
Abstract
Cell type annotation is an essential step in single-cell RNA-sequencing analysis, and numerous annotation methods are available. Most require a combination of computational and domain-specific expertise, and they frequently yield inconsistent results that can be challenging to interpret. Large language models have the potential to expand accessibility while reducing manual input and improving accuracy, but existing approaches suffer from hyperconfidence, hallucinations, and lack of reasoning. To address these limitations, we developed CASSIA for automated, accurate, and interpretable cell annotation of single-cell RNA-sequencing data. As demonstrated in analyses of 970 cell types, CASSIA improves annotation accuracy in benchmark datasets as well as complex and rare cell populations, and also provides users with reasoning and quality assessment to ensure interpretability, guard against hallucinations, and calibrate confidence.
Similar content being viewed by others


Introduction
Accurate cell type annotation is essential for understanding the cellular composition of tissues, characterizing the functional states of distinct cell types, and identifying dynamic changes in response to disease. Reference-based annotation methods typically rely on existing single-cell RNA-seq datasets with known cell-type labels. Among these, classifier-based methods train supervised machine learning models such as random forests1, support vector machines2, or deep learning models like transformer-based architectures3,4 on labeled reference datasets to classify unknown query cells. Alternatively, correlation-based methods annotate query cells by comparing their expression profiles to labeled reference data without explicit training5,6,7. Reference-based approaches generally perform well given representative reference datasets having features and cell subpopulations that closely match the query data. However, the performance of reference-based methods is compromised when suitable reference datasets are not available8,9.
Reference-free approaches have been developed that leverage prior knowledge of marker genes, thus avoiding reliance on matched reference datasets10,11,12. Whether reference-based or reference-free methods are initially employed, manual annotation often follows to refine those clusters having no or low-confidence annotations, and/or those showing discrepant results across methods9,13. While most investigators consider manual annotation by an expert to be the gold standard, it is time consuming, labor intensive, and requires a combination of computational and domain-specific expertise13. Large language models (LLMs) address a number of these challenges and are being used to streamline and improve computational analyses of genomic data in many domains14,15,16, including cell annotation. Most of these LLMs are built on transformer-based neural networks trained on vast text corpora, including scientific literature. Unlike domain-specific models trained on gene expression profiles3,4, general-purpose LLMs benefit from continuous improvement through intense competition among leading technology companies. They also possess sophisticated reasoning capabilities that approximate human expert-level analysis, making them particularly valuable for repetitive biological tasks. The popular LLM-based approach GPTCelltype, for example, demonstrates the potential of LLM-based models to reduce the effort and expertise required for cell type annotation17. Indeed, with a single-prompt, Hou and Ji demonstrate that GPTCelltype provides annotations in “strong concordance with manual annotations”. In spite of the advantages brought forth by simplifying cell annotation analysis, GPTCelltype has several limitations. A major one is its potential to hallucinate. In addition, the approach does not explain the reasoning underlying any given annotation, nor does it provide quality scores. As a result, it is often not possible for a user to distinguish hallucinations or low-quality annotations from high-quality ones. Finally, in an effort to reduce the likelihood of hallucinations, GPTCelltype uses relatively few markers, which may be sufficient for annotating general cell types but does not allow for accurate annotation of distinct subpopulations. See Supplementary Note 1 for details.
To address these limitations, we developed a collective agent system for single-cell interpretable annotation (CASSIA). In short, CASSIA is a multi-agent LLM framework consisting of five interconnected LLMs for annotation, validation, formatting, quality scoring, and reporting. For applications requiring highly detailed annotations, CASSIA also includes a retrieval-augmented generation (RAG) agent that retrieves external information. Additionally, for cases exhibiting signs of low-quality annotations, CASSIA provides an advanced annotation boosting agent to further refine and correct results. Optional LLMs are also available for subclustering and uncertainty quantification.
Benchmark analyses in 970 cell populations show that CASSIA increases cell annotation accuracy, improves interpretability, and provides for robust quality assessment of annotations. In addition to improving the accuracy and ease with which cell annotation can be performed, the utility of CASSIA is further demonstrated in analyses of complex datasets, rare cell types, and mixed cell populations. CASSIA is also used here to identify errors in gold-standard datasets, demonstrating its potential for refining existing reference annotations.
Results
Overview of CASSIA
CASSIA is a modular, multi-agent LLM framework for accurate, interpretable, and adaptable cell type annotation in scRNA-seq data. The framework requires a user to specify species, tissue type, groups of cells (cell types) to be annotated, and a collection of markers associated with each cell type, if known. We expect that most analyses will use markers from FindAllMarkers in Seurat or tl.rank_genes_groups in Scanpy, although markers from other sources may be used if desired. The default CASSIA workflow consists of five main steps:
-
1.
Annotation: Analyze marker expression patterns to generate cell type labels with detailed reasoning.
-
2.
Validation: Iteratively check annotations for consistency in marker-cell type alignment (up to three refinement cycles).
-
3.
Quality Assessment: Assign quality scores (0–100) based on scientific accuracy and marker balance.
-
4.
Refinement: Flag low-scoring or mixed clusters for additional refinement.
-
5.
Reporting: Provide full interpretability with detailed reasoning, quality scores, and refinements.
A schematic illustrating this multi-agent LLM system and workflow is provided in Fig. 1. Users provide input information such as species, tissue type, and markers (Fig. 1a), which is then processed to create input for annotation, validation, formatting, scoring, and reporting (Fig. 1b), culminating in comprehensive output (Fig. 1c). For cell types requiring additional refinement, CASSIA offers optional specialized agents (Fig. 1d). Specifically, the Annotation Boost agent leverages complete differential expression results such as p-values and fold-changes to improve low-scoring annotations; and the Subclustering agent resolves mixed cell populations with subtle phenotypic differences. For novel or complex datasets where markers are mentioned in the literature relatively infrequently, and therefore are not well represented in GPT and similar LLMs, users can augment the default Annotation agent with a RAG agent that integrates external knowledge from databases like CellMarker18 and ontologies19, while maintaining the same validation, quality assessment, and reporting workflow.

a A user interacts directly with the Onboarding platform by specifying species, tissue type, and a collection of markers associated with cell subtypes within that tissue, if known. Any information associated with experimental conditions, interventions, or other sample specific information may be provided. Created in BioRender. Shireman, J. (2025) https://BioRender.com/e55de3z. b Together, this input is used to create the user prompt given to the Annotator agent. The Annotator agent performs a comprehensive annotation of the single-cell data using a zero-shot chain-of-thought approach that mimics the standard workflow that a computational biologist would typically follow for cell annotation. Results are then passed to the Validator agent to check marker and cell type consistency; results failing validation are passed back to the Annotator and this iterative process continues until results pass validation (or the maximum number of iterations is reached). Results are then moved to the Formatting agent, which summarizes each cell annotation; this summary along with the full conversation history is provided to the Scoring agent for quality scoring. The Reporter agent then generates a comprehensive report documenting the complete annotation process, including agent conversations, quality evaluation reasoning, and validation decisions with supporting evidence to facilitate transparent interpretation of results. The output from default CASSIA is shown in (c). Optional agents include those shown in (d). Source data are provided as a Source Data file. Created in BioRender. Shireman, J. (2025) https://BioRender.com/9gzyoe5.
CASSIA improves annotation accuracy in benchmark datasets
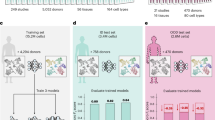
CASSIA was compared with state-of-the-art cell annotation methods on 970 cell types from five classic benchmark datasets for which gold-standard annotations are available: GTEx, Tabula Sapiens (TS), Human Cell Landscape (HCL), Mouse Cell Atlas (MCA), and Azimuth. To assess annotation accuracy, we utilize the hierarchical evaluation framework used to evaluate GPTCelltype17. The framework is based on the Cell Ontology tree structure, classifying annotations as fully correct, partially correct, or incorrect based on their taxonomic distance from reference annotations. A fully correct annotation matches the reference exactly while an annotation one step away from the reference on the ontology tree is considered partially correct (e.g., predicting “T-cell” for a CD8 + T-cell); annotations more than one step away are classified as incorrect (e.g., predicting “immune cell” for a CD8 + T-cell). See Methods. Fig. 2a, b show that CASSIA increases fully correct annotations by 12–41% relative to existing approaches and combined correct annotations (fully or partially correct) by 9–20% relative to the next-best approach across benchmark datasets. Performance averaged across all annotations shows that CASSIA increases annotation accuracy by over 20% for most datasets (Fig. 2c).

a Fully correct annotation rates across 8 datasets where CASSIA (blue) increases annotation accuracy by 12–41% over the next best performing method. b Combined correct annotation rates (fully or partially correct annotations) across the same datasets where CASSIA maintains a 9–20% advantage over the next-best approach (typically GPTCelltype4/4o). c Heatmap of average accuracy across cell types for each dataset (rows) and method (columns), with values ranging from 0 (white) to 1 (dark red). Average accuracy is computed by assigning 1 to fully correct, 0.5 to partially correct, and 0 to incorrect annotations, then averaging these scores across all clusters within each dataset. CASSIA consistently achieves the highest accuracy scores. The “Cancer” and “Immune” rows correspond to the specialized datasets discussed in the Results subsection “CASSIA performance is maintained in more specialized applications.” d Effect of marker set size on annotation accuracy across benchmark datasets. Line plots show the average accuracy (1 = fully correct, 0.5 = partially correct, 0 = incorrect) achieved by CASSIA when the top 10, 30, 50, 70, or 100 ranked marker genes are used as input. For each benchmark dataset—GTEx, Tabula Sapiens (TS), Human Cell Landscape (HCL), Mouse Cell Atlas (MCA), and Azimuth—accuracy is averaged across all clusters. Performance generally plateaus at around 50 markers, suggesting this number offers an effective trade-off between information content and efficiency. e Performance benchmarking of CASSIA with different LLMs versus GPTCelltype implementations on 100 cell types sampled from five benchmark datasets. CASSIA-Claude-3.5 achieves superior performance (0.92), followed by CASSIA-GPT4.0 (0.88) and CASSIA-LLAMA-3.2 (0.82), demonstrating the framework’s robustness to underlying model selection. Source data are provided as a Source Data file.
We systematically evaluated the optimal number of marker genes and determined that using 50 marker genes per cell type provides the best balance of accuracy and efficiency. When analyzing clusters with complex cellular compositions or those requiring detailed annotation, it may be useful to include more than 50 markers. Fig. 2d demonstrates that CASSIA maintains robust performance even with larger marker sets (e.g., top 100 markers), while competing methods such as GPTCelltype show significant performance degradation due to context window limitations (Supplementary Note 1a). CASSIA’s performance is also robust across different underlying language models, including Claude 3.5 Sonnet, GPT-4o, and open-source LLaMA-3.2-90B (Fig. 2e).
Beyond accuracy and robustness, CASSIA provides interpretable results through structured analysis reports that document CASSIA’s reasoning process from functional marker evaluation through cell type marker identification and database validation. Each annotation includes validation steps with marker consistency checks and pathway verification, along with a confidence score. This transparent process allows researchers to understand the basis for annotations and assess their reliability. Fig. 3 provides an example where CASSIA analyzes a colorectal cancer cluster, identifying it as intestinal epithelial cells with a predominant colorectal cancer phenotype. Having established a strong baseline in standard benchmark datasets, we next investigated CASSIA’s capabilities in more challenging biological contexts.

a The report presents a comprehensive analysis including functional and cell type marker identification, database cross-referencing, cell type determination, and subtype classification. The validation section confirms marker consistency and pathway verification, while the quality assessment provides a numerical score (92/100) with identified strengths and areas for improvement. Source data are provided as a Source Data file.
CASSIA performance is maintained in more specialized applications
To systematically assess CASSIA’s performance in more specialized biological contexts, we considered datasets from cancer biology, immunology, and non-model organisms. We first focused on evaluating CASSIA’s performance in cancer datasets composed of two primary cancer samples and three metastatic samples. CASSIA outperforms other methods in assigning correct cell types, achieving 79% accuracy across all cancer datasets (Fig. 2c). When tasked with specifically distinguishing cancer from non-cancer cells, CASSIA correctly identifies 72.5% of cancer cells on average across multiple datasets, compared with 20% for GPTCelltype (Fig. 4a, b). A simple enhancement to CASSIA - adding a single prompt that reads “You should carefully distinguish between cancer cells and normal cells with reasoning” - further improves detection accuracy to 88–100% across all cancer datasets (Fig. 4b and Supplementary Data 1). To assess whether the improvement from cancer-specific prompting reflects true robustness or prompt sensitivity, we conducted a prompt variation analysis on one representative cancer dataset. Four cancer-aware prompts, differing slightly in wording, all yielded similarly high cancer cell recall (Supplementary Data 1). Beyond cell type identification, we also investigated CASSIA’s ability to detect metastasis signals in three datasets. Each consists of single-cell RNA-seq data profiled from metastatic brain tumors with primaries in lung. CASSIA successfully identifies both the metastatic nature of the cells and the tissue of origin in all three datasets (Supplementary Data 1), whereas GPTCelltype accurately detected metastatic cells in only one sample.

a UMAP visualizations of cancer cell classifications in brain metastasis samples. Left: Gold standard annotations showing cancer cells (red) and non-cancer cells (blue). CASSIA annotations closely match the gold standard (middle). GPTCelltype4 annotations show significant misclassification of cancer cells as non-cancer (right). Note: In the gold standard panel, all cancer cells are labeled according to a single broad cluster defined by the original study. In contrast, CASSIA and GPTCelltype operate on subclusters derived from this broad cancer cluster, which results in partially annotated cancer regions. See Supplementary Note 4 for details. b Heatmap quantifying tumor cell detection accuracy across five cancer datasets (rows) for five annotation methods (columns). CASSIA achieves 57–100% accuracy, while CASSIA-Enhanced reaches 88–100% accuracy. Other methods (GPTCelltype4, GPTCelltype4o, ScType) show substantially lower performance (0–40%). c Radial plots comparing immune cell subtype classification in PBMC samples. Gold standard annotations (top) compared with CASSIA (middle) and GPTCelltype4 (bottom). Color-coding indicates annotation accuracy: green (correct), yellow (partially correct), and red (incorrect). CASSIA accurately identifies most immune cell subtypes and activation states, while GPTCelltype4 shows substantial misclassifications (red sectors). d Bar chart comparing annotation performance across non-model organisms. CASSIA (blue) consistently achieves >85% accuracy across shark, cat, tiger, and pangolin datasets, while GPTCelltype4 (red) and GPTCelltype4o (orange) show variable and significantly lower performance, particularly in evolutionarily distant species like shark. Source data are provided as a Source Data file.
We next examined whether this robust performance extends to complex immune cell landscapes, which present distinct annotation challenges due to their hierarchical differentiation patterns and functional plasticity. We considered the PBMC68k dataset, which contains major peripheral blood mononuclear cell populations representing a generic immune cell landscape, and the ProjecTILs dataset, a specialized manually curated reference capturing subtle transcriptional differences between T cell functional states. For these immune cell datasets, CASSIA improved average annotation accuracy by 27% over the next best performing method (Fig. 2c). While most existing methods perform reasonably well at broad cell type assignments (such as distinguishing T cells from B cells), CASSIA stands out for its ability to preserve high accuracy when annotating fine-grained functional states. Notably, it is the only method that consistently resolves subtle distinctions among naive, memory, and exhausted T cell subsets (Fig. 4c and Supplementary Fig. 1).
Having demonstrated CASSIA’s capabilities in annotating cancer and specialized immune cell populations, we next sought to determine whether its annotation framework could extend beyond well-characterized human and mouse systems to evolutionarily diverse species with limited reference data. Specifically, we tested CASSIA on datasets from diverse vertebrate species, including cartilaginous fish (shark) and several mammals (domestic cat, tiger, and pangolin). CASSIA demonstrates robust cross-species annotation capabilities, correctly identifying 20 of 22 cell types in shark, 57 of 64 in domestic cat, 66 of 79 in tiger, and 41 of 54 in pangolin, increasing annotation accuracy over other methods by 14–77% (Fig. 4d). These results further demonstrate CASSIA’s ability to transfer knowledge across phylogenetically diverse species despite the lack of comprehensive reference databases.
CASSIA provides robust annotation-specific quality scores
LLMs almost always provide answers to queries, and they typically do so with uniform confidence. In addition, while generally accurate, LLMs are known to hallucinate, even in recent models like GPT-4o20,21. To address these concerns, guidelines for the safe and effective use of LLMs in practice strongly suggest that results be cross-checked and validated for accuracy22,23. To address this need and thereby allow users to distinguish hallucinations and/or low-quality annotations from high-quality ones, CASSIA employs a two-tiered quality assessment framework. Specifically, CASSIA reports annotation-specific quality scores ranging from 0 to 100. As the quality score is derived from a single run of CASSIA, an optional Consensus Similarity (CS) score can also be obtained to quantify agreement across multiple CASSIA runs (the CS score is optional as it requires additional computational time due to multiple CASSIA runs; see Methods).
To evaluate CASSIA’s quality assessment framework, we consider quality scores for more than 500 cell types across five reference datasets: GTEx, TS, HCL, MCA, and Azimuth. For the more computationally intensive CS score calculation, we randomly selected a total of 95 cell types, drawing from each of the five datasets. As shown in Fig. 5a, there is a significant association between quality scores and annotation correctness. Annotations with scores below 75% were predominantly partially correct or incorrect, indicating that low scores reliably flag uncertain or erroneous annotations. (20/29). The 75% cutoff was selected through threshold testing to optimize overall classification performance (see “Methods”). Scores between 75% and 90% reflect intermediate confidence, while scores above 90% are strongly associated with correct classifications.

a Box plots demonstrating the relationship between CASSIA’s quality scores and annotation accuracy (n = 530). Scores are higher for correct annotations, intermediate for partially correct annotations, and lower for incorrect annotations. We tested whether score distributions differed across correctness categories using the Kruskal-Wallis rank sum test (two-sided, p = 2.56 × 10⁻⁸). No adjustment for multiple comparisons was applied. b Box plots showing CASSIA’s Consensus Similarity (CS) stratified by annotation accuracy (n = 100). CS scores effectively discriminate between correct or partially correct (median ~90) and incorrect annotations (median ~60), providing reliable confidence estimation. Five runs are used to compute the CS score to balance runtime and confidence. For both panels, box boundaries represent the 25th and 75th percentiles with the median indicated by the center line, whiskers extend to 1.5× IQR, and dots represent individual data points beyond the whiskers or all observations. c Schematic illustration of CASSIA’s Annotation Boost framework, which can be used to refine low scoring annotations. The system works iteratively: the Hypothesis Agent generates initial hypotheses based on CASSIA’s output, then the Query Agent identifies marker genes for validation and retrieves relevant information from FindAllMarkers output files. Results are fed back to the Hypothesis Agent to generate refined hypotheses. This cycle continues through multiple iterations, with the final output from Annotation Boost providing enhanced cell type classifications. Annotation Boost agent reports for challenging cell types including proximal tubule cells from kidney (d) and inflammatory stromal support cells (e). Each report presents a structured analytical workflow including initial dataset overview and highly expressed markers; multi-phase decision points with evidence-based reasoning; marker validation steps with expression thresholds; final annotation with comprehensive supporting evidence; confidence assessment with positive/negative markers; key biological implications of the identified cell type. The reports demonstrate CASSIA’s transparent, systematic approach to complex cell type identification with explicit reasoning chains. Source data are provided as a Source Data file. Created in BioRender. Shireman, J. (2025) https://BioRender.com/9gzyoe5.
To further assess the generalizability of this 75% threshold, we evaluated it on an independent validation set of 132 cell type annotations from non-model organisms (cat, tiger, and pangolin). This validation confirmed the robustness of our threshold: among annotations scoring below 75%, 80% (16/20) were incorrect or partially correct, while 97% (128/132) of correct annotations scored above the threshold. These results demonstrate that CASSIA’s quality scoring framework generalizes well across diverse species.
Figure 5b illustrates the relationship between CS scores and annotation accuracy. While the difference in CS scores between partially correct and fully correct annotations was not statistically significant, incorrectly annotated cell types exhibit significantly lower CS scores compared to both of the other categories. This finding suggests that CS scores are particularly effective at identifying incorrect annotations. Quality scores, CS scores, and detailed scoring reasoning for all evaluated cell types are provided in Supplementary Data 5.
The Annotation Boost agent refines low-scoring annotations to improve accuracy
When a cell type is returned with low quality and/or CS scores, the Annotation Boost agent may prove useful in refining the annotation. Unlike the default workflow which relies only on ranked marker lists, the Annotation Boost agent leverages all statistical metrics from the full FindAllMarkers file (including p-values, percentage expression, and log2 fold changes) to generate and test specific hypotheses about cell identity. As illustrated in Fig. 5c, the Annotation Boost agent employs an iterative query-and-verify approach, with each cycle refining the annotation hypothesis until convergence or a maximum iteration limit. We considered 42 annotations (out of 586) with quality scores at or near the low-confidence threshold (≤ 78%) across our benchmark datasets; 27 were incorrectly annotated by CASSIA and 15 were correctly annotated. While the predefined threshold for low quality was 75% (see Methods), we included a small number of annotations with slightly higher scores (up to 78%) to broaden the evaluation set. When applied to these 42 annotations, the Annotation Boost agent successfully corrected 24 of the 27 (89%) previously incorrect annotations while preserving all 15 annotations that were originally correct. This improvement was consistent across all evaluated datasets, with the most substantial gains observed in complex tissues with closely related cell types. Raw results are provided in Supplementary Data 7, and failure case analyses (for the 3 of 27 annotations that were not successfully corrected) are detailed in Supplementary Note 5.
A representative example of the Annotation Boost agent is shown in Fig. 5d for a collection of proximal tubule cells. CASSIA, GPTCelltype, and SingleR incorrectly classify these cells as hepatocytes, but CASSIA’s low quality score (75%) suggests further attention. We applied the Annotation Boost agent, which identified a tissue context mismatch and ultimately provided a detailed annotation of “proximal tubule cells (mixed segments)”. The sequential reasoning pattern of the agent, visible in the chat history excerpts in Fig. 5d, e, demonstrates how CASSIA systematically builds evidence for its revised annotation by identifying and then querying the expression patterns of additional marker genes that were not in the initial ranked list.
Another notable refinement involved cells labeled as “stromal cell_PLA2G2A high” that were misidentified as epithelial cells by CASSIA and all other methods. Given CASSIA’s quality score for this cell type was 75%, the Annotation Boost agent was applied and correctly identified these cells as “inflammatory stromal support cells” by first ruling out epithelial characteristics through additional stromal marker identification, then conducting a specialized functional assessment that revealed the inflammatory state associated with elevated PLA2G2A expression (Fig. 5e). These examples demonstrate how the Annotation Boost agent extends CASSIA’s capabilities beyond initial annotation and facilitates the refinement of challenging annotations through expert-like reasoning processes.
CASSIA’s quality assessment framework identifies heterogeneous cell populations and errors in gold standard datasets
To further evaluate CASSIA’s quality assessment framework, we evaluated cell types with low quality scores as well as those with high quality scores that contradicted the gold standard annotations. First, we considered two datasets - the small intestine and eye TS datasets- as these had low quality scores on average. For these datasets, cell type identification is particularly challenging due to poorly separated clusters and unbalanced cell counts. In the small intestine dataset, we identified annotations with low CS scores, including goblet cells (75%) and mature enterocytes (30%). Further investigation revealed the goblet cell cluster contained mixed cell types, while the mature enterocytes cluster showed an unusually high mitochondrial ratio (> 60%) (Fig. 6a). Similar patterns were observed in the TS eye dataset (Supplementary Note 2). These analyses demonstrate that low CS scores are often derived from low-quality cell populations or heterogeneous populations that require additional subclustering.

a Scatter of quality scores vs. CS scores for cell-type annotations, colored by annotation accuracy. The mature enterocyte cluster shows an abnormally low CS score (~ 30%). The accompanying distribution of mitochondrial gene content for that cluster indicates most cells have >60% mitochondrial reads, consistent with poor data quality and the low CS. b Heatmaps compare marker expression between CASSIA and gold-standard labels. Column 1: cells CASSIA calls Schwann, but the gold standard calls monocytes show strong Schwann markers and minimal monocyte markers, indicating the monocyte label is likely incorrect. Column 2 (positive controls): agreed-upon monocytes (top) and Schwann cells (bottom) show expected marker patterns, confirming marker sets. Column 3: cells CASSIA calls macrophages, but the gold standard calls neutrophils express definitive macrophage markers and lack neutrophil markers, again indicating a gold-standard error. Column 4 (positive controls): agreed-upon neutrophils (top) and macrophages (bottom) show expected expression. c Bar plots report detection accuracy for 50:50 (blue) and 80:20 (red) mixtures across methods. “CASSIA default” uses CASSIA’s mixed-type flag plus sub-type inspection; “CASSIA scoring” considers the reasoning of the quality score agent. “CASSIA consensus” runs CASSIA five times with majority voting and CS thresholds. All CASSIA modes outperform GPTCelltype approaches in both mixture settings. d Schematic of CASSIA’s Retrieval-Augmented Generation (RAG) pipeline. The system integrates specialized agents accessing the CellMarker2.0 database for validated markers and the Owlready ontological database for tissue-specific hierarchies. These components provide contextual knowledge to enhance CASSIA’s default annotation pipeline. Created in BioRender. Shireman, J. (2025) https://BioRender.com/9gzyoe5. e Across mouse motor cortex (n = 23), mouse cerebellum (n = 18), and human lung (n = 41), CASSIA with RAG (light blue) consistently attains the highest annotation accuracy versus CASSIA without RAG (dark blue), GPTCelltype4, GPTCelltype4o, ScType, and SingleR. f Hierarchical classification schema for mouse motor cortex neurons, illustrating the multi-level annotation challenge from basic neuron identity to excitatory/inhibitory classification, layer specification, projection subtype determination, and inhibitory marker profiling. g In neuronal subtype tasks, CASSIA with RAG achieves the best accuracy for projection neurons (~ 65%), inhibitory neurons (~ 72%), and non-neurons (~ 85%), improving identification of specialized subtypes over CASSIA without RAG and other methods. Source data are provided as a Source Data file.
We also investigated high-confidence annotations that contradicted established reference labels. Specifically, we first examined 15 annotations (out of over 500 total) where CASSIA’s quality scores exceeded 90%, but CASSIA’s annotation did not match the gold standard. For each annotation, we built an evaluation system using three LLMs. Each LLM was given the marker gene profile of the cell type and asked to determine which label—CASSIA’s prediction or the gold standard—the profile more closely resembled, assigning a score to each option. When all three LLMs consistently favored CASSIA’s annotation, we manually analyzed and visualized the key classical markers.
Of the 15 annotations reviewed, 11 (73.3%) agreed with CASSIA’s annotation, while only 1 (6.7%) disagreed. The remaining 3 cases (20%) yielded conflicting or inconclusive results across the different LLMs (see Supplementary Note 2c). Figure 6b shows two representative examples where the three LLMs agree with CASSIA’s annotations: cells identified by CASSIA as “Schwann cells” but labeled in the reference as “monocytes,” and cells identified as “macrophages” but labeled in the reference as “neutrophils.” To investigate these discrepancies, we identified canonical cell type markers with high discriminatory power for each contested cell type pair (monocytes vs. Schwann cells; neutrophils vs. macrophages). Fig. 6b shows that the marker gene expression profiles strongly support CASSIA’s annotations. Additional examples are provided in Supplementary Fig. 4 and Supplementary Data 9.
CASSIA identifies mixed cell types
To evaluate CASSIA’s ability to recognize mixed cell types, we synthetically generated input profiles by combining the top 50 marker genes from two distinct cell types in defined ratios (50:50 and 80:20). We compared results from running CASSIA in both default mode and consensus mode with 5 repetitions, the latter providing CS scores, consensus cell type annotations, and potential mixed cell type identifications.
In the balanced 50:50 mixing scenario, default CASSIA directly identified mixed populations in 6 out of 10 cases. An additional 3 cases were flagged by CASSIA’s scoring agent as potentially mixed, yielding an overall detection accuracy of 90%. The consensus mode performed similarly, detecting one additional mixed situation (7/10), though it misidentified the specific mixed cell types in this additional case. In contrast, GPTCelltype4 failed to detect any mixed populations, and GPTCelltype4o identified only 3 out of 10 mixed cases (Fig. 6c).
In the more challenging 80:20 mixing scenario, default CASSIA directly identified mixed populations in 11 out of 20 cases, with the scoring agent detecting 5 additional mixed profiles. This resulted in an 80% overall detection accuracy. The consensus mode demonstrated superior performance, correctly identifying mixed populations in 14 out of 20 cases. Both default CASSIA and consensus mode CASSIA achieved an average dominant cell type annotation accuracy of 0.9, correctly identifying the cell type contributing 80% of the markers. By contrast, GPTCelltype4 and GPTCelltype4o showed substantially lower performance, detecting only 0 and 2 mixed populations, respectively, and achieving average dominant cell type annotation accuracies of 0.35 and 0.45.
To further test sensitivity and robustness, we repeated the simulation with 60:40 and 70:30 mixtures and with smaller (top 30) and larger (top 70) marker sets. CASSIA’s performance remained stable across all conditions (Supplementary Fig. 8 and Supplementary Data 11).
CASSIA’s RAG agent enhances annotation of complex tissues
While CASSIA performs well on standard datasets with its core functionality, some tissues with complex hierarchical organization require additional domain-specific knowledge for optimal annotation. Toward this end, CASSIA employs a RAG agent that leverages tissue-specific markers from external databases and ontologies to refine annotations and accurately subdivide major cell types into specialized ones.
Across three such challenging tissues—mouse motor cortex, mouse cerebellum, and human lung—the RAG agent yields consistent gains in average accuracy over both core CASSIA (7–13%) and four strong baselines (Fig. 6e; see also Supplementary Data 2 for evaluation rubric and Supplementary Data 3 for full annotation outputs and hierarchical breakdowns).
To further illustrate the contribution of RAG in resolving fine-grained cell identities, we use the mouse motor cortex dataset from the Azimuth atlas as a representative case study. Neurons make up the largest cell type in this dataset, and the general class of neurons is relatively straightforward to annotate. However, a detailed classification requires annotation of major type (excitatory, inhibitory), as well as numerous subtypes (local vs. projection, and within projection intratelencephalic (IT), extratelencephalic (ET), corticothalamic (CT), or near projection (NP)), inhibitory subtype (Pvalb+, SST+, VIP+, and Lamp5+), and layer location (2–6). Figure 6f summarizes this hierarchical structure and highlights the depth of annotation required in the motor cortex. We use the RAG agent here to provide additional background information to CASSIA related to cell types and their corresponding markers. In this analysis, the Marker Database agent identified 43 markers based on seven related cell types, including glutamatergic neurons, GABAergic neurons, astrocytes, dopaminergic neurons, oligodendrocytes, microglia, and vascular cells. The Ontology Database agent identified and filtered ontology trees to ensure specificity to the mouse cortex; the initial trees identified were rooted by neurons, glial cells, vascular cells, and meningeal cells and then restricted to only contain mouse cortex cells, resulting in four trees overall. The Hierarchical Feature agent performed a hierarchical analysis on the four ontology trees selected from the second module to identify key discriminative features and associated markers. For example, in the neuron-rooted tree, there are three sublevels that discriminate features within the cortex ontology: inhibitory vs. excitatory, laminar location, and projection patterns. Markers associated within each of the sublevels are then identified by the RAG agent and provided to CASSIA. As shown in Fig. 6g, CASSIA with RAG is the only method capable of precise annotation of neuronal subtypes.
Discussion
Accurate cell type annotation is an essential step in the majority of single-cell RNA-sequencing studies, and numerous methods are available. Most methods require computational expertise, domain-specific knowledge, and/or reference datasets for training, which limit their applicability in practice. LLMs have the potential to expand accessibility and improve performance, but the few existing LLM-based approaches available for annotation are limited by a lack of quality scores and internal reasoning. To address these limitations, we developed CASSIA, the first multi-agent LLM framework designed specifically for cell annotation. As demonstrated, CASSIA provides users with reasoning and quality assessment to ensure interpretability and calibrate confidence, while at the same time substantially improves annotation accuracy in benchmark datasets as well as complex and rare cell populations.
CASSIA’s multi-agent structure consists of Annotation, Validation, Formatting, Quality Scoring, and Reporter agents, with optional agents for subclustering, uncertainty quantification, and annotation boosting. A RAG agent is also available for applications requiring highly detailed annotations. Comprehensive quality control is provided via the self-reflective Annotation and Validation agents, the Quality Scoring agent, and the CS agent. Results from these agents proved useful not only in flagging low-confidence annotations but also, as demonstrated, in identifying errors in gold-standard datasets. In addition to providing annotation-specific quality assessment, CASSIA also provides the full annotation history, which allows a user to interpret the reasoning behind each annotation.
Taken together, CASSIA exemplifies how LLM frameworks can move beyond sequential prompting to perform adaptive, expert-like analyses, which leads to improvements in overall accuracy. In particular, as demonstrated in benchmark analyses of 970 cell types, CASSIA improves annotation accuracy by 12–41% over existing methods. CASSIA is also able to detect mixed cell types, including those containing cancer signatures. To understand the relative contributions of each agent to CASSIA’s enhanced performance, we conducted systematic ablation analyses across our benchmark datasets (Supplementary Note 6). These analyses quantify how each agent contributes to overall performance gains.
A number of CASSIA’s agents introduce novel designs. The Annotation Boost agent, for example, performs a hypothesis-driven breadth-then-depth search, autonomously generating candidate cell types that might not have been considered by a user, and then testing those cell types against structured marker statistics in a way that mimics expert reasoning. Furthermore, the RAG agent expands the model’s search space by retrieving plausible candidate cell types and markers from external sources, then distills tissue-specific ontologies into functional axes through a PCA-like “feature prompt” approach. Another novel aspect is the CS agent, which introduces a principled framework for quantifying annotation uncertainty via multi-run consensus, which allows CASSIA to flag unstable annotations with high precision.
Despite these advances, limitations remain. First, CASSIA’s performance depends on the quality of input marker genes, which can be challenging to derive in datasets with poorly defined clusters or continuous trajectories. To assess this, we evaluated CASSIA on the Mouse Organogenesis Cell Atlas, a large-scale developmental time-course dataset profiling ~2 million cells across 61 embryonic stages. As expected, the average accuracy was lower compared to well-separated benchmarks, yet CASSIA achieved an average accuracy of 0.53 compared to 0.15 for GPTCelltype, indicating that while performance is reduced in this setting, the multiagent architecture retains a clear advantage (Supplementary Data 13). To ensure that marker profiles reflect true single-cell identities, we recommend performing standard quality control (QC) procedures, including basic QC, doublet detection, and ambient RNA correction prior to marker gene identification. Second, the initial marker gene calculation is both time-consuming and potentially suboptimal. Future work will explore adaptive marker selection strategies that dynamically adjust the number and types of markers based on cluster characteristics, as well as methods that incorporate gene set enrichment results or other functional signals to better utilize CASSIA’s reasoning capacity. Third, CASSIA currently assumes that clustering is performed externally. While this enables flexibility with upstream workflows, it limits the opportunity for deeper integration between clustering and annotation. We envision future versions of CASSIA that incorporate clustering directly into the multi-agent framework.
While CASSIA currently focuses on cell type annotation, its multi-agent architecture provides a foundation that could be extended to other single-cell analysis tasks. The self-reflection loop, quality scoring, and uncertainty scoring framework, and external knowledge integration via RAG represent modular components that could support tasks such as clustering, trajectory inference, or multimodal integration. However, we believe that achieving optimal performance will require task-specific adaptations of agents and prompts, similar to how the single-cell field has developed specialized methods for each analytical challenge rather than relying on one-size-fits-all solutions. Future work could explore developing specialized multi-agent systems for other core analytical tasks, each optimized for its specific requirements while building on the architectural principles demonstrated here.
In conclusion, CASSIA leverages the reasoning capabilities of LLMs for robust validation, interpretability, and quality assessment to provide accurate and transparent cell type annotation across diverse biological contexts. Importantly, CASSIA provides a user with the logical reasoning behind each annotation along with annotation-specific quality scores, reducing the black-box24 nature of LLM-based approaches and guarding against hallucinations. Ultimately, CASSIA lowers the computational expertise needed to conduct single-cell annotation, while also providing more biological-specific expertise through the RAG agent. Taken together, CASSIA increases both the accuracy and accessibility of cell type annotation in practice.
Methods
CASSIA is a multi-agent LLM consisting of an Onboarding platform and five interconnected LLMs: an annotation agent, a validation agent, a formatting agent, a quality scoring agent, and a reporter agent. Optional LLMs are also available for subclustering, uncertainty quantification, and annotation boosting. For applications requiring highly detailed annotations, CASSIA includes a RAG agent. Each agent is described in detail below.
CASSIA implementation
We provide CASSIA as an R package, a Python package, and a web-based user interface (www.cassia.bio and www.cassiacell.com). Both programming packages support all CASSIA functionalities, including optional agents, with the default pipeline executable via a single command.
Onboarding
A user provides information to an Onboarding platform by specifying species, tissue type, and a collection of markers associated with cell subtypes within that tissue, if known. Most analyses will use markers from FindAllMarkers in Seurat or tl.rank_genes_groups in Scanpy, although markers can be manually added if desired. We suggest 50 markers be used if available (see the “Identification of Marker Genes” section below). Any information associated with experimental conditions, interventions, or other sample-specific information may be provided, and will be used to create the user prompt given to the Annotator agent. The specific user prompt reads: “Your task is to annotate a single-cell [species] dataset from [tissue type]. Please identify the cell type based on this ranked marker list: [marker list]. Below is some additional information about the dataset: [additional information].” In some cases (e.g., multiple mixed cell types from an atlas), tissue type is not known. A tissue-blind version can also be used (see Supplementary Note 3).
Annotation
The Annotator agent performs a comprehensive annotation of the single-cell data. To minimize hallucinations and prevent oversimplification of complex biological concepts, we implemented multiple prompt engineering techniques: a zero-shot chain-of-thought approach that structures the model’s reasoning to follow the same systematic analytical steps a professional computational biologist would take25,26, role specification where the Annotator agent acts as a “professional computational biologist with expertise in single-cell RNA-sequencing,” and audience framing as “an expert in the field.” These role-playing strategies have been shown to improve the likelihood of receiving detailed and technically accurate responses27. Finally, we included a hypothetical reward statement: “You will be rewarded $10,000 if you do a good job” as such reward modeling in LLMs has been shown to prime the model for maximum effort and attention to detail28.
Taken together, the system prompt to the Annotator Agent reads: “You are a professional computational biologist with expertise in single-cell RNA-sequencing (scRNA-seq). A list of highly expressed markers ranked by expression intensity from high to low from a cluster of cells will be provided, and your task is to identify the cell type. You must think step-by-step, providing a comprehensive and specific analysis. The audience is an expert in the field, and you will be rewarded $10000 if you do a good job. Steps to follow:
-
1.
List the Key Functional Markers: Extract and group the key marker genes associated with function or pathway, explaining their roles.
-
2.
List the Key Cell Type Markers: Extract and group the key marker genes associated with various cell types, explaining their roles.
-
3.
Cross reference Known Databases: Use available scRNA-seq databases and relevant literature to cross reference these markers.
-
4.
Determine the Most Probable General Cell Type: Based on the expression of these markers, infer the most likely general cell type of the cluster.
-
5.
Identify the Top 3 Most Probable Sub Cell Types: Based on the expression of these markers, infer the top three most probable sub cell types. Rank them from most likely to least likely. Finally, specify the most likely subtype based on the markers.
-
6.
Provide a Concise Summary of Your Analysis.
Always include your step-by-step detailed reasoning. You can say “FINAL ANNOTATION COMPLETED” when you have completed your analysis. If you receive feedback from the validation process, incorporate it into your analysis and provide an updated annotation.”
Once FINAL ANNOTATION COMPLETED is reached, annotations are provided to the Validator agent to ensure accuracy and consistency.
Validation
The Validator agent is designed to iteratively cross-check the results provided by the Annotator agent, forming a feedback loop that significantly enhances the robustness of the final annotations. This iterative design is inspired by self-verification techniques, which allow for critical evaluation and continual refinement of annotations29. The Validator agent primarily ensures marker and cell type consistency, verifying that the key markers identified by CASSIA are present in the provided marker list and accurately represent the identified cell type. This step is crucial to confirm that the markers used for annotation are both appropriate and reliable. If the validation fails, the Validator agent provides detailed feedback and sends the results back to the Annotator agent for revision. This process iterates up to three times to ensure the highest quality annotation. Regardless of whether validation finally passes or fails, the results are then forwarded to the Formatting agent for further processing. The full system prompt for the Validator agent is provided in Supplementary Note 3.
Formatting
A formatting agent was implemented to process annotation outputs from the previously mentioned two agents. The agent operates in two distinct states: a standard mode that extracts and structures validated cell type annotations, including mixed population information, and a diagnostic mode that activates upon validation failure to analyze error sources and generate recommendations for resolution. The full system prompt is provided in Supplementary Note 3.
Quality scoring
The Scoring agent functions as an objective evaluator that analyzes the complete annotation conversation history to generate a quantitative assessment score (0–100%). It focuses on scientific accuracy of the annotations along with balanced use of multiple markers where high-ranked markers contribute most to the annotation and no single marker drives the annotation. The full system prompt for the Quality Scoring agent is provided in Supplementary Note 3.
To establish thresholds for the quality scores, we implemented a weighted cost optimization approach defined as Total cost = (w × FP) + FN where FP represents false positives (correct annotations incorrectly flagged as low quality), FN represents false negatives (incorrect annotations misclassified as high quality), and w is the incorrect weight parameter (default = 2). We assigned greater weight to false positives to minimize the risk of unnecessarily flagging correct annotations for manual review. Supplementary Fig. 5 shows that the lower threshold minimizing total cost is ~75% when evaluated on more than 500 cell type annotations from the five benchmark datasets. To further evaluate the generalizability of this threshold, we tested it on an independent set of 132 cell type annotations. The evaluation confirmed that the 75% threshold maintains high classification performance across species, with 80% of low-scoring annotations (< 75%) being incorrect or partially correct, and 97% of correct annotations scoring above the threshold (Supplementary Data 5). Given this, annotations with quality scores below 75% are flagged as low-quality. In these cases, the (optional) Annotation Boost agent could be used to improve the annotation.
Reporter agent
The Reporter agent generates a comprehensive HTML report documenting the complete annotation process, including agent conversations, quality evaluation reasoning, and validation decisions with supporting evidence. The full system prompt is provided in Supplementary Note 3.
Subclustering agent
CASSIA provides an optional subclustering agent designed to optimize the analysis of specialized cell populations (e.g., CD4 + T cells cluster). Rather than processing individual subclusters separately, this agent analyzes all subclusters within a specific cell population simultaneously. This concurrent analysis strategy improves computational efficiency, enabling better detection of subtle phenotypic differences between closely related cell states. The full system prompt is provided in Supplementary Note 3.
Uncertainty quantification
CASSIA provides an optional feature to assess annotation reliability by quantifying consensus across multiple runs. By default, five runs are recommended to compute the CS score for a balance between runtime and confidence.
To ensure consistent comparisons across CASSIA runs, cell type nomenclature is standardized using two complementary approaches. The first approach applies basic text normalization (converting to lowercase, replacing hyphens with spaces, removing trailing “s“), then maps each normalized label to standardized Cell Ontology terms via the CL API19. This mapping matches cell types to their formal ontological definitions, ensuring consistent terminology. After unifying cell type names, we employ a CS score to measure the similarity across the n CASSIA runs. Recall that the Annotator agent identifies the most probable general cell type and the three most probable sub-cell types, ranked from most likely to least likely. Letting R denote the set of all annotation results, each result r ϵ R is a pair \(\left(g,s\right)\) where g represents the general cell type and s represents the most likely sub-cell type. Then
where \({g}_{c}\) is the most frequent general cell type in R; \({s}_{c}\) is the most frequent cell sub-type in R; \({w}_{g}\) and \({w}_{s}\) specify the weights given to matches of the general cell type and cell sub-type, respectively. \(I\left(x\right)\) denotes the indicator function where \(I\left(x\right)=1\) if x is true and 0 otherwise; \(\left|R\right|\) denotes the number of annotations in R. We use CSCL to emphasize the fact that this CS score was calculated using terms unified by the Cell Ontology CL API. The CSCL score takes into account both the general and sub-cell types, giving more weight to matches in the general type while still considering sub-type matches and potential type inversions.
To address potential API mapping inconsistencies, a second approach employs an LLM-based unification agent that harmonizes synonyms and slight variations in cell type nomenclature. A CSLLM based score is calculated based on this unification. Finally, to enhance robustness and mitigate potential errors from unification steps, a consensus agent independently estimates the consensus general cell type and subtype. Taken together, three measures of consensus are obtained (CSCL, CSLLM, and the consensus agent’s estimated consensus score). The minimum score is taken as the final CS score. Annotations with a CS score below 75% are flagged as uncertain and recommended for further validation using the Annotation Boost agent.
Although CASSIA uses consensus voting primarily to quantify annotation uncertainty, the same mechanism can—in principle—increase prediction accuracy when individual runs perform above chance (i.e., p > 0.5). This idea draws on recent work in LLM ensemble methods, which shows that repeated or multi-model responses often yield better outputs via majority voting or similar aggregation strategies30,31.
Under the assumption that model outputs are independent, the probability of obtaining a correct consensus after n runs is given by the binomial tail:
For example, a model with 70% base accuracy achieves ≈84% majority accuracy after 5 runs and ≈95% after 15 (Supplementary Fig. 6). These values represent theoretical upper bounds based on the assumption of independent outputs and serve to illustrate how consensus can improve reliability in stochastic LLM settings. Empirical validation on the Azimuth bone marrow dataset confirms that the majority voting improves annotation accuracy and the gains plateau at 5–10 runs (Supplementary Data 14), supporting our default setting of n = 5 as an optimal balance between computational efficiency and performance.
Annotation Boost agent
CASSIA provides an optional Annotation Boost agent to improve low-confidence annotations (those with quality scores below 75%). Unlike the default pipeline, which uses ranked marker lists, this agent leverages the full statistical output from Seurat or Scanpy’s FindAllMarkers file, including adjusted p-values, average log₂ fold change, and expression percentages in the target cluster (pct.1) and background (pct.2). These metrics capture the core features used by human experts when evaluating markers visually (e.g., via FeaturePlot or VlnPlot), enabling expert-like assessment in a compact, LLM-friendly format. Since FindAllMarkers is already computed during the default CASSIA workflow, no additional input or access to Seurat objects is required, making the approach modular and efficient. The agent builds on the ReAct agent framework, extending it in two main ways. Specifically, the Annotation Boost agent autonomously generates multiple hypotheses about a cluster’s identity, allowing the agent to consider cell types not specified in the user query; the agent also dynamically proposes new hypotheses during iterative reasoning. Each hypothesis is evaluated against marker-level statistics, and the agent either confirms or refines the hypothesis based on the data. This process defines a hybrid search structure that combines breadth-first exploration with depth-first refinement. The loop continues until a high-confidence annotation is reached or a maximum of five iterations is completed. Full system prompts and implementation details are provided in Supplementary Note 3.
Retrieval-Augmented Generation (RAG) pipeline
To improve annotation of cell types that are underrepresented in the literature and therefore not well encoded in LLMs, CASSIA includes an optional multi-agent RAG pipeline. This module expands the model’s reasoning space by integrating curated biological knowledge from external sources, including marker databases and structured ontologies, and is particularly valuable for complex tissues or non-model organisms.
The pipeline begins with a Marker Database agent that uses tissue and species information to search the CellMarker database, retrieving canonical markers for related broad and tissue-specific cell types. This agent infers plausible candidate cell identities and supplies their associated markers to CASSIA, effectively expanding the annotation space while preserving the user-provided input. The Ontology Database agent constructs a tissue-specific ontology by querying the Owlready ontological framework. It identifies relevant cell type categories based on anatomical context, developmental origin, and functional role, then filters and restructures the resulting ontology trees to retain only tissue-appropriate branches. These curated hierarchies are passed to the Hierarchical Feature agent, which performs a reasoning-based dimensionality reduction analogous to PCA. Rather than relying on numeric vectors, this agent identifies conceptual feature axes—such as lineage, spatial compartmentalization, or activation state—and associates each axis with markers that discriminate between subtypes along that dimension. This enables principled subtyping of complex populations such as cortical neurons or liver-resident macrophages. In cases involving non-model organisms, an optional Cross-Species agent compares the target organism to reference species, assesses marker gene conservation and divergence, and adjusts marker interpretation accordingly to reflect evolutionary differences.
The outputs of all four agents are consolidated into structured text and incorporated into the default CASSIA workflow as additional context for the Annotator agent. This augmentation provides biologically grounded guidance that enhances both the accuracy and interpretability of LLM-based annotations. Additional details on evaluation procedures, accuracy scoring, and full system prompts are provided in Supplementary Data 2 and 3.
Fully correct vs. partially correct annotations
We adopted the three-level evaluation framework introduced by GPTCelltype (Hou & Ji, Nat Methods 2024), establishing a hierarchical evaluation framework based on the Cell Ontology tree structure to assess annotation accuracy. Annotations are classified as fully correct, partially correct, or incorrect based on their taxonomic distance from reference annotations. An annotation one step away from the reference on the ontology tree is considered partially correct (e.g., predicting “T-cell” for a CD8 + T-cell), while annotations multiple steps removed are classified as incorrect (e.g., predicting “immune cell” for a CD8 + T-cell). For rare or highly specific cell types, we adapted this framework to consider annotations as partially correct when they accurately identify the general cell type category, such as classifying “epithelial cell” as partially correct for Bestrophin 4 (BEST4) + cells. See Supplementary Data 2 for more evaluation examples. A similar, but more detailed, scoring system was used for the RAG agent applied to the mouse motor cortex data (Supplementary Data 2).
Average accuracy
To calculate average accuracy, cell type annotations are evaluated on a three-tier scale: fully correct annotations are assigned a score of 1, partially correct a score of 0.5, and incorrect a score of zero. Average accuracy for a dataset is the average score across all annotated cell types for that dataset.
Identification of marker genes
We evaluated CASSIA’s performance using varying numbers of top-ranked marker genes per cluster (n = 10, 30, 50, 70, 100) across all five benchmark datasets: GTEx, TS, HCL, MCA, and Azimuth. In total, 2,670 CASSIA runs were performed. Across datasets, average annotation accuracy increased with more markers up to approximately 50, after which performance typically plateaued or declined (Fig. 2d). This trend suggests that most canonical signal is captured within the top ~50 markers. Adding more markers often introduces noise or redundant context, though certain clusters may benefit from longer marker lists when early-ranked markers include contaminants or uninformative genes. Based on this comprehensive analysis, we recommend using the top 50 marker genes as the default setting for most applications, balancing accuracy with computational and prompt efficiency.
To further rule out trivial gains from longer model inputs, we also analyzed accuracy versus exact input token length. Accuracy plateaued at ~50 markers (corresponding to ~200–250 tokens) and remained flat or slightly decreased despite increases in token length up to 70 or 100 markers (Supplementary Data 15). These results confirm that CASSIA’s performance is driven by biological signal quality rather than input length.
Parallel computing and computational time
CASSIA utilizes Python’s concurrent.futures module with ThreadPoolExecutor for efficient parallel processing. Cell type analyses are distributed across a user-defined thread pool (default: 10 workers), enabling concurrent annotation of multiple cell types. This parallel architecture scales efficiently, processing each cell type in about 30 s per CPU core with pre-prepared marker files. On a standard 8-core computer, CASSIA completes the annotation workflow for typical single-cell datasets with 20 clusters in under 2 min.
Memory management
CASSIA implements selective memory access across its agent network to optimize annotation processing. The annotation and validation agents maintain access to their shared conversation history for iterative refinement, while the scoring agent evaluates the complete dialogue record. For successful annotations, the formatting agent processes only the final validated exchange, whereas for failed cases, it analyzes the full conversation history to generate comprehensive error reports. This targeted memory management strategy ensures efficient information flow while maintaining contextual isolation between distinct cell type analyses.
Model selection
To evaluate CASSIA’s multi-agent framework across different LLMs, we conducted comprehensive benchmarking using three distinct models: Claude 3.5 Sonnet (2024-10-22), GPT-4o (2024-08-06), and LLaMA-3.2-90B. We created a benchmark dataset consisting of 100 cell types drawn from five randomly selected tissues within our benchmark datasets to ensure diverse representation. Performance was evaluated against GPTCelltype implementations using both GPT-4 and GPT-4o. Our analysis revealed that CASSIA’s architecture enhances annotation accuracy regardless of the underlying language model. Notably, even when using LLaMA-3.2-90B, an open-source model that typically demonstrates lower performance metrics compared to proprietary models, CASSIA achieved superior results compared to GPTCelltype implementations. While Claude 3.5 Sonnet achieved the highest accuracy, we selected GPT-4o as the default model due to its optimal balance between performance and computational efficiency. All runs were conducted under uniform decoding settings (temperature = 0; top-p, frequency penalty, and presence penalty = provider defaults; max tokens = 4096). Because random seed control is not exposed by API providers, reproducibility was assessed through repeated runs and quantified using the CS framework. Details of model build IDs across optional CS runs, LLM voting, and the Annotation Boost agent are provided in Supplementary Note 3.
Model implementation and costs
CASSIA supports flexible API integration through three options: OpenRouter API, OpenAI API, or Anthropic API. While direct API connections to OpenAI and Anthropic are supported, the OpenRouter API option enables users to access most of the commercially available LLMs without code modifications, including but not limited to GPT-4, Claude, and LLaMA variants. Based on our benchmarking results and cost analysis as of October 2024, we established a tiered recommendation system. GPT-4o serves as the default model, costing approximately $0.02 per annotation ($2.50/M input tokens, $10.00/M output tokens). For applications requiring maximum accuracy, Claude 3.5 Sonnet provides enhanced performance at $0.03 per annotation. For large-scale analyses (> 10000 clusters) or cost-sensitive applications, LLaMA-3.2-90B offers a cost-effective alternative at $0.003 per annotation through OpenRouter.
To further characterize computational efficiency, we also benchmarked the three optional agents (CS, Annotation Boost, and RAG) on representative datasets. Median wall-clock times, input/output token usage, and costs are summarized in Supplementary Data 16.
Data preprocessing
Datasets were processed using a standard pipeline. Raw count matrices were normalized using the NormalizeData function in Seurat v5, and differentially expressed genes were identified with Seurat’s FindAllMarkers function, using a minimum cell expression threshold (min.pct) of 0.1, an adjusted p-value cutoff of 0.05, and a log2 fold change threshold of 0.25. For datasets where raw data were not confidently available, we used the marker sets provided in the corresponding publications. Details for each dataset are provided in Supplementary Table 1.
Statistics and reproducibility
No statistical method was used to predetermine sample size. We analyzed 970 cell types across multiple benchmark datasets, which provides ample power, and observed consistent performance gaps across datasets; accordingly, we did not perform hypothesis tests on the leaderboard metrics. No data was excluded from the analysis. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. All analyses are scripted and version-controlled. To mitigate inherent non-determinism in LLM APIs, we pinned model versions and used deterministic decoding wherever supported.
Comparative evaluation of existing cell type annotation methods
For comprehensive benchmarking, we compared CASSIA with established annotation methods, including GPTCelltype (version 1.0, using GPT4 and GPT4o), ScType11 (version 1.0), SingleR6 (version 2.4.1), CellTypist32 (version 1.6.3), and scCATCH10 (version 3.2.2). Full analysis results are detailed in Supplementary Data 4. The optional agents in CASSIA were not used for benchmarking and so all CASSIA benchmarking results demonstrate performance of CASSIA’s default model. As described in the text, the uncertainty quantification agent was used for further analysis of the TS intestinal dataset; the RAG agent was used to analyze the neuronal dataset; and the subclustering agent was applied to the ProjecTILs dataset.
Analysis with each package followed the author’s standard workflow. A few methods required us to specify preferences as follows: for analysis with GPTCelltype, we set the marker size parameter to 10, as recommended; tissue specificity was configured according to each dataset’s context. Analysis with ScType was restricted to tissues with available marker references; gold-standard annotations were used as reference cluster labels. We used majority voting for cluster annotations within SingleR. Special considerations were required for two key datasets: the HCL analysis was restricted to marker-based approaches due to annotation-metadata discrepancies, while the mixed-tissue nature of the MCA limited our comparison to CASSIA and GPTCelltype.
Immune system analyses
We evaluated CASSIA using two immune datasets. For the PBMC68k dataset, we applied the default CASSIA workflow. For the ProjecTILs dataset, we created a challenging test case for the subclustering agent by integrating specialized human CD4+ and CD8 + T cell reference datasets that capture T cells in various functional states. Since the dataset contains exclusively T cells with subtle state-specific transcriptional differences rather than distinct cell types, it provided an ideal scenario to evaluate CASSIA’s subclustering agent for fine-grained annotation, aligning with our hierarchical annotation design.
Cancer dataset analyses
We evaluated CASSIA’s performance on cancer datasets, focusing on three aspects: annotation accuracy in cancer-altered transcriptional states, cancer cell detection, and metastatic signal identification. CASSIA successfully identified cancer-specific clusters across all datasets. In three metastasis datasets, CASSIA detected and correctly classified carcinoma cells in non-epithelial anatomical sites (brain and lymphoid tissues). This tissue-discordant detection of carcinoma cells, which originate exclusively from epithelial tissues, demonstrates CASSIA’s ability to identify metastatic signatures through the recognition of cells in developmentally distinct anatomical locations.
Mixed population simulation analysis
To systematically evaluate CASSIA’s performance in detecting and annotating mixed cell populations, we developed a controlled benchmarking framework using the GTEx lung dataset, selected for its high-quality cell type annotations and well-defined clusters. We generated synthetic mixed populations through a structured marker-mixing strategy. For each simulation, we selected pairs of distinct cell types and created hybrid marker profiles using two mixing ratios: balanced (50:50) and dominant (80:20). In the balanced scenario, we randomly selected 50% of markers from each cell type and interleaved them in alternating order (A₁B₁A₂B₂…An₂Bn₂). For the dominant scenario, we selected 80% of markers from one cell type and 20% from the other, maintaining the same interleaving pattern. We evaluated these synthetic populations using both CASSIA, GPTCelltype4, and GPTCelltype4o. In the balanced mixing condition, we assessed each method’s ability to detect the presence of mixed populations. For the dominant scenario, we evaluated both the detection of mixed populations and the accurate identification of the predominant cell type (defined by 80% marker representation). This framework provides a rigorous assessment of each method’s capacity to handle complex cellular compositions while maintaining annotation accuracy.
Brain metastases tissue collection and scRNA-seq profiling
Tissue samples from patients with confirmed brain metastatic lesions were obtained during the published clinical trial duration of NCT03398694 and processed as detailed in Shireman et al. 33. Briefly, patients with up to 4 brain metastatic lesions with at least one symptomatic lesion were consented and enrolled in the clinical trial which was approved and monitored by the IU Simon Cancer Center institutional review board and data safety monitoring committee. Confirmation of brain metastatic lesion was done based on MRI imaging, IHC analysis under pathologist supervision, and previous known diagnosis of systemic primary cancer. For this study, patients with primary diagnosis of non-small cell lung cancer and an associated brain metastatic lesion needing resection were taken to the OR and a tissue specimen was obtained by the consulting surgeon. The resulting tissue was dissociated and subject to single-cell sequencing analysis using the 10× Chromium method according to manufacturer’s protocol. For single-cell annotation of tumor tissue, gold standard reference for presence of tumor cells and categorization of brain metastasis or primary CNS tumor was confirmed by patient health record and pathologist determination upon biopsy/resection. For reference genes within the tumor samples, the outputs of FindAllMarkers at the cluster level were used as input to CASSIA.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The single-cell RNA-sequencing data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession code GSE307976. The processed data are also available under the same accession. Additional publicly available datasets used in this study are listed in the Supplementary Information file. Source data are provided with this paper.
Code availability
The code used to develop the model, perform the analyses, and generate results in this study is publicly available and has been deposited in CASSIA at https://github.com/ElliotXie/CASSIA, under the MIT license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://doi.org/10.5281/zenodo.1726168934
References
Tan, Y. & Cahan, P. SingleCellNet: a computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst. 9, 207–213 e202 (2019).
Alquicira-Hernandez, J., Sathe, A., Ji, H. P., Nguyen, Q. & Powell, J. E. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 20, 264 (2019).
Yang, F. et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 4, 852–866 (2022).
Cui, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods 21, 1470–1480 (2024).
Kiselev, V. Y., Yiu, A. & Hemberg, M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 (2018).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
de Kanter, J. K., Lijnzaad, P., Candelli, T., Margaritis, T. & Holstege, F. C. P. CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 47, e95 (2019).
Fu, Q. et al. A comparison of scRNA-seq annotation methods based on experimentally labeled immune cell subtype dataset. Brief Bioinform 25 https://doi.org/10.1093/bib/bbae392 (2024).
Abdelaal, T. et al. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 20, 194 (2019).
Shao, X. et al. scCATCH: automatic annotation on cell types of clusters from single-cell RNA sequencing data. iScience 23, 100882 (2020).
Ianevski, A., Giri, A. K. & Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 13, 1246 (2022).
Zhang, Z. et al. SCINA: a semi-supervised subtyping algorithm of single cells and bulk samples. Genes (Basel) 10. https://doi.org/10.3390/genes10070531 (2019).
Clarke, Z. A. et al. Tutorial: guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 16, 2749–2764 (2021).
Toufiq, M. et al. Harnessing large language models (LLMs) for candidate gene prioritization and selection. J. Transl. Med. 21, 728 (2023).
Kim, J., Wang, K., Weng, C. & Liu, C. Assessing the utility of large language models for phenotype-driven gene prioritization in the diagnosis of rare genetic disease. Am. J. Hum. Genet. 111, 2190–2202 (2024).
Gill, J. K., Chetty, M., Lim, S. & Hallinan, J. Large language model based framework for automated extraction of genetic interactions from unstructured data. PLoS ONE 19, e0303231 (2024).
Hou, W. & Ji, Z. Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis. Nat. Methods 21, 1462–1465 (2024).
Zhang, X. et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728 (2019).
Diehl, A. D. et al. The Cell Ontology 2016: enhanced content, modularization, and ontology interoperability. J. Biomed. Semant. 7, 44 (2016).
Farquhar, S., Kossen, J., Kuhn, L. & Gal, Y. Detecting hallucinations in large language models using semantic entropy. Nature 630, 625–630 (2024).
Huang, L. et al. A survey on hallucination in large language models: Principles, taxonomy, challenges, and open questions. ACM Trans. Inf. Syst. 43, 1–55 (2025).
Gallifant, J. et al. The TRIPOD-LLM reporting guideline for studies using large language models. Nat. Med. 31, 60–69 (2025).
Huttenlocher, D., Ozdaglar, A. & Goldston, D. Technical Report MIT Schwarzman College of Computing. https://computing.mit.edu/wp-content/uploads/2023/11/AIPolicyBrief.pdf (2023).
ChatGPT is a black box: how AI research can break it open. Nature 619, 671–672 (2023).
Kojima, T., Gu, S. S., Reid, M., Matsuo, Y. & Iwasawa, Y. Large language models are zero-shot reasoners. Adv. Neural Inf. Process. Syst. 35, 22199–22213 (2022).
Wang, L. et al. Plan-and-Solve Prompting: Improving Zero-Shot Chain-of-Thought Reasoning by Large Language Models. In Proc. 61st Annual Meeting of the Association for Computational Linguistics, 2609–2634 (ACL, 2023).
Shanahan, M., McDonell, K. & Reynolds, L. Role play with large language models. Nature 623, 493–498 (2023).
Bsharat, S. M., Myrzakhan, A., Shen, Z. Principled instructions are all you need for questioning LLaMA-1/2, GPT-3.5/4. Preprint at https://arxiv.org/abs/2312.16171 (2023).
Weng, Y. et al. Large Language Models are Better Reasoners with Self-Verification. Findings of the Association for Computational Linguistics: EMNLP 2550–2575 (2023).
Li, J., Zhang, Q., Yu, Y., Fu, Q. & Ye, D. More Agents Is All You Need. Transactions on Machine Learning Research (ICLR, 2024).
Wang, X. et al. Self-Consistency Improves Chain-of-Thought Reasoning in Language Models. In Proc. Int. Conf. Learn. Represent. (ICLR 2023).
Dominguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 376, eabl5197 (2022).
Shireman, J. M. et al. Genomic analysis of human brain metastases treated with stereotactic radiosurgery reveals unique signature based on treatment failure. iScience 27, 109601 (2024).
Xie, E., Cai, Y., Liu, J., Cheng, L. & Shireman, J. ElliotXie/CASSIA: CASSIA 1.2.0 (v1.2.0). Zenodo https://doi.org/10.5281/zenodo.17261689 (2025).
Acknowledgements
This work was supported by NIH GM102756 (C.K.), NIH K08NS092895 (M.D.), and the IU Value Research grant (M.D.).
Author information
Authors and Affiliations
Contributions
E.X. conceived the study and led the method development, implementation, validation, and case study analyses. C.K. co-led the project. L.C. contributed to the method development and developed the RAG agent. L.C., Y.C. and J.L. contributed to case study analyses. M.D. collected the brain tumor samples and assisted with interpretation of results; J.S. processed the brain tumor samples for scRNA-seq profiling, contributed to method development, and assisted with interpretation of results from all case study analyses. C.M. contributed to figure preparation and visualization. C.K., E.X., L.C. and J.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jesper Tegner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, E., Cheng, L., Shireman, J. et al. CASSIA: a multi-agent large language model for automated and interpretable cell annotation. Nat Commun 17, 389 (2026). https://doi.org/10.1038/s41467-025-67084-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67084-x
