
Abstract
Manganese-rich layered transition-metal oxides are of great interests as positive electrodes for sodium-ion batteries considering their high specific capacity and operating voltage. However, it remains challenging to synthesize layered transition-metal oxides with high stability and rapid ion-migration kinetics. Herein, we develop a straightforward and universal synthesis approach for P2-type layered transition-metal oxides by simply updating the conventional solid-state reaction with only additional dicyandiamide introduced. We reveal the ammonia released successfully restrains the irreversible oxygen redox for stabilizing cationic migration and decreases the crystallization temperature for high-quality structure formation. The effectively suppressed Jahn-Teller distortion and facilitated Na+ transport kinetics therefore endow Na1/2MnO2 with overall performance enhancement, including a 6.7-fold improvement in rate capability and enhanced cycling stability. Similar performance enhancements are also found in other manganese-based layered transition-metal oxides, presenting great strategy universality. This work marks a key step forward in the synthesis-by-design of high-performance sodium-ion batteries electrode materials.
Similar content being viewed by others

Introduction
Recently, sodium-ion batteries (SIBs) are emerging as one of the most promising alternatives to its lithium counterparts for next-generation energy storage devices due to their advantages of sodium availability and reliable performance advantages for fast charging1,2. The design of positive electrodes with excellent redox reversibility and high Na+ mobility is the key focus for promoting the commercialization of SIBs3,4. P2-type Mn-based layered transition-metal oxides (LTMOs) attract broad attentions because of their high theoretical capacity, compositional flexibility and structure adaptability5. Their structural similarity to Li-LTMOs also enables the great potential for feasible transformation of current manufacturing lines for lithium ion batteries6,7. However, most materials still suffer from unstable cyclic performances and low reversible capacity8,9, while the development of high-performance LTMOs positive electrodes for SIBs still remains challenging.
It is always accepted for Mn-rich LTMOs that undistorted [MnO6] octahedral unit is formed by the presence of Mn4+ for stabilizing structure, while high-spin Mn3+ always results in a Jahn-Teller distortion that causes the irreversible multiphase transition and thus rapid capacity decay10,11. Meanwhile, the synthesis of high-quality LTMOs with stable ion tunnels can not only stabilize the structure but also accelerate Na+ desodiation with high reaction kinetics12,13. To date, the solid state reaction is viewed as the primary synthesis strategy for LTMOs in practical use14,15. However, its limited ion diffusion rate and the difficulty for crystal structure control in solid phase retards its use for high-performance positive electrode synthesis. Currently, the development of Mn based LTMOs with high stability and rapid ion migration kinetics faces the critical challenges and it is highly imperative to propose effective synthesis strategies that offer new possibilities for SIB positive electrodes.
In this work, we develop a universal synthesis strategy for P2-type LTMOs positive electrodes for SIBs. Na1/2MnO2 (NM) that is one of the most commonly studied LTMO benchmarks was selected as the modeled material. By introducing dicyandiamide (DA) into the conventional solid-state processing without additional treatment, we synthesize the optimized Na1/2MnO2 (NMD) with all-around performance elevation compared with its pristine counterpart NM. Ammonia released from thermal decomposition of DA was found to facilitate the solid-state reaction kinetics by creating oxygen vacancies to activate intermediate bondings and decrease thermodynamic barriers of ion migration. Meanwhile, the presence of NH3 gas phase can also decrease the interaction force between atoms/ions in solid reactants and lower the diffusion resistance, which reduces the crystallization temperature and promotes the formation of high-quality layered structure. In addition to the different working mechanism of gas-phase assisted solid state reaction revealed in this strategy, its broad universality for the synthesis of highly active P2-type LTMOs positive electrodes with various composition attracts our interests. Furthermore, the simple and direct processing with only DA additive introduced into the current solid-state synthesis route avoids complicated facility and hardware updates. The aforementioned merits of different mechanism, universal approach and simple synthesis routes enable this strategy with great potential for large-scale production of P2-type LTMOs. A detailed design principle for this strategy is illustrated in Fig. 1a.

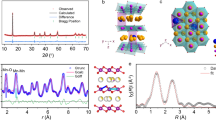
a Design principle for ammonia induced synthesis approach of Na1/2MnO2. Rietveld refinements of X-ray diffraction measurements for (b) NM and (c) NMD. d Schematic diagram of the corresponding structural changes from NM to NMD. High-resolution TEM images of (e) NM and (g) NMD with the insets showing the corresponding FFT patterns. f, h The corresponding strain distributions of (e) and (g). Scanning transmission electron microscopy equipped with (i) high-angle annular dark field (STEM-HAADF) and (j) annular bright field (ABF) for NMD. k The selected area electron diffraction (SAED) images for NMD. l EDS mappings of NMD: Na, Mn, O.
Results
Synthesis and structural characterizations of NM and NMD
NM and NMD were synthesized by the classic solid-state reaction with the only difference in the addition of dicyandiamide as the ammonia release source. The X-ray diffraction (XRD) patterns along with highly fitted Rietveld refinement results (Fig. 1b–c) confirm the successful synthesis of P2-type Na1/2MnO2 with a space group of P63/mmc for both samples16. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis confirms their similar stoichiometry which is consistent with the designed reactant ratios (Supplementary Table 1). The further analysis of refined crystallographic data (Supplementary Table 2) shows the reduced a/b lattice parameter of NMD from 2.990 Å to 2.920 Å while c-axis increases from 11.127 Å to 11.163 Å, attributed to the weakening of O-O repulsion due to the formation of oxygen vacancies. This crystallographic change results in its enhanced structural stability compared with NM. Meanwhile, the decreased d(O-TM-O) and increased d(O-Na-O) values of NMD compared with NM are beneficial for inhibiting Mn dissolution and MnO6 layer gliding (Fig. 1d). Meanwhile, the primary peak (002) of NMD shows an apparent shift to the lower angle (Supplementary Fig. 1a), indicating its enlarged d(O-Na-O) spacing which can reduce Na+ diffusion resistance during insertion/extraction. Furthermore, NMD shows significantly enhanced crystallinity with sharp and high-intensity (002) peak compared with NM (Supplementary Fig. 1a), confirming this proposed strategy can effectively overcome the bottleneck of conventional solid-state reaction for forming the P2-type LTMOs with high crystallinity. Furthermore, the presence of impurity phases is observed in NM, while a pure phase is confirmed for NMD (Supplementary Fig. 1b). The low-magnification scanning electron microscopy (SEM) image shows a higher proportion of hexagonal stacked layer structure for NMD compared with the irregular micron-particles of NM (Supplementary Figs. 2 and 3). Further high-resolution transmission electron microscope (HRTEM) analysis (Fig. 1e, g) reveals their crystalline nature with clear lattice strips and meanwhile enlarged spacing of (002) plane for NMD (0.574 nm) compared with that of NM (0.564 nm)17. The corresponding line profiles are presented in Supplementary Fig. 4. Furthermore, the fast Fourier transformation (FFT) patterns (insets of Fig. 1e, g) indicate the formation of single-crystalline structure that can be indexed to P2 phase within NMD, while NM patterns with diffraction rings demonstrate its localized polycrystalline structure. Geometric phase analysis (GPA) reveals the large stress fluctuations of NM (Fig. 1f) while the relatively homogenous stress distribution of NMD (Fig. 1h) indicates the mild phase transition in a wide temperature range effectively relieves the internal stress that is beneficial for enhancing the stability of crystal structure18,19. Scanning transmission electron microscopy (STEM) equipped with high-angle annular dark field (HAADF) (Fig. 1i) and annular bright field (ABF) (Fig. 1j) is employed to analyze the atomic-level crystal structure of NMD. In the HAADF mode, the light dots signify the transition metal atoms while the O and Na atoms are revealed within the dark columns, whereas the manifestation is reversed in the ABF mode17,20. According to the STEM-HAADF image, the TM-TM distance is 0.273 nm corresponding to the (100) plane of the P2 structure. Selected area electron diffraction (SAED) pattern along the [001] axis substantiates the single-crystalline hexagonal structure of the bulk phase of NMD (Fig. 1k). Correspondingly, energy-dispersive spectroscopy (EDS) analysis demonstrates the homogeneous distribution of Na, Mn, and O within NMD (Fig. 1l).
Structural growth analysis
To investigate the detailed influence of DA addition on the growth mechanism of LTMOs, the structural evolution from precursor to the final crystalline form was analyzed with in-situ XRD technique from room temperature to 500 °C (Fig. 2a–c). The (002) peak of sodium manganite at 15.6° can be detected at a low temperature of 320 °C for NMD (vs. 400 °C for NM), demonstrating this strategy significantly decreases the formation temperature of Na1/2MnO2 structure and allows the fully growth of crystals with more ordered atomic arrangement during wide temperature range. The (001) peak of Na2CO3 at 14.9° progressively disappears while the (222) peak of Mn2O3 at 33° also gradually diminishes with increasing temperatures, indicating the transformation into Na1/2MnO2. Meanwhile, a gradual sharpening of the (002) peak with the increase of the half-peak full width is also observed (Supplementary Fig. 5). The extended phase transition temperature region with the earlier appearance of crystalline structure endows NMD with alleviated internal structure stress, resulting in a more robust structure for stabilized Na+ storage. In addition, since non-uniform strains within the material will lead to variations in lattice spacing, the lattice strains of both samples were also calculated based on the neutron diffraction data (discussed later) using the Williamson-Hall equation (Supplementary Fig. 6)21. The calculated lattice strain is 7.5 × 10−2 % for NM and 5 × 10−2 % for NMD, indicating that NMD shows a robust crystal structure with alleviated internal strain.

a Y-shifted stacked line plots of NMD/NM at different temperatures for in-situ XRD data. b Magnified XRD data of the selected region in (a) with the highlighting (002) peaks. c Corresponding contour plots. d Mn 2p spectra. e O 1 s spectra of NM and NMD. f Mn K-edge XANES spectra and (g) the corresponding Fourier-transformed EXAFS spectra. h, i Wavelet transform (WT) contour plots of NMD and NM.
The decomposition of Na1/2MnO2 was then analyzed through thermogravimetric (TGA) curves to investigate the detailed role of DA (Supplementary Fig. 7). DA undergoes a dramatic weight loss at 210–385 °C for NH3 release and this process is consistent with the weight losses within NMD considering the high content of DA in the precursor (~40 wt%), confirming the primary contribution of DA for ammonia release to the phase formation. The decomposition of DA also takes the primary weight change for the phase transition between 170-470 °C, in which ammonia facilitates the diffusion kinetics of Na+ and Mn3+ in the solid-phase reaction for promoting the early formation of intermediate phases. Meanwhile, the released ammonia can seize lattice O atoms by forming N-containing gases, leading to the formation of oxygen vacancies and accelerated solid-reaction kinetics. This is confirmed by the higher content of oxygen vacancies within NMD as characterized through its sharper electron paramagnetic resonance (EPR) signal than that of NM at g = 2.003 (Supplementary Fig. 8)17.
X-ray photoelectron spectroscopy (XPS) was then performed and the results (Supplementary Fig. 9, Supplementary Table 3 and Fig. 2c) show the content of Mn4+ within NMD decreases from 9.76 to 5.16 at.% whereas Mn3+ increases from 3.89 to 10.86 at.%, which is attributed to the introduction of oxygen vacancies in NMD22. High resolution O profiles (Fig. 2d) can be deconvoluted into peaks associated with lattice oxygen (Olat) at 529.5 eV, oxygen vacancy (Ovac) peak at 530.9 eV and chemisorbed oxygen (Oads) at 531.5 eV23. The reduced Oads content of NMD from 27.44 to 19.84 at.% (Fig. 2d) confirms its high air stability and great inertness to H2O and CO2, which is highly desirable for stabilized positive electrodes24. Compared to NM, Olat content in NMD decreases from 13.25 to 12.97 at.% due to the partial oxygen seized by the produced ammonia during low-temperature sintering to create oxygen vacancies. The content of Ovac in NMD also increases and this is consistent with EPR results, induced by O capture from ammonia. The introduction of oxygen vacancies can lead to an unbalanced charge distribution through non-saturated coordination sites, creating a local electric field that enhances conductivity and provides extra Na+ storage sites. Furthermore, it can also effectively suppress oxygen release and retard the irreversible oxygen chemistry, which is generally accepted as one of the primary reasons leading to structural instability and voltage fading of LTMO positive electrodes25.
Ex situ X-ray absorption fine structure (XAFS) spectra were collected to reveal the change of valance states for metal elements between samples. The X-ray absorption near edge structure (XANES) at the Mn K-edge and the corresponding Fourier transformed of extended X-ray absorption fine structure (FT-EXAFS) spectra were presented in Fig. 2e–f. Compared to NM, NMD shows a left-shift to the lower energy for its Mn K-edge (Fig. 2e), indicating the oxidation of Mn with the decreased valence state, which coincides with the enlarged interatomic distances within the first Mn-O coordination shell that is presented with the increased Mn-O peak in Fig. 2f21. This observation is also consistent with the XPS result of NMD that presents the increased Mn3+/Mn4+ ratio compared with NM. Meanwhile, the decreased second Mn-Mn coordination shell peak of NMD (Fig. 2f) suggests the reduced interatomic distances of Mn, which demonstrates the contraction of MnO6 unit. The wavelet transform (WT) contour plot of EXAFS spectra of NMD (Fig. 2g) exhibits two distinct scattering peaks of Mn-O and Mn-Mn coordination shells located at (5.6 Å⁻1, 1.4 Å) and (5.6 Å⁻1, 2.3 Å), respectively. Correspondingly, the center of these scattering peaks is shifted with the same trend of Mn-O and Mn-Mn peaks of FT-EXAFS for those in NM (5.4 Å⁻1, 1.4 Å and 5.7 Å⁻1, 2.3 Å) (Fig. 2h)22. The Mn-Mn coordination shell in NMD with low R-value demonstrates the locally disordered Mn-Mn coordination environment that is caused by the introduction of oxygen vacancies. Meanwhile, the well-maintained shape without change from NM to NMD suggests the uniform introduction of oxygen vacancies into NMD bulk. The aforementioned analysis confirms the introduction of oxygen vacancies into NMD along with electronic/structural changes that are the basis for its electrochemical performance enhancement.
Electrochemical performance evaluation
The electrochemical performances of samples were continuously evaluated in half-cell configurations with galvanostatic charge/discharge (GCD) curves recorded (Supplementary Figs. 10 and 11). NMD presented significantly enhanced rate capabilities with 187.2, 178.6, 163.3, 142.9, 123.8 and 102.1 mAh g⁻1 delivered at 0.05, 0.1, 0.2, 0.5, 1.0 and 2.0 A g⁻1 compared with those of NM (144.3, 91.5, 61.9, 29.0 and 18.4 mAh g⁻1 at 0.05, 0.1, 0.2, 0.5 and 1.0 A g⁻1, respectively) (Fig. 3a). The capacity of NMD is even 6.7 times of NM at 1.0 A g⁻1, further highlighting its significantly enhanced rate capability. The addition content of DA was also optimized while NMD samples with different contents of DA always show the enhanced performance compared with NM (Supplementary Fig. 12). Its fully overlapped cyclic voltammetry (CV) curves within the initial 3 cycles (Supplementary Fig. 13a) also confirm the highly reversible redox reactions within NMD, which is not achieved in NM (Supplementary Fig. 13b). The current area for NMD is significantly larger than that of NM, indicating a higher Na+ storage capacity in NMD (Supplementary Fig. 14). Based on the CV, NMD also presents higher diffusion rate than NM (Supplementary Figs. 15–17) and narrow/sharp peaks with high intensities in dQ/dV curves (Fig. 3b), indicating its rapid redox reaction kinetics and great reversibility26. Electrochemical impedance spectroscopy (EIS) analysis (Fig. 3c, Supplementary Table 4) demonstrates that NMD shows the apparently increased Na+ diffusion kinetics with the effectively suppressed internal resistance and robust interfacial stability to deliver a fast ionic response, resulting in rate performance improvement27. Correspondingly, a 2.6-fold increase of derived Na+ diffusion coefficient (DNa+) value for NMD (2.41 × 10⁻13 cm2 s⁻1) compared with NM (8.46 × 10⁻14 cm2 s⁻1) (Fig. 3c and Supplementary Fig. 18) also confirms the rapid Na+ diffusion rate within NMD that is consistent well with CV results, facilitating its high discharge capacity under elevated specific currents. Galvanostatic intermittent titration technique (GITT) was also conducted to further probe into the ion diffusion kinetics of positive electrodes (Supplementary Fig. 19). Consistent with EIS results, NMD also demonstrated higher DNa+ values than that of NM, confirming its efficient Na+ transport kinetics. In addition, the in-situ EIS and the distribution of relaxation times (DRT) analysis were performed to reveal the evolution of the internal states of NM and NMD at different potentials. In-situ EIS analysis reveals that the charge transfer resistance initially decreases and then increases (Supplementary Fig. 20), fluctuating with the Na concentration in the electrode during the early charge-discharge process28. Notably, NMD exhibits a lower transfer resistance after charge-discharge cycles (Supplementary Fig. 21), highlighting its great ion transport kinetics. Further DRT analysis (Supplementary Fig. 22) shows lower intensity values across all regions for NMD compared to NM, indicating its reduced electrochemical impedance29,30. The reduced impedance and enhanced ion transport kinetics in NMD can be ascribed to oxygen vacancies that weaken the Na-O electrostatic interaction, thereby expanding interlayer spacing and promoting both Na-ion diffusion and electronic conductivity.

a Discharge capacities at different specific currents. b dQ/dV curves. c Impedance fitting results with the inset showing the calculated Na+ diffusion coefficients and equivalent circuit model. Cycling performances of samples at (d) 0.5 A g⁻1 and (e) 1.0 A g⁻1. f Charge-discharge curves of NMD and NM for 300 cycles at 1.0 A g⁻1. g Comparison of the capacity and cycling performances of NMD with reported results. h Schematic of full cell assembly. i Rate capabilities at different specific currents. j Cycling performances of full cells at 0.2 A g⁻1.
The long-term stability of samples was then evaluated at different specific currents (Supplementary Fig. 23, Fig. 3d–e). NMD presents excellent cycling performances with a capacity retention rate of 67.3% and 74.0% after 300 cycles at 0.2 and 0.5 A g⁻1, far surpassing those of NM (54.5% and 63.7%). Besides, NMD also exhibits 74.1 and 60.3 mAh g⁻1 after 500 and 1000 cycles at 1.0 A g⁻1, with the capacity retention of 80.0% and 64.1%. Correspondingly, the capacity attenuation of NMD is 0.036% per cycle, much lower than that of NM (0.084%) at 1.0 A g⁻1 (Fig. 3f). A direct comparison of overall electrochemical performances between NMD and other state-of-the-art LTMOs is also listed in Fig. 3g and Supplementary Table 5, showing appealing combination of positive electrode features4,8,11,23,31,32,33,34,35,36. Full cells were then evaluated with commercial hard carbon (HC) as the negative electrode (Fig. 3h). HC | | NMD full cell exhibits an initial discharge capacity of 130.7 mAh g⁻1 at 0.05 A g⁻1 and excellent rate capability of 75.3 mAh g⁻1 at 1.0 A g⁻1 with a high specific energy (based on the mass of the active material) of 244.4 W h kg⁻1 achieved (Fig. 3i and Supplementary Fig. 24). Additionally, it also demonstrates excellent cycling performance, maintaining a discharge capacity of 77.8 mAh g⁻1 with a retention rate of 66.7% after 300 cycles at 0.2 A g⁻1 (Fig. 3j and Supplementary Fig. 25). In contrast, HC | | NM showed an overall-weakening performance with a retention rate of only 47.4% (23.7 mAh g⁻1). The HC | | NMD full batteries present better performance advantages in capacities and specific energy (based on the mass of the active material) over the recently reported representative full cells (Supplementary Fig. 26 and Supplementary Table 6). Moreover, pouch cells (single layer) were assembled by using presodiated HC and NMD materials to assess their practical cycling stability within 1.5-3.6 V (Supplementary Fig. 27). HC | | NMD pouch cell presents a reverse capacity of 140.0 mAh g⁻1 at 0.1 A g⁻1, which is higher than HC | | NM pouch cell (86.9 mAh g⁻1). The corresponding GCD curves in Supplementary Fig. 27b further shows the excellent electrochemical reversibility of the NMD electrode. The HC | | NMD pouch cell also presents high capacity retention of 82.5% for 200 cycles and high specific energy (based on the mass of the active material) of 210.3 Wh kg⁻1 at 0.5 A g⁻1, verifying its great practical potential as the SIB positive electrodes.
Structural evolution analysis
In-situ XRD analysis was then performed to reveal the phase transition changes during the initial charge/discharge cycles. The extraction of Na+ weakens the attraction between sodium and oxygen layers, expanding interlayer spacing. Both samples therefore show (002) and (004) peaks shift towards lower angles during charging and the opposite trend during discharging (Fig. 4a, d). During the high-voltage charging stage, the disappearance of the P2 phase (002) peak is accompanied by the emergence of a new diffraction peak at 17.4°, which is attributed to the (001) crystal plane of the OP4 phase. The original lattice symmetry and periodicity are disrupted due to the presence of oxygen vacancies, which are more favorable for the transition to the OP4 phase during Na+ extraction. As visualized in contour plot (Fig. 4b), NMD maintained a small peak shift of 0.52° for (002) plane and 1.09° for (004). The calculated c-axis value is also increased by only 2.56% of the pristine cell parameter due to the expansion of Na layers (Fig. 4c), confirming its small volume change during discharging. Furthermore, after subsequent charging and discharging, the diffraction peak returns to its original position with no new phases detected, indicating high reversibility of the P2-OP4 phase transition within NMD34. In contrast, NM exhibits more pronounced structure changes with a larger shift in both (002) (0.71°) and (004) peaks (1.22°) as well as c-axis value (2.87%) in Fig. 4e, f. The incompletely recovered diffraction peaks of NM suggest its poor structural reversibility. A detailed structural change comparison of samples during discharging/charging is illustrated in Fig. 4g. NMD achieves minimum structural changes and robust structural reversibility over NM during Na+ de-intercalation due to oxygen vacancies to buffer interlayer stresses in NMD.

In-situ XRD results investigation with Swagelok cell corresponding to the charge/discharge curves between 1.8-4.3 V at 50 mA g⁻1 and 25 °C for NMD and NM. a, d Patterns with the highlight of (002) and (004) peaks. b, e The corresponding contour plots. c, f Lattice parameter variations. g Schematic of crystal structure changes.
Analysis of Na+ transport mechanism
To further elucidate the impact of ammonia-induced strategy on Na+ transfer kinetics within Na1/2MnO2, neutron powder diffraction (NPD) combined with maximum entropy method (MEM) was performed to visualize the transport path of Na+. Based on the NPD Rietveld refined data (Fig. 5a and Supplementary Table 7), a/c axis of NMD is shrunk that is consistent with XRD results. Furthermore, the corresponding three-dimensional nuclear density distribution calculated by MEM (Fig. 5b) shows Na+ with positive scattering lengths migrate along the Z path through oxygen sites or vacancies in NMD, which provides shorter Na+ diffusion distances and more active sites than NM37. This indicates enhanced Na+ transport kinetics along a-axis with an optimized diffusion pathway and low energy barrier, contributing to improved capacity and rate performance in NMD.

a Neutron powder diffraction (NPD) refinement. b Three-dimensional nuclear density distributions calculated by MEM using NPD data for NM and NMD. Total density of states (TDOS), partial density of states (PDOS) of Mn 3 d and O 2p for (c) NM and (d) NMD, and (e) schematic PDOS with X = 0. f Na+ migration energy barrier for NM and NMD. g Schematic diagram of Na+ migration.
Density-functional theory (DFT) calculations were performed to further investigate the diffusion mechanism of Na+ using a computational model that introduces oxygen vacancies for NMD (Supplementary Fig. 28). Based on the analysis for electronic density of states (DOS) at different desodiation states (Fig. 5c–e and Supplementary Fig. 29), NMD demonstrates the dramatically increased number of electronic states at the Fermi level and significantly reduced band gap compared to NM, even in different desodiation states38,39, which confirms its improved electronic conductivity. The partial density of states (PDOS) analysis demonstrates that this conductivity enhancement originates from the synergistic contribution of Mn 3 d and the O 2p orbitals surrounding the oxygen vacancies, which create localized states that facilitate electron hopping throughout the structure. This finding solidifies the critical role of oxygen vacancies that engineered by our synthesis approach in optimizing the electronic structure of Mn-based layered oxides. Furthermore, the calculated Na+ transport barrier for NMD (ΔENMD = 0.258 eV) is also lower than that for NM (ΔENM = 0.369 eV) (Fig. 5f) owing to the slight expansion in the c-axis direction caused by oxygen vacancies, which directly reduces the electrostatic repulsion and the spatial potential resistance of Na⁺ migration in NMD40. A detailed schematic is correspondingly illustrated in Fig. 5g.
General applicability investigation
Considering the aforementioned results, we prepared three sets of manganese based layered oxides (Na1/2Mn6/7Ni1/7O2 (NMN), Na1/2Mn6/7Cu1/7O2 (NMC), Na1/2Mn6/7Mg1/7O2 (NMM)) for investigating the broad applicability of this strategy (Supplementary Figs. 30–35 and Supplementary Tables 8–11). All samples were P2-type structure with the space group P63/mmc. The detailed rate and cycling performances of samples with and without DA addition are listed in Fig. 6a–f with the summary of performance improvements presented in Fig. 6g. All samples synthesized through the proposed strategy showed an apparent performance enhancement compared with their pristine counterparts, even by >300%. Notably, the other two ammonia-releasing sources including melamine and 5-aminotetrazole also presented similar performance-enhancing effect to DA (Supplementary Figs. 36–38), although their improvements were not as pronounced as those of DA. Therefore, this synthesis strategy can be considered as a widely applicable approach for comprehensively improving the electrochemical performances of Mn-rich P2-type LTMOs as the SIB positive electrodes.

a, c, e Rate capabilities and b, d, f cycling performances of different manganese based layered oxides and their optimized counterparts treated with the proposed strategy presented in this work (NMN: Na1/2Mn6/7Ni1/7O2; NMND: Na1/2Mn6/7Ni1/7O2 with DA; NMC: Na1/2Mn6/7Cu1/7O2; NMCD: Na1/2Mn6/7Cu1/7O2 with DA; NMM: Na1/2Mn6/7Mg1/7O2; and NMMD: Na1/2Mn6/7Mg1/7O2 with DA). g The performance enhancement summary of samples.
Discussion
We report an ammonia-induced general synthesis strategy for manganese based layered oxides. Na1/2MnO2 as the demo-type material was synthesized through the classic solid-state reaction with the introduction of dicyandiamide. The release of ammonia leads to the formation of oxygen vacancies and dramatic decrease of crystallization temperatures, which alleviates the internal stress during material growth and activates ion migration through sintering. The material therefore presents high quality structures with excellent Na+ storage activity. These oxygen vacancies effectively suppress the Jahn-Teller distortion of Mn3+ octahedra by electron delocalization, as confirmed by DFT calculations, which reveal a reduced band gap and a significantly increased density of states near the Fermi level upon desodiation. NPD data combined with MEM analysis visualizes the shortened and optimized Na+ migration pathways via oxygen vacancies. Consequently, these advantageous structural attributes endow NMD with excellent capacity of up to 187.2 mAh g⁻1 and a high rate capability of 123.8 mA h g⁻1 at 1.0 A g⁻1, which is 6.7 times of the pristine material. The assembled full cell also presents a 160 % capacity increase to 130.7 mAh g⁻1 and retention of 65.0% after 300 cycles at 0.2 A g⁻1. This work provides new insights for the development of high-performance battery technologies.
Methods
Materials synthesis
Na1/2MnO2 (NM) with dicyandiamide (DA, 99.9%, Sinopharm Chemical Reagent) addition (NMD) was synthesized directly by high temperature solid-state reaction. Specifically, Na2CO3 (99.9%, Macklin), Mn2O3 (99.9%, Aladdin) and DA with the fixed stoichiometric proportions were mixed, following with the ball-milling. A planetary ball milling was performed with a 100 mL agate jar and balls (4–12 mm diameter) at a 20:1 ball-to-powder mass ratio. The milling process consisted of 30 minutes of forward rotation, a 5-min pause, and then 30 min of reverse rotation.
The obtained precursor after drying was pressed into the pellet, following with heating at 570 °C for 2 h and then 970 °C for 10 h to achieve complete phase formation. The obtained powder is denoted as NMD. The same procedure but without the addition of DA was used to synthesize Na1/2MnO2 (NM). All other samples Na1/2Mn6/7Ni1/7O2 (NMN), Na1/2Mn6/7Cu1/7O2 (NMC), Na1/2Mn6/7Mg1/7O2 (NMM) were synthesized with the same procedure but different added oxides while their counterparts with the addition of DA were denoted as NMND, NMCD, NMMD, etc. In addition, as a universal validation, NM with the addition of melamine (99.9%, Macklin) (NMME) and 5-aminotetrazole (99.9%, Macklin) (NMAT) were synthesized by the same procedure, respectively. All the prepared samples were quickly transferred to an argon-filled glove box (H2O, O2 < 0.01 ppm, 25 °C) for storage.
Materials characterizations
The relative proportions of various elements were analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES, prodigy XP). X-ray diffraction (XRD) patterns were collected through Bruker D8-Advance diffractometer (λ = 1.54 Å, Cu Kα radiation) for identifying phase composition. GSAS II software was used to refine the result for the corresponding Rietveld data. High-resolution transmission electron microscopy (HRTEM, JEM-ARM200, JEOL) was used to check the detailed structure of materials at 300 kV. Scanning transmission electron microscopy (STEM) equipped with high-angle annular dark field (HAADF) and annular bright field (ABF) were performed on JEM-ARM200. Scanning electron microscope (SEM, Hitachi S-4800) equipped with an energy dispersive spectrometer (EDS, Oxford) was used to determine the surface morphology of samples. In-situ high-temperature XRD patterns were employed to observed the phase formation of precursors through Smart lab diffractometer (λ = 1.54 Å, Cu Kα radiation) at a heating rate of 5 °C/min from room temperature to 500 °C and a scanning rate of 20 °/min with 10° ~ 50°. A simultaneous thermal analyzer (TGA, DSCI/1600LF) was used to monitor the pyrolysis process of samples in the temperature range of 30-1000 °C with a temperature increase rate of 10 °C min⁻1. Electron paramagnetic resonance spectrometry (EPR, Bruker EMX Plus) with the spins of 1.111 × 1012 was used to analyze the oxygen vacancies of samples at room temperature. Raman spectral analysis was performed using a confocal Raman micro spectrometer (Raman, XPLORA) with an excitation wavelength of 532 nm in the range of 400–2000 cm⁻1. X-ray photoelectron spectrometer (XPS, Thermo Scientific K-Alpha) with a monochromatic Al Kα X-ray source was used to analyze the elemental valence states on the material surface. The X-ray absorption fine structure (XAFS) spectra of Mn K-edge were recorded on a commercial Laboratory-Based XAFS spectrometer (Table XAFS-500A, Specreation Instruments Co., Ltd.) with the voltage and current of 20 kV and 15 mA. The Ge (531) spherically bent crystal analyzers and the R250 mm Rowland circle were used to provide monochromatized X-ray beam. To clarify the phase transition during charging/discharging, in-situ XRD tests were performed in a specially designed cell with a Be window at 0.05 A g⁻1. For further analysis of the Na+ diffusion pathway, time-of-flight neutron powder diffraction data (NPD) were recorded by the Spallation Neutron Source Science Center in Dongguan, China (CSNS, Dongguan, China). The original data were then investigated through Rietveld analysis and maximum entropy (MEM) calculations using Z-Rietveld software. Three-dimensional nuclear density distributions are visualized by the VESTA program.
Electrochemical measurements
Coin cells (CR2025, 15.4 × 1.1 mm shrapnel, 16.1 × 0.8 mm washer) were assembled in an argon-filled glove box (H2O, O2 < 0.01 ppm, 25 °C) to evaluate the electrochemical performances of samples. The active material, polyvinylidene fluoride (PVDF, Guangdong Canrd New Energy Technology Co., Ltd., > 99.5%, 50,000 molecular weight) and conductive charcoal black (Super P, TIMCAL Graphite & Carbon, > 99%) were weighed in a mass ratio of 8:1:1, and then were mixed with N-methyl pyrrolidone (NMP, Shanghai Macklin Biochemical Technology Co., Ltd., > 99%) for a slurry that was coated using a 150 μm squeegee on a carbon coated aluminum foil (16 µm thick with 1 µm carbon layers on both sides, 99.65% purity) and heated at 75 °C for 12 h. The mass loadings of active materials were 2–3 mg cm⁻2. Before battery assembly, sodium metal electrodes (Sinopharm Chemical Reagent Co., Ltd., chemical purity) were prepared directly in an argon-filled glove box (H2O, O2 < 0.01 ppm, 25 °C), cutting with a 14 mm diameter cutter at a thickness of approximately 0.5–0.6 mm. For half-cell testing, Na metal foil was used as the counter electrode, glass fiber (GF-A2916, Olegeeino (Chongqing) New Materials Co., Ltd., aperture is 1.63, thickness is 0.29, porosity is > 90%) as the separator and 1.0 M NaPF6 dissolved in 1,2-dimethoxyethane (DME) (80 µL, Nanjing Modges Energy Technology Co., Ltd.) as the electrolyte. For full-cell assembly, hard carbon after activation was used as the counter electrode. The hard carbon negative electrode was pre-sodiated first at 20 mA g⁻1 within 0.01–3.0 V for 3 cycles to form a strong solid electrolyte interphase (SEI) film in the half-cell for further testing. This coin cell was disassembled in an argon-filled glove box to obtain the activated negative electrode, and then assembled with the positive electrodes to form the full cell. The theoretical capacity ratio between negative and positive electrodes was set to be 1.2. The galvanostatic charge-discharge (GCD) tests were carried out on a Land CT2001A battery test system (Wuhan LAND Electronic Co., Ltd) with a voltage range of 1.8-4.3 V at 300 K. Cyclic voltammetry (CV) curves were recorded with a CHI 760E electrochemical workstation (Chenhua Shanghai, China) at 1.8–4.3 V at the scan rates of 0.1, 0.08, 0.05, 0.03, 0.01 mV s−1 and electrochemical impedance spectroscopy (EIS) was measured in constant-potential mode with a frequency of 10⁻2 ~ 105 Hz after leaving the battery in an open-circuit state for 2 h with a total of >80 data points recorded. The full battery and pouch cells tests were also conducted with Land CT2001A battery test system for charge/discharge testing during 1.9–4.2 V. Pouch cells (single layer) were assembled by using NM/NMD and presodiated HC as the positive/negative electrodes with a N/P capacity ratio in the range of 1.02 ~ 1.15. The mass loadings of the positive and negative electrodes are 23 ~ 25 mg cm−2 and 13 ~ 15 mg cm−2, respectively, with corresponding sizes of 6.0 × 7.0 cm2 and 6.5 × 7.5 cm2. Electrolyte injection (3 g/Ah) was performed in a glove box (H2O, O2 < 0.01 ppm, 25 °C) and a vacuum sealing machine (MSK-115-III, Hefei Kejing Material Technology Co., Ltd) was used to remove gas and seal the pouch cells. The pouch cells were cycled in the potential windows within 1.5–3.6 V.
Theoretical calculations
The computational methodology in this work was based on spin-polarized density functional theory (DFT)41,42 as implemented in the Vienna ab initio simulation package (VASP)43,44. The projector augmented-wave (PAW) method was used with a plane-wave cutoff energy of 450 eV, and the Perdew-Burke-Ernzerhof (PBE)45 functional was selected for the generalized gradient approximation (GGA). A vacuum layer of 15 Å was applied to suppress inter-slab interactions. Structural optimizations for NM and NMD were conducted with a 3 × 3 × 2 k-point mesh, with convergence reached when the energy difference was less than 10⁻5 eV and the maximum force on any atom was below 0.02 eV Å⁻¹. The computational structural information files for NM and NMD are provided in Supplementary Data 1 and 2, respectively.
Data availability
All of the data supporting the conclusions of this study are available within the article and its Supplementary Information. Source data are provided with this paper.
References
Yang, Y. et al. Decoupling the air sensitivity of Na-layered oxides. Science 385, 744–752 (2024).
Rong, X. et al. Anionic redox reaction-induced high-capacity and low-strain cathode with suppressed phase transition. Joule 3, 503–517 (2019).
Sun, L. et al. Unraveling and suppressing the voltage decay of high-capacity cathode materials for sodium-ion batteries. Energy Eviron. Sci. 17, 210–218 (2024).
Gao, A. et al. Topologically protected oxygen redox in a layered manganese oxide cathode for sustainable batteries. Nat. Sustain. 5, 214–224 (2022).
Deng, T. et al. Interfacial-engineering-enabled practical low-temperature sodium metal battery. Nat. Nanotechnol. 17, 269–277 (2021).
Ren, M. et al. Homeostatic solid solution in layered transition-metal oxide cathodes of sodium-ion batteries. J. Am. Chem. Soc. 145, 224–233 (2023).
Zhao, C. et al. Rational design of layered oxide materials for sodium-ion batteries. Science 370, 708–711 (2020).
Ding, F. et al. Tailoring planar strain for robust structural stability in high-entropy layered sodium oxide cathode materials. Nat. Energy 9, 1529–1539 (2024).
Wang, J. et al. Routes to high-performance layered oxide cathodes for sodium-ion batteries. Chem. Soc. Rev. 53, 4230–4301 (2024).
Xiao, Z. et al. Suppressing the Jahn–Teller effect in Mn-based layered oxide cathode toward long-life potassium-ion batteries. Adv. Funct. Mater. 32, 2108244 (2022).
Tang, Y. et al. Sustainable layered cathode with suppressed phase transition for long-life sodium-ion batteries. Nat. Sustain. 7, 348–359 (2024).
Jian, Z. et al. Solid-state synthesis of low-cost and high-energy-density sodium layered-tunnel oxide cathodes: dynamic structural evolution, Na+/vacancy disordering, and prominent moisture stability. Nano Energy 125, 109528 (2024).
Gan, L. et al. The synergy of dis-/ordering ensures the superior comprehensive performance of P2-type Na-based layered oxide cathodes. Carbon Neutralizat 2, 235–244 (2023).
Wang, Q. et al. Fast-charge high-voltage layered cathodes for sodium-ion batteries. Nat. Sustain. 7, 338–347 (2024).
Wang, X. et al. Achieving a high-performance sodium-ion pouch cell by regulating intergrowth structures in a layered oxide cathode with anionic redox. Nat. Energy 9, 184–196 (2024).
Zou, P. et al. Regulating cation interactions for zero-strain and high-voltage P2-type Na2/3Li1/6Co1/6Mn2/3O2 layered oxide cathodes of sodium-ion batteries. Angew. Chem. Int. Ed. 62, e202304628 (2023).
Wang, Y. et al. Unexpected elevated working voltage by Na+/vacancy ordering and stabilized sodium-ion storage by transition-metal honeycomb ordering. Angew. Chem. Int. Ed. 63, e202409152 (2024).
Feng, J. et al. La3+ doped nickel-manganese oxide as high-capacity cathode for sodium-ion batteries guided by Bayesian optimization. Angew. Chem. Int. Ed. 64, e202424572 (2025).
Gao, Z. et al. Kirkendall effect-induced uniform stress distribution stabilizes nickel-rich layered oxide cathodes. Nat. Comm. 15, 1503 (2024).
Jian, Z. et al. Accelerating lattice oxygen kinetics of layered oxide cathodes via active facet modulation and robust mechanochemical interface construction for high-energy-density sodium-ion batteries. Energy Environ. Sci. 18, 7995–8008 (2025).
Wang, H. et al. Multicationic interactions mitigating lattice strain in sodium layered cathodes. Nat. Comm. 16, 4409 (2025).
Jin, J. et al. Annealing in argon universally upgrades the Na-storage performance of Mn-based layered oxide cathodes by creating bulk oxygen vacancies. Angew. Chem. Int. Ed. 62, e202219230 (2023).
Qiu, L. et al. Structural reconstruction driven by oxygen vacancies in layered Ni-rich cathodes. Adv. Energy Mater. 12, 2200022 (2022).
Zuo, W. et al. The stability of P2-layered sodium transition metal oxides in ambient atmospheres. Nat. Comm. 11, 3544 (2020).
Li, Q. et al. Improving the oxygen redox reversibility of Li-rich battery cathode materials via Coulombic repulsive interactions strategy. Nat. Comm. 13, 1123 (2022).
Li, Y. et al. Competing mechanisms determine oxygen redox in doped Ni–Mn based layered oxides for Na-ion batteries. Adv. Mater. 36, 2309842 (2024).
Ren, H. et al. Impurity-vibrational entropy enables quasi-zero-strain layered oxide cathodes for high-voltage sodium-ion batteries. Nano Energy 103, 107765 (2022).
Fan, S. et al. Recrystallization-driven quasi-spherical Prussian blue analogs with high tap density and crystallinity for sodium-ion batteries. ACS Energy Lett. 10, 1751–1761 (2025).
Jing, Z. et al. Rational design of cobalt-based prussian blue analogues via 3 d transition metals incorporation for superior Na-ion storage. Angew. Chem. Int. Ed. 64, e202423356 (2025).
Huang, Z. et al. Multifunctional and radii-matched high-entropy engineering toward locally-regulable metal oxide layers in sodium-layered oxide cathode. Angew. Chem. Int. Ed. 64, e202505367 (2025).
Fu, F. et al. Entropy and crystal-facet modulation of P2-type layered cathodes for long-lasting sodium-based batteries. Nat. Comm. 13, 2826 (2022).
Yu, Y. et al. Triggering reversible anion redox chemistry in O3-type cathodes by tuning Na/Mn anti-site defects. Energy Eviron. Sci. 16, 584–597 (2023).
Shi, Q. et al. Niobium-doped layered cathode material for high-power and low-temperature sodium-ion batteries. Nat. Comm. 13, 3205 (2022).
Liu, Z. et al. Achieving a deeply desodiated stabilized cathode material by the high entropy strategy for sodium-ion batteries. Angew. Chem. Int. Ed. 63, e202405620 (2024).
Wang, Y. et al. Boosting the reversibility and kinetics of anionic redox chemistry in sodium-ion oxide cathodes via reductive coupling mechanism. J. Am. Chem. Soc. 145, 22708–22719 (2023).
Dong, H. et al. Lithium orbital hybridization chemistry to stimulate oxygen redox with reversible phase evolution in sodium-layered oxide cathodes. J. Am. Chem. Soc. 146, 22335–22347 (2024).
Gao, G. et al. Direct regeneration of spent LiCoO2 cathodes with Ca2+-assisted molten salt strategy. Acta Mater. 273, 119969 (2024).
Zhang, T. et al. Negative lattice expansion in an O3-type transition-metal oxide cathode for highly stable sodium-ion batteries. Angew. Chem. Int. Ed. 63, e202316949 (2024).
Zhang, K. et al. Regulating phase transition and oxygen redox to achieve stable high-voltage O3-type cathode materials for sodium-ion batteries. Adv. Energy Mater. 13, 2302793 (2023).
van der Lubbe, S. C. C., Wang, Z., Lee, D. K. J. & Canepa, P. Unlocking the inaccessible energy density of sodium vanadium fluorophosphate electrode materials by transition metal mixing. Chem. Mater. 35, 5116–5126 (2023).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phy. Rev. 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phy. Rev. 140, A1133–A1138 (1965).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phy. Rev. B 54, 11169–11186 (1996).
Blöchl, P. Projector augmented-wave method. Phy. Rev. B 50, 17953–17979 (1994).
Perdew, J., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phy. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
This work was supported by National Natural Science Foundation of China (22378055), Applied Basic Research Program of Liaoning (2024JH2/101900010), Guangdong Basic and Applied Basic Research Foundation (2022A1515140188), Fundamental Research Funds for the Central Universities (N2225044). We appreciate the instrumental analysis from Analytical and Testing Center, Northeastern University and Multi-Physics Instrument (MPI) granted from the China Spallation Neutron Source (CSNS). We acknowledge Specreation Instruments Co., Ltd. for the support related to the XAFS measurements.
Author information
Authors and Affiliations
Contributions
L.L. conceived and supervised the project. Y.Z. designed and performed the synthesis, characterization and analysis of materials. Z.G. performed the partial synthesis and electrochemical characterizations. J.Zeng carried out DFT calculations. J.Zhao, H.L., G.G. and S.D. helped for data analysis. S.W. helped edit the work. Y.Z. drafted the manuscript and L.L. finished the writing and editing of this paper. All authors checked the paper and agreed with its content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Anh Vu, Lai Chen and Juan Wang for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Y., Guo, Z., Zeng, J. et al. An ammonia-induced universal synthesis approach for manganese based layered oxides. Nat Commun 17, 263 (2026). https://doi.org/10.1038/s41467-025-66960-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66960-w
