
Abstract
Enhancing the hydrogen embrittlement (HE) resistance of alloys caters to the urgent needs of engineering safety and long-distance hydrogen transportation. Highly dense precipitates in the alloys act as H traps, however, some of them cannot strongly trap H thus failing to prevent its accumulation at the critical regions. Experimentally, it is challenging to expeditiously identify and generate phases causing strengthening and acting as strong H traps. Here, we demonstrate a computation-based design strategy to generate precipitates strongly trapping H. Based on the quantum machine learning Al-Sc-Cu potential, the optimal processing parameters of strong H trapping phases are determined, even though they are metastable in nature. Elemental mapping in electron microscope and atom probe tomography confirms the presence of Cu in Al3Sc and its strong interaction with H. Hence, we envisage the proposed strategy will accelerate the design of HE-resistant microstructures of various technologically relevant materials via identification of desirable phases.
Similar content being viewed by others


Introduction
Trapping hydrogen (H) that widely exists in metals and discerning its effects have been extensively reported for almost a century1. The hydrogen embrittlement (HE) is common in high-strength steel2,3, Al alloys4, Ti alloys5, and Ni alloys6,7, where the HE triggering concentration can be as low as a few ppm. A typical failure mechanism is H-enhanced decohesion (HEDE) in which fracture, like cleavage cracking, is mainly attributed to H aggregation at crack tips, thereby lowering the fracture energy by weakening atomic cohesion near the crack front8. In addition, the accumulated H suppresses the emission of dislocations at the crack tips, and weakens the ability of the material to blunt cracks9. Another HE mechanism is H-enhanced local plasticity (HELP)10, H decreases the critical stress for screw dislocation initiation in α-Fe by ~ 27–43%11. These two mechanisms are not mutually exclusive, but are strongly correlated with the underlying microstructure of the material12.
The distribution of H in the microstructure encompasses the lattice, dislocations, vacancies, interfacial defects, and secondary phases13. For Al alloys, the solubility of H in the lattice is lowest among all trapping sites14 with the H diffusion coefficient for lattice having frequency factor of D0 = 1.75 ± 0.15 × 10−8 m2 s–1. Using atom probe tomography (APT), it was observed that even in a non-H-charged environment, the H concentration at the Al grain boundaries (GBs) was ~ 12 at.% under stress-corrosion deformation, while the corresponding concentration at the dislocation was 23 at.%15. With the occurrence of dislocation slip and partial annihilation at the GBs, the H concentration increased at the GBs owing to H carried by the dislocations16. The computational based studies revealed that the aggregation of H at the GBs significantly lowered their cohesive energy (~ 86% reduction in 7xxx Al alloys with MgZn2 phase), which accelerated intergranular cracking17. Unless there is a pronounced density of dislocation (> 1011 cm−2), the effect of dislocations on the effective diffusivity of H in Al was found to be weak14. Vacancies (e.g., C or N) in phases (e.g., carbides, nitrides, carbonitrides in steel, etc) have been found to exhibit strong H trapping ability depending upon the composition of these phases18.
Identifying phases with superior H trapping ability is an important topic that is being actively discussed19,20. High-resolution X-ray tomography confirmed that the pre-existing intermetallic particles, precipitates, and vacancies strongly affect the H distribution in the microstructure21. Several studies were performed to probe the H trapping ability of various phases with the intention of designing HE-resistant Al alloys22,23. With the aid of APT and isotope deuterium (D) imaging (Fig. 1a), it was established that the Al3Zr precipitate can strongly trap H24,25. Furthermore, the T-Al2Mg3Zn3 phase, whose structure differs from η-MgZn2, was reported to exhibit high H trapping ability25,26. Therefore, it can be deduced from the above-mentioned work that small structural or compositional changes in the precipitates can greatly affect their H trapping ability. Examination of the H-trapping phases in steel yields that the available range of precipitates remains limited, with carbides still being the predominant choice as H traps (Fig. 1a, green circle)3,19,23,25,26,27,28,29. The experimental determination of the potential H trapping phases in Al, however, remains difficult owing to the complex underlying microstructure of Al alloys.

a Currently identified H-trapping phases in steel and Al alloys. The red stars highlight phases (formed under stringent conditions) exhibiting high H trapping ability. b Combining ab-initio calculations, machine learning interatomic potential, molecular dynamics, and experiments to identify H trapping phases.
In this regard, atomistic simulations can be performed, which provide insights into H interactions with individual microstructural features. For instance, the H trapping ability of various phases that are thermodynamically stable has been probed using density-functional theory (DFT), and the Al11Mn3Zn2 phase is found to have good H trapping ability23. Apart from the stable phases, a multitude of metastable precipitates are simultaneously generated in Al alloys, i.e., GP zone – θ″– θ′ – θ-Al2Cu (in 2xxx series), or GP zone – η′ – η-MgZn2 (in 7xxx series, at least 15 types of η-phases)30. The precipitation of some metastable phases, potentially serving as strong H-trapping sites, poses challenges in accurately simulating and subsequently controlling their formation to maximize their volume fraction or number density, primarily due to the narrow range of conditions under which they form. Specifically, Cu doping resulted in significant optimization of Al3Sc phases, which showed poor performance with regard to H trapping in the absence of Cu20,31. However, the volume fraction of this Cu-doped Al3Sc precipitate might be low in the matrix, and it might be metastable in nature. For simulating the precipitation dynamics, an embedded atom model-type Al-Sc potential has been used to obtain Al/Al3Sc phase boundary energy32. The nucleation of the Al3Sc precipitates has also been investigated using the kinetic Monte Carlo (MC) method, which requires the free energies of the interface and for nucleation energies along with the diffusion coefficients33. Given the lack of the Cu element, these methods cannot be directly used for studying the Al-Sc-Cu system. Therefore, whether the metastable phases can strongly trap H, and how to fabricate Al alloys with more of these metastable phases, are critical conundrums.
Here, we address the above questions and explore the vast HE-resistant phase space by proposing a robust strategy (Fig. 1b) combining DFT calculations and molecular dynamics (MD) simulations to probe the H trapping ability of Al3-xCuxSc precipitates (x ranging from 0 to 3), and recommend appropriate heat treatment to generate metastable precipitates that can strongly trap H. At the first stage, the formation energy (Ef), H dissolved Gibbs free energy change at 0 K (\({\Delta G}_{{\rm{soln}}}^{0}\), ignoring entropy), and the strength of the H bond with Al/Cu/Sc in Al3-xCuxSc precipitates were calculated by DFT. To overcome the shortcoming of DFT calculations restricted to 0 K and to simulate heat treatment, an Al-Sc-Cu interatomic potential was trained by active learning based on the DFT dataset. After verifying the applicability of the potential, the precipitation of the Al3-xCuxSc was simulated using MC/MD. The optimal heat treatment process to obtain the metastable phase was determined statistically based on the above-mentioned approach. Finally, experiments including H-charging slow strain rate testing, elemental energy-dispersive spectroscopy (EDS) using X-ray spectroscopy in scanning transmission electron microscopy (STEM), APT, and H thermal desorption mass-spectroscopy (TDS) were performed within this study to validate the proposed strategy of having a metastable phase in the microstructure to significantly improve the HE resistance of Al alloys.
Results
Analysis of H trapping capability
The Ef in Fig. 2a shows that the Al3Sc with space group \({{\rm{Pm}}}\bar{3}{{\rm{m}}}\) is most thermodynamically stable (Ef = − 0.48 eV atom−1). When the Al atoms in this phase are substituted by Cu atoms, the Ef of the corresponding phases are observed to increase. In the extreme case when all the Al atoms are replaced by Cu forming Cu3Sc, the corresponding Ef is found to be ~ − 0.07 eV atom-1. Thus, the Cu-doped precipitates are deemed as metastable phases as compared to the pure Al3Sc, and hence the precipitates with higher Cu content are less likely to form. Another typical Cu-rich precipitate (Ω phase, Fmmm, the Ef is − 0.16 eV atom−1) has been reported to be interstitially doped with Sc34 (V phase, Cu/Sc ≈ 4:1), which can decrease its Ef to − 0.27 eV atom–1. It is illustrated that the Al-Sc-Cu cluster with Cu/Sc ratio > 2.3:1 (Cu substitution ratio in Al3Sc greater than ~ 0.76) is more inclined to transform into V phase. Therefore, when the Cu content is strictly controlled, the alloy has the potential to form \({{\rm{Pm}}}\bar{3}{{\rm{m}}}\) structure containing Cu.

a Formation energies Ef of Al3-xCuxSc precipitates (\({{\rm{Pm}}}\bar{3}{{\rm{m}}}\)) where Al is substituted by Cu. The red symbols indicate the Ef of the most stable phases with specific Cu content, while the gray symbols are for phases with Cu at different positions whose corresponding Ef are larger than the ones indicated by the red symbols. The green dashed oval is to indicate the metastable region where it is observed that more or less Cu increases the Ef of precipitates. The \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to the dissolved H trapped at the (b) vacancies and (c) interstices of phases at 0 K. The bottom of the blue bar indicates \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapped at Al-vacancy while the top of the blue bar indicates \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapped at the Sc-vacancy. The bottom of the pink bar indicates \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapped at the Cu-vacancy. The orange bar in (c) indicates there are no thermodynamically favorable sites for H trapping by Al3Sc. d Charge density difference of H with respect to neighboring atoms; The blue region indicates that electrons are concentrated at this region, while the red one indicates electron depletion. For a clear representation of the charge transfer, the maximum of the legend for all the systems is set to 0.0026, and the minimum of the legend changes with the system, e.g., the minimum charge density difference of Al3Sc is − 0.004. e Bond strength and bond length of H with Sc, Al, and Cu. f The \({\Delta G}_{{\rm{intf}}}^{0}\) and (g) the energy barrier of H diffusion across the {011} interface. The yellow region in (f) highlights the fact that the lowest values of \({\Delta G}_{{\rm{intf}}}^{0}\) correspond to H trapped by the bulk of the precipitate. All error bars are standard deviations.
Next, the \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapped at the vacancies and interstices of different precipitates are computed and compared (Supplementary Fig. 1). It can be seen from Fig. 2b, c that the \({\Delta G}_{{\rm{soln}}}^{0}\) of defective Al3Sc is 0.30 eV H−1 (Al-vacancy) and 0.01 eV H−1 (interstices), whereas the \({\Delta G}_{{\rm{soln}}}^{0}\) of defective Al2CuSc (with one Cu atom substituting an Al atom) decreases to − 0.62 eV H−1, and their interstices exhibit − 0.29 eV H−1. The trapping of H atoms is stronger when there are more Cu doped into the phase, especially for the interstitial sites. The charge density difference results (Fig. 2d) reveal that more charge density concentrates on the Cu atoms (gain in electrons) when H occupies the tetrahedral interstitial site (T-site) with lower corresponding \({\Delta G}_{{\rm{soln}}}^{0}\). However, when all Al atoms in the precipitates are replaced by Cu, the H atom occupies the octahedral interstice site (O-site), and its \({\Delta G}_{{\rm{soln}}}^{0}\) significantly decreases (− 4.27 eV H−1). To demonstrate the bond strength of H with Al, Cu, and Sc atoms, the negative integrated crystal orbital Hamilton population (-ICOHP), which quantitatively determines the strength of a bond, is calculated (Fig. 2e). For the H-Cu bond, the corresponding -ICOHP value is found to be 0.088 eV, and its length is the lowest (1.73 Å). To briefly summarize, the H trapping ability of Al3Sc could be significantly enhanced with Cu doping of these precipitates.
In addition to the lattice diffusion, the diffusion of H from the Al matrix to the bulk of precipitates happens through their interfaces, thereby making it important to be investigated (Fig. 2f, g). Although H trapping in the bulk of Al3Sc is thermodynamically unfavorable, their interface (0\(\bar{1}\)1) is observed to trap H (corresponding \({\Delta G}_{{\rm{intf}}}^{0}\) reaches − 0.230 eV H−1) and that of (100) is − 0.360 eV H−1. The \({\Delta G}_{{\rm{soln}}}^{0}\) of 1/6 < 112> Shockley dislocation is − 0.310 eV H−1, indicating that the H trapping ability of Al3Sc (100) interface is slightly stronger (~ 0.05 eV H−1 lower) than the H trapping ability of the dislocation. Further, the chemical potentials of H inside the Cu-doped precipitates are significantly lower (ranging from − 0.147 ~ − 6.791 eV H−1) than that at the dislocation. This implies that the H carried by the dislocation35 would be strongly trapped by the interface when the dislocation interacts with the precipitates. From the diffusion analysis, it is observed that H needs to overcome an energy barrier of 0.75 eV to diffuse into the Al3Sc precipitate. The corresponding energy barrier for H diffusion into the Cu-doped precipitate is found to be lesser i.e., 0.31 eV. Therefore, considering the diffusion barriers, the Cu-doping of the Al3Sc precipitate is found to facilitate the diffusion of H into the bulk of the precipitates.
Development of interatomic potential to simulate phase transition
The MD and MC methods are employed to simulate the precipitation of Al3Sc at different temperatures. Given the lack of Al-Sc-Cu interatomic potential, we employed 5,166 DFT-calculated configurations and D-optimality criterion-based active learning (AL, Supplementary Fig. 2) to train a moment tensor-based machine learning interatomic potential (MLIP). As shown in Fig. 3a, the mean of absolute value error (MAE) and root mean squared error (RMSE) decrease with the AL iteration. The reliability of the developed potential is established by comparing the interfacial energies γintf obtained using DFT and MLIP (Fig. 3b). The interatomic potentials trained with a static dataset (without new configurations supplemented) cannot precisely predict the interface energy, and it is found to not being able to describe well the force (overestimated) when atoms are extremely close. Therefore, the usage of AL to include the temperature effects is mandatory. However, when the AL is further continued, it is found that the error in energy difference increases to 19% which shows that excessive high-temperature AL (600–1500 K) introduces anomalous structures which are not conducive in predicting energies.

a AL generates new configurations corresponding to different temperatures and deformations, which further lowers the training error in energy. The new configurations transitorily increase the RMSE to 4.46% but the AL lowers the RMSE quickly. b The interfacial energy (γintf) differences obtained using DFT and the ones predicted by potentials trained using different datasets to verify their generalization ability. The error is the standard deviation. The raw-dataset (Raw) comprises of the ideal configurations, while the deform-dataset (Deform) comprises of the raw configurations with compressive or tensile strain of 5%–30%. The defect-dataset (Defect) comprises of configurations with vacancies, and the all-dataset (All) includes all the above-mentioned datasets. Finally, the 1st AL and 2nd AL are trained on the all-dataset with first-time AL and second-time AL, respectively. The supplemented configurations corresponding to high temperatures and deformation obtained with 1st AL not only lower the energy RMSE (7.20) compared with the All potential (7.40), they also improve the γintf prediction accuracy. The γintf error bar is calculated from 150 interfaces. c The comparison of energy calculated by DFT and MLIP, and the accuracy of the selected potential with 1st AL. The accuracy of 80% configurations (red dashed line) is lower than 8.22 meV atom−1, and 90% configuration (blue dashed line) is lower than 11.70 meV atom−1.
Since the purpose of the majority of the simulations is to study phase precipitation (which has mostly to do with the formation energy and interface energy of phases), the first-time AL (1st AL, the red dashed oval symbol in Fig. 3b) is selected for the study as it exhibits higher energy accuracy than the potential obtained using second-time AL (2nd AL). As shown in Fig. 3c, the 1st AL potential achieves a quantum-accurate force field, enabling reliable modeling of phase transition in Al-Sc-Cu systems with accuracies of 4.8 and 7.2 meV atom−1 in the energy for training and validation, respectively. In the case of force, the accuracy indexes for training and validation are 40.2 and 87.1 meV Å−1, respectively. It exhibits better reliability for high-temperature simulations than the static potential (Supplementary Table 1).
Using the developed Al-Sc-Cu potential, three 12 nm-sized systems containing 1.2 at% Cu and 0.2 at% Sc are employed to study the phase precipitation at different temperatures. The initial simulation is to simulate solution treatment at temperatures of 470, 485, 500, 515, and 530 °C. During the solution treatment, the ratio of the Cu-containing cluster with respect to all Al3-xCuxSc cluster (RCu_cluster) is shown in Fig. 4a. Initially, this ratio at different temperatures is higher than 30% which suddenly drops to ~ 23% at 0.25 ps and beyond. This is attributed to the utilization of the MC method, which cannot represent the real-time well.

a The fluctuation of RCu_cluster with time at different temperatures. b The number of clusters at different temperatures of solution treatment. The error bar is calculated from the three systems under consideration. c The RCu_cluster at different solution treatment temperatures. d Microstructure showing several precipitates containing Cu atoms as indicated by the dashed circles, and Al3-xCuxSc precipitates nucleation and growth at aging treatment. The specific morphology is the result at 120 °C-aging temperature. The extraction criterion of the precipitate is the 12 nearest neighbor atoms of the FCC structure. The black dashed circle (in the figure showing nucleation and growth) highlights the largest phase, and the orange one is to highlight the phase containing Cu atoms. e Total energy converges within the simulation time. The entire simulation can be divided into aging treatment and peak-aging treatment according to the size of the phases. Fitted using an exponential association function. f Phase transition energy at different aging temperatures. g The RCu_ppt at different aging temperatures. The standard error bars are calculated from three systems subjected to different solution treatment temperatures.
It is observed from the simulated microstructure that the Sc atoms are dispersed within the matrix, and the RCu_cluster fluctuates. However, there are still several Al3Sc precipitates that contain Cu atoms (Fig. 4d). Statistically, the number of Al-Cu-Sc clusters increases with the increase in solution treatment temperature, except for the case of 500 °C (Fig. 4b), the number of Al-Cu-Sc clusters forming at 500 °C is close to the number obtained at 470 °C (approx. 155). Upon further analysis, it is found that the 470 °C-solution treatment shows the lowest RCu_cluster (only 22.50%), while the corresponding value is 23.08% for solution treatment at 500 °C (Fig. 4c). As discussed before, the Al3-xCuxSc exhibit high H trapping ability, which gets stronger with an increase in Cu content. The Cu proportion in the Cu-containing precipitates (ξpptCu) is illustrated in Supplementary Fig. 3a. The result shows that employing a solution treatment temperature of 500 °C is not the optimal strategy since the ξpptCu for this case (3.37 at%) is lower than that of 485 °C (3.59 at%).
As a consequence of the elevated temperatures employed, the atoms consistently exhibit a state characterized by erratic and disordered motion during solution treatment. Aging treatments at temperatures lower than solution treatment temperatures (i.e., 120, 140, and 160 °C) provide an environment conducive to the formation of Al3-xCuxSc precipitates. The total energy of all systems experiences an initial decrease and then gets stabilized. The initial decrease is strongly dependent on the initial solution treatment temperature (Supplementary Fig. 4). For instance, the total energy illustrated in Fig. 4e is for the system subjected to 500 °C-solution treatment, whose initial drop in the total energy is observed to be − 110.13 eV. Combined with the increment in the number of atoms present in the largest phase (Supplementary Fig. 3b), we attribute this energy reduction to the latent heat of phase transition.
Further analysis of the latent heat energy at different aging temperatures (Fig. 4f) reveals that the lower aging temperature (120 °C) can release more latent heat, which is about 14.97 ± 4.85% larger than the higher aging temperature (160 °C). This in turn implies that the systems with a higher number density of small-size Al3-xCuxSc precipitates are more energetically stable as compared to systems with large-size Al3-xCuxSc precipitates (Supplementary Fig. 3c). The higher aging temperatures destabilize the precipitates and provide sufficient energy for Sc atoms to fully diffuse, leading to precipitation of large-size precipitates, which cannot trap H effectively.
In contrast to the decreasing trend of RCu_cluster when subjected to solution treatment (Fig. 4c), the ratio of the Cu-containing precipitates with respect to all Al3-xCuxSc precipitates (RCu_ppt) with aging treatment (Fig. 4g) gradually increases, and the final value is stabilized between 52.66% to 63.81%. This high value can be attributed to the decrease in the number of precipitates and significant atomic motion at high temperatures. Ideally, the RCu_ppt corresponding to 160 °C-aging treatment should be higher than that at 120 °C owing to the large-size Cu-containing precipitates present at 160 °C. However, we found the RCu_ppt at 120 °C to be 7.46% higher than that at 160 °C, not only at the aging stage but also at the peak-aging stage. Although the peak-aging treatment is conducive to maintain the maximum RCu_ppt, the ξpptCu should also be kept under consideration. Owing to the high diffusion coefficient of Cu in Al and the metastable nature of Al3-xCuxSc, Al is not readily substituted by Cu in the precipitates, thereby resulting in a decrease of ξpptCu during aging treatment (Supplementary Fig. 3d). However, the minimum ξpptCu is still found to be higher than 1 at% at the peak-aging stage.
Implementation of HE-resistant design and experimental verification
We fabricated the Al-0.51 at.% Cu-0.12 at.% Sc alloy and subjected these samples to different heat treatments to implement our strategy to achieve HE-resistance. When the samples are subjected to the slow strain rate testing (SSRT) in 3.5 wt.% NaCl solution with electrochemical H-charging, their elongation (loss) can reflect their resistance to HE. As shown in Fig. 5a, the best outcome in terms of lowest HE susceptibility is observed for the sample exposed to solution treatment at 500°C and aging treatment at 120 °C, with an elongation loss of only 0.15. Without aging treatment, simple solution treatment dissolves alloying elements back into the Al matrix, showing obvious HE susceptibility (0.7). Aging treatment can greatly reduce the susceptibility, while excessively high or low precursor solution temperature increases sensitivity.

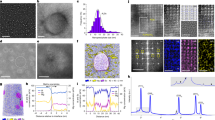
a HE susceptibility parameter: total elongation loss of SSRT samples. b Dense distribution of Al3Sc precipitates (green arrows) and its diffraction pattern in aging treated samples. The (c) HAADF and (d) EDS mapping to show Cu and Sc distribution. The blue square in (c) indicates the region rich in Cu, while the yellow square indicates the region low in Cu content. e Elemental atomic mapping showing Cu-doped in the interior of the precipitate. The arrows indicate strong H trapping sites similar to the atomic arrangements shown on the left corner of the figure. f The lattice d-spacing difference obtained by HAADF and corresponding FFT images. g APT detection of the density fluctuation of H, Sc, and Cu. It is clearly evident that the H atoms are aggregated in Sc-Cu rich areas. h The H desorption rate of samples treated at 500 °C solution treatment and 120 °C aging treatment. Peaks 1–6 are ordered from left to right.
As shown in Supplementary Fig. 5, three samples exposed to 120°C aging treatment exhibit higher elongation than the sample undergoing 500°C-solution treatment without aging treatment, whose elongation is only 5.81%. Comparing the effect of the solution treatment temperature subjected to samples (exposed to 120 °C aging treatment), we observe that their strength is not affected (~ 250.61 ± 0.97 MPa). The best elongation in the H-charging environment is observed for the sample exposed to 500 °C-solution treatment and 120 °C-aging treatment, whose elongation reaches 22.20%. Comparing the samples treated at 470 °C and 530 °C, the elongation of the sample at 470 °C (20.60%) is observed to be higher than that of 530 °C (18.51%).
This phenomenon is attributed to the smaller size of the Al3Sc precipitates for the sample exposed to 470°C solution treatment than that at 530 °C, which results in more interfaces with the Al matrix for the former case that can trap the H (\({\Delta G}_{{\rm{soln}}}^{0}\) for H is − 0.36 eV). From Fig. 5b, it can be seen that there are numerous Al3Sc precipitates distributed within the matrix, and its diffraction pattern also confirm the structure. The density and size of the precipitates are summarized in Supplementary Fig. 6a, similar to the aging treatment, higher solution treatment temperatures can promote the growth of Al3Sc precipitates. Their number and size at 530 °C solution treatment and 120 °C aging treatment are 12 and 174.4 nm (measured within a 4 × 4 μm2 observation area), respectively. For the lower solution treatment temperature (470 °C), the number of the Al3Sc precipitates increases to 20 with an average size of 111.0 nm.
To confirm the presence of Cu atoms within the Al3Sc precipitates and explain the HE-resistance of the sample at 500 °C-solution treatment and 120 °C-aging treatment, we conducted the high-angle annular dark-field (HAADF) STEM and EDS mapping. As shown in Fig. 5c–f, there are multiple positions inside the Al3Sc precipitate that are found to be rich in Cu (indicated by a blue square in Fig.5c). This confirms that the Cu atoms enter the precipitates for the heat treatment applied in our study. Owing to the metastable nature of the Cu-doped Sc phase, not all of the Al3Sc precipitates transform to Al3-xCuxSc precipitates.
Despite the fact that not all Al3Sc precipitates are doped with Cu, the ones which have Cu exhibit the strongest H trapping ability. We performed the APT experiments to obtain insights into the distribution of Sc, Cu, and H atoms in samples with 500 °C-solution treatment and 120 °C-aging treatment, the results are shown in Fig. 5g. There is a total of three H aggregations, but the distinctly concentrated one (marked by “pole”) is perhaps the combined effects of the Al pole, zone line and Cu/Sc enrichment, so we cannot determine that H atoms are primarily trapped in the Cu-rich region. Gault et al. also demonstrated that the APT-measured distribution of H and Cu can be influenced by local electrostatic field variations at the specimen surface, hence, the density correction in our work was performed36,37. In the right area (marked by “rich”), it can be found that there is obvious Sc-Cu enrichment, and its size is consistent with the H distribution range. Supplementary Figs. 7–9 display the density fluctuation of Al, Sc, Cu, and H over varying thicknesses and regions, which consistently confirm H enrichment. Therefore, the APT measurements suggest that the H atoms can be trapped by Al3-xCuxSc precipitates.
In Fig. 5h, the H desorption rates are presented, which show the intensity of different H trapping sites, for instance, the lattice, which corresponds to the temperature of 129°C. In addition, there is another trapping site, which is the dislocation (Peak 3) corresponding to 334 °C. At a slightly higher temperature than 334 °C, the sample releases H again (Peak 4) whose peak temperature reaches 405 °C. For the peak 2 whose H rate temperature is 268 °C, we deem it belong to the dislocation at the Al/Al3Sc phase boundary which \({\Delta G}_{{\rm{soln}}}^{0}\) is − 0.157 eV H−1 (Supplementary Table 2). According to our DFT calculations, the \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapped by 1/6 < 112> Shockley dislocation is − 0.31 eV and that of H trapped at the Al3Sc (100) interface is − 0.36 eV. Therefore, we reckon that the peak 4 belongs to H trapped by the Al3Sc (100) interface. Furthermore, the peak 5 corresponding to H trapped by the Cu-doped Al3Sc (whose ΔG is significantly more negative than the ones corresponding to dislocation and the interface) is observed at 485 °C. As for the remaining peak (Peak 6, 525 °C), it is usually deemed as to be corresponding to H trapped by the vacancy cluster in Al alloys22. The Cu-doped Al3Sc precipitates (\({\Delta G}_{{\rm{soln}}}^{0}\) < − 1 eV) may also release H at this elevated temperature, and hence it is difficult to discern them. Compared with other heat treatments samples, the sample with 500 °C solution and 120 °C aging treatment exhibit biggest H rates in the high temperature range (Supplementary Fig. 10).
The length of the primitive lattice of \({{\rm{Pm}}}\bar{3}{{\rm{m}}}\) Al3Sc is 4.14 Å, which is also the d-spacing of {100} plane. As shown in Fig. 5f, the d-spacing of {011} plane is found to be 2.91 Å. However, the length of the lattice of the Cu-doped system changes, such as the d-spacing of {011} and {111} are obtained to be 2.96 Å and 2.43 Å respectively, from which the lattice parameter of the Cu-doped phase is deduced to be 4.38 Å. Yet this modification does not change the relationship between the precipitate and matrix such that the (100) interface is coherent to the Al matrix, and there is one edge dislocation inside the (0\(\bar{1}\)1) interface (Supplementary Fig. 11). From the elemental atomic mapping (Fig. 5e), we confirm that Cu is doped in the interior of the precipitates and the T-site (− 0.85 eV) and O-site (− 4.27 eV) surrounded by Cu and Sc atoms can significantly trap H in the bulk of the precipitates as discussed before.
Discussion
The predominant focus of H-related research involves precise investigations into H distribution around specific defects such as dislocations11, precipitates2 or GB3. Limited studies have been dedicated to engineering microstructures aimed at selectively trapping H, for instance, by transforming the traditional η phase in 7xxx series Al alloys to the T-phase26. Owing to the infinite chemical space for alloy designing, it is difficult to choose a system which possesses strong H trapping ability (Fig. 1a). ML has the potential to solve this problem38, however, this approach does not allow understanding of the underlying physical mechanisms. To this end, DFT and MD-based simulations can help us to better understand and design HE-resistant microstructures. The computation-based study presented here showcases the feasibility of designing alloys with low HE susceptibility via cross-scale computational techniques. Furthermore, our computation-based approach guides the experiment so as to generate strong H traps in the form of Cu-doped Al3Sc precipitates in Al alloys with a notable reduction in elongation loss sensitivity (0.15).
With this strategy, a phase with extremely strong H trapping ability is discovered, but it might be metastable in nature with stringent formation conditions. This poses a challenge in sustaining the materials’ excellent performance and their long-term service. Increasing the Cu content can understandably increase the probability of the appearance of Cu in the precipitates. Despite of the fact that the Cu doping of the Al3Sc is beneficial for HE resistance, it does not mean that Cu can be added infinitely. According to the \({\Delta G}_{{\rm{Cu}}}^{0}\) values corresponding to Cu distributed at the interfaces and GBs (Fig. 6a), various coverages of Cu are thermodynamically favorable. For the typical (111) Al GBs, their \({\Delta G}_{{\rm{Cu}}}^{0}\) value significantly decreases with the increase in the Cu content, especially for Σ3(111) GB, for which \({\Delta G}_{{\rm{Cu}}}^{0}\) reaches − 1.17 eV Cu−1 when the coverage is 0.66. The Cu atoms tend to diffuse towards the Σ7 GBs when the Cu coverage is less than 0.45.

a The thermodynamics of Cu distribution at the precipitate-matrix interface and the GBs. b The \({\Delta G}_{{\rm{soln}}}^{0}\) values corresponding to H trapping by the two interfaces as a function of Cu coverage. c H decorated dislocation cuts through the Al3Sc causing the H atoms to be trapped in the bulk of the Cu-doped precipitate. d Cu concentration at the (100) interface via atomic elemental mapping. e The HAADF image showing the orientation between the Al matrix and the Al3Sc precipitate.
However, Fig. 6a also reflects the reality that although Cu in the bulk of the Al3-xCuxSc is metastable, it will transform to Al3Sc during the long-term service of the Al alloys, with the rejected Cu segregating at the Al/Al3Sc interfaces. Similar to Cu coverage at the (111) GBs, the segregation of Cu at Al3Sc interfaces is also thermodynamically favorable, whose corresponding \({\Delta G}_{{\rm{Cu}}}^{0}\) values are lower than − 0.4 eV Cu−1 (Fig. 6a). The tendency of the two Al3Sc-matrix interfaces towards Cu coverage are observed to be different. For instance, the \({\Delta G}_{{\rm{Cu}}}^{0}\) corresponding to Al3Sc (100) is observed to decrease (becomes more negative) with the increase in the Cu coverage, whereas it increases (becomes less negative) for the case of Al3Sc (0\(\bar{1}\)1) interface. The increase in ΔG value for Al3Sc (0\(\bar{1}\)1) interface with the increase in the Cu coverage indicates that the drive for trapping H goes down. If we compare the \({\Delta G}_{{\rm{Cu}}}^{0}\) values corresponding to the interfaces, we observe that the Cu atoms segregate at the (0\(\bar{1}\)1) interface until 0.3 Cu coverage, above which the (100) interface gets decorated with the Cu atoms. Thus, it can be concluded from this study that the metastable Cu atoms present in the bulk of the Al3-xCuxSc precipitates might segregate at the interfaces. However, these segregation can increase the stress of dislocation slip, so as to improve the strength of alloys (Supplementary Fig. 14).
Although a small amount of Cu can improve the cohesive energy of the Al GBs39, the excessive Cu segregation at the GBs can result in galvanic corrosion40. In a practical multi-alloyed system, Cu can interact with elements to form other precipitates, such as Mg(AlZnCu)2 phase distributed at the GB in 7xxx Al alloys. Whereas, in a single grain, there are numerous finely dispersed Al3Sc precipitates, which is favorable as the Cu atoms get distributed at their interfaces since these interfaces are more accessible to the Cu atoms as compared to the GBs. The high density of Cu trapping sites within the grains similarly affects its kinetic diffusion coefficient, thereby retarding Cu segregation at the GBs (Supplementary Fig. 15). Computing the \({\Delta G}_{{\rm{soln}}}^{0}\) values corresponding to H trapping (Fig. 6b) show that the \({\Delta G}_{{\rm{soln}}}^{0}\) for (100) interface increases to − 0.25 eV H−1 as a function of Cu coverage whereas it decreases significantly (becomes more negative) to ~ − 0.65 eV H−1 (corresponding to Cu coverage of 0.9) for (0\(\bar{1}\)1) interface. When the (0\(\bar{1}\)1) interface is fully occupied by Cu atoms, the \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapping sharply increases (becomes less negative) to − 0.33 eV H−1. This implies that the full Cu coverage at the Al3Sc-Al interface lower their H trapping ability.
In addition, it is experimentally revealed that multiple H atoms move with dislocation15 while the Cu-rich region, which is a strong H trap, is in the bulk of the Al3-x CuxSc precipitate. Thus, we performed MD simulations of dislocation slip based on which we could confirm that the dislocation cuts through the Al3Sc phase (Fig. 6c and Supplementary Fig. 16), making it possible for the bulk of the Al3-x CuxSc precipitates to trap the H atoms carried by the dislocation. For Al3Sc, the \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapping in the bulk of the precipitate is higher (less negative) than that of dislocation, and the trapping at the interface is only 0.05 eV H−1 lower than trapping by the dislocation. However, when the Cu atoms are doped into the Al3Sc, the \({\Delta G}_{{\rm{soln}}}^{0}\) corresponding to H trapping in this precipitate ranges from − 0.147 to − 6.791 eV H−1 (Fig. 2b, c, f), which can strongly trap H.
MD simulations provide deeper atomic insights into H diffusion kinetics within the Al-Sc-Cu alloys. As illustrated in Supplementary Fig. 17a, H exhibits significantly reduced diffusivity when trapped within Al3-xCuxSc precipitates compared to the Al matrix, demonstrating effective H trapping by these precipitates that depletes solute H concentration in the matrix. In contrast, Cu-free precipitates (500 °C treated sample in Supplementary Fig. 18) exhibit diminished trapping capacity20, allowing H accumulation at crack tips and GBs. This localized H enrichment at the crack tip facilitates dislocation emission (HELP) and promotes intergranular crack propagation (Supplementary Fig. 19).
In conclusion, we present a HE resistant microstructure comprising of a metastable Cu-doped Al3Sc precipitates in Al alloy based on a computational study employing DFT and developed quantum machine learning potential used within MD, which is validated by experiments. Our proposed strategy clearly reveals the optimal precipitate that can strongly trap H atoms (i.e., Cu-doped Al3Sc) in Al alloy, and provides insights into the underlying mechanism behind the precipitation of Cu-free and Cu-doped Al3Sc phases via heat treatment. With the aid of the developed Al-Sc-Cu interatomic potential and MC simulations, despite the metastable nature of the Cu-doped Al3Sc precipitates, we still can readily determine the optimal heat treatment parameters to generate desirable microstructural features. The feasibility of designing cross-scale modeling-guided HE-resistant microstructure in Al alloys is validated by experiments, which can be generalized to other technologically relevant materials in order to make them immune to HE. The development of MLIP approaching DFT accuracy enables simulation of arbitrary elemental combinations (encompassing all precipitates) for both steel and Al alloys, transcending conventional search limitations to achieve targeted design structures.
Methods
Ab-initio calculations
All DFT equations are summarized in Supplementary Methods, and their computations are performed using Vienna Ab-initio Simulation Package (VASP 5.4)41 with generalized gradient approximation and Perdew-Burke-Ernzerhof exchange-correlation functional. The electron and force convergence accuracy are set to 10−5 eV and 0.01 eV Å−1, respectively. The setting of k-points is based on the spatial length (0.2 Å−1) in each direction, and the cutoff energy is 450 eV. The bond strength calculation is performed using Local Orbital Basis Suite Towards Electronic-Structure Reconstruction (LOBSTER 4.1)42, which uses the Hamiltonian matrix to quantitatively calculate the bond strength. The diffusion barriers are obtained using the Nudged Elastic Band (NEB) method43. The VESTA (Version 3.5.8) is used for visualization.
Interatomic potential
The interatomic Al-Sc-Cu potential is developed based on the Machine Learning Interatomic Potentials (MLIP 2)44. The training dataset is built by DFT calculations, which includes energy, force, and stress information. All DFT-calculated configurations are the bulk systems, which derivatize the raw, compress/strained, and vacancy-containing structures. The configurations are randomly divided into a training set (0.8) and the testing set (0.2). Data leakage was avoided by ensuring that structurally or physically related configurations are not simultaneously included in both the training and test sets. The weights of energy, force and stress are 1, 0.1, and 0.001, respectively. For the active learning, the cutoff distance is set to be 5 Å, and the accuracy index is 16 whose radial basis size is 8 and the radial function is 4. The extrapolation grade is 2.1 and the active learning is continuously performed until no new configuration is generated45.
MD simulations
The system for MD simulations comprises of 108,000 atoms including 1.2% Cu and 0.2 Sc atoms with a size of ~ 12.1 nm which is built by ATOMSK (Version 0.13)46. The Open Visualization Tool (OVITO 3.9.4) is used to visualize the MD data47. Three random models are considered for the system to enhance the statistics. These systems are initially heated to 743, 758, 773, 788, and 803 K to simulate solution treatments, and the Nosé–Hoover barostat and thermostat method is used to maintain the temperature. At the same time, the positions of less than 1% atoms are swapped every 5 fs to carry out MC diffusion48. For the aging treatment simulations, the system is initially cooled down from the solution treatment temperature in an interval of 10 ps, and then MC is performed. The diffusion time for the solution treatment is 5 ps, while that of the aging treatment is 10 ps. The size of the Sc-phase is determined using the nearest neighbor analysis for the FCC structure.
In view of the RCu_ppt and ξpptCu, 120 °C is found to be the optimal aging temperature, which can precipitate small-size Al3-xCuxSc phases with a high probability of Cu doped into the phase. Besides, the RCu_ppt in the aging stage is significantly higher than the peak-aging stage, hence, the experimental treatment stage is chosen to be the aging stage. Using the experimental diffusion coefficient of Sc49 and its mean square displacement, the aging time is calculated as 6.96 h (Supplementary Fig. 6). As discussed before, the solution treatment temperature of 500 °C and 120 °C aging treatment exhibit the highest probability of Cu doping of the Al3Sc precipitates.
Microstructure observations
An FEI Talos F200X TEM (Thermo Fisher Scientific) operating at 200 kV is performed to observe the structure of precipitates, and energy-dispersive X-ray spectroscopy (Super X G2) is used for the elemental mapping. The HAADF-STEM and the atomic-resolution elemental mapping are carried out on the FEI Themis Z advanced probe aberration-corrected analytical TEM/STEM equipped with a Super-X EDS detector operated at 300 kV. Samples for HRTEM were 3 mm diameter discs with an initial thickness of 0.2 mm. They were mechanically ground to 5000#, and the thickness reduced to 60 µm which then were electropolished. The twin-jet solution was a 5% perchloric acid in ethanol at − 30 °C, and the voltage was 15 V. The post-processing software is GATAN (Version 3.53.4031.2).
Tensile experiments
To study the HE of the samples, pre-hydrogen charging is conducted using the electrochemical method on SSRT samples whose gauge dimension of 3 × 3 × 15 mm3. All samples are successively ground with waterproof paper and acetone up to 2500 grit. Then, 0.05 M NaCl + 0.03 wt.% thiourea and the cathodic galvanic polarization are applied on specimens for hydrogen charging, the charging time is 72 h with a current density of 1 mA cm−2. The strain rate is 3.4 × 10−6 s−1 at room temperature. To evaluate the susceptibility of HE, the total elongation loss for each sample can be calculated as \(HE=({\delta }_{W}-{\delta }_{H})/{\delta }_{W}\), where δW and δH are the elongations of the samples without and with H, respectively. The relevant experiments were independently repeated six times by different research groups.
APT experiments
The APT specimens were prepared by PFIB from bulk sample with the size of 10 × 10 × 1 mm3 on an FEI Helios 5 PFIB instrument operated with an Xe source. A wedge-shaped lamella with dimensions of approximately 7 × 3 × 7 µm3 was extracted using a 30 kV, 4 nA PFIB beam and subsequently mounted onto a silicon micro-post. The specimen was then shaped into an APT needle by sequential annular milling at 30 kV with progressively reduced beam currents of 0.3 nA, 0.1 nA, and 30 pA to achieve the final tip geometry. Atom probe measurement was performed on the local electrode atom probe (CAMECA LEAP 5000XS) at a cryogenic temperature of 50 K under UHV conditions of 10−11 Torr. All APT measurements were carried out using voltage pulsing with a 15% pulse fraction and a 250 kHz pulse rate.
TDS measurements
For TDS measurements, specimens with 13 × 14 × 1.5 mm3 dimension are electrochemically charged as SSRT in tensile experiments. The pre-charged specimens are immediately cleaned with ethanol and dried before TDS. H trap sites are determined using Bruker G8 GALILEO ONH Analyzer with mass spectroscopy at a heating rate of 100 °C h−1. All samples were repeated for three times.
Data availability
The authors declare that the data supporting the findings of this study are provided in the included Source Data file and accessible at https://doi.org/10.5281/zenodo.17576724. Additional data are available upon request from the corresponding author. Source data are provided in this paper.
Code availability
The training algorithm and dataset of machine learning interatomic potential can be available in https://www.jiyucheng.group or https://github.com/yucheng-ji/Hydrogen-resistant-Materials-design (https://doi.org/10.5281/zenodo.17576850).
References
Cotterill, P. The hydrogen embrittlement of metals. Prog. Mater. Sci. 9, 205–301 (1961).
Gong, P., Nutter, J., Rivera-Diaz-Del-Castillo, P. E. J. & Rainforth, W. M. Hydrogen embrittlement through the formation of low-energy dislocation nanostructures in nanoprecipitation-strengthened steels. Sci. Adv. 6, eabb6152 (2020).
Chen, Y. et al. Observation of hydrogen trapping at dislocations, grain boundaries, and precipitates. Science 367, 171–175 (2020).
Gest, R. & Troiano, A. Stress corrosion and hydrogen embrittlement in an aluminum alloy. Corrosion 30, 274–279 (1974).
Kim, J. et al. Roughening improves hydrogen embrittlement resistance of Ti-6Al-4V. Acta Mater. 220, 117304 (2021).
Tarzimoghadam, Z. et al. Multi-scale and spatially resolved hydrogen mapping in a Ni–Nb model alloy reveals the role of the δ phase in hydrogen embrittlement of alloy 718. Acta Mater. 109, 69–81 (2016).
Seita, M., Hanson, J. P., Gradečak, S. & Demkowicz, M. J. The dual role of coherent twin boundaries in hydrogen embrittlement. Nat. Commun. 6, 6164 (2015).
Troiano, A. R. The role of hydrogen and other interstitials in the mechanical behavior of metals. Trans. Am. Soc. Met. 52, 54–80 (1960).
Song, J. & Curtin, W. A. Atomic mechanism and prediction of hydrogen embrittlement in iron. Nat. Mater. 12, 145–151 (2013).
Birnbaum, H. K. & Sofronis, P. Hydrogen-enhanced localized plasticity—a mechanism for hydrogen-related fracture. Mater. Sci. Eng. A 176, 191–202 (1994).
Huang, L. et al. Quantitative tests revealing hydrogen-enhanced dislocation motion in α-iron. Nat. Mater. 22, 710–716 (2023).
Dong, X. et al. Hydrogen-associated decohesion and localized plasticity in a high-Mn and high-Al two-phase lightweight steel. Acta Mater. 239, 118296 (2022).
Pundt, A. & Kirchheim, R. Hydrogen in metals: microstructural aspects. Annu. Rev. Mater. Res. 36, 555–608 (2006).
Young, G. A. & Scully, J. R. The diffusion and trapping of hydrogen in high purity aluminum. Acta Mater. 46, 6337–6349 (1998).
López Freixes, M. et al. Revisiting stress-corrosion cracking and hydrogen embrittlement in 7xxx-Al alloys at the near-atomic-scale. Nat. Commun. 13, 4290 (2022).
Kacher, J., Eftink, B. P., Cui, B. & Robertson, I. M. Dislocation interactions with grain boundaries. Curr. Opin. Solid State Mater. Sci. 18, 227–243 (2014).
Ji, Y. et al. Discontinuous model combined with an atomic mechanism simulates the precipitated η′ phase effect in intergranular cracking of 7-series aluminum alloys. Comput. Mater. Sci. 166, 282–292 (2019).
Hammer, P., Romaner, L. & Razumovskiy, V. I. Hydrogen trapping in mixed carbonitrides. Acta Mater. 268, 119754 (2024).
Liu, P.-Y. et al. Engineering metal-carbide hydrogen traps in steels. Nat. Commun. 15, 724 (2024).
Jiang, S. et al. Structurally complex phase engineering enables hydrogen-tolerant Al alloys. Nature 641, 358–364 (2025).
Su, H. et al. Assessment of hydrogen embrittlement via image-based techniques in Al–Zn–Mg–Cu aluminum alloys. Acta Mater. 176, 96–108 (2019).
Safyari, M. et al. New insights into hydrogen trapping and embrittlement in high strength aluminum alloys. Corros. Sci. 223, 111453 (2023).
Xu, Y. et al. Suppressed hydrogen embrittlement of high-strength Al alloys by Mn-rich intermetallic compound particles. Acta Mater. 236, 118110 (2022).
Safyari, M., Moshtaghi, M., Hojo, T. & Akiyama, E. Mechanisms of hydrogen embrittlement in high-strength aluminum alloys containing coherent or incoherent dispersoids. Corros. Sci. 194, 109895 (2022).
Zhao, H. et al. Hydrogen trapping and embrittlement in high-strength Al alloys. Nature 602, 437–441 (2022).
Wang, Y. et al. Switching nanoprecipitates to resist hydrogen embrittlement in high-strength aluminum alloys. Nat. Commun. 13, 6860 (2022).
Zhang, B. et al. Atomic-scale insights on hydrogen trapping and exclusion at incoherent interfaces of nanoprecipitates in martensitic steels. Nat. Commun. 13, 3858 (2022).
Lin, Y.-C. et al. Hydrogen trapping and desorption of dual precipitates in tempered low-carbon martensitic steel. Acta Mater. 196, 516–527 (2020).
Chen, Y.-S. et al. Direct observation of individual hydrogen atoms at trapping sites in a ferritic steel. Science 355, 1196–1199 (2017).
Kim, H. et al. Elucidating the role of a unique step-like interfacial structure of η4 precipitates in Al-Zn-Mg alloy. Sci. Adv. 9, eadf7426 (2023).
Ji, Y. et al. Artificial intelligence combined with high-throughput calculations to improve the corrosion resistance of AlMgZn alloy. Corros. Sci. 233, 112062 (2024).
Hyland, R. W., Asta, M., Foiles, S. M. & Rohrer, C. L. Al(f.c.c.):Al3Sc(L12) interphase boundary energy calculations. Acta Mater. 46, 3667–3678 (1998).
Clouet, E., Nastar, M. & Sigli, C. Nucleation of Al3Zr and Al3Sc in aluminum alloys: From kinetic Monte Carlo simulations to classical theory. Phys. Rev. B 69, 064109 (2004).
Xue, H. et al. Highly stable coherent nanoprecipitates via diffusion-dominated solute uptake and interstitial ordering. Nat. Mater. 22, 434–441 (2023).
Dadfarnia, M., Martin, M. L., Nagao, A., Sofronis, P. & Robertson, I. M. Modeling hydrogen transport by dislocations. J. Mech. Phys. Solids 78, 511–525 (2015).
Gault, B. et al. Towards establishing best practice in the analysis of hydrogen and deuterium by atom probe tomography. Microsc Microanal. 30, 1205–1220 (2024).
Gault, B., Moody, M. P., Cairney, J. M. & Ringer, S. P. Atom probe crystallography. Mater. Today 15, 378–386 (2012).
Sasidhar, K. N. et al. Enhancing corrosion-resistant alloy design through natural language processing and deep learning. Sci. Adv. 9, eadg7992 (2023).
Ji, Y., Dong, C., Chen, L., Xiao, K. & Li, X. High-throughput computing for screening the potential alloying elements of a 7xxx aluminum alloy for increasing the alloy resistance to stress corrosion cracking. Corros. Sci. 183, 109304 (2021).
Li, N. et al. Insight into the localized strain effect on micro-galvanic corrosion behavior in AA7075-T6 aluminum alloy. Corros. Sci. 180, 109174 (2021).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558 (1993).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Novikov, I. S., Gubaev, K., Podryabinkin, E. V. & Shapeev, A. V. The MLIP package: moment tensor potentials with MPI and active learning. Mach. Learn Sci. Technol. 2, 025002 (2020).
Ji, Y. Hydrogen resistant Materials design. Zenodo https://doi.org/10.5281/zenodo.16880825 (2025).
Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commun. 197, 212–219 (2015).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2009).
Sadigh, B. et al. Scalable parallel Monte Carlo algorithm for atomistic simulations of precipitation in alloys. Phys. Rev. B 85, 184203 (2012).
Marquis, E. A. & Seidman, D. N. Nanoscale structural evolution of Al3Sc precipitates in Al(Sc) alloys. Acta Mater. 49, 1909–1919 (2001).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 52125102 for C.D. and U23A20392 for Y.J.), the China Postdoctoral Science Foundation (No. 2024M750172 for Y.J.). This work was sponsored by Nederlandse Organisatie voor WetenschappelijkOnderzoek (The Netherlands Organization for Scientific Research, NWO) domain Science for the use of supercomputer facilities (P.D.). The authors also acknowledge the use of the DelftBlue supercomputer, provided by Delft High Performance Computing Center (https://www.tudelft.nl/dhpc). The authors would like to acknowledge Prof. Alexander Shapeev (from Skolkovo Institute of Science and Technology) for the helpful discussions on MLIP, Prof. Jianghua Chen and Yuxiang Lai from Hainan University for their help in APT and FIB, and Prof. Weimin Zhao from China University of Petroleum (East China) for TDS.
Author information
Authors and Affiliations
Contributions
Y.J. performed high-throughput DFT calculations and developed the interatomic potential, performed the experimental verification, and wrote the first draft of the manuscript. Y.J., P.D., and C.D. conceptualized the project and designed the research. X.F. performed sample fabrication and SSRT. M.S. and M.M. performed the SSRT and TDS experiments. C.Y. and F.S. analyzed the modeling data. P.D., M.M., X.C., X.Y., X.L., and C.D. analyzed the results and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, Y., Fu, X., Safyari, M. et al. Tailoring precipitates for enhanced hydrogen trapping in aluminum alloys. Nat Commun 17, 279 (2026). https://doi.org/10.1038/s41467-025-66998-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66998-w
