
Abstract
Functionalization of all three distinct C–H bonds (para, meta, and ortho) within a particular arene has remained a long-standing challenge in chemical synthesis. However, overcoming this challenge would significantly benefit medicinal chemistry and drug discovery research. It would also streamline lengthy synthetic pathways, making them more efficient for total synthesis and industrial applications. The present study aims to address the challenge of functionalizing all three distinct C–H bonds (para, meta & ortho) within diverse classes of arenes. Here, we report, a general concept demonstrating the ability of a single ligand framework to selectively activate all three electronically distinct C-H bonds (para, meta & ortho) of a given arene just by varying the additives under iridium catalysis. The proposed mechanistic aspects are validated by experimental results and DFT computations. In addition, the developed method enables the selective installation of a deuterium atom or other functional groups at any desired C–H bond of arenes using boron as the linchpin. Finally, the application of the developed method using a single ligand scaffold is demonstrated in the short synthesis of an indole-derived combretastatin molecule.
Similar content being viewed by others
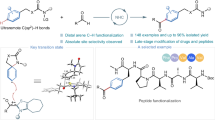
Introduction
Transition metal-catalyzed C-H bond functionalization1,2,3,4,5,6,7,8 is a key synthetic tool for selectively transforming specific C–H bonds9,10,11,12,13,14 (either ortho or meta or para) in aromatic compounds. Despite its significance, direct functionalization of different types of C–H bonds by a single catalyst remains a long-standing challenge in chemical synthesis. However, if successfully implemented, it could rapidly generate diverse molecular scaffolds, potentially advancing medicinal chemistry and drug discovery15 by streamlining multiple synthetic steps and exploring the molecular properties related to the drug discovery program16,17. For example, the different positional isomers of aspirin (o/m/p-NSOH-aspirins)18 exhibit strong inhibition of HT29 human colon cancer cell proliferation and possess unique characteristics that could be utilized in the development of potent drug candidates (Fig. 1A). Thus, to improve the structure activity relationship (SAR), functionalization of different C-H bonds of arenes in a regioselective manner would be highly beneficial. In this context, aromatic amides19, phenols20 and related molecular scaffolds that are important in biology and prevalent in living organisms, provide significant versatility for functionalization in organic chemistry21,22. These molecular scaffolds are crucial for developing biologically active molecules, including peptides, proteins, and drugs for cancer and infectious diseases, significantly impacting drug discovery, medicinal chemistry, and related fields. As a result, enabling C–H bond functionalization of these important molecular scaffolds would be a major development in chemical synthesis. But difficulties rely in designing an appropriate catalyst system for each type of C-H bond. For example, ortho C-H functionalization23,24,25,26,27,28 typically relies on the directing ability29 of the functional group that usually forms five- or six-membered cyclic transition state30 with the metal. In contrast, meta and para C–H functionalization31 require specific catalyst designs or substrate modifications for optimal geometry30,32. Moreover, the approaches for meta and para selective C–H bond functionalization33,34,35,36,37,38,39,40,41,42,43,44,45,46,47 often involve distinct strategies, such as substrate engineering or using different set of ligands/catalysts11. Consequently, for each type of C–H bond functionalization, a different set of ligands/catalysts is necessary because of the requirement of specific distance and geometry for preferred cyclic transition state13. Several approaches can drive remote C-H functionalization, including sterically controlled48,49, template design30, transient mediator50 or transient directing group24 attached with the substrate (Fig. 1B). However, these methods often require additional steps for substrate modification and the design of new templates. Besides these, there is no general method available to functionalize all three distinct C–H bonds (para–meta–ortho) of a given molecular scaffold or a pharmacophore. Typically, the activation of these different C–H sites necessitate distinct ligand scaffolds and optimized reaction conditions, owing to their intrinsic electronic properties and geometric constraints.

A Representative example for functionalization of all three C–H bonds of arene and their effects in medicinal chemistry; B Previous development of C-H functionalization; C This Work: One ligand–all three functionalizable C–H bonds; D Working model of all three functionalizable C–H bonds.
In this work, we address the aforementioned challenges by introducing a single ligand strategy, wherein site-selectivity can be precisely modulated through the rational variation of additives. This approach establishes a versatile platform that enables controlled activation of electronically distinct C–H bonds within a single catalytic framework (Fig. 1C). In the field of iridium-catalyzed C-H borylation8,51, ligands have been pivotal in achieving excellent reactivity, and site-selectivity. Innovative ligand designs are crucial for advancing borylation chemistry, requiring a diimine-type backbone (e.g., bipyridine) for Ir binding, and a noncovalent procreator for guiding selectivity. Drawing inspiration from diverse weak interactions52,53,54,55, we have developed 7-Aza-BPY (L1) for the iridium-catalyzed C-H borylation. We anticipated that azaindole being an electronically rich scaffold and bioisostere of indole ring56, could independently or synergistically with other additives facilitate the desired site-selectivity through noncovalent interactions within the realm of C-H borylation (Fig. 1D). Having remarkable importance of the catalytic C-H borylation57,58,59,60,61 in organic synthesis, here we report a concept by the design of a single ligand scaffold (L1: 7-Aza-BPY) that produces an efficient catalytic system, which selectively activate and functionalize all three distinct C–H bonds (para, meta & ortho). Notably, our strategy is versatile, spanning across a wide range of substrate classes, encompassing aromatic amides, phenols, and other diverse types of substrates.
Results
Reaction Development
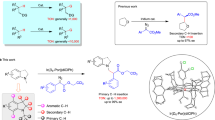
Initially, our comprehensive study using the developed ligand (L1: 7-Aza-BPY) in presence of [Ir(cod)(OMe)]2 with diverse classes of arenes revealed predominantly non-selective borylations (See Fig. S1 in SI for details). Subsequently, using aromatic amide (1a) as the model substrate, we introduced various additives. Consequently, we incorporated several alkali metal salts in quantities equivalent to the ligand loading (3.0 mol%). This revealed very surprising and intriguing results. The incorporation of various alkali metal salts, including KOtBu, NaOtBu, KOPiv, KOAc, NaOAc and KF was observed to exert a notable impact on the borylation reaction (entries 3-8, see Table S1 in SI for details) (Fig. 2A). Notably, the pronounced para-selective (82% para selectivity) borylation was achieved using KOtBu (3.0 mol%) with almost quantitative conversion (Fig. 2A, entry 3). Then, we were curious about the outcomes if we vary the loading of KOtBu under the same reaction conditions. Accordingly, an extensive KOtBu titration was carried out up to 19.0 mol% loading (Fig. 2B), which disclosed an unusual switching of selectivity between the para and meta C-H bonds of the substrate (1a). We observed from this titration experiment that while the best para selectivity (para/meta/ortho = 97/3/0) is obtained with 2.0 mol% KOtBu, the best meta selectivity (para/meta/ortho = 4/96/0) is achieved with 14.0 mol% KOtBu (Fig. 2B). After achieving notable switchable selectivity, we hypothesized that varying the KOtBu loading might form different in situ complexes, contributing to the switchable selectivities. Subsequently, inspired by Marder’s pioneering work62, we envisioned that B2pin2, in conjunction with KOtBu and ligand L1, alongside with iridium metal, might form diverse catalytic systems that are crucial for achieving flexible remote selectivity switches.

A Screening of different additives with amide as model substrate; B Titration with KOtBu and tuning of selectivity; C Initial investigation; D Catalytic activity of Complex-A (Optimized conditions for para borylation); E Development of ortho selective borylation; GC/MS ratios and conversions are given using dodecane as internal standard. aConversions and selectivities were measured by analysis of crude 1H NMR. See SI for details.
Following this hypothesis, we prepared Complex-A (Fig. 2C), which confirmed that the two boron atoms are completely different in nature (one boron atom is sp2, giving 37.9 ppm in 11B NMR and the other one is sp3 coordinated by tert-butoxide that resulted in 5.3 ppm in 11B NMR) (Fig. 2C). Next, to understand the role of Complex-A, we performed a control experiment using Complex-A as an additive instead of the KOtBu, which afforded 98% para selectivity that is even better than the outcomes of the 2.0 mol% KOtBu result (Fig. 2D). Thus, this control experiment indicated that the in situ generated Complex-A with lower KOtBu loading is primely responsible for the para selectivity. Hence, the para borylation was performed using Complex-A (1.3 mol%) as our optimized additive and the meta borylation was conducted using 14.0 mol% KOtBu as the optimized additive.
After conducting the borylation reaction employing various alkali metal salts, we then examined the borylation reaction across a spectrum of organic bases (Fig. 2E, see Table S4 in SI for details). We found that while the inorganic base (KOtBu) afforded remote para and meta selective borylation depending on the loadings of additive and developed ligand (L1), surprisingly, replacing the inorganic additive KOtBu with organic base DABCO (3.0 mol%, entry 1) changed the whole scenario from remote to proximal ortho selectivity (97% ortho selectivity with 86% conversion). In this study, we found that organic bases as additives differed significantly from inorganic bases in terms of outcomes. Notably, the inclusion of 3.0 mol% urotropine also yielded a remarkable 98% ortho-selectivity (entry 2). Seeking further enhancements, we conducted experiments by varying the loading of DABCO and urotropine. With 5.0 mol% DABCO, we achieved even higher ortho selectivity (99%) and better conversion (92%). Similarly, using 5.0 mol% urotropine led to comparable selectivity, albeit with a slightly reduced conversion (See SI for other additive screening).
Substrate scope: switchable C−H borylation of diverse classes of arenes
Achieving the optimized reaction conditions (Fig. 2) for the functionalization of all three distinct C–H bonds (para, meta & ortho) of arenes using a single ligand scaffold (L1: 7-Aza-BPY), we then studied the applicability of these reactions. Our initial focus involved exploring the viability of the developed conditions for arenes containing various unsubstituted aromatic amides (Fig. 3A). To our delight, the catalytic conditions (Conditions A–C) displayed remarkable efficiency for all three C–H bonds (para, meta, ortho) (entries 2a−2o) across a broad range of different amides with varying N-substitutions.

A Scope of unsubstituted arenes bearing numerous amide functionalities; B Scope of unsubstituted arenes bearing numerous other classes of functionalities; Reactions are carried out in 0.2 mmol scale. Isolated yields are reported based on the substrate as limiting agent. GC/MS ratios are given using dodecane as internal standard. aConversions and selectivities were measured by analysis of crude 1H NMR. bIsolated along with di-meta isomer. cAdditional di-meta isomer was found. nr = no reaction, nos = no ortho selectivity. For detailed isolation of the products and isomer ratios, see the Supporting Information.
Notably, our method’s effectiveness extended beyond N,N-di-substituted amides; even mono N-substituted amides (1p & 1q), which possess an available N-H proton, successfully underwent switchable borylation. Moreover, the method proved effective for homologous N,N-disubstituted amides (1r), resulting in switchable para and meta selective borylation. This inclusive applicability across various N-substituted amides underlines the potency of the developed approach, which relies on a singular ligand framework (L1) achieved through strategic additive interactions.
We then expanded the scope of the reaction using various other classes of substrates, with a specific focus on phenols. Among them, phenols bearing carbamate (1s-1u), phosphonate (1x) and phosphonamide (1z) displayed excellent reactivity under the developed conditions, affording switchable para, meta and ortho borylation (Fig. 3B). However, the substrates (1v & 1x) did not react under the ortho borylation conditions. Notably, our strategy extends beyond amides and phenols, demonstrating excellent tolerance for diverse substrates, such as 1-methyl-3-phenylimidazolidin-2-one (1y), wherein we achieved switchable C-H borylation of all three C-H bonds. Moreover, sulfonamides (1aa, 1bb) and phenyl(triisopropylsilyl)methanone (1cc) were also found to be well tolerated under our optimized reaction conditions.
We subsequently explored the potential of our approach to achieve switchable remote (para and meta) C-H borylation in multi-substituted aromatic amides (Fig. 4). We found that a large number of ortho-substituted aromatic amides smoothly underwent borylations affording switchable para and meta borylated products (entries 4a-4i). For instance, electronically diverse ortho substituents like Me, OMe, F, Cl, Br, CF3, OCF3, Ph, and p-tolyl, which can significantly alter the electronic nature of the substrates, resulted in para and meta borylated products with excellent regioselectivities. Similarly, the remote para and meta-C–H bonds of 2,5-, 2,3- and 2,6-di-fluoro-substituted substrates (3k, 3l & 3p) can selectively be functionalized without touching other C–H bonds. Sterically congested substrates (3m & 3n) were also found to be compatible to provide the switchable remote borylation. Notably, from a competitive study between two different functional groups in the same substrate (3o), it was observed that the amide functionality (91% para & 93% meta selectivity) is dominating over the ester group. Next, we were curious for the remote borylation for those substrates bearing a substituent at the para position (since, meta borylation in presence of a para-substituent is still an extraordinary challenge because of the steric reason). Accordingly, we performed the reactions under the developed meta borylation conditions and found that regardless of the substituents present at the para position of the arenes, all the substrates (3r-3x) afforded excellent meta borylation. Furthermore, the versatility of our strategy has been successfully demonstrated with various other classes of substituted arenes (Fig. 4). For instance, a wide range of electronically distinctive ortho-substituted arenes underwent borylation providing switchable para and meta products. Additionally, we extended our reaction to 3-substituted arenes and found that even for these substrates, switching of selectivity between meta and para can also be achieved. This further strengthens the broad applicability and effectiveness of our developed approach.

Reactions are carried out in 0.2 mmol scale. Isolated yields are reported based on the substrate as limiting agent. GC/MS ratios are given using dodecane as internal standard. aConversions and selectivities were measured by analysis of crude 1H NMR. bYields referred to the isolated materials with the para isomers. cBased on GC/MS analysis of the crude reaction mixture using dodecane as the internal standard. For detailed isolation of the products and isomer ratios, see the Supporting Information.
To this end, the method was examined further for the scope of ortho C–H bonds of substituted arenes (Fig. 5). For example, we first tested the borylation with the meta-substituted aromatic amides employing the same reaction conditions in presence of DABCO as additive, which afforded exclusively the ortho selective borylation (entries 5ao–5fo). Notably, for para-substituted aromatic amides, despite the presence of other competitive C–H bonds, the ortho borylation conditions were found to be so selective that it did not produce any other borylations (entries 5go–5no). Moreover, it has been observed that the reaction is highly selective for various 3,4-, 2,3-, 2,5-, 3,5- and 2-substituted aromatic amides (entries 5oo–5to). Here it should be mentioned that few substrates underwent ortho borylation in presence of urotropine instead of DABCO (entries 5qo–5to). Furthermore, we expanded the scope of ortho borylation for the 4-substituted carbamate-protected phenols that provided high ortho selectivity and yields of the products (entries 5uo–5zo).

Reactions are carried out in 0.2 mmol scale. Isolated yields are reported based on the substrate as limiting agent. GC/MS ratios are given using dodecane as internal standard. a10 mol% DABCO was used. b5 mol% DABCO was used. c10 mol% Urotropine was used. dConversions and selectivities were measured by analysis of crude 1H NMR. For detailed isolation of the products and isomer ratios, see the Supporting Information.
Synthetic Applications
Whereas the late-stage C–H functionalization17 is always an important synthetic application for the innovation of new drugs and drug-like molecules, but it is also challenging to selectively functionalize a particular C−H bond of a complex molecule. Pleasingly, our developed approach demonstrated that it can effectively be applied toward the late stage borylation of various molecules containing biologically important scaffolds (Fig. 6A). For example, we showed that even in the presence of so many similar C−H bonds, seasamol (6a), piperonylic acid (6b), mefenamic acid (6c), borneol (6d), naproxen (6e), monobenzone (6f) and tocopherol (6g) derived amides can be selectively activated to put a boron functionality at the desired position with high yields.

A Late-stage borylation; B Switchable deuteration and arylation, C Regioselective switchable borylation of Indole moiety; D Short synthesis of indole-derived combretastatin (tubulin inhibitor) using a single ligand (7-Aza-BPY); Reactions are carried out in 0.2 mmol scale for Fig. 6 A, B, C. For Fig. 6D, see SI for details. Isolated yields are reported based on the substrate as limiting agent. GC/MS ratios are given using dodecane as internal standard. aConversions and selectivities were measured by analysis of crude 1H NMR. See SI for details. bOrtho borylation result of 3j: conversion: 90%, selectivity (ortho/others = 99/01); two ortho isomers detected with C2/C6 = 30/70. cYields are based on isolated borylated products. For detailed isolation of the products and isomer ratios, see the SI.
The synthetic utilities of this approach have been further demonstrated by the diversification of the functionalized products. We first selected a substrate (3j) having a fluorine atom at the meta position, which is a challenging substrate for the borylation because of the presence of electronically indistinguishable C–H bonds that often produces a mixture of products. Our aim was to functionalize all three different types of C–H bonds of this substrate using the developed single ligand (L1: 7-Aza-BPY). Accordingly, we performed the reactions and found that all three C-H bonds of this substrate (3j) can selectively be activated and functionalized by installing a boron functionality, which eventually can further be functionalized by deuteration and arylation to achieve three different isomers in pure form (Fig. 6B). This application demonstrated the powerfulness of the developed strategy as well as the developed ligand framework (L1) that may find wide application in the context of the medicinal chemistry, to cut the long steps into shorter for the total synthesis, industrial deployment and other C–H bond functionalization chemistry.
For further application of our developed method using a single ligand (7-Aza-BPY), we targeted N-amide indole (16, due to the prevalence of C3-functionalized indole-containing drugs) for the regioselective borylation (Fig. 6C). We observed that N-amide indole (16) under our developed para borylation conditions yielded 93% C5 selectivity and complete (100%) C2 selectivity under ortho borylation conditions (Fig. 6C). Notably, while C5-substituted indoles are common in natural products, borylation at the C5 position is underexplored due to multiple challenges63. Moreover, achieving C2 borylation at such a sterically hindered position presents challenges. But our developed single ligand strategy has successfully demonstrated highly regioselective switchable proximal and remote borylation of N-amide indole moiety.
Finally, we have shown the application of our method for the short synthesis of an indole-derived combretastatin molecule64 using a single ligand L1 (7-Aza-BPY). We showed that this important molecule (23) can be synthesized following three consecutive C–H borylations as the key synthetic steps in good yield using our developed methods (Fig. 6D). For example, the first intermediate (20) can be achieved from N-amide indole (19, R = H) by the C3 borylation followed by the arylation. The second intermediate (21) can be prepared by the C5 selective borylation followed by a routine methoxylation in good yield. Subsequently, the third intermediate (22) can be prepared by the C2 selective borylation followed by the heteroarylation. The target molecule (23) can be achieved in 95% yield by a simple deprotection from the intermediate (22).
Preliminary mechanistic studies
Next, we focused our attention toward the preliminary understanding of the switchable selectivities of all three functionalizable C–H bonds (para, meta & ortho) by a single ligand scaffold (L1).
Origin of para selective borylation
After identifying the additive (Complex-A) (Fig. 2D) for the para borylation, we then became interested in to know the plausible intermediate generated in situ that might be responsible for the observed para selectivity. Since the Complex-A has two different types of boron atoms (sp2 and sp3) (Fig. 2C), thus we envisioned that the sp2 boron atom having a vacant p-orbital may engage in weak interaction (if so, then the boron value should be altered in 11B NMR) with either the ligand (L1) or with the substrate (1a). Thus, a control experiment was performed where stoichiometric ratio of L1 and Complex-A was stirred at room temperature in MTBE solvent that resulted in an unknown complex (Complex-B), which showed an upfield boron value at around 31.4 ppm from 37.9 ppm in 11B NMR (Fig. 7A). The upfield shift in the 11B NMR signal indicates that the boron atom is likely participating in a weak interaction with the ligand (L1) (see SI for more detailed control experiments).

A Role of Complex-A & aza indole ring in L1 to guide the para-selectivity; B Catalytic activity of Complex-B & Complex-A-II; C Ligand effect toward para selectivity; D Role of 7-aza nitrogen in L1.
Since the ligand L1 contains two distinct parts capable of interacting with the sp2 boron of Complex-A: a bipyridine unit and a 7-aza-indole unit, thus to verify the probable interacting site of the ligand (L1), we then performed an experiment between stoichiometric ratio of 2,2’-bipyridine and Complex-A. The absence of a boron NMR shift suggested that the bipyridine unit of L1 likely does not interact with the boron atom. Instead, it indicated that the 7-azaindole part of L1 may engage in a weak interaction. Moreover, it was also confirmed that the substrate (1a) does not coordinate with the sp2 boron of the Complex-A as similar set of reactions gave no shift in the boron NMR (Fig. 7A). Next, the catalytic activity of the Complex-B was tested by conducting a control experiment. To our delight, 90% para selectivity was observed, which further indicated the role of the in situ generated Complex-B toward para selectivity (Fig. 7B). For further understanding the role of the Complex-B, having two different boron atoms, we performed borylation using the Complex-A-II having no such sp2 boron atom, which resulted in nonselective reaction. This experiment indicated that the sp2 boron of Complex-A has a significant role for the para borylation. Similar observations have been found using other types of diboron complexes. Moreover, a detailed NMR experiment using 1-methyl-aza-indole and Complex-A at varied temperatures indicated the engagement of weak interaction between the Complex-A and the L1 ligand (See Fig. S53 in SI for details).
Next, we prepared two more ligands, such as: L4 (without the N atom at the 7-position and without π-electron) and L2 (reduced 7-azaindole ring), tested these ligands under our optimized reaction conditions and analyzed the experimental results along with the results of 2,2’-bipyridine ligand and the optimized ligand (L1) (Fig. 7C). We observed that in presence of the Complex-A, while the borylation using ligand 2,2’-bipyridine provided 42% para selectivity, attaching an octahydro indole unit (L4) also gave 42% para selectivity indicating its ineffectiveness toward a weak interaction with Complex-A. Interestingly, modifying the ligand structure from L4 to L2 that has an additional N atom at the 7-position, the para selectivity increased from 42% to 62%. This observation underscores the pivotal role of the nitrogen atom in enhancing para selectivity through a weak interaction. Remarkably, Ligand L1, differing only in π-electron density compared to Ligand L2, provided enormous improvement of the para selectivity from 62% to 98%, emphasizing the significance of the π-system of the 7-aza indole ring. Consequently, the progression of ligands from L4 to L2 to L1 indicates that the 7-aza-indole ring, with its nitrogen atom and electron-rich π-electron density, likely engages in a weak interaction with the non-coordinated sp2 boron atom of Complex-A, thereby directing the para selectivity. Furthermore, we noticed that without additive, all these ligands provided comparable para selectivity, which indicated the comparable combining electronic and steric effect of these ligands with the substrate (1a) (Fig. 7C). We also designed the ligands L5 and L6 to know the important role of the N atom at the specific 7-position. We envisioned that if the N atom at this specific 7-position has profound role for controlling the para selectivity, then moving the N atom around the ring should lead to a dramatic change in the para selectivity. Accordingly, we performed the reactions using these two ligands L5 (N atom at the 6-position) and L6 (N atom at the 4-position) and found that the para selectivity went down dramatically from 98% (L1) to 54% (L5) to 40% (L6), which is comparable to the result with the 2,2’-bipyridine (42% para selectivity) (Fig. 7D). Based on all aforementioned control experiments, we conclude that, a weak interaction between sp2 boron atom of Complex-A and the 7-azaindole unit of L1 may be the guiding factor for such enormous para selectivity. Though the control experiments revealed some intriguing phenomena, the complicity of the reaction prevented us to determine a precise mechanism. Several models have been explored through DFT calculations, but none was found to adequately explain the para selectivity, which is the matter of our future studies.
Origin of Meta Borylation
To gain insight into the reaction mechanism of meta borylation, at first, we performed reactions between KOtBu and B2pin2, incrementally adding KOtBu (1.0 to 5.0 equivalent) that resulted in only Complex-A, confirmed by 11B-NMR (Fig. 8A). Despite an excess of KOtBu, only a monoadduct was isolated, indicating limited or non-existent formation of a dianionic sp3-sp3 adduct due to the unfavorable charge buildup and diminished Lewis acidity of the Complex-A, excluding higher coordination boron complexes in the reaction mixture. Subsequently, we conducted a stoichiometric experiment involving Complex-B and KOtBu (Fig. 8B). Upon incremental addition of KOtBu to Complex-B, it was observed from the 11B NMR that the interaction between Complex-A and 7-azaindole, as present in Complex-B, progressively diminished. This led to a shift in the upfield boron signal of Complex-B from 31.4 ppm to around 37–38 ppm, which corresponds to the signal observed for Complex-A.

A KOtBu titration with B2pin2; B KOtBu titration with B2pin2 and L1 by 11B NMR; C Authentic Complex-A-II; D Control experiments with Complex-A-II having one boron atom: Role of K+ ion; E Role of K+ ion; F Role of bipyridine unit to guide meta selectivity; G Ligand effect toward meta selectivity; H DFT-computed C-H activation transition stated for meta borylation: Computed at SMD(iPr2O)-M06/6-311+G(d,p)[SDD for Ir]//SMD(iPr2O)-M06/6-31 G(d)[LANL2DZ for Ir] level of theory. Relative Gibbs free energies at 298 K, 1 atm are given in kcal/mol. Distances are given in Angstroms. Atom colors: gray, C; white, H; blue, N; red, O; pink, B; purple, K; brown, Ir; I Support of primary origin of meta selectivity.
This shift was associated with a change in selectivity from non-selective (at 4.0 mol% KOtBu) to meta-selective (at 14.0 mol% KOtBu, 99% meta selectivity) as indicated by the KOtBu titration experiment (Fig. 2B). This stoichiometric experiment (Fig. 8B) revealed that at higher concentration of KOtBu (conditions for meta borylation), the interaction within Complex-B is lost. For further understanding the role of the higher concentration of the K+ ion, we performed borylations at varied concentration of the Complex-A-II having only one sp3 boron atom (Fig. 8C). We postulated that if the origin of the meta selectivity is solely dependent on the concentration of the K+ ion, then meta selectivity should be enhanced at higher concentration of the K+ ion. Following this postulation, we conducted a series of experiments altering the amount of Complex-A-II, and found that upon increasing the concentration of the K+ ion, meta selectivity increased drastically, which indicated that the origin of the meta selectivity solely dependent on the higher concentration of the K+ ion (Fig. 8D). Next, we performed our optimized reaction in presence of 18-crown-6 and observed that the borylation underwent nonselective (Fig. 8E), which further supports the active participation of K+ ion.
Next, we investigated the effects of ligands under our meta borylation conditions. Since our optimized ligand L1 comprises two components: a bipyridine unit and an aza indole ring linker—we began by examining the effects of each component individually at higher concentrations of KOtBu. For para borylation, the outcomes with or without 1.3 mol% Complex-A (indicative of very low K+ concentration) were similar for the 2,2’-bipyridine ligands, which suggests that the bipyridine unit in L1 does not significantly influence para selectivity. In contrast, under meta borylation conditions with higher KOtBu concentrations, the 2,2’-bipyridine ligand exhibited a marked effect. Specifically, the meta selectivity increased from 59% without the additive to 79% in the presence of 14 mol% KOtBu. Furthermore, the introduction of 18-crown-6, which scavenges K+, resulted in a decreased selectivity of 60% for 2,2’-bipyridine (Fig. 8F). These findings indicate that higher concentrations of K+ ions play a crucial role in enhancing meta selectivity with the bipyridine core. Next, we conducted control experiments with two more ligands: L4 (containing an octahydro indole unit with bipyridine) and L2 (a reduced version of L1). It was observed that regardless of linker structure L2, L4 and other bipyridine based ligands provided similar selectivities to 2,2’-bipyridine, indicating that these linkers do not have a special role for meta selectivity (See Figure S70 in SI for details). However, the incorporation of a 7-azaindole unit into the bipyridine framework (L1) significantly enhanced the meta selectivity from 79% to 96%, indicates that the 7-azaindole unit is also essential in attaining the optimal selectivity for the meta borylation (Fig. 8G). Based on the collective evidences, we realized that the π-system of the bipyridine core and the 7-azaindole ring in L1 may interact with higher concentrations of K+ ions, resulting in the optimal meta selectivity for L1.
To elucidate the origin of meta-selectivity and the role of K+ ion in a comprehensible manner, we next took help of DFT calculations. Within the framework of our DFT calculations, C-H activation transition states based on the classical Ir(III)/Ir(V) mechanism61,65,66 were located (Fig. 8H). In this computational model, cation-π interaction and dispersion interaction between ligand skeleton and Complex-A-binded substrate are predominant, which are likely to contribute most to the 2.1 kcal/mol of energetic preference for meta-borylation. The ligand N atom also binds to Complex-A in the case of L1, which is believed to be a secondary factor to enhance the regioselectivity. Several variants of this model also lead to meta-selectivity (See Figure S96 in SI for details). Moreover, the predicted interactions by the DFT calculations keep largely unchanged when add Complex A-II instead of Complex-A, which is consistent with the experimental findings (Fig. 8D).
Notably, after identifying the primary (K+– π) and secondary (K+–N) interactions by DFT computations, we were curious about the outcomes of the meta borylation by moving the position of the N atom from the original ligand structure (L1). We envisioned that moving the N atom around the aza-indole ring should not lead to a dramatic change in the meta selectivity as here the primary origin of meta selectivity will remain intact i.e., the proposed bipyridine π–K+ interaction and substrate·Complex-A-bipyridine dispersion. Accordingly, the meta borylation reactions were performed with the ligands L5 (N atom at the 6-position) and L6 (N atom at the 4-position) and observed no dramatic change in meta selectivity (L1: 96%, L5: 77%, L6: 79%), which is comparable to the result with 2,2’-bipyridine (79% meta selectivity) (Fig. 8I). The slight decrease in meta selectivity in L5 and L6 may be attributed to the diminishing influence of the secondary governing factor of meta selectivity: K+–N interaction, present in L1.
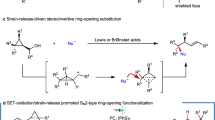
Mechanistic Studies of Ortho Borylation
Next, we aimed to understand the role of the developed ligand scaffold (L1: 7-Aza-BPY) for the DABCO-mediated ortho borylations and thus performed a series of control experiments in presence of DABCO (Fig. 9A). We observed that while the 4,4’-substituted ligands (dtbpy) afforded no ortho borylation, the ligand (bpy) without any substitution resulted in 65% ortho selectivity, although the conversion was moderate relative to the 7-Aza-BPY ligand (L1). Interestingly, incorporation of an aryl group to the 5-position of bpy (L8) remarkably improved the ortho selectivity (though the conversion was still moderate). Moreover, the rigid ligand systems (L9 & L10) gave no ortho borylations.

A Role of L1 ligand for ortho borylation; B DFT supported rollover cyclometallation; C DFT computed mechanism for ortho borylations; D Mechanistic evidences of rollover cyclometallation with model ligand L8.
These results indicated that in presence of DABCO, the mode of metal binding may be different (N,C-Ir binding) than the standard C–H borylation reactions with N,N-Ir binding, which reminded us our previously reported7 C–H ortho borylation via the generation of a monoanionic ligand, as well as other reports in the literature on N,C-Ir binding67,68. (A series of control experiments were performed with several other ligands; See Fig. S71 in SI for details). To gain further insight into reaction mechanism, a detailed DFT calculations was conducted. The results revealed that in the presence of DABCO, the neutral bipyridine ligand-bearing INT0 is transformed into the anionic ligand-bearing INT1 that eventually leads to the directed ortho-borylation (Fig. 9B). We have located a transition state representing such transformation as TS0, which gives a barrier of only 23.5 kcal/mol. Therefore, INT0 can effectively be transformed to INT1 before meta-/para-borylation take place (for meta-borylation via TS1-1, ΔG‡ = 27.2 kcal/mol). Owing to its (sp3)-N lone pair, the additive DABCO exerts strong coordination to Ir, which is likely to allow the ligand to undergo roll-over cyclometallation. After the active catalyst becomes INT1, exclusive directed ortho-borylation will occur (Fig. 9C), without forming any meta- or para- borylated products (See Fig. S94 in SI for details). For bipyridine as ligand, some meta- and para-borylation remained as competitive pathways (o:m:p = 65:23:12 as observed), while for the optimum ligand L1, the transformation was efficient enough for exclusive ortho-selectivity.
To validate the generation of monoanionic ligand (N,C-Ir binding) and to identify the active catalysts of the ortho-selective reaction, we conducted a stoichiometric reaction between the L8 ligand, [Ir(cod)OMe]2 and B2pin2 in presence of DABCO in THF-d8 (Fig. 9D). To our delight, we have detected the ligand borylated intermediate that was also characterized by spectroscopic data and further isolated it as the arylated product. Importantly, the ligand borylation can only be accounted if the reaction follows via the rollover cyclometallation i.e., N,C-Ir binding. The generation of the ligand borylated product serves as conclusive evidence for rollover cyclometallation and the N,C-Ir binding. Our stoichiometric reactions also revealed that in presence of DABCO, a rollover cyclometallation (formation of a five-membered metallacycle, which is the most common metallacycle for iridium-catalyzed borylation) occurs from the standard Ir(tris)(boryl)complex. This transformation proceeds via intramolecular C–H activation, accompanied by the release of HBpin through the reductive elimination, leading to the formation of INT1-Ph, consistent with our computational analysis (Fig. 9B). Subsequently, in the presence of the substrate, exclusive directed ortho-borylation takes place, as corroborated by detailed computational studies (Fig. 9C). Experimentally, we found that the released HBpin is consumed by DABCO, forming a DABCO•HBpin adduct, which plays a crucial role in driving the reaction forward. Confirmation of the DABCO•HBpin adduct’s presence is achieved through thorough analysis, including 1H NMR, 11B NMR, and 1D selected NOESY experiments. We have additionally conducted numerous control experiments to further solidify our proposed mechanism of ortho-borylation through N,C-Ir binding (See the Mechanistic Investigations for the switchable ortho selective borylation section in SI for details).
Discussion
In summary, we have developed a general method where all three different types of C–H bonds (para, meta & ortho) of diverse classes of arenes can be functionalized using a single ligand scaffold (7-Aza-BPY). The designed ligand directed to the development of all three modes of C–H bond activation and functionalization for the rapid diversification of a wide number of arenes. It has also been demonstrated that a single ligand scaffold in presence of three different additives can produce a robust catalytic system that can be used directly to functionalize all three C–H bonds without the requirement of substrate engineering. The mechanistic details have been discussed both by experiments and DFT computations. The utility of the method has been demonstrated by the late-stage C–H bond functionalization of various molecules containing biologically important scaffolds including the replacement of all three C–H bonds toward the C–D bonds and C–C bonds. Moreover, it has been shown that the method can be applied toward the short synthesis of an indole-derived combretastatin molecule by using sequential –C3, –C5, and –C2 borylation/arylation using our developed methods. Further developments of the present method for the C–H bond activation using this ligand are underway. Considering the predominance of bipyridine ligand frameworks in the transition metal catalysis, we anticipate that the developed ligand scaffold (7-Aza-BPY) will also empower reactivity and selectivity in various other catalytic C–H bond functionalization reactions.
Methods
Experimental procedure: para C-H borylation
In an argon-filled glove box, a 5.0 mL Wheaton microreactor was charged with Complex-A (0.9 mg, 1.3 mol%), ligand L1 (1.6 mg, 3.0 mol%) and dry MTBE (2.0 mL) sequentially. The reaction mixture was stirred for 5 minutes at room temperature. Then, B2pin2 (45.7 mg, 0.9 equiv.) was added to it and stirred for another 5 min. Thereafter [Ir(cod)OMe]2 (2.0 mg, 1.5 mol%) was added. Finally stirring for another 5 min substrate (0.2 mmol) was added. The microreactor was capped with a Teflon pressure cap and stirred at 40 °C for 12 h. After completion (judged by GC-MS), MTBE was removed under reduced pressure and chromatographic separation with silica gel gave the para borylated product.
Experimental procedure: meta C-H borylation
In an argon-filled glove box, a 5.0 mL Wheaton microreactor was charged with KOtBu (3.0 mg, 14.0 mol%), ligand L1 (1.6 mg, 3.0 mol%) and dry MTBE (2.0 mL) sequentially. The reaction mixture was stirred for 5 minutes at room temperature. Then B2pin2 (50.8 mg, 1.0 equiv.) was added to it and stirred for another 5 min. Thereafter [Ir(cod)OMe]2 (2.0 mg, 1.5 mol%) was added. Finally stirring for another 5 min substrate (0.2 mmol) was added. The microreactor was capped with a Teflon pressure cap and stirred at 40 °C for 12 h. After completion (judged by GC-MS), MTBE was removed under reduced pressure and chromatographic separation with silica gel gave the meta borylated product.
Experimental procedure: ortho C-H borylation
In an argon-filled glove box, a 5.0 mL Wheaton microreactor was charged with DABCO (1.2 mg, 5.0 mol%), ligand L1 (1.6 mg, 3.0 mol%) and dry MTBE (2.0 mL) sequentially. The reaction mixture was stirred for 5 min at room temperature. Then B2pin2 (101.6 mg, 2.0 equiv.) was added to it and stirred for another 5 min. Thereafter [Ir(cod)OMe]2 (2.0 mg, 1.5 mol%) was added. Finally stirring for another 5 minutes substrate (0.2 mmol) was added. The microreactor was capped with a Teflon pressure cap and stirred at 40 °C for 12 h. After completion (judged by GC-MS), MTBE was removed under reduced pressure and chromatographic separation with silica gel gave the ortho borylated product.
Data availability
All data, including experimental details, characterization data, NMR spectra are available in the Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Center, under deposition numbers CCDC 2295075 (L1) and CCDC 2294882 (L2). Coordinates of the optimized structures are provided as Supplementary Dataset 1. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C−H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010).
Davies, H. M. L., Bois, J. D. & Yu, J. Q. C–H functionalization in organic synthesis. Chem. Soc. Rev. 40, 1855–1856 (2011).
Sambiagio, C. et al. A comprehensive overview of directing groups applied in metal catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 47, 6603–6743 (2018).
Crabtree, R. H. & Lei, A. Introduction: CH activation. Chem. Rev. 117, 8481–8482 (2017).
Shilov, A. E. & Shul’pin, G. B. Activation of C−H bonds by metal complexes. Chem. Rev. 97, 2879–2932 (1997).
Rogge, T. et al. C–H activation. Nat. Rev. Methods Prim. 1, 43 (2021).
Hoque, M. E., Hassan, M. M. M. & Chattopadhyay, B. Remarkably efficient iridium catalysts for directed C(sp2)–H and C(sp3)–H borylation of diverse classes of substrates. J. Am. Chem. Soc. 143, 5022–5037 (2021).
Bisht, R. et al. Metal-catalysed C–H bond activation and borylation. Chem. Soc. Rev. 51, 5042–5100 (2022).
Davies, H. M. L. & Morton, D. Recent advances in C–H functionalization. J. Org. Chem. 81, 343–350 (2016).
Dalton, T., Faber, T. & Glorius, F. C–H Activation: toward sustainability and applications. ACS Cent. Sci. 7, 245–261 (2021).
Sinha, S. K. et al. Toolbox for distal C–H bond functionalizations in organic molecules. Chem. Rev. 122, 5682–5841 (2022).
Roudesly, F., Oble, J. & Poli, G. Metal-catalyzed C-H activation/functionalization: the fundamentals. J. Mol. Catal. A Chem. 426, 275–296 (2017).
Meng, G. et al. Achieving site-selectivity for C–H activation processes based on distance and geometry: a carpenter’s approach. J. Am. Chem. Soc. 142, 10571–10591 (2020).
Haldar, C., Hoque, M. E., Chaturvedi, J., Hassan, M. M. M. & Chattopadhyay, B. Ir-catalyzed proximal and distal C–H borylation of arenes. Chem. Commun. 57, 13059–13074 (2021).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Zhang, L. & Ritter, T. A perspective on late-stage aromatic C–H bond functionalization. J. Am. Chem. Soc. 144, 2399–2414 (2022).
Guillemard, L., Kaplaneris, N., Ackermann, L. & Johansson, M. J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 5, 522–545 (2021).
Vannini, F., Chattopadhyay, M., Kodela, R., Rao, P. P. N. & Kashfi, K. Positional isomerism markedly affects the growth inhibition of colon cancer cells by NOSH-aspirin: COX inhibition and modeling. Redox Biol. 6, 318–325 (2015).
de Mattos-Shipley, K. Greener amide bonds. Nat. Chem. Biol. 18, 238 (2022).
Scott, K. A., Cox, P. B. & Njardarson, J. T. Phenols in pharmaceuticals: analysis of a recurring motif. J. Med. Chem. 65, 7044–7072 (2022).
Sun, W. et al. Chemodivergent transformations of amides using gem-diborylalkanes as pro-nucleophiles. Nat. Commun. 11, 3113 (2020).
Bartolomei, B., Gentile, G., Rosso, C., Filippini, G. & Prato, M. Turning the light on phenols: new opportunities in organic synthesis. Chem. Eur. J. 27, 16062–16070 (2021).
Ros, A., Fernandez, R. & Lassaletta, J. M. Functional group directed C–H borylation. Chem. Soc. Rev. 43, 3229–3243 (2014).
Gandeepan, P. & Ackermann, L. Transient directing groups for transformative C–H activation by synergistic metal. Catal. Chem. 4, 199–222 (2018).
Rej, S., Ano, Y. & Chatani, N. An efficient tool in C–H bond functionalization chemistry for the expedient construction of C–C bonds. Chem. Rev. 120, 1788–1887 (2020).
Kawamorita, S., Ohmiya, H., Hara, K., Fukuoka, A. & Sawamura, M. Directed ortho borylation of functionalized arenes catalyzed by a silica-supported compact phosphine−iridium system. J. Am. Chem. Soc. 131, 5058–5059 (2009).
Boebel, T. A. & Hartwig, J. F. Silyl-directed, iridium-catalyzed ortho-borylation of arenes. A one-pot ortho-borylation of phenols, ar-ylamines, and alkylarenes. J. Am. Chem. Soc. 130, 7534–7535 (2008).
Ghaffari, B. et al. & Smith, M. R. Silyl phosphorus and nitrogen donor chelates for homogeneous ortho borylation catalysis. J. Am. Chem. Soc. 136, 14345–14348 (2014).
Engle, K. M., Mei, T., Wasa, M. & Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 45, 788–802 (2012).
Leow, D., Li, G., Mei, T.-S. & Yu, J.-Q. Activation of remote meta-C–H bonds assisted by an end-on template. Nature 486, 518–522 (2012).
Lam, N. Y. S. et al. Empirical guidelines for the development of remote directing templates through quantitative and experimental analyses. J. Am. Chem. Soc. 144, 2793–2803 (2022).
Bag, S. et al. Remote para-C–H functionalization of arenes by a D-shaped biphenyl template-based assembly. J. Am. Chem. Soc. 137, 11888–11891 (2015).
Zhang, Z., Tanaka, K. & Yu, J.-Q. Remote site-selective C–H activation directed by a catalytic bifunctional template. Nature 543, 538–542 (2017).
Bisht, R. & Chattopadhyay, B. Formal Ir-catalyzed ligand-enabled ortho and meta borylation of aromatic aldehydes via in situ-generated imines. J. Am. Chem. Soc. 138, 84–87 (2016).
Yang, L., Uemura, N. & Nakao, Y. Meta-selective C–H borylation of benzamides and pyridines by an iridium–lewis acid bifunctional catalyst. J. Am. Chem. Soc. 141, 7972–7979 (2019).
Yang, L., Semba, K. & Nakao, Y. Para-selective C−H borylation of (hetero)arenes by cooperative iridium/aluminum catalysis. Angew. Chem. Int. Ed. 56, 4853–4857 (2017).
Davis, H. J., Madalina, M. T. & Phipps, R. J. Ion pair-directed regiocontrol in transition-metal catalysis: a meta-selective C–H borylation of aromatic quaternary ammonium salts. J. Am. Chem. Soc. 138, 12759–12762 (2016).
Ramadoss, Y., Jin, S., Asako, S. & Ilies, L. Remote steric control for undirected meta-selective C–H activation of arenes. Science 375, 658–663 (2022).
Kuninobu, Y., Ida, H., Nishi, M. & Kanai, M. A meta-selective C-H borylation directed by a secondary interaction between ligand and substrate. Nat. Chem. 7, 712–717 (2015).
Saito, Y., Segawa, Y. & Itami, K. Para-C–H borylation of benzene derivatives by a bulky iridium catalyst. J. Am. Chem. Soc. 137, 5193–5198 (2015).
Chaturvedi, J., Haldar, C., Bisht, R., Pandey, G. & Chattopadhyay, B. Meta selective C−H borylation of sterically biased and unbiased substrates directed by electrostatic interaction. J. Am. Chem. Soc. 143, 7604–7611 (2021).
Chang, W. et al. Computationally designed ligands enable tunable borylation of remote C-H bonds in arenes. Chem 8, 1775–1788 (2022).
Wang, Y. et al. Diversification of aryl sulfonyl compounds through ligand controlled meta- and para-C-H borylation. Angew. Chem. Int. Ed. 61, e202206797 (2022).
Lu, S. et al. Para-selective C−H borylation of aromatic quaternary ammonium and phosphonium salts. Angew. Chem. Int. Ed. 61, e202201285 (2022).
Genov, G. R., Douthwaite, J. L., Lahdenperä, A. S. K., Gibson, D. C. & Phipps, R. J. Enantioselective remote C–H activation directed by a chiral cation. Science 367, 1246–1251 (2020).
Bastidas, J. R. M. et al. Para-selective, iridium-catalyzed C–H borylations of sulfated phenols, benzyl alcohols, and anilines directed by ion-pair electrostatic interactions. J. Am. Chem. Soc. 141, 15483–15487 (2019).
Hoque, M. E., Bisht, R., Haldar, C. & Chattopadhyay, B. Noncovalent interactions in Ir-catalyzed C–H activation: L-shaped ligand for para-selective borylation of aromatic esters. J. Am. Chem. Soc. 139, 7745–7748 (2017).
Cheng, C. & Hartwig, J. F. Rhodium-catalyzed intermolecular C–H silylation of arenes with high steric regiocontrol. Science 343, 853–857 (2014).
Cho, J. Y., Tse, M. K., Holmes, D., Maleczka, R. E. & Smith, M. R. Remarkably selective iridium catalysts for the elaboration of aromatic C-H bonds. Science 295, 305–308 (2002).
Shi, H., Herron, A. N., Shao, Y., Shao, Q. & Yu, J.-Q. Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 558, 581–585 (2018).
Hassan, M. M. M., Guria, S., Dey, S., Das, J. & Chattopadhyay, B. Transition metal–catalyzed remote C─H borylation: an emerging synthetic tool. Sci. Adv. 9, eadg3311 (2023).
Hobza, P. & Řezáč, J. Introduction: noncovalent Interactions. Chem. Rev. 116, 4911–4912 (2016).
Jin, M. Y. et al. Engineered non-covalent π interactions as key elements for chiral recognition. Nat. Commun. 13, 3276 (2022).
Yamada, S. Cation−π interactions in organic synthesis. Chem. Rev. 118, 11353–11432 (2018).
Fanourakis, A., Docherty, P. J., Chuentragool, P. & Phipps, R. J. Recent developments in enantioselective transition metal catalysis featuring attractive noncovalent interactions between ligand and substrate. ACS Catal. 10, 10672–10714 (2020).
Immadi, S. S. et al. Exploring 6-azaindole and 7-azaindole rings for developing cannabinoid receptor 1 allosteric modulators. Cannabis Cannabinoid Res. 3, 252–258 (2018).
Iverson, C. N. & Smith, M. R. Stoichiometric and catalytic B-C bond formation from unactivated hydrocarbons and boranes. J. Am. Chem. Soc. 121, 7696–7697 (1999).
Ishiyama, T. et al. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions, and isolation of a potential intermediate. J. Am. Chem. Soc. 124, 390–391 (2002).
Mkhalid, I. A. I., Barnard, J. H., Marder, T. B., Murphy, J. M. & Hartwig, J. F. C−H Activation for the construction of C−B bonds. Chem. Rev. 110, 890–931 (2010).
Hartwig, J. F. Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev. 40, 1992–2002 (2011).
Boller, T. M. et al. Mechanism of the mild functionalization of arenes by diboron reagents catalyzed by iridium complexes. intermediacy and chemistry of bipyridine-ligated iridium trisboryl complexes. J. Am. Chem. Soc. 127, 14263–14278 (2005).
Pietsch, S. et al. Synthesis, structure, and reactivity of anionic sp2–sp3 diboron compounds: readily accessible boryl nucleophiles. Chem. Eur. J. 21, 7082–7099 (2015).
Prabagar, B., Yang, Y. & Shi, Z. Site-selective C–H functionalization to access the arene backbone of indoles and quinolines. Chem. Soc. Rev. 50, 11249–11269 (2021).
Medarde, M., Ramos, A. C. & Caballero, E. Peláez-Lamamié de Clairac, R., López, J. L., Grávalos, D. G. & Feliciano, A. S. Synthesis and pharmacological activity of diarylindole derivatives. Bioorg. Med. Chem. Lett. 9, 2303–2308 (1999).
Tamura, H., Yamazaki, H., Sato, H. & Sakaki, S. Iridium-catalyzed borylation of benzene with diboron. Theoretical elucidation of catalytic cycle including unusual iridium(V) intermediate. J. Am. Chem. Soc. 125, 16114–16126 (2003).
Green, A. G., Liu, P., Merlic, C. A. & Houk, K. N. Distortion/interaction analysis reveals the origins of selectivities in iridium-catalyzed C−H borylation of substituted arenes and 5-membered heterocycles. J. Am. Chem. Soc. 136, 4575–4583 (2014).
Kuleshova, O., Asako, S. & Ilies, L. Ligand-enabled, iridium-catalyzed ortho-borylation of fluoroarenes. ACS Catal. 11, 5968–5973 (2021).
Zhang, M., Wu, H., Yang, J. & Huang, G. A computational mechanistic analysis of iridium-catalyzed C(sp3)–H borylation reveals a one-stone–two-birds strategy to enhance catalytic activity. ACS Catal. 11, 4833–4847 (2021).
Acknowledgements
The authors thank their previous institute, Center of Biomedical Research (CBMR) and their present institute, Indian Institute Science Education and Research (IISER) Pune for providing research facility. We thank the High-Performance Computing Center (HPCC) of Nanjing University for doing the numerical calculations in this paper on its blade cluster system. This work was supported by SERB-CRG grant (CRG/2022/002733) and SERB-TETRA grant (TTR/2022/000007). J.D., S.G. and S.D. thank CSIR for their SRF and M.M.M.H. thanks for RA fellowship.
Author information
Authors and Affiliations
Contributions
B.C. & J.D. conceived the concept. J.D. and M.E.H. developed ligand. J.D. and M.M.M.H. established the reaction mechanism. J.D., M.M.M.H., S.G., and S.D. performed all other experimental works. J.M., Z.D., and Y.L. did the theoretical calculations. All authors contributed to writing and proofreading of manuscript and SI.
Corresponding authors
Ethics declarations
Competing interests
J.D., M.E.H., M.M.M.H., S.G., and B.C. declare that they have filled an Indian Patent (Patent Application No: 202311008149) and an international patent (Patent Application No: PCT/IN2024/050122) based on this work (including the designed ligands). S.D., J.M., Z.D., and Y.L. do not have any competing interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Das, J., Hoque, M.E., Hassan, M.M.M. et al. Iridium catalyzed regiodivergent functionalization of all three C–H bonds of arenes by a single ligand. Nat Commun 17, 451 (2026). https://doi.org/10.1038/s41467-025-67138-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67138-0
