S faecal microbiome data from 18,340 individuals across 24 cohorts to further understand the relationship between host genetics and microbiota composition23.
In this analysis, we incorporated 1104 GWAS summary statistics for GM taxa from the four abovementioned studies20,21,22,23, spanning five taxonomic levels from phylum to species, substantially improving the resolution of the taxa. The inclusion of large publicly available genetic summary statistics on GM, featuring the largest sample size to date, has significantly enhanced our ability to investigate the complex relationship between GM and diseases. By leveraging these datasets, all derived from individuals of European ancestry, we investigated the role of genetically predicted abundance of specific bacterial taxa on BMD. This exploration was conducted through an assessment of the polygenic SNP heritability (\({h}_{{SNP}}^{2}\)) employing linkage disequilibrium (LD) score regression method24. We then estimated the pleiotropic genetic effects between GM and BMD at two genetic levels including genome-wide and LD-independent regions. Furthermore, we employed MR and multivariable MR approaches to evaluate the causal effect and the mediating effect between bacterial taxa and BMD traits.
Results
The estimation of heritability of the GM taxa
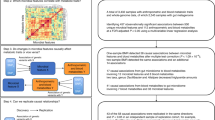
An overview of the study design was depicted in Fig. 1. A total of 1104 GM taxa GWAS summary data were obtained from four studies (FINRISK, DMP, German and MiBioGen) (Fig. 1 and Supplementary Data 1)20,21,22,23, and the studies encompassed a sample size ranging from 5959 to 18340 participants (Supplementary Data 1). As most GM taxa are strongly influenced by environmental factors, and a few taxa are primarily shaped by host genetic factors17,25, the first step was to identify GM taxa exhibiting significant polygenicity to ensure that subsequent analyses reflected genetically informed relationships.

Step 1: To estimate to the heritability of the 1,104 gut microbiota (GM) taxa with publicly available GWAS summary datasets from four studies (FINRISK, DMP, German and MiBioGen) in European population. Step 2: To explore the pleiotropy genetic effect between 96 unique GM taxa and eBMD at two levels, [i] genome-wide pleiotropy (by estimating Pearson correlation between LD-independent SNP effects), [ii] genetic loci pleiotropy (by estimating GWAS-PW), identifying 14 candidate taxa. Step 3: To estimate the causal effects of the 14 candidate GM taxa on BMD using two-sample Mendelian randomisation. Three unique GM taxa (family Bifidobacteriaceae; genus Bifidobacterium and species Bifidobacterium adolescentis) showed robust causal effects on BMD. Step 4: Multivariable MR analyses were conducted to estimate the mediation effects of blood stearidonate on the associations between three bifidobacterial taxa and eBMD.
We estimated the SNP heritability (\({h}_{{SNP}}^{2}\)) for all GM taxa using LD score regression method (LDSC). The gut taxon species Bifidobacterium catenulatum exhibited the highest SNP-based heritability (\({h}_{{SNP}}^{2}\) = 0.9845) (Supplementary Data 2). The Z scores of \({h}_{{SNP}}^{2}\) in all GM taxa exhibited a significant pearson correlation with both the number of sample size (r = 0.116, P = 0.0001093) (Fig. 2A) and the number of SNPs in GWASs (r = 0.112, P = 0.0001848) (Fig. 2B). This implies that the heritability of gut taxa could be attributed to genome-wide polygenicity and larger participant numbers in GM GWAS. A total of 108 GM taxa with a Z score of \({h}_{{SNP}}^{2}\) great than 1.64 (\({P}_{{h}_{{SNP}}^{2}}\) < 0.05; one-side) were considered to exhibit significant polygenicity, and their SNP-based heritability values were both statistically significant and meaningful (\({h}_{{SNP}}^{2}\) ≥ 0.0445). For taxa with multiple datasets available, we retained the dataset with the highest Z score of \({h}_{{SNP}}^{2}\) for subsequent analyses.

A The scatter plot with linear regression line showing the relationship between the bacterium estimated heritability and sample size. Statistical analysis was performed using two-sided Pearson correlation and linear regression; the Pearson correlation coefficient (r), exact two-sided P value, and 95% confidence interval (shown as the shaded area around the red regression line) are reported. The red regression line represents the fitted linear model. B The scatter plot with linear regression line showing the relationship between the bacterium estimated heritability and the number of detected SNPs. Statistical analysis was performed using two-sided Pearson correlation and linear regression; the Pearson correlation coefficient (r), exact two-sided P value, and 95% confidence interval (shown as the shaded area around the red regression line) are reported. The red regression line represents the fitted linear model. C The bar graph presents the GM taxa with significant SNP-heritability polygenicity, relative to the proportion of GM taxa within each taxonomic classification level. D The bar chart displaying the 96 selected GM taxa with significant estimated heritability through LDSC in six different taxonomic level.
Consequently, 96 unique GM taxa were selected for further analyses, representing approximately 8.7% of all tested taxa (96/1104) (Fig. 1, Fig. 2C, Fig. 2D and Supplementary Data 3). These taxa are categorised into one phylum, four classes, four orders, 11 families, 33 genera, and 43 species (Fig. 2C and Fig. 2D). The name of included gut taxon for further analysis was reported by the lowest bacteria taxa level name for consistency. The estimated median \({h}_{{SNP}}^{2}\) of those 96 unique GM taxa from the four studies distributed from 0.072 to 0.1735 (Table 1). Additionally, we observed that eBMD GWAS exhibited a Z score for \({h}_{{SNP}}^{2}\) of 14.96 (Supplementary Data 3).
Genome-wide pleiotropic association between GM taxa and eBMD
We then aimed to identify the shared genetic pleiotropic effects between GM and bone mass (Fig. 1). We employed two complementary approaches (pleiotropic association at genome-wide level or LD-independent regions) to identify the candidates GM taxa out of the 96 selected GM taxa with significant polygenicity.
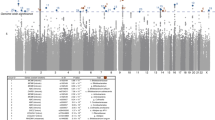
In order to identify gut taxa that exhibited a concordant genetic effect across the entire genome (genome-wide pleiotropic) with eBMD, we employed an approach to estimate the similarity in SNP effects at the genome-wide level, similar to a previous study26. We evaluated the correlation of Z score of the LD-independent SNPs between the 96 unique GM taxa and eBMD. The LD-independent SNPs from the eBMD GWAS were selected and matched with the 96 unique GM taxa GWASs, leaving us with a range of 31,712 to 58,953 SNPs for estimating genome-wide effect similarity. The pearson correlation coefficients (r) were calculated using the Z scores of the overlapped SNPs, which were harmonised by the effect allele. Among the 96 unique GM taxa, two GM taxa displayed significant negative SNP effects correlations with eBMD at a false discovery rate (FDR) less than 0.1 (Fig. 3A), including Haloplasmatales (r = -0.00896; p = 0.02965) and Bifidobacterium adolescentis (r = -0.0153; p = 0.0055) (Supplementary Data 4).

A The volcano plot of pearson correlation results between the SNP Z-scores from the eBMD GWAS and gut microbiota taxa GWASs. Statistical analysis was performed using two-sided Pearson correlation; exact two-sided P values are reported. P values were adjusted for multiple comparisons using the false discovery rate (FDR), and gut microbiota taxa with FDR ≤ 0.1 are highlighted in red. B Genetic loci pleiotropy within 1703 LD-independent loci identified by GWAS-PW analysis. An independent locus on chromosome 2 (135,159,453–137,041,842) was identified as being associated with both seven gut microbiota and BMD. PPA_3 represents the posterior probability that the locus influence both eBMD and gut microbiota via the SNP rs2090660 (hg19 position: chr2:136,818,719; effect allele: T). The forest plot illustrates the effect sizes (beta) of SNP rs2090660 for seven gut microbiota taxa: Bifidobacteriales (N = 7,252 participants); Bifidobacteriaceae (N = 7252 participants); Bifidobacterium (N = 7244 participants); Bifidobacterium adolescentis (N = 6328 participants); Bifidobacterium pseudocatenulatum (N = 5959 participants); Negativibacillus sp000435195 (N = 5959 participants); Turicibacter (N = 5959 participants) and eBMD (N = 426,824 participants). Effect sizes (beta) in each GWAS are shown as point estimates with 95% confidence intervals calculated as beta ± 1.96 × SE. Error bars therefore represent 95% confidence intervals. The effect sizes are colour-coded according to significance: red (P < 5 × 10–8), green (P < 1 × 10–5) and black (P < 1 × 10–3). C The forest plot displays the results of three MR analyses (Inverse-variance weighted, weighted median, and MR-Egger.) assessing the causal effects of four gut microbiota taxa: Bifidobacterium adolescentis (N = 6,328 participants), Bifidobacteriales (N = 7252 participants), Bifidobacteriaceae (N = 7252 participants) and Bifidobacterium (N = 7244 participants) on eBMD. All MR analyses were conducted as two-sided statistical tests, and exact two-sided P values are reported. Point estimates represent causal effect beta values, and horizontal error bars indicate the 95% confidence intervals calculated as beta ± 1.96 × SE. Instrumental variables were selected using three significance thresholds: 1 × 10-5, 5 × 10-6 and 5 × 10-8.
Pleiotropic genetic loci influencing association between GM taxa and eBMD
Using the pairwise GWAS (GWAS-PW) method27, we have identified independent genetic loci which were associated with both eBMD and 96 unique GM taxa (Fig. 1). The independent loci with third posterior probability (PPA3) ≥ 0.9 was suggested the pleiotropic associated SNP between BMD and GM taxa in this analysis. Specifically, we have identified five pleiotropic genetic loci that are jointly associated with estimated eBMD and 13 distinct GM taxa, all through the influence of shared SNPs (Supplementary Data 5). Among these, one locus located on chromosome 2 with the lead SNP rs2090660 positioned near the MCM6/LCT locus, affecting both estimated eBMD and five bifidobacterial taxa (order Bifidobacteriales; family Bifidobacteriaceae, genus Bifidobacterium and species Bifidobacterium adolescentis and Bifidobacterium pseudocatenulatum) with opposite effect (Fig. 3B and Supplementary Data 5). This locus also had pleiotropic effect on eBMD and the phylum Bacillota (consisting of genus Negativibacillus and genus Turicibacter) (Supplementary Data 5). Additionally, we uncovered another locus, situated on chromosome 9 with the lead SNP rs635634 near the ABO gene (Supplementary Fig. 1) that was associated with eBMD and order Ruminococcales (including species Faecalicatena torques and genus Faecalibacterium).
At last, we identified 14 candidates out of the 96 tested GM taxa with shared genetic pleiotropic effects on eBMD based on two complementary approaches (Fig. 1).
Candidate GM taxa with a causal effect on eBMD
Next, we tested for the causal relationships between eBMD and these 14 candidate GM taxa by employing the inverse variance-weighted (IVW) method, applying a Bonferroni-corrected p-value threshold 0.00357 ( = 0.05/14) (Fig. 1). Additionally, we utilised the weighted median method to enhance the robustness of our results and the MR-Egger regression method to assess directional (unbalanced) pleiotropy. Finally, leave-one-out IVW regression analyses were performed to further evaluate the robustness of the causal relationships.
We have identified the 11 specific GM taxa abundances with a causal relationship with eBMD, including Bifidobacteriales (βIVW = -0.02922, p = 0.000981); Bifidobacteriaceae (βIVW = -0.02921, p = 0.000981); Bifidobacterium (βIVW = -0.02921, p = 0.000981); Bifidobacterium adolescentis (βIVW = -0.03142, p = 3.585×10-5) (Table 2); Bifidobacterium pseudocatenulatum (βIVW = -0.02584, p = 0.00301); Clostridiales CHKCI006 sp900018345 (βIVW = 0.4172, p = 1.317×10-31); Merdibacter massiliensis (βIVW = 0.3241, p = 4.036×10-20); Negativibacillus sp000435195 (βIVW = 0.0542, p = 9.944×10-5); Turicibacter (βIVW = 0.3241, p = 4.036×10-20); Faecalibacterium (βIVW = -0.1692, p = 1.305×10-19); Haloplasmatales (βIVW = 0.04885, p = 0.00138) (Table 2 and Supplementary Data 6). In additional sensitivity analyses, 5 of those 11 specific GM taxa robustly demonstrated causal effects eBMD using IVs at both 1×10-5 and 5×10-6 P value thresholds. These GM taxa include Bifidobacteriales, Bifidobacteriaceae, Bifidobacterium, Bifidobacterium adolescentis, and Bifidobacterium pseudocatenulatum (Fig. 3C and Supplementary Data 7). In leave-one-out IVW regression analyses, several estimates for the association between eBMD and Bifidobacterium pseudocatenulatum crossed the null line, indicating instability in the causal inference and suggesting the absence of a robust and consistent effect. In contrast, no outlying genetic variants were detected in the causal relationships between eBMD and the other four GM taxa (Bifidobacterium adolescentis, Bifidobacteriales, Bifidobacteriaceae, and Bifidobacterium) (Supplementary Data 8 and Supplementary Fig. 2). Finally, robust causal effects on eBMD were observed for four of the 14 GM taxa: Bifidobacteriales, Bifidobacteriaceae, Bifidobacterium and Bifidobacterium adolescentis (Fig. 3C).
The validation of three bifidobacterial taxa associated with BMD
As three bifidobacterial taxa (family Bifidobacteriaceae, genus Bifidobacterium, species Bifidobacterium adolescentis) were measured across two independent studies (DMP21 and FINRISK20), we defined the GM taxa from DMP GWAS dataset as the discovery samples, thus, we verified the causality association between the three bifidobacterial taxa and eBMD with independent GM GWAS dataset from FINRISK study (Fig. 1). Specifically, MR analyses indicated that the abundance of family Bifidobacteriaceae (βIVW = -0.021512, p = 0.0221), the genus Bifidobacterium (βIVW = -0.0194, p = 0.01543) and the species Bifidobacterium adolescentis (βIVW = -0.0174, p = 0.0082) was causally associated with a decrease on eBMD in replication samples (Table 2). Furthermore, GWAS-PW analyses yielded consistent results in both the discovery and replication samples (Supplementary Data 9).
To further validate these associations, we performed additional inverse-variance weighted MR analyses using three bifidobacterial taxa and total body BMD (TB-BMD) GWAS datasets. We found that the abundance of three GM taxa Bifidobacteriaceae (βIVW = -0.0597, p = 0.046), Bifidobacterium (βIVW = -0.06, p = 0.045) and Bifidobacterium adolescentis (βIVW = -0.0632, p = 0.013) was causally associated with a decrease on TB-BMD (Table 2).
The mediating effect of three bifidobacterial taxa on BMD through the metabolite
Here, we conducted multivariable MR analysis to investigate the mediating role of blood metabolites on the effect of the GM taxa (Bifidobacteriaceae, Bifidobacterium and Bifidobacterium adolescentis) on eBMD. Initially, we identified 41 out of 453 blood metabolites that could be causally influenced by the species Bifidobacterium adolescentis, as determined by IVW analysis with a Bonferroni-corrected p-value < 0.05 (PIVW < 0.05/453) (Supplementary Data 10). Subsequently, we performed IVW analyses between these 453 metabolites and eBMD, and found that 45 metabolites showed a causal effect on eBMD at significant level (PIVW < 0.05/257), considering only 257 metabolites that contained at least one SNP at genome-wide significance (p < 5 × 10-8) (Supplementary Data 11). Among these, stearidonate (18:4n3) were causal associated with eBMD (βIVW = -0.3924, p = 3.808 × 10-12) (Supplementary Data 11). We found that Bifidobacterium adolescentis was causally associated with blood stearidonate level (βIVW = 0.0621, p = 1.04×10-4) (Fig. 4A). Moreover, we observed that the direct effect of the species Bifidobacterium adolescentis on eBMD disappeared after adjusting for stearidonate (Fig. 4B), suggesting that stearidonate plays an important mediating role in the causal relationship between gut and eBMD. Mediation analysis revealed that the indirect effect of the species Bifidobacterium adolescentis on BMD through stearidonate was -0.02432 (95% CI: -0.038, -0.0106), with a mediation proportion of 77.53% (Table 3). Similar results were observed for the family Bifidobacteriaceae and the genus Bifidobacterium, with mediation proportion of 87.24% (Fig. 4A, B, Table 3 and Supplementary Fig. 3).

A The forest plot displays the results of two MR analyses (Inverse-variance weighted and weighted median) assessing the causal effects of three gut microbiota taxa: Bifidobacterium adolescentis (N = 6328 participants), Bifidobacterium (N = 7244 participants) and Bifidobacteriaceae (N = 7252 participants) on stearidonate (N = 7824 participants). All analyses were conducted as two-sided statistical tests, and exact two-sided P values are reported. Point estimates represent causal effect beta values, and horizontal error bars indicate 95% confidence intervals calculated as beta ± 1.96 × SE. B he forest plot displays the results of three MR analyses (Inverse-variance weighted; weighted median and multivariable mendelian randomisation with adjusting stearidonate) assessing the causal effects of three gut microbiota taxa: Bifidobacterium adolescentis (N = 6328 participants), Bifidobacterium (N = 7244 participants) and Bifidobacteriaceae (N = 7252 participants) on eBMD (N = 426,824 participants). All analyses were conducted as two-sided statistical tests, and exact two-sided P values are reported. Point estimates represent causal effect beta values, and horizontal error bars indicate 95% confidence intervals calculated as beta ± 1.96 × SE.
The genetic determinants of three bifidobacterial taxa and the stearidonate
It is worth noting that the genetically determined abundance of three gut taxa (Bifidobacteriaceae, Bifidobacterium, and Bifidobacterium adolescentis) was derived by a series of SNPs located within the MCM6/LCT locus (Fig. 5A and Supplementary Fig. 4A, B). The SNP rs4988235 is a well-established predictor of LCT/MCM6 function28, which was the significant SNP in these gut taxa GWAS in DMP cohort (Effect allele: G; Bifidobacterium adolescentis: beta = 0.1545, p = 1.29×10-13; Bifidobacterium: beta = 0.1515, p = 1.48 × 10-14; Bifidobacteriaceae: beta = 0.1504, p = 1.01 × 10-14) (Fig. 5B). Furthermore, we found that blood stearidonate levels were influenced by several significant SNPs located within the FADS1/FADS2 locus (Fig. 5C), with the significant SNP rs174547 in stearidonate GWAS (Effect allele: T; beta = 0.0328, p = 1.63 × 10-15) (Fig. 5D). However, this association was not observed for DHA (Supplementary Fig. 4C-D). Notably, chi-square analyses revealed significant differences in the distribution of rs174547 genotypes between individuals with the rs4988235-GG genotype and those with rs4988235-GA/AA genotypes (p < 0.001) in the 469,133 participants from the UK Biobank (Supplementary Data 12). The proportion of individuals carrying the rs174547-TT genotype was significantly higher among those with the rs4988235-GG genotype than among those with the rs4988235-GA/AA genotypes (Fig. 5E).

A Manhattan plot displaying GWAS results for Bifidobacterium adolescentis in DMP cohort (N = 6328 participants). Each point represents a SNP with its −log10(two-sided P value) derived from linear regression. Red and blue horizontal lines indicate genome-wide significance (p = 5 × 10⁻⁸) and suggestive significance (p = 1×10⁻⁶), respectively. Inset shows the corresponding QQ plot of observed versus expected −log10(p) values. B Locuszoom plot showing the regional association of Bifidobacterium adolescentis GWAS around SNP rs4988235 (MCM6) on chromosome 2 (13.4–13.8 Mb) in the DMP cohort. Each point represents a SNP with its −log10(two-sided P value) from linear regression. SNPs are coloured according to their linkage disequilibrium with the lead SNP. Statistical analysis was performed using two-sided linear regression. C Manhattan plot displaying GWAS results for Stearidonate (18:4n3) (N = 7824 participants). Each point represents a SNP with its −log10(two-sided P value) derived from linear regression. Red and blue horizontal lines indicate genome-wide significance (p = 5×10⁻⁸) and suggestive significance (p = 1 × 10⁻⁶), respectively. Inset shows the corresponding QQ plot of observed versus expected −log10(p) values. D Locuszoom plots showing the regional association of Stearidonate (18:4n3) GWAS around SNP rs174547 (FADS1) on Chromosome 11 61.4–61.8 Mb. Each point represents a SNP with its −log10(two-sided P value) from linear regression. SNPs are coloured according to their linkage disequilibrium with the lead SNP. Statistical analysis was performed using two-sided linear regression. E Proportional distribution of rs174547 genotypes across the three rs4988235 genotypes (values represent percentage within each rs4988235 group).
Discussion
The GM is a complex group colonised in the human gut, in adults, the gut bacteria were mainly consisting of Bacteroidetes and Firmicutes, whereas Actinobacteria and Proteobacteria were secondary components29. In this study, we employed genetic summary data for GM taxa from four studies20,21,22,23, focusing on gut taxa exhibiting significant polygenicity, therefore, we refined the analysis to 96 unique taxa with significant heritability from the original 1104 GM taxa. The study provided evidence of both pleiotropic and causal associations between specific GM taxa and BMD. Specifically, we identified the GM species Bifidobacterium adolescentis, along with its parent taxon family Bifidobacteriaceae and genus Bifidobacterium, exhibited genetic associations with BMD. Lastly, we identified the mediating effect of these three gut taxa on BMD through the stearidonate (18:4n3) level in blood.
Previous study suggested that host genetics had a minor role in determining gut microbiome30. Therefore, we first estimated the SNP-based heritability of the 1104 gut taxa, the results suggested that most of the taxa exhibited low heritability, only 96 (8.7%) taxa consistently met the criteria for statistical significance (\({Z}_{{h}_{{SNP}}^{2}}\,\)> 1.64) and biological relevance (0 <\({h}_{{SNP}}^{2}\) < 1), with estimated median of the 96 GM taxa distributed only from 0.072 to 0.1735 in different studies. We also revealed the association between the heritability and the number of included SNPs, suggesting that the heritability of gut taxa could be attributed to genome-wide polygenicity and could be enhanced by larger participants in GM GWAS.
Besides the GM Clostridiales, Turicibacter and Faecalibacterium which were previously reported to be associated with bone metabolism15,31,32,33,34,35, more importantly, we discovered the pleiotropic and casual association of three GM taxa (Bifidobacteriaceae, Bifidobacterium and Bifidobacterium adolescentis) and BMD. Notably, Bifidobacterium adolescentis is a species within the genus Bifidobacterium, and genus Bifidobacterium belongs to the family Bifidobacteriaceae. Our results revealed a potential causal association between Bifidobacterium adolescentis and decreasing BMD. This finding contrasts with a previous study reporting that bifidobacterial supplementation enhanced skeletal protection following fractures36. In addition, Bifidobacterium used as probiotics have been reported to benefit bone health, as indicated in a prior review37.
To explain why genetically informed abundance of the identified gut taxa (Bifidobacteriaceae, Bifidobacterium, and Bifidobacterium adolescentis) had a negative association with BMD, we checked the genetic determinants of these taxa. We noticed that the abundance of the three gut taxa was derived by a series of significant SNPs located within the MCM6/LCT locus. The lactase (LCT) gene encodes lactase-phlorizin hydrolase, an enzyme that hydrolyses lactose into glucose and galactose for intestinal absorption. The minichromosome maintenance complex component 6 (MCM6) gene, located upstream of LCT, functions as a transcriptional enhancer for LCT38. Interestingly, multiple studies have demonstrated that the abundance of Bifidobacterium is associated with functional genetic variants near the LCT, making this locus the most validated finding thus far39. The functional variant rs4988235 within in locus is a robust predictor of lactase persistence, with the GG genotype indicating lactase non-persistence28. Consistent with previous European studies17,20,23, we observed the GG genotype was associated with increased abundance of three gut taxa. Considering Bifidobacterium have the ability to degrade lactose40,41, it is proposed that individuals with lactase non-persistence genotype GG typically show higher gut abundance of bifidobacterial taxa during consumption of milk in Europe23,39,42. The genetically informed increase in Bifidobacterium abundance may represent an adaptive microbial response to lactose intolerance. In European cohorts, individuals carrying the lactase non-persistence genotype exhibited higher Bifidobacterium abundance when consuming milk42, this claim replicated across multiple microbiota GWAS from UK17, Dutch21, Canadian43, Finnish20 populations, as well as in the meta-analysis of the MiBioGen consortium23. Taken together, these findings support the hypothesis that host–microbe interactions at the LCT locus represent a form of co-adaptation, whereby lactase persistence evolved as a genetic solution to milk consumption, while in lactase non-persistent individuals, milk sugar digestion is instead facilitated by the GM, particularly Bifidobacterium44. Importantly, individuals with lactose intolerance are at increased risk of reduced BMD45,46,47,48, suggesting that the negative association between these three gut taxa and bone mass might reflect host–microbe interactions at the LCT/MCM6 locus.
Notably, the human GM produces a wide array of metabolites, some of which enter the circulation and can significantly influence host physiology49. Recent advances in metabolomics have begun to uncover links between circulating metabolites and BMD50. To explore this connection, we conducted multivariable MR and mediation analyses and found that bifidobacterial taxa may reduce BMD by increasing circulating blood stearidonate levels (mediating proportion from 77.5 to 87.2%). Further, the direct effect of Bifidobacterium adolescentis on eBMD disappeared after adjusting for stearidonate, suggesting that its influence on bone density is likely mediated through this metabolite. A cross-sectional study in Swedish cohort reported a positive association between Bifidobacterium adolescentis abundance and circulating stearidonate levels49. Stearidonate was the intermediate products from alpha-linolenic acid (ALA) (ALA, 18:3n-3) to eicosapentaenoic acid (EPA)/docosahexaenoic acid (DHA)51. Delta-6 desaturase, encoded by fatty acid desaturase 2 (FADS2) gene, participates in the n-3 polyunsaturated fatty-acid biosynthetic pathway and catalyzes its first rate-limiting step in which ALA (ALA, 18:3n-3) is converted to stearidonate (18:4n-3)52. Delta-5 desaturase, encoded by fatty acid desaturase 2 (FADS1) gene, mediates the subsequent rate-limiting step, generating EPA/DHA from stearidonate acid53. We found that blood stearidonate levels were influenced by significant SNPs located within the FADS1 locus. The SNP rs174547, a variant widely associated with FADS1/FADS2 activity54,55, could enhance the conversion of ALA (ALA, 18:3n-3) to stearidonate (18:4n-3). However, this association was not observed for DHA, suggesting that elevated blood stearidonate may occur at the expense of EPA/DHA synthesis.
Along with the lactose intolerance SNP (rs4988235), we found that the proportion of individuals carrying the rs174547-TT genotype was significantly higher among those with the rs4988235-GG genotype, suggesting that individuals with lactose intolerance (rs4988235-GG) would also exhibit genetically determined higher gut Bifidobacterium abundance. Therefore, these people may show different FADS1/FADS2 activity, leading to elevated blood stearidonate levels, but reduced EPA/DHA levels, and ultimately lower BMD. Such genetically determined changes in these gut taxa may reflect an adaptive microbial response to lactose intolerance, which limited access to fat and protein from dairy products and thereby increases reliance on the metabolism of plant-derived fatty acids56. A previous study showed that higher circulating stearidonate concentrations are inversely associated with eBMD and positively associated with fracture risk, which were driven by genetic locus near FADS1/FADS257. Another report identified FADS2 as a biomarker of osteoporosis, a disease characterised by low bone-mineral density58. We therefore propose that elevated blood stearidonate levels may indicate reduced capacity to convert stearidonate into EPA/DHA owing to limited FADS1 activity, which could contribute to bone loss, as observed in spaceflight conditions59, thereby posing significant risks to human health60.
In conclusion, our systematic investigation assessed pleiotropic relationships between 96 GM taxa with significant polygenic heritability and BMD traits. We identified 14 GM taxa exhibiting shared genetic effects with eBMD. Specifically, an increased abundance of three GM taxa (Bifidobacteriaceae, Bifidobacterium and Bifidobacterium adolescentis) is associated with an elevated risk of low BMD, the negative association may reflect the host–microbe interactions at the LCT/MCM6 locus and may represent an adaptive microbial response to lactose intolerance. In addition, this adverse effect on bone mass appears to be mediated by elevated circulating stearidonate levels and could potentially be mitigated through supplementation with bifidobacterial probiotics and n-3 polyunsaturated fatty acids (e.g., DHA). Collectively, these results provide new insights into the GM–bone axis and highlight the role of blood metabolites level in skeletal health.
Methods
Data resources
We collected GWAS summary statistics for 1104 GM taxa abundance quantitative trait loci from four abovementioned studies (FINRISK, DMP, German and MiBioGen) (Table 1, Supplementary Data 1 and Fig. 1) conducted between January 2021 and February 202220,21,22,23. The majority of participants in these studies were of European ancestry. However, it should be noted that there are about 2900 individuals were included in both studies (German and MiBioGen)22,23. The SNPs information from the human genome build 37 (hg19) was utilised to annotate the GWAS summary statistics, addressing missing rsIDs information.
For BMD traits, we sourced two GWAS summary datasets61,62 from the Genetic Factors for Osteoporosis (GEFOS) Consortium, a large-scale international collaboration involving multiple research groups (http://www.gefos.org).al This study included estimated heel quantitative ultrasound BMD (eBMD) data from the UK Biobank collected in 201861. The eBMD summary-statistic data was derived from the UK10K/1000 G combined imputation panel (hg19) and included 14 million SNPs with a minor allele frequency (MAF) ≥ 5%, involving up to 426,824 participants (Supplementary Data 1). We also used total body BMD (TB-BMD) assessed by dual-energy X-ray absorptiometry (DXA) to strengthen the robustness of casual association results. The TB-BMD summary-statistic comprised data from 30 studies (66,628 individuals)62. Each study performed genotype imputation using the 1000 G reference panel included approximately 30,000,000 SNPs for analysis62.
For blood metabolites, we collected GWAS summary statistics for 453 metabolites in human blood from 7,824 adults with approximately 2.1 million SNPs, which were genotyped or imputed from the HapMap 2 panel63.
SNP heritability estimation of GM taxa GWAS
We calculated the polygenic SNP heritability (\({h}_{{SNP}}^{2}\)) of the 1104 GM taxa and eBMD GWAS datasets using LD score regression software (LDSC, https://data.broadinstitute.org/alkesgroup/LDSCORE)24. SNPs from approximately 1.2 million common SNPs in European populations from the HapMap3 reference panel (excluding the HLA region) were used. The LD scores of these SNPs referenced the European 1000 Genomes Project phase 3. We established filtering criteria requiring the Z score of \({h}_{{SNP}}^{2}\) is > 1.64 (\({P}_{{h}_{{SNP}}^{2}}\) < 0.05; one-side) to filter the GM taxa. Additional, we adjusted the criteria to a more lenient level to filter the GM GWAS datasets with \({h}_{{SNP}}^{2}\) between 0 and 1 for further analysis (\({h}_{{SNP}}^{2}\) > 1 and \({h}_{{SNP}}^{2}\) < 0 is not meaningful) in replication analysis.
Pearson correlation for LD-independent SNPs between gut taxa and BMD
We employed a genome-wide pleiotropy approach to estimate overall concordant or discordant genetic effects, following the approach used by Tanha et al.26 (Fig. 1). The Z scores of the LD-independent SNPs derived from GM taxa and eBMD were used to calculate the Pearson correlation (r) for trait pairs, including GM taxa and eBMD. We retained independent SNPs shared between the gut taxon and eBMD. These SNPs were then used for the subsequent Pearson correlation analysis. To identify LD-independent SNPs, we employed the P-value-informed LD-clumping method using PLINK 1.9 software with --clump-kb 10000, --clump-p1 0.05, --clump-r2 0.1 flags, along with the European 1000 Genome Project reference panel for each gut taxon GWAS. The Pearson correlation coefficients were computed after ensuring the harmonisation of the effect alleles among these independent SNPs between the gut taxon and BMD traits. To address the multiple testing problem, we applied FDR correction (Benjamini-Hochberg method) to adjust P-values for four studies and each gut microbiome level separately (FDR < 0.1) (Fig. 1).
Pairwise GWAS analysis for LD-independent regions between gut taxa and BMD
We performed the pairwise GWAS-PW analysis27 involving the GM taxa GWASs and eBMD. The goal was to identify shared genetic variants affecting both gut taxon GWAS and eBMD within any of the 1703 LD-independent regions (GRCh37)64. For each of the 1703 loci, we calculated four posterior probabilities (PPA) using Bayesian analysis via GWAS-PW software: PPA1: The probability of containing a genetic variant which was only associated to BMD; PPA2: The probability of containing a genetic variant which was only associated to gut taxon; PPA3: The probability of sharing a genetic variant which was both associated to BMD and gut taxon; PPA4: The probability of containing two distinct associations which were associated to BMD and gut taxon respectively. When PPA3 exceeded 0.9 for a locus, it was defined as a pleiotropic-associated locus (Fig. 1). If a gut taxon had at least one pleiotropic-associated locus, it was identified as demonstrating genetic pleiotropy with BMD at the genetic variant level.
Mendelian randomisation analysis
We evaluated the causal association between exposure and outcome through two-sample mendelian randomisation (MR) analyses. We employed three complementary MR methods including inverse variance-weighted (IVW) test; MR-Egger and the weighted median method to investigate the causal association. The IVW test was prioritised for exposure screening results due to its increased power under specific conditions, where the intercept is set to zero in the linear regression between exposure and outcome effects65. The weighted median method was employed to enhance the robustness of IVW results, contributing to the overall reliability of the findings66. The MR-Egger Regression was utilised to examine directional (unbalanced) pleiotropy67, adding an additional layer of analysis to test potential biases. For each exposure, we selected instrumental variants (IVs) with a significance level at P < 5×10-8 for the MR analysis. In cases where two significant SNPs within a distance of 250 kb (windows) had an LD score (r2) greater than 0.1 (r2 < 0.1), we selected the independent SNP with the most statistically significant association, as indicated by the lowest p-value. When a trait included only one instrumental variable, we applied the Wald ratio method to estimate the causal effect, as an alternative to the IVW method. Traits without any eligible IVs (i.e., no SNPs with P < 5×10-8) were excluded from subsequent analyses. In additional sensitivity analyses, we employed relaxed significance thresholds (1 × 10-5 and 5 × 10-6) to select instruments for GM taxa exposure by using IVW analyses. We also conducted the leave-one-out IVW regression analyses, sequentially excluding each IV SNP with the significance thresholds at 1×10-5 from GM taxa, to identify any potential dominance of specific variant in casual estimation. All analyses, including sensitivity and MR analyses, were conducted in R version 4.2.1, using the R package “MendelianRandomization” and “TwoSampleMR”.
In this study, we conducted MR analyses to evaluate the causal relationship between candidate GM taxa exhibiting genetic correlation with BMD. The GM taxa were identified that could potentially causally affect eBMD at Bonferroni corrected P value below 0.05 in IVW analyses (IVW-P < 0.05/N, N = 14, the number of candidate GM taxa). Blood metabolites GWAS summary statistics were obtained from 7824 adults in two European population cohorts63. Two-sample MR analyses were performed to assess whether metabolites causally influence eBMD.
Multivariable MR and mediation analysis
We conducted multivariable MR analyses68 to investigate the mediation effect of specific metabolites on the relationship between the GM taxon and eBMD. First, we performed the IVW MR analyses to assess the causal effect of each GM taxon (exposure) on metabolites (outcome) (using genetic instruments selected at genome-wide significance P < 5×10-8 and r2 < 0.1 in a 250 kb window). Metabolites were considered to be potentially causally influenced by the GM taxon with the Bonferroni-corrected P value below 0.05 (IVW-P < 0.05/453; the number of metabolites). Second, we performed the IVW MR analyses to evaluate the causal association between metabolites (exposure) and eBMD (outcome) (using genetic instruments selected at genome-wide significance P < 5×10-8 and r2 < 0.1 in a 250 kb window). Metabolites were considered to have a causal effect on eBMD with the Bonferroni corrected P value below 0.05 (IVW-P < 0.05/257; only 257 metabolites had genome-wide significant SNPs and were included, excluding those without eligible instruments). Third, we identified metabolites that met the above criteria in both two steps, considering these as candidate mediators of the relationship between GM taxa and eBMD. For these candidate mediators, we extracted genetic association estimates for both the GM taxon and the candidate metabolite using genome-wide significance P < 5 × 10-8 and r2 < 0.1 in a 250 kb window for instrumental variables in these multivariable MR analyses. We then estimated the effect (β1) of the GM taxon on the mediator metabolite using the generalised IVW method. Next, we estimated the effect (β2) of the mediator metabolite on eBMD, adjusting for the effects of the gut taxon, through multivariable MR analysis. The indirect (mediated) effect of the GM taxon on eBMD was estimated by β1 × β2. The proportion of the total effect mediated by each metabolite was estimated by dividing the indirect effect (β1 × β2) by the total effect (the IVW effect of the GM taxon on eBMD)69.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All datasets utilised in this study were publicly accessible. The GWAS summary statistics for eBMD was available at http://www.gefos.org/?q=content/data-release-2018. The GWAS summary statistics for TB_BMD was available at http://www.gefos.org/?q=content/gefos-lifecourse-tb-bmd-gwas-results. The GWAS summary statistics for 1,104 GM taxa were from four studies: the FINRISK and German datasets were available at https://www.ebi.ac.uk/gwas/ from GCST90032172 to GCST90032644 and GCST90011301 to GCST90011730; the DMP datasets were available at https://dutchmicrobiomeproject.molgeniscloud.org; the MiBioGen datasets were available at www.mibiogen.org. The GWAS summary statistics for 453 metabolites were available at https://metabolomics.helmholtz-muenchen.de/gwas/. The GWAS summary statistics for eBMD, TB_BMD, stearidonate (18:4n-3) (SDA) and three gut taxa (Bifidobacteriaceae, Bifidobacterium and Bifidobacterium adolescentis) were also available at China Precision BioBank, could be downloaded from http://cpbb.cn/resources/BMDGWASgut. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
All analyses were performed using publicly available software. GWAS summary statistics data processing and Pearson correlation analyses were conducted using R (v4.2.1; https://www.r-project.org/). SNP heritability was estimated using LDSC (v1.0.1; https://github.com/bulik/ldsc). Plink 1.9 (https://www.cog-genomics.org/plink/) was used to identify LD-independent SNPs. Pairwise GWAS analyses were performed using GWAS-PW software (v0.21; https://github.com/joepickrell/gwas-pw). MR analyses were conducted using the R packages “MendelianRandomization” (v0.10; https://github.com/cran/MendelianRandomization) and “TwoSampleMR” (v0.6.17; https://github.com/MRCIEU/TwoSampleMR).
References
Zhu, X. & Zheng, H. Factors influencing peak bone mass gain. Front. Med. 15, 53–69 (2021).
Burge, R. et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J. Bone Min. Res. 22, 465–475 (2007).
Ralston, S. H. Genetic determinants of osteoporosis. Curr. Opin. Rheumatol. 17, 475–479 (2005).
Zhai, G., Andrew, T., Kato, B. S., Blake, G. M. & Spector, T. D. Genetic and environmental determinants on bone loss in postmenopausal Caucasian women: a 14-year longitudinal twin study. Osteoporos. Int. 20, 949–953 (2009).
Zhu, X., Bai, W. & Zheng, H. Twelve years of GWAS discoveries for osteoporosis and related traits: advances, challenges and applications. Bone Res. 9, 23 (2021).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
Yan, J. et al. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 113, E7554–E7563 (2016).
Li, J. Y. et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Invest. 126, 2049–2063 (2016).
Rodrigues, F. C. et al. Yacon flour and Bifidobacterium longum modulate bone health in rats. J. Med Food 15, 664–670 (2012).
Clarke, G. et al. Minireview: gut microbiota: the neglected endocrine organ. Mol. Endocrinol. 28, 1221–1238 (2014).
van Wijngaarden, J. P. et al. Vitamin B12, folate, homocysteine, and bone health in adults and elderly people: a systematic review with meta-analyses. J. Nutr. Metab. 2013, 486186 (2013).
Villa, J. K. D., Diaz, M. A. N., Pizziolo, V. R. & Martino, H. S. D. Effect of vitamin K in bone metabolism and vascular calcification: a review of mechanisms of action and evidences. Crit. Rev. Food Sci. Nutr. 57, 3959–3970 (2017).
Lucas, S. et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 9, 55 (2018).
Tang, W. H., Kitai, T. & Hazen, S. L. Gut microbiota in cardiovascular health and disease. Circ. Res. 120, 1183–1196 (2017).
Ni, J. J. et al. Assessing causal relationship from gut microbiota to heel bone mineral density. Bone 143, 115652 (2021).
Cheng, S. et al. Assessing the relationship between gut microbiota and bone mineral density. Front. Genet. 11, 6 (2020).
Goodrich, J. K. et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19, 731–743 (2016).
Burgess, S., Small, D. S. & Thompson, S. G. A review of instrumental variable estimators for mendelian randomization. Stat. Methods Med. Res. 26, 2333–2355 (2017).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Qin, Y. et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 54, 134–142 (2022).
Lopera-Maya, E. A. et al. Effect of host genetics on the gut microbiome in 7738 participants of the Dutch microbiome project. Nat. Genet. 54, 143–151 (2022).
Ruhlemann, M. C. et al. Genome-wide association study in 8956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 53, 147–155 (2021).
Kurilshikov, A. et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165 (2021).
Bulik-Sullivan, B. K., Loh, P. R., Finucane, H. K., Ripke, S. & Yang, J. Schizophrenia Working Group of the Psychiatric Genomics C et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159, 789–799 (2014).
Tanha, H. M. International Headache Genetics C, Nyholt DR. Genetic analyses identify pleiotropy and causality for blood proteins and highlight Wnt/beta-catenin signalling in migraine. Nat. Commun. 13, 2593 (2022).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Chin, E. L. et al. Association of lactase persistence genotypes (rs4988235) and ethnicity with dairy intake in a healthy U.S. population. Nutrients 11, 1860 (2019).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215 (2018).
Wan, X. et al. Effects of (R)-ketamine on reduced bone mineral density in ovariectomized mice: a role of gut microbiota. Neuropharmacology 213, 109139 (2022).
Xu, Z. et al. Gut microbiome reveals specific dysbiosis in primary osteoporosis. Front. Cell Infect. Microbiol. 10, 160 (2020).
Chen, L. et al. The gut microbiome is associated with bone turnover markers in postmenopausal women. Am. J. Transl. Res. 13, 12601–12613 (2021).
Wang, Q. et al. Linking gut microbiome to bone mineral density: a shotgun metagenomic dataset from 361 elderly women. GigaByte 2021, gigabyte12 (2021).
Sun, M. et al. Characterization of intestinal flora in osteoporosis patients based on 16S rDNA sequencing. Int J. Gen. Med. 17, 4311–4324 (2024).
Roberts, J. L. et al. Bifidobacterium adolescentis supplementation attenuates fracture-induced systemic sequelae. Biomed. Pharmacother. 132, 110831 (2020).
Malmir, H. et al. Probiotics as a new regulator for bone health: a systematic review and meta-analysis. Evid. Based Complement Altern. Med. 2021, 3582989 (2021).
Anguita-Ruiz, A., Aguilera, C. M., Gil A. Genetics of lactose intolerance: an updated review and online interactive world maps of phenotype and genotype frequencies. Nutrients 12, 2689 (2020).
Sanna, S., Kurilshikov, A., van der Graaf, A., Fu, J. & Zhernakova, A. Challenges and future directions for studying effects of host genetics on the gut microbiome. Nat. Genet. 54, 100–106 (2022).
Hove, H., Norgaard, H. & Mortensen, P. B. Lactic acid bacteria and the human gastrointestinal tract. Eur. J. Clin. Nutr. 53, 339–350 (1999).
Viborg, A. H. et al. A beta1-6/beta1-3 galactosidase from Bifidobacterium animalis subsp. lactis Bl-04 gives insight into sub-specificities of beta-galactoside catabolism within Bifidobacterium. Mol. Microbiol. 94, 1024–1040 (2014).
Luo, K. et al. Variant of the lactase LCT gene explains association between milk intake and incident type 2 diabetes. Nat. Metab. 6, 169–186 (2024).
Turpin, W. et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48, 1413–1417 (2016).
Suzuki, T. A., Ley R. E. The role of the microbiota in human genetic adaptation. Science 370, eaaz6827 (2020).
Gorczynska-Kosiorz, S. et al. Bone mineral density and the risk of type-2 diabetes in postmenopausal women: rs4988235 polymorphism associated with lactose intolerance effects. Nutrients 16, 3002 (2024).
Obermayer-Pietsch, B. M. et al. Genetic predisposition for adult lactose intolerance and relation to diet, bone density, and bone fractures. J. Bone Min. Res. 19, 42–47 (2004).
Falbova, D. et al. Association between self-reported lactose intolerance, additional environmental factors, and bone mineral density in young adults. Am. J. Hum. Biol. 37, e24202 (2025).
Treister-Goltzman, Y., Friger, M. & Peleg, R. Does primary lactase deficiency reduce bone mineral density in postmenopausal women? A systematic review and meta-analysis. Osteoporos. Int. 29, 2399–2407 (2018).
Dekkers, K. F. et al. An online atlas of human plasma metabolite signatures of gut microbiome composition. Nat. Commun. 13, 5370 (2022).
Panahi, N. et al. Metabolomic biomarkers of low BMD: a systematic review. Osteoporos. Int. 32, 2407–2431 (2021).
Mathias, R. A., Pani, V. & Chilton, F. H. Genetic variants in the FADS gene: implications for dietary recommendations for fatty acid intake. Curr. Nutr. Rep. 3, 139–148 (2014).
Ge, L., Gordon, J. S., Hsuan, C., Stenn, K. & Prouty, S. M. Identification of the delta-6 desaturase of human sebaceous glands: expression and enzyme activity. J. Invest Dermatol. 120, 707–714 (2003).
Cho, H. P., Nakamura, M. & Clarke, S. D. Cloning, expression, and fatty acid regulation of the human delta-5 desaturase. J. Biol. Chem. 274, 37335–37339 (1999).
Wang, Y. et al. Association between FADS1 rs174547 and levels of long-chain PUFA: a meta-analysis. Br. J. Nutr. 126, 1121–1129 (2021).
Loukil, I., Mutch, D. M. & Plourde, M. Genetic association between FADS and ELOVL polymorphisms and the circulating levels of EPA/DHA in humans: a scoping review. Genes Nutr. 19, 11 (2024).
Stock, J. T. & Wells, J. C. K. Dairying and the evolution and consequences of lactase persistence in humans. Anim. Front. 13, 7–13 (2023).
Yuan, S., Lemming, E. W., Michaelsson, K. & Larsson, S. C. Plasma phospholipid fatty acids, bone mineral density and fracture risk: evidence from a Mendelian randomization study. Clin. Nutr. 39, 2180–2186 (2020).
Qiu, C. et al. Multi-omics data integration for identifying osteoporosis biomarkers and their biological interaction and causal mechanisms. iScience 23, 100847 (2020).
Zwart, S. R., Pierson, D., Mehta, S., Gonda, S. & Smith, S. M. Capacity of omega-3 fatty acids or eicosapentaenoic acid to counteract weightlessness-induced bone loss by inhibiting NF-kappaB activation: from cells to bed rest to astronauts. J. Bone Min. Res. 25, 1049–1057 (2010).
Jiang, X., Xue, Y., Mustafa, M. & Xing, Z. An updated review of the effects of eicosapentaenoic acid- and docosahexaenoic acid-derived resolvins on bone preservation. Prostaglandins Other Lipid Mediat. 160, 106630 (2022).
Morris, J. A. et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 51, 258–266 (2019).
Medina-Gomez, C. et al. Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am. J. Hum. Genet. 102, 88–102 (2018).
Shin, S. Y. et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 46, 543–550 (2014).
Berisa, T. & Pickrell, J. K. Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285 (2016).
Burgess, S., Dudbridge, F. & Thompson, S. G. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat. Med. 35, 1880–1906 (2016).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Carter, A. R. et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur. J. Epidemiol. 36, 465–478 (2021).
Sanderson, E. Multivariable mendelian randomization and mediation. Cold Spring Harb. Perspect. Med. 11, a038984 (2021).
Acknowledgements
We thank the UK Biobank database, the GEnetic Factors for OSteoporosis (GEFOS) Consortium and Microbial composition. We also thank the Westlake University Supercomputer Centre for the facility support and technical assistance. This work was supported by Beijing GuoKe biotechnology co., LTD (Beijing, China) and China National GeneBank (CNGB) (Shenzhen, China). This work was supported by the National Key R&D Programme of China (#2024YFC3405703), and by the National Natural Science Foundation of China (#82370887).
Author information
Authors and Affiliations
Contributions
H.-F.Z. conceptualised and designed the study. P.-L.G., M.-Y.H., C.-D.Y., and Y.-H.F. conducted the data analysis. Y.Q., C.-F.Y., P.-P.Z., J.-W.X., P.W., C.-R.L., M.-Y.Y., C.-L.C., and W.X. contributed to the data collection, processing and preliminary data analysis. P.-L.G. draughted the manuscript, H.-F.Z., S.-Y.X., X.-L.R., and F.-S.Z. reviewed and edited manuscript. All authors contributed, discussed and approved manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lei Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guan, PL., Yuan, CD., Han, MY. et al. Genetically informed causal links between gut microbiota and bone mass: pleiotropy and metabolic mediation. Nat Commun 16, 11711 (2025). https://doi.org/10.1038/s41467-025-66881-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66881-8
