
Abstract
Wide-bandgap (WBG) perovskite solar cells (PSCs) are employed for tandem solar cells. Understanding the crystallization mechanism of mixed-halide WBG perovskite will contribute to achieving the high-performance photovoltaics. Herein, we demonstrate that the asynchronous halide insertion is accompanied by random crystal facets and orientations, restricting the efficient carrier extraction. Guided by density functional theory calculations, we construct a π-conjugated molecular wall structure using o-phenylenediamine (OPD). The molecular wall at the grain boundary induces templated perovskite crystallization through the ortho-diamine group, enabling synchronous [PbBr6]4- and [PbI6]4- halide insertion. The OPD-treated perovskite film exhibits a preferred (100) facet and a highly vertical orientation. Benefited from the improved carrier extraction, the resulting WBG PSC (1.69 eV) achieves a power conversion efficiency of 24.13% (certified 23.43%), representing one of the highest values among WBG PSCs. Meanwhile, the improved perovskite crystal quality ensures the enhanced operational stability of the PSCs.
Similar content being viewed by others


Introduction
Wide-bandgap (WBG) perovskite materials show advantages for employment as top cells in tandem solar cells (TSCs)1,2,3,4. Nevertheless, WBG perovskite solar cells (PSCs) suffer from severe power conversion efficiency (PCE) loss of more than 20%, which is significantly higher than that of common formamidinium lead iodide (FAPbI3)-dominated perovskite systems5,6. WBG perovskites are usually constructed with mixed halides to achieve a tunable optical bandgap7, and bromine (Br) contents above 20 mol% are usually used8. Since Br possesses a smaller ionic radius and lower solubility than iodine (I), Br and I halides exhibit large differences in crystallization kinetics9. The crystallization behavior of mixed-halide anions plays a crucial role in preparing high-quality WBG perovskite films10. The differentiated crystallization properties of halides can induce phase separation, residual lattice strain, random facet orientation, etc.11,12,13,14, which restrict efficient carrier extraction and crystal structure stability. Unfortunately, researchers have not yet fully understood the crystallization mechanism of mixed-halide perovskite films.
For conventional solution-processed methods, a large number of perovskite grains crystallize unevenly during their independent nucleation and growth processes, and thus spontaneously self-assemble into various crystal facets and orientations15. The preferential crystallization of Br-based components has been widely demonstrated in WBG perovskite systems10, and the subsequent insertion of I components will restructure the unit cells to form mixed-halide perovskite crystals. The asynchronous halide insertion will exacerbate the randomness of crystal facets and orientations16. The macroscopically disordered crystal arrangement induces defects such as atomic dislocation and ion vacancies, resulting in reduced carrier mobility and shortened carrier diffusion length17. Previous works have reported some methods to control crystal orientation and halide distribution by molecular additives. Jiao et al. and Shi et al. are committed to achieving pure α phase in the FAPbI3 perovskite system18,19. They introduce additives to achieve strong coordination forces with the Pb-I skeleton, thereby inducing oriented crystallization of perovskite films. Yan et al. achieves oriented nucleation of I-based Pb-Sn perovskite by reducing the surface energy of the (100) plane20. Crystal growth control of mixed-halide WBG perovskites is more difficult than that of these pure I halide perovskite systems, due to the natural asynchronous crystallization nucleation of I and Br components. Although Li et al. achieve the synchronous phase transition of I-based and Br-based components through intermediate phase regulation, this phase transition at the annealing stage cannot achieve a homogeneous mixed-halide perovskite phase with preferred crystal facets and orientations11. Therefore, understanding the mixed-halide insertion behaviors and achieving controllable halide insertion is the basis for obtaining the WBG perovskite films with efficient carrier transport.
In-situ grazing-incidence wide-angle X-ray scattering (GIWAXS) characterization is an important technique to reveal the evolution of crystallographic information in perovskite films21. For perovskite films with asynchronous insertion of halide components, experimental results show that each crystal facet exhibits distinguishable structural evolution characteristics. Combined with density functional theory (DFT) analysis, it is found that the post-crystallization I component prefers to be inserted along the (100) facet when entering the perovskite lattice. Unfortunately, this directed insertion of the I component does not improve the diversity of crystal facets and orientations. Inspired by the tendency of the I component to be inserted along the (100) facet, we envision realizing the synchronous halide (I and Br) insertion along the (100) facet. Considering that the strong interaction of functionalized π-conjugated molecules can build a molecular wall structure, the amine groups exposed on the same side of the molecular wall can serve as nucleation sites for perovskite crystallization along the (100) facet. It is expected to achieve synchronous halide insertion, thus obtaining preferred crystal facets and orientations.
Guided by DFT calculations, the π-conjugated ortho-diamine cation, o-phenylenediamine (OPD), is identified as an ideal candidate for building a robust molecular wall and regulating halide insertion. As expected, the ortho-diamine groups of OPD molecules show the strongest interaction with the (100) facet of the perovskite crystal, and almost the same binding energy exists between OPD molecules and [PbX6]4- octahedra ([PbBr6]4- and [PbI6]4-). The templated crystallization induced by the OPD-constructed molecular wall ensures the synchronous insertion of mixed-halide components, forming a preferred (100) facet and an orientation perpendicular to the substrate. The resulting perovskite films exhibit reduced defects and prolonged carrier diffusion length, guaranteeing efficient carrier extraction and suppressed nonradiative recombination. As a result, the OPD-treated WBG PSC (1.69 eV) achieves a PCE of 24.13% (certified 23.43%), representing one of the highest PCEs among WBG PSCs. Based on a semi-transparent (ST) PSC, the resulting four-terminal (4T) perovskite/copper indium gallium selenide (CIGS) tandem solar cell (TSC) achieves a PCE of 29.34%. Benefiting from the highly vertical crystal orientation and the stable perovskite structure, the encapsulated WBG PSC still maintains more than 90% of the initial PCE after maximum power point tracking (MPPT) for 800 h under the ISOS-L-1 testing protocol.
Results
Crystallographic evolution mechanism of asynchronous halide insertion
To investigate the crystallographic evolution of WBG perovskite films, we perform in-situ GIWAXS characterization of FA0.8MA0.1Cs0.1Pb(I0.8Br0.2)3 film at the spin-coating stage (Fig.1a)21. As the antisolvent drips, three common perovskite characteristic crystal facets gradually appear, including (100), (110), and (111) facets. The q-value variation corresponding to each facet is extracted (Fig.1b, Supplementary Table 1), and all crystal facets exhibit decreased q values. It is noteworthy that a significant q-value decrease (Δq, −0.115 nm−1) from 10.150 nm−1 to 10.035 nm−1 is observed on the (100) facet, compared with that on the (110) facet (−0.038 nm−1) and (111) facet (−0.042 nm−1). According to the Bragg equation, a reduced q value means an increased interplanar spacing (d)22. Moreover, almost at the same time as the q-value decreases, all the crystal facets show completely random orientation (Fig.1c). It is not until 30 s that the perovskite film gradually self-assembles to form a partial orientation trend (Supplementary Fig. 1). Nevertheless, even though the crystallinity is enhanced after annealing, the control film still exhibits random crystal orientation (Fig.1d). To exclude the influence of solvent on perovskite nucleation and orientation, we introduce a common solvent, N-methyl-2-pyrrolidone (NMP), into the perovskite precursor solution, resulting a mixed solvent system (25 V% of NMP). As shown in the GIWAXS patterns (Supplementary Fig. 2), the control sample still exhibit random crystal facets and orientations after NMP solvent introduction.

a In-situ GIWAXS patterns of control perovskite film at the spin-coating stage. b Extracted q-value evolution of (100), (110), and (111) facets. c Extracted crystal orientation evolution of (100) facet. d GIWAXS pattern of the control perovskite film after annealing. e Perovskite crystal models and DFT-calculated interplanar spacings of (100), (110), and (111) facets before and after I ion insertion.
Such lattice evolution is usually accompanied by asynchronous crystallization of halides in perovskite films. Asynchronous crystallization of mixed halides has been reported, and preferential crystallization of Br components has been demonstrated9,10. We thus speculate that the q-value decrease is caused by the post-crystallization I ion insertion. As shown in the in-situ GIWAXS data, the (100) facet exhibits a larger change in q value compared to the other two facets, which may be related to the way I components are inserted into the perovskite lattice. Therefore, we perform DFT calculations to simulate the mechanism of asynchronous halide insertion. For perovskite crystals with a cubic ABX3 structure, the atomic layer arrangement shown on the (100) facet is …AX-BX2-AX-BX2…. The atomic arrangements corresponding to the (110) and (111) facets are …X2-ABX-X2-ABX… and …AX3-B-AX3-B…, respectively23. We attempt to insert I element into the ABBr3 lattice along the (100) facet, and the intervening Br ion layers are replaced by I ion layers. As shown in Fig. 1e, the atomic arrangement after I ion insertion is shown on the AX(100), ABX(110), and AX3(111) terminations, respectively24. Changes in the interplanar spacing (Δd) of the (100), (110), and (111) facets can be obtained, which are 0.19 Å, 0.03 Å, and 0.02 Å, respectively. Theoretical simulation results for the I element insertion along the (100) facet show the same trend as the in-situ GIWAXS results. The (100) facet exhibits a sharp interplanar spacing change compared to the other two crystal facets due to the post-crystallization I ion insertion.
To further confirm whether the specific I ion insertion direction determines the above-mentioned change in interplanar spacing, we also simulate a random I ion insertion model. As shown in Supplementary Fig. 3, 80% of the Br atoms are randomly replaced by I atoms at the X sites of each crystal facets, which results a APb(Br0.2I0.8)3 crystal structure. According to the DFT calculation results, almost the same Δd variation is observed at all three crystal facets, which is inconsistent with the in-situ GIWAXS experiments. Therefore, we conclude that the post-crystallization I ion insertion direction shows a preference for the (100) facet. This asynchronous halide insertion is accompanied by random crystal facets and orientations, which are undoubtedly detrimental to efficient carrier extraction. Considering the crystal facets’ selectivity of halide insertion and the dominance of the (100) facet, we are committed to inducing synchronous halide insertion, obtaining a preferred (100) facet and a perpendicular crystal orientation19.
Crystallographic modulation of synchronous halide insertion
We propose to construct a functionalized π-conjugated molecular wall structure to induce the synchronous mixed-halide insertion (Fig. 2a). It has been demonstrated that large-sized amine cations can effectively anchor the [PbX6]4- octahedron20. We therefore design a series of benzene-based monoamine and diamine cations, namely aniline (AD), o-phenylenediamine (OPD), m-phenylenediamine (MPD), and p-phenylenediamine (PPD). Strong intermolecular π-π stacking is crucial for constructing a stable molecular structure25. We simulate three stacking models of π-conjugated molecules using DFT calculations, including parallel (0°), perpendicular (90°), and antiparallel (180°)26. All four molecules exhibit close intermolecular forces in both the perpendicular and antiparallel models, as well as monoamines and diamines (Fig. 2b). In the parallel stacking model, the intermolecular force of the OPD is 2–20 times that of the other three molecules (Supplementary Fig. 4). Moreover, the parallel stacking model of OPD exhibits a larger binding energy (0.163 eV) between molecules, compared with their perpendicular stacking model (0.081 eV) and antiparallel stacking model (0.073 eV) (Fig. 2c). In contrast, the AD with monoamines exhibits close and weak π-π interaction force in both perpendicular and antiparallel modes, making it difficult to form aligned molecular wall. Therefore, the parallel stacking of OPD should be the most stable structure, and OPD with ortho-diamine exhibits advantages in constructing a stable molecular wall structure.

a Schematic diagram of the OPD molecular wall-induced synchronous halide insertion and templated crystallization of perovskite film. b Binding energies of OPD, MPD, PPD, and AD with different stacking models by DFT simulations. c Binding energy of OPD with parallel, perpendicular, and antiparallel models. d ESP distribution of four molecules. e Binding energy between OPD and different crystal facets of perovskite by DFT simulation. In-situ GIWAXS patterns of f AD-treated and j OPD-treated perovskite films at the spin-coating stage. g Extracted q-value evolution of (100), (110), and (111) facets for AD-treated and OPD-treated perovskite films. k Extracted signal intensity of (100) facet for control, AD-treated, and OPD-treated perovskite films. Extracted crystal orientation evolution of (100) facet for h AD-treated and l OPD-treated perovskite films. GIWAXS patterns of i AD-treated and m OPD-treated perovskite film after annealing.
This parallel stacking allows all ortho-diamine groups to be located on the same side of the molecular wall structure. The OPD also exhibits the largest electrostatic potential (ESP) distribution in the ortho-diamine region compared to the other three molecules, driving the perovskite nucleation (Fig. 2d). Since a large number of ortho-diamine groups are exposed on the same side, the increased nucleation sites and the strong electrostatic potential guarantee the templated crystallization. In general, halide components are present in the form of [PbX6]4- octahedra in the perovskite precursor solution before the perovskite crystals are formed. The binding energy between OPD molecules and [PbX6]4- octahedra is crucial for controlling the crystallization kinetics of perovskites. To evaluate whether the OPD-constructed molecular wall is selective for the crystal facet of perovskite nucleation, we employ DFT calculations to simulate the binding energy between OPD cations and [PbI6]4- octahedra of different perovskite crystal facets (Fig. 2e, Supplementary Figs. 5–7). As shown, the maximum binding energy (1.20 eV) is observed at the (100) face than at the (110) facet (0.94 eV) and (111) facet (1.06 eV). Moreover, the binding energies for OPD with [PbBr6]4- at the (110) and (111) facets are lower than that at the (100) facet. This provides a theoretical basis for the OPD-constructed molecular wall to induce a preferred (100) facet18. More importantly, OPD cation exhibits similar binding energies with both [PbI6]4- and [PbBr6]4- octahedra, suggesting that the synchronous insertion of I and Br halides is possible during the templated crystallization process.
We then monitor the crystallization dynamics of OPD molecular wall-induced mixed-halide WBG perovskite films through in-situ GIWAXS characterization (Fig. 2f, j). All perovskite films transform directly from the solvent phase to the perovskite phase, and no additional transitional phase is observed (Supplementary Fig. 8). As shown in the extracted q-value evolution, the (100) facet of the AD-treated film still exhibits a large q-value decrease from 10.112 nm−1 to 10.058 nm−1 (Fig. 2g). This may be because the monoamine molecules cannot exert strong forces to induce the simultaneous insertion of halides and templated crystallization of the perovskite film. Therefore, the preferred (100) facet and vertical crystal orientation are not fully achieved (Fig. 2h, i). Due to the indiscriminate anchoring of [PbBr6]4- and [PbI6]4-halides by the stable molecular wall structure, the OPD-treated perovskite film exhibits great potential to achieve synchronous halide insertion. The OPD-treated film also exhibits the fastest increase in GIWAXS signal intensity for the (100) facet (Fig. 2k), implying that the nucleation sites provided by the ortho-diamine accelerate the crystallization rate of the perovskite film27. The same conclusion can also be obtained from in-situ laser scanning confocal microscopy (LSCM) images (Supplementary Fig. 9). As expected, the resulting film obtains nearly constant q values (10.088 nm−1) for the (100) facet. Within a very short time (1 s) after the antisolvent is added, the preferred (100) facet and the crystals perpendicular to the substrate are formed (Fig. 2l Supplementary Fig. 10). We also monitor the GIWAXS evolution of the films at the annealing stage (Supplementary Fig. 11). All films exhibit nearly constant q values on all facets, which suggests that the halide insertion-induced perovskite crystal structure is formed at the spin-coating stage.
To further substantiate the synergistic halide insertion, we measure in-situ UV-Vis absorption spectra of perovskite films at the spin-coating stage. As shown in Supplementary Fig. 12, the absorption edge of the control perovskite film gradually red-shifts with the addition of antisolvent, indicating that the Br-rich component is formed preferentially, and the subsequent I insertion narrows the bandgap. Although the AD-treated film accelerates the red shift of the absorption edge, subsequent I halide insertion still exists and synchronous halide insertion cannot be successfully achieved. In contrast, the OPD-treated film exhibits a constant absorption edge almost immediately after the antisolvent is added. This constant bandgap also implies the simultaneous halide insertion, which is consistent with the results of in-situ GIWAXS characterization. As a result, the strong vertical orientation of (100) facet is dominant in the OPD-treated film (Supplementary Fig. 13). After OPD treatment with the optimal concentration of 1.0 mol% (Supplementary Fig. 14), the perovskite film shows the preferred (100) facet and the vertical crystal orientation (Fig. 2m). We extract the GIWAXS signal of the perovskite films on the (100) facet, and perform deconvolution of the GIWAXS peaks (Supplementary Fig. 15). Control and AD-treated samples exhibit additional fitting peak components near the main peak of (100) facet. This may be related to the inhomogeneous WBG perovskite phase. In contrast, a single (100) peak and the smallest full width at half maximum (FWHM) value can be observed in the OPD-treated films. The narrow FWHM of the (100) peak can also rule out contributions from grain size variation (Supplementary Fig. 16), indicating that the synchronized halide insertion leads to a high-quality homogeneous WBG perovskite phase.
We then perform the depth-profiling X-ray photoelectron spectroscopy (XPS), to investigate the halogen element distribution in the resulting perovskite films. As displayed in XPS spectra of I and Br elements (Supplementary Fig. 17), the signal of the Br element in the control film gradually decreases as the etching depth increases, while the signal of the I element gradually increases. The Br-rich component formed first on the surface is distributed on the top of the perovskite film, while the I-rich phase formed later is distributed on the bottom. Due to the accelerated crystallization rate, the inhomogeneous halide distribution in the AD-treated film is alleviated to a certain extent, but it is still not negligible. In contrast, the OPD-treated film exhibited an almost homogeneous halide distribution from top to bottom. The same conclusion can be obtained from EDS spectra of the cross-sectional PSCs (Supplementary Fig. 18). The XPS peak at 291 eV shows that π-conjugated OPD molecules exist in the OPD-treated perovskite film28,29 (Supplementary Fig. 19). The signal gradually disappears with the increase of etching depth (Supplementary Fig. 20), which indicates that the OPD molecules are mainly concentrated in the top region of the perovskite film. The OPD molecular wall located on the top film will induce top-down templated crystallization of the perovskite film (Fig. 2a). The OPD molecular wall can also be demonstrated by the anisotropic GIWAXS pattern of OPD-treated perovskite film (Fig. 2m). The π-π interactions signal is observed at 15.3 nm−1, which corresponds to the distance (4.1 Å) between OPD molecules in the stacked structure (Supplementary Fig. 21). The OPD molecular wall structure at the grain boundary will form a strong interaction with the perovskite precursor through the ortho-diamine group on the same side, as confirmed by Fourier transform infrared (FTIR) spectra and XPS spectra (Supplementary Figs. 22–25). This strong anchoring effect determines synchronous halide insertion and templated crystallization. Therefore, the resulting preferred (100) facet and highly vertical crystal orientation will ensure efficient carrier extraction.
Efficient carrier extraction of perovskite films
According to the atomic force microscopy (AFM) images, we can obtain distinguishable grain boundaries of the perovskite films (Supplementary Fig. 26). Low surface roughness is observed in the OPD-treated films, and the smooth surface morphology may be related to the preferred (100) facet and highly vertical orientation. As shown in Fig. 3a–c, height and the contact potential difference (VCPD) line-scan spectra are extracted from the region marked with horizontal lines in AFM and Kelvin probe force microscopy (KPFM) images. Combined with AFM images, we can detect surface electrical information at the perovskite grain interior (GI). Control film exhibits nonuniform surface potential at GIs, resulting in a large VCPD difference (ΔVCPD, GI) of 101 mV. The ΔVCPD, GI (71 mV) of the AD-treated film is reduced, and the OPD-treated film shows the smallest ΔVCPD, GI (48 mV). Moreover, the lowest average surface potential can also be observed in the OPD-treated film compared to the control and AD-treated samples. The reduced and uniform VCPD at GIs may be attributed to the preferred (100) facets, and similar results have been obtained in previous work30. In general, a low surface potential means a low work function, resulting in a Fermi level closer to the conduction band, which will be more conducive to electron extraction31.

The spatial VCPD maps and extracted VCPD line-scan spectra of a control, b AD-treated, and c OPD-treated perovskite films. PL intensity mapping of d control, e AD-treated, and f OPD-treated perovskite films with and without ETL. g TAS spectra of control, AD-treated, and OPD-treated perovskite films. h Electron mobility and trap density of control, AD-treated, and OPD-treated perovskite films obtained from SCLC curves. i Carrier diffusion length for both electron and hole calculated from the optical method.
We perform confocal fluorescence microscopy (TCFM) mapping on perovskite films with an electron transport layer (ETL) to evaluate the electron extraction capability (Fig. 3d–f). As expected, the lowest photoluminescence (PL) signal intensity is evenly distributed in the OPD-treated film compared to the control and AD-treated films. The reduced PL intensity can also be observed in the steady-state PL spectrum of OPD-treated film (Supplementary Fig. 27)32. Similarly, for the perovskite films with hole transport layer (HTL), the OPD-treated sample exhibits the lowest PL intensity compared to control and AD-treated samples (Supplementary Fig. 28). Considering that defect-induced carrier recombination in the perovskite film may also lead to the reduced PL intensity, we also measure the TCFM mapping of the bare perovskite film. The strongest PL intensity and longest PL lifetime are exhibited on the OPD-treated film (Supplementary Fig. 29), indicating the lowest defect-induced carrier recombination. The defect states (tDOS) of perovskite films can be measured by thermal admittance spectroscopy (TAS)33. The density of defect states (tDOS) of the OPD-treated film is nearly an order of magnitude lower than that of the control film (Fig. 3g). Therefore, we can conclude that the reduced PL intensity in the perovskite/ETL and perovskite/HTL samples should be attributed to the efficient charge extraction, while excluding the defect-induced carrier recombination. Efficient and uniform carrier extraction is benefited from the preferred (100) facet and highly vertical orientation.
We further evaluate the crystalline quality and carrier transport behavior of the perovskite films. The space-charge-limited current (SCLC) model with electron-only device is often used to extract trap density (Nt) and carrier mobility (μ) in perovskite films (Supplementary Fig. 30). As displayed in Fig. 3h, OPD-treated films show the lowest Nt of 2.31 × 1015 cm−3, compared to control (1.04 × 1016 cm−3) and AD-treated films (3.02 × 1015 cm−3). This is consistent with the conclusion obtained from the TAS results. Moreover, an increased μ of 1.77 × 10−3 cm2 V−1 S−1 is obtained in the OPD-treated sample, which is tens of times higher than that of the control (7.21 × 10−5 cm2 V−1 S−1) and AD-treated samples (7.48 × 10−4 cm2 V−1 S−1). Low defects and high carrier mobility should be beneficial to prolonging the carrier diffusion length (LD). An approximation of the LD can be estimated by the following equation34:
Where d is the perovskite film thickness, τ2 and τ2,quench is the bimolecular recombination lifetime of the perovskite film with and without a charge transport layer, respectively. The LD-e and LD-h are the diffusion lengths of electrons and holes, which can be calculated by the time-resolved PL (TRPL) decay (Supplementary Fig. 31)35. As expected, the OPD-treated film exhibits enhanced carrier diffusion capabilities for both electrons and holes, specifically, LD-e increased from 389 nm to 648 nm, and LD-h increased from 299 nm to 687 nm (Fig. 3i). The prolonged carrier diffusion length can facilitate the carrier extraction from the perovskite layer to the charge transport layer. Moreover, LD-e close to LD-h also ensure balanced charge extraction for electrons and holes, which is beneficial for achieving high-performance WBG PSCs.
Improved performances and stability of WBG PSCs
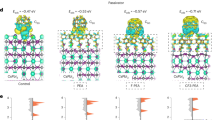
We further analyze the carrier transport characteristics at the device level. According to the Mott-Schottky equation, the built-in potential (Vbi) can be extracted from the capacitance-voltage (C-V) curves of PSCs (Fig. 4a). The OPD-treated device shows a higher Vbi (1.01 V) than the control and AP-treated devices (0.89 V and 0.95 V). The strong built-in electric field will drive the efficient separation of electrons and holes. To evaluate the carrier recombination behavior in the PSCs, we measure the current density-voltage (J-V) curves of PSCs under dark conditions. As shown in Fig. 4b, the OPD-treated device exhibits the lowest reverse saturation current density (J0) (3.30 × 10−8 mA cm−2). A low J0 means that leakage current caused by carrier recombination is effectively suppressed. The results of electrochemical impedance spectroscopy (EIS) also support the same conclusion (Supplementary Fig. 32), and the charge recombination resistance (Rrec) of the device treated with OPD is increased from 811 Ω to 2526 Ω. The ideality factor (n) is usually used to quantitatively measure the carrier recombination of PSCs. It can be obtained from the light intensity-dependent VOC relationship by fitting the slope (Fig. 4c). The n value (1.53) closes to 1 indicates that free-carrier-induced radiative recombination is dominant in the OPD-treated device, and thus defect-induced non-radiative recombination is reduced.

a Mott-Schottky plots, b Dark J-V curves of control, AD-treated, and OPD-treated PSCs. c VOC evolution of the PSCs under different illumination intensities. d EQEEL spectra of PSCs. Inset: EL image of OPD-treated devices. e SCAPS-simulated J-V curves and f experimental J-V curves of PSCs. g PCE comparison of our work and the reported PCE of WBG PSCs (1.65–1.71 eV). h Steady-state PCE of PSCs. i J-V curves of 4-T perovskite/CIGS TSC.
Suppressed non-radiative recombination is generally accompanied by reduced VOC losses in PSCs. When a solar cell is operated as a light-emitting diode, the VOC loss of the device can be calculated using the electroluminescence external quantum efficiency (EQEEL). When the injected current density is approximate to the short-circuit current density (JSC) under 1-sun illumination, the corresponding EQEEL values can be used to evaluate the VOC loss of PSCs (Fig. 4d). The potential VOC of PSCs can be obtained by the previously reported equation as follows:36,
where VOC,rad, k, T, and e are the radiative limit of VOC, Boltzmann’s constant, thermodynamic temperature, and elementary charge. The second term in the equation represents the VOC loss induced by non-radiative recombination. The OPD-treated device achieves an EQEEL of 0.78%, and thus a VOC loss of 0.126 V could be obtained. According to the Shockley-Queisser (S-Q) limit theory37, the perovskite with bandgap of 1.69 eV corresponds to theoretical radiative limit VOC of 1.400 V. Therefore, a potential VOC of 1.274 V should be obtained in the OPD-treated devices.
Solar cell capacitance simulator (SCAPS−1D) is a professional one-dimensional simulation tool for solar cells38. It is often used to simulate and analyze key physical phenomena within solar cells, such as charge transport and carrier recombination. This can guide optimizing device structure design and improving device performance. Based on the preferred (100) facet and highly vertical orientation, the improved perovskite material properties are employed in SCAPS simulations (Fig.4e, Supplementary Table 2). As a result, the OPD-treated PSC achieves a high PCE of 24.60%, with a VOC of 1.275 V. Guided by SCAPS simulation, we fabricate 1.69 eV-bandgap PSCs with an inverted device structure of ITO/NiOx/SAM/Perovskite/ PEAI/C60/BCP/Ag. The optimized OPD-treated PSC with negligible hysteresis exhibits an improved PCE from 21.12% to 24.13% (Supplementary Figs. 33, 34), which is accompanied by a high fill factor (FF) of 83.78% and JSC of 22.72 mA cm−2 (Fig. 4f). The resulting PCE of reaches 82.8% of the Shockley-Queisser (S-Q) limit, which represents one of the highest PCEs among all WBG PSCs (Fig. 4g, Supplementary Table 3). The resulting device exhibits reproducibility (Supplementary Fig. 35), demonstrating the reliability of the synchronous halide insertion strategy. The external quantum efficiency (EQE) spectra of PSC are used to evaluate the efficiency of utilizing light of different wavelengths, which is closely related to the transmittance of ITO glass and the reflectance of perovskite films (Supplementary Figs. 36, 37). The JSC extracted from the EQE spectrum is also close to that obtained from the J-V curve (Supplementary Fig. 38). The bandgap of the perovskite can be also determined to be 1.69 eV according to the slope of the EQE absorption edge39 (Supplementary Fig. 39). It is worth noting that a high VOC of 1.274 V is obtained in the OPD-treated PSC, which is close to the EQEEL prediction and SCAPS simulation results.
We then measure the champion device at the National Measurement and Test Centre for Photovoltaic Industry (Fujian Institute of Metrology and Science). The OPD-treated PSC achieves a certified PCE of 23.42% under reverse scan conditions (Supplementary Fig. 40). Moreover, the resulting device exhibits power output characteristics under MPPT conditions, achieving a steady-state PCE of 23.80% (certified 23.28%) (Fig. 4h, Supplementary Fig. 41). Furthermore, we increase the Br content of the perovskite film to 30 mol%, obtaining a PSC with bandgap of 1.75 eV. The resulting OPD-treated PSC achieves a PCE of 20.45% (Supplementary Fig. 42). This demonstrates that the use of ODP to direct templated crystallization via a molecular wall is generalizable for mixed-halide WBG PSCs. Benefited from the high-quality perovskite film and the carrier transport properties, the OPD-treated ST WBG PSC (1.69 eV) also achieves a PCE of 22.28%. Integrated with a narrow-bandgap CIGS solar cell, the resulting 4T Perovskite/CIGS TSC achieves a PCE of 29.34% (Fig. 4i, Supplementary Table 4).
The crystal structure stability of perovskite films is crucial to achieving robust operational stability of photovoltaics40. We perform accelerated aging experiments on perovskite films at 65 °C and 45% relative humidity (RH), investigating the crystallographic evolution by in-situ GIWAXS characterization. As shown in Fig. 5a, a recognizable lead iodide (PbI2) signal quickly appears in the control film, indicating the decomposition of the perovskite film. AD treatment can suppress the decomposition of the perovskite film to a certain extent, but the PbI2 signal still appears in the film after 25 h. In contrast, the OPD-treated film exhibits a constant perovskite phase with negligible decomposition even after 100 h. The decomposition of the perovskite film is also reflected in the crystal orientation evolution as displayed in Fig. 5b. The crystal orientation of the control sample and AD-treated films is destroyed at 25 h and 60 h, respectively. The OPD-treated film still maintains well-defined orientation on the preferred (100) facet.

a In-situ GIWAXS patterns of control, AD-treated, and OPD-treated perovskite film at 65 °C and 45% RH. b Extracted crystal orientation evolution of (100) facet. c In-situ PL spectra of perovskite films under continuous 1-sun illumination. d EL spectra of perovskite films at various bias voltages. e The PCE evolution of encapsulated devices with the ISOS-L-1 testing protocol.
We evaluate the light stability of perovskite films under continuous 1-sun illumination by in-situ PL measurements (Fig. 5c). Rapidly decreasing PL signals occur in the control and AD-treated films, while the OPD-treated film exhibits a constant PL intensity after 400 h. We also monitor the stability of the perovskite films under electric fields by EL spectra at various bias conditions (Fig. 5d). Control and AD-treated films exhibit a split EL peak at a bias of ~3.5 V, implying that the ion migration-induced phase separation is amplified under a strong electric field. In contrast, the OPD-treated film still maintains a stable perovskite crystal structure, and its EL peak remains constant even under a bias of 5 V. Based on the preferred facet and the vertical orientation, the stability of the perovskite films and the resulting PSCs is significantly improved. The OPD-treated PSC exhibits improved long-term stability under continuous 1-sun illumination conditions (Fig. 5e). According to the ISOS-L-1 test standard, we monitor the PCE evolution of the encapsulated devices under MPPT conditions41. The OPD-treated device still maintains 90% of the initial PCE (T90) for more than 800 h, while the control and AD-treated devices quickly decay to 40% and 66% of their initial PCE in a very short time. Meanwhile, we monitor the PCE evolution of the encapsulated PSCs under the conditions of damp heat at 85 °C and 45% RH according to ISOS-D-2 protocol (Supplementary Fig. 43). The OPD-treated device still maintains 90% of its initial PCE after 800 h, while the control device and the AD-treated device only maintains 50% and 75% of their initial PCE. We further strictly implement the stability test conditions of PSCs at 85 °C and 85% RH according to ISOS-D-3 protocol. The OPD-treated device still maintains 88% of its initial PCE after 600 h, and the improved device stability is attributed to the enhanced perovskite crystal quality due to the OPD treatment.
Discussion
In summary, we demonstrate efficient and stable mixed-halide WBG perovskite photovoltaics by applying a halide insertion management strategy. Based on a comprehensive analysis of in-situ GIWAXS monitoring and DFT calculations, we have demonstrated that asynchronous halide insertion is accompanied by random crystal facets and orientations, which are detrimental to carrier extraction. Guided by DFT calculations, we design functionalized amine cations to construct a π-conjugated molecular wall structure. The ortho-diamine groups exposed on the same side of the molecular wall can anchor halides uniformly, enabling their synchronous halide insertion. The OPD-treated perovskite film exhibits a preferred (100) facet and a vertical crystal orientation. Benefited from the improved carrier extraction characteristics, the OPD-treated WBG PSC (1.69 eV) achieves a PCE of 24.13% (certified 23.43%), representing one of the highest PCEs among WBG PSCs. The encapsulated WBG PSC still maintains more than 90% of the initial PCE after MPPT for 800 h under the ISOS-L-1 testing protocol. This work provides an alternative strategy for the crystallization regulation of mixed-halide WBG perovskite, promoting the development of efficient WBG PSCs and TSCs.
Methods
Materials
Indium tin oxide (ITO) glass, [(3,6-dimethoxy-9H-carbazol-9-yl)ethyl] phosphonic acid (MeO-2PACz, 98.0%), [4-(3,6-dimethyl-9H-carbazol-9-yl)butyl] phosphonic acid (Me-4PACz, 99.0%), PbI2 (99.9%), lead bromide (PbBr2, 99.9%), formamidinium iodide (FAI, 99.5%), methylammonium bromide (MABr, 99.5%), cesium iodide (CsI, 99.99%), 2-phenylethylamine hydroiodide (PEAI, 99.5%), fullerene-C60 (C60, 99%), tin oxide (SnO2, 99.5%), aniline hydrochloride (ADCl, 99.4%), o-phenylenediamine hydrochloride (OPDCl, 99%), m-phenylenediamine hydrochloride (MPDCl, 99%), and p-phenylenediamine hydrochloride (PPDCl, 99%) were purchased from Advanced Election Technology Co. Bathocuproine (BCP, 99%) was obtained from Xi’an Yuri Solar Co. N, N-dimethylformamide (DMF, 99.9%), dimethyl sulfoxide anhydrous (DMSO, 99.9%), N-methyl-2-pyrrolidone (NMP, 99.9%), ethyl alcohol (EtOH, 99.5%), and chlorobenzene (CB, 99.5%) were supplied by Sigma-Aldrich. Nickel oxide (NiOx, 99.999%) was from Xi ‘an E-light New Material Co. All materials and solvents used in this work have not been purified again and are used directly after purchasing from the manufacturer.
Fabrication of single-junction WBG PSCs
The ITO glass was first cleaned and then treated using a plasma machine for 90 s. NiOx solution was deposited on the ITO substrate by spin-coating at 3000 rpm for 30 s, and the NiOx film was annealed at 100 °C for 15 min. Next, a mixed SAM materials of MeO-2PACz and Me-4PACz were dissolved in EtOH. SAM layer was deposited by spin-coating at 3000 rpm for 30 s, and then annealed at 100 °C for 10 min. For FA0.8MA0.1Cs0.1Pb(I0.8Br0.2)3 and FA0.8MA0.1Cs0.1Pb(I0.7Br0.3)3 (1.68 eV and 1.75 eV) perovskite precursor solution, OPD, MABr, CsI, PbBr2, PbI2, and FAI were dissolved by DMF: DMSO (4:1) or DMF: NMP (4:1). The precursor solution was spin-coated at 5000 rpm for 40 s, and CB (100 ul) was added at 6 s. Perovskite film was then annealed in a nitrogen glove box at 100 °C for 50 min. After cooling, the PEAI (3 mg ml−1, dissolved in IPA) solution was dynamically spin-coated on the perovskite film at 3000 rpm for 30 s, and then annealed at 100 °C for 10 min. Finally, C60 (30 nm), BCP (8 nm), and Ag (100 nm) were deposited by thermal evaporation.
Fabrication of ST WBG PSCS
Preparation of ITO/NiOx/SAM/Perovskite/PEAI/C60 films as shown above. Then, SnO2 (20 nm) was deposited by atomic layer deposition (ALD), and IZO (100 nm) was subsequently deposited by sputtering to form the transparent electrode.
Fabrication of CIGS solar cells
The preparation of CIGS solar cells adopts the following standard process. Firstly, a molybdenum layer (1 μm) was deposited on soda-lime glass by sputtering as the back electrode. Subsequently, a three-stage co-evaporation process was adopted to deposit copper, indium, gallium, and selenium, forming a CIGS light-absorbing layer (2.3 μm). Subsequently, a CdS buffer layer (50 nm) was deposited by chemical bath deposition, while an i-ZnO layer (50 nm) and an Al-doped ZnO window layer (300 nm) were deposited by radio frequency sputtering. To enhance electron collection, a Ni/Al/Ni metal mesh was deposited by electron beam evaporation.
Device characterization
J-V curves characterization was carried out under standard AM 1.5 G spectral (100 mW cm-²) illumination, using a calibrated solar simulator and a standard silicon reference cell to ensure light intensity accuracy. Tests were performed in forward (−0.1 V to 1.3 V) and reverse (1.3 V to −0.1 V) voltage scanning modes with the scan rate controlled at 100 mV s−1. The devices were connected with four probes to eliminate the effect of wire resistance. A shading mask (0.0867 cm−2) was used to ensure that the effective light area. PSCs were encapsulated with cover glass and butyl rubber for MPPT stability testing. EQE spectra were obtained by using a QE system (Enli Tech).
Other characterizations
GIWAXS patterns of perovskites were measured at BL 17B1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF). Incident-angle-dependent measurements were achieved by X-ray with a wavelength of 1.238 Å. GIWAXS data were collected every 2 s for in-situ GIWAXS patterns. TCFM images were acquired using a customized scanning confocal microscope equipped with TCSPC (PicoHarp 300) functionality. The microscope was based on a Nikon TE-2000 inverted microscope and is fitted with an infinity-corrected ×50 dry objective lens (Nikon L Plan, NA 0.7, CC 0-1.2). Excitation was performed using a 470 nm pulsed diode laser (PDL-800 LDH-P-C-470B, pulse width 350 ps), with a repetition rate of 1 MHz for time-resolved fluorescence spectroscopy measurements and 40 MHz for fluorescence image acquisition.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data supporting the findings of this study are available within this article and its Supplementary Information. Any additional information can be obtained from corresponding authors upon request. Source data are provided with this paper.
References
Chen, Y. et al. Nuclei engineering for even halide distribution in stable perovskite/silicon tandem solar cells. Science 385, 554–560 (2024).
Brinkmann, K. O. et al. Perovskite-organic tandem solar cells with indium oxide interconnect. Nature 604, 280–286 (2022).
Luo, C. et al. Perovskite tandems: the next big leap in photovoltaic technology. Adv. Mater. e08331 (2025).
Wang, S. et al. Suppressed recombination for monolithic inorganic perovskite/ silicon tandem solar cells with an approximate efficiency of 23%. eScience 2, 339–346 (2022).
Fu, P. et al. Synergistic cooperation between photovoltaic and thermoelectric effects in solar cells. Energy Environ. Sci. 18, 7082–7088 (2025).
Zhang, Y. et al. Nonalloyed α-phase formamidinium lead triiodide solar cells through iodine intercalation. Science 387, 284–290 (2025).
Wang, Z. et al. Regulation of wide bandgap perovskite by rubidium thiocyanate for efficient silicon/perovskite tandem solar cells. Adv. Mater. 36, 2407681 (2024).
Li, Z. et al. Nanocrystal-nucleus template strategy for efficient wide-bandgap perovskite solar cells with enhanced homogeneity and energy-level alignment. Adv. Mater. 37, 2509202 (2025).
Song, Z. et al. Optimization of crystallization dynamics in wide-bandgap bromine-iodine perovskite films for high-performance perovskite-organic tandem solar cells. Energy Environ. Sci. 18, 4883–4892 (2025).
Zheng, J. et al. Selective crystallization delay in wide-bandgap perovskites enables initial homogeneous phase for square centimeter perovskite/organic tandem solar cells. Adv. Mater. 37, e10437 (2025).
Li, Y. et al. Synchronous phase transformation for efficient wide-bandgap perovskite photovoltaics. Adv. Mater. 37, e05694 (2025).
Muscarella, L. et al. The influence of strain on phase stability in mixed-halide perovskites. Joule 6, 2016–2031 (2022).
Wu, Z. et al. Enhancing photovoltaically preferred orientation in wide-bandgap perovskite for efficient all-perovskite tandem solar cells. Adv. Mater. 37, 2412943 (2025).
Wu, Y. et al. Tailoring tin-based perovskite crystallization via large cations and pseudo-halide anions for high mobility and high stable transistors. Sci. Adv. 11, eadv4138 (2025).
Bi, H. et al. Selective contact self-assembled molecules for high-performance perovskite solar cells. eScience 5, 100329 (2024).
Xiang, W. et al. Intermediate phase engineering of halide perovskites for photovoltaics. Joule 6, 315–339 (2022).
Liu, Z. et al. All-perovskite tandem solar cells achieving >29% efficiency with improved (100) orientation in wide-bandgap perovskites. Nat. Mater. 24, 252–259 (2025).
Jiao, B. et al. Realizing stable perovskite solar cells with efficiency exceeding 25.6% through crystallization kinetics and spatial orientation regulation. Adv. Mater. 36, 2313673 (2024).
Shi, P. et al. Oriented nucleation in formamidinium perovskite for photovoltaics. Nature 620, 323–327 (2023).
Yan, N. et al. Ligand-anchoring-induced oriented crystal growth for high-efficiency lead-tin perovskite solar cells. Adv. Funct. Mater. 32, 2201384 (2022).
Yang, Y. et al. Synchrotron radiation-based in situ GIWAXS for metal halide perovskite solution spin-coating fabrication. Adv. Mater. 11, 2403778 (2024).
Tan, W. L. & McNeill, C. R. X-ray diffraction of photovoltaic perovskites: Principles and applications. Appl. Phys. Rev. 9, 021310 (2022).
Ma, C., Michael, G. & Park, N. G. Facet engineering for stable, efficient perovskite solar cells. ACS Energy Lett. 7, 3120–3128 (2022).
Wang, J. et al. Exposing binding-favourable facets of perovskites for tandem solar cells. Energy Environ. Sci. 15, 7680–7694 (2025).
Zhao, T. et al. π-conjugation-induced in situ nanoscale ordering of Spiro-OMe TAD boosts the efficiency and stability of perovskite solar cells. J. Am. Chem. Soc. 146, 30893–30900 (2024).
Wang, R. et al. Efficient wide-bandgap perovskite photovoltaics with homogeneous halogen-phase distribution. Nat. Commun. 15, 8899 (2024).
Li, X. et al. Strain regulation of mixed-halide perovskites enables high-performance wide-bandgap photovoltaics. Adv. Mater. 36, 2401103 (2024).
Enkvist, C., Lunell, S., Sjögren, B., Brühwiler, P. A. & Svensson, S. The C1s shakeup spectra of buckminsterfullerene, acenaphthylene, and naphthalene, studied by high resolution X-ray photoelectron spectroscopy and quantum mechanical calculations. J. Chem. Phys. 103, 6333–6342 (1995).
Onoe, J., Nakao, A. & Takeuchi, K. XPS study of a photopolymerized C60 film. Phys. Rev. B 55, 10051 (1997).
Ma, C. et al. Photovoltaically top-performing perovskite crystal facets. Joule 6, 2626–2643 (2022).
Li, X. et al. Modulating buried passivation and aligned interface toward high-performance perovskite modules. Adv. Mater. 35, 2506718 (2025).
Kirchartz, T., Jose, A. M., Stolterfoht, M. & Unold, T. Photoluminescence-based characterization of halide perovskites for photovoltaics. Adv. Energy Mater. 10, 1904134 (2020).
Wu, W. et al. Molecular doping enabled scalable blading of efficient hole-transport-layer-free perovskite solar cells. Nat. Commun. 9, 1625 (2018).
Stranks, S. D. et al. Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 342, 341–344 (2013).
Ross, R. T. Some thermodynamics of photochemical systems. J. Chem. Phys. 46, 4590–4593 (1967).
Rau, U. Reciprocity relation between photovoltaic quantum efficiency and electroluminescent emission of solar cells. Phys. Rev. B 76, 085303 (2007).
Miller, O. D., Yablonovitch, E. & Kurtz, S. R. Strong internal and external luminescence as solar cells approach the Shockley-Queisser limit. IEEE J. Photovolt. 2, 303–311 (2012).
He, Y. et al. Unveiling recombination in top cells: SCAPS-1D simulations for high-efficiency bifacial planar perovskite/silicon tandem solar cells. Sol. Energy 28, 112921 (2024).
Almora, O. et al. Quantifying the absorption onset in the quantum efficiency of emerging photovoltaic devices. Adv. Energy Mater. 11, 2100022 (2021).
Hee, J. K., Gill, S. H. & Hyun, S. J. Managing the lifecycle of perovskite solar cells: Addressing stability and environmental concerns from utilization to end-of-life. eScience 4, 100243 (2022).
Khenkin, M. V. et al. Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures. Nat. Energy 5, 35–49 (2020).
Acknowledgements
We acknowledge the National Natural Science Foundation of China (No. 52403373, T.H.), the Natural Science Foundation of Hebei Province (No. E2024201002, T.H.), the Science and Technology Project of Hebei Education Department (No. BJ2025025, T.H.), and the Baoding Science and Technology Plan Project (No. 2472P001, T.H.). The authors thank the staff at BL17B1 beamline of the National Facility for Protein Science in Shanghai (NFPS), Shanghai Advanced Research Institute, CAS, for providing technical support in X-ray diffraction data collection and analysis.
Author information
Authors and Affiliations
Contributions
Z.S., N.M., and Y.T. contributed equally to this work. T.H. conceived the idea and supervised the work. Z.S., N.M. and Y.T. fabricated devices and analyzed the data. Z.S. and J.L. performed a DFT simulation. Z.S., H.Y. and A.M. guided the GIWAXS and XRD measurements. Z.S. and Y.T. co-wrote the paper. C.S., Z.L., S.Y. and T.H. revised the paper. All authors read and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rui Wang, Yi Hou, Xi Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, Z., Meng, N., Tian, Y. et al. Halide insertion regulation for efficient and stable wide-bandgap perovskite photovoltaics. Nat Commun 17, 288 (2026). https://doi.org/10.1038/s41467-025-66970-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66970-8
