
Abstract
Immune checkpoint inhibitors (ICIs) have improved survival in advanced non-small cell lung cancer (NSCLC), yet resistance remains a major challenge. Here, we report the results from cohort A of a multi-cohort, phase II, open-label trial [NCT04777084], evaluating the efficacy and safety of IBI318 (a bispecific anti-PD-1/PD-L1 antibody, 300 mg intravenous every 2 weeks) plus lenvatinib (a receptor tyrosine kinase inhibitor, 8 mg orally daily). Forty patients with advanced NSCLC and acquired resistance to first-line ICIs were enrolled and received at least 1 cycle of the study regimen. The primary endpoint of 12-week objective response rate was 40.0% (95% CI: 24.9–56.7), satisfying prespecified efficacy threshold. Secondary endpoints included other efficacy endpoints and safety. Median progression-free and overall survival were 6.9 months (95% CI: 4.8–9.5) and 18.2 months (95% CI: 10.7–29.1), respectively. The most common treatment-related adverse event was increased thyroid-stimulating hormone (n = 11). Grade ≥3 adverse events occurred in 12.5% (5/40) of patients. Further, we performed a post-hoc exploratory analysis and developed an XGBoost model (scPro-X) using scRNA-seq data from pre-treatment tumor samples to predict 12-week ORR identified CD4_Tfh_CXCL13 and CD8_Tex_CTLA4 as potential biomarkers predictive of response. These findings demonstrate that IBI318 plus lenvatinib exhibit promising clinical activity and a manageable safety profile in patients with advanced NSCLC.
Similar content being viewed by others

Introduction
For patients with non-small cell lung cancer (NSCLC) without actionable driver mutations, immune checkpoint inhibitors (ICIs), with or without chemotherapy, represent the current standard of care1. However, resistance to ICIs frequently arises, limiting their long-term efficacy. ICI resistance is generally categorized as either primary, where tumors fail to respond from the outset or acquired, where initial responses are followed by disease progression2. Over 60% of patients with NSCLC who showed an initial objective response to ICIs eventually develop acquired resistance, representing a critical unmet medical need3,4.
Second-line treatment options for patients with acquired resistance to ICI remain limited5. Chemotherapy, such as docetaxel, is currently recommended by the National Comprehensive Cancer Network and Chinese Society of Clinical Oncology guidelines, but offers only modest clinical benefit, with a median OS of 8–12 months6,7,8. Preclinical studies suggest that combining ICIs with anti-angiogenic agents may yield synergistic effects by enhancing CD8+ T cell infiltration, promoting macrophage polarization, and reducing immunosuppressive cell populations9,10,11. Our previous real-world study demonstrated that a history of ICI failure does not diminish the effectiveness of subsequent treatment with anlotinib combined with PD-1 blockade12. Recent phase III randomized trials, including CONTACT‑01, LEAP‑008, and SAPPHIRE, reported that combining ICIs with anti-angiogenic agents did not significantly improve clinical outcomes compared to docetaxel in patients with NSCLC who progressed after prior ICI therapy13,14,15. In contrast, a randomized phase II trial demonstrated that ramucirumab plus pembrolizumab improved OS in patients with ICI-refractory advanced NSCLC compared to standard of care16. A phase III trial evaluating this combination is currently ongoing. Additionally, the recently published phase 3 HARMONi-2 trial revealed that the PD-1/VEGF bispecific antibody ivonescimab significantly prolonged progression-free survival (median PFS: 11.1 vs. 5.8 months; HR 0.51, 95% CI 0.38–0.69; p < 0.0001) compared with pembrolizumab in treatment-naïve patients with PD-L1–positive advanced NSCLC17. These results highlight the potential of incorporating anti-angiogenic strategies with immunotherapy for NSCLC treatment. In parallel, bispecific immunotherapy antibodies have emerged as a promising therapeutic modality distinct from conventional ICIs. Ongoing exploratory studies, including AK104-IIT-018 and AK104-307, are investigating the efficacy of bispecific antibodies in combination with anti-angiogenic agents in ICI-refractory NSCLC. However, conclusive evidence is still lacking regarding the effectiveness of combining ICIs and anti-angiogenic therapy as a second-line treatment strategy for ICI-resistant NSCLC. In particular, the therapeutic potential of bispecific antibodies in this setting remains poorly understood and requires further investigation.
Bispecific antibodies have emerged as a novel modality that may overcome the limitations of conventional agents by targeting two distinct antigens simultaneously to enhance immune cell engagement and cytotoxicity. IBI318 is a recombinant bispecific IgG1 antibody that targets PD-1 and PD-L118,19. Unlike standard monoclonal ICIs, IBI318 disrupts PD-1/PD-L1 interactions while simultaneously bridging cytotoxic T-cells and tumor cells, potentially enhancing T-cell-mediated antitumor responses20,21,22. In humanized mouse tumor models reconstituted with human immune cells, IBI318 (LY3434172) demonstrated significantly enhanced and robust antitumor activity compared with monospecific PD-1 and PD-L1 antibodies, suggesting its potential efficacy in patients who developed resistance to anti-PD-1/PD-L1 therapies19.
Lenvatinib, a receptor tyrosine kinase inhibitor targeting VEGFR1–3 and FGFR1–4, has established anti-angiogenic and immunomodulatory properties23,24,25. By normalizing tumor vasculature and alleviating hypoxia-induced immune exclusion, VEGF inhibition through lenvatinib facilitates CD8⁺ T cell infiltration and activation, reduces immunosuppressive cell populations, and synergizes with ICIs to restore effective antitumor immunity9,26,27. These combined activities provide strong preclinical and translational evidence for its use in combination with ICIs, including bispecific antibodies. Given the complementary mechanism of action of IBI318 and lenvatinib, we hypothesized that their combination could overcome resistance to ICIs and improve clinical outcomes for patients with NSCLC who acquired resistance to ICI.
In this work, we evaluate the efficacy and safety of IBI318 combined with lenvatinib as a second-line treatment in patients with advanced NSCLC who developed acquired resistance to first-line ICI therapy. Using single-cell RNA sequencing (scRNA-seq) analysis, we compare the tumor microenvironment (TME) composition between responders and non-responders. We show that IBI318 plus lenvatinib achieves promising clinical efficacy with manageable safety. Moreover, we develop a single-cell proportion–based XGBoost model (scPro-X) that identifies potential biomarkers predictive of treatment response to IBI318 plus lenvatinib, providing insights for patient selection in clinical practice.
Results
Patients and treatment
Between December 2021 and June 2023, a total of 59 patients were screened, of whom 40 patients were enrolled in this study (Fig. 1). All enrolled patients had stage IV NSCLC with documented acquired resistance after initial clinical benefit from first-line ICI therapy. Detailed inclusion and exclusion criteria are provided in the Methods section. Baseline clinical and pathological characteristics of the study cohort are summarized in Table 1. The median age of the cohort was 59.5 years (range 35–75 years). The majority of the cohort were male (85.0%, n = 34), current or former smokers (75.0%, n = 30), and had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 1 (82.5%, n = 33). Brain metastases were detected in eight patients (20.0%), bone metastases in 14 patients (35.0%), and liver metastases in seven patients (17.5%). Histologically, 21 patients (52.5%) had squamous cell lung carcinoma, while 18 (45.0%) had lung adenocarcinoma. All patients received at least four cycles of first-line ICI-based combination regimen, with a median of 10 treatment cycles and an ORR of 67.5% (n = 27), including two patients achieving complete response (CR), 25 partial responses (PR), and 13 stable disease (SD). Among the 35 patients with available PD-L1 tumor proportion score (TPS) data from post-ICI-acquired resistance tumor samples, 70.0% had a TPS of <1%, 17.5% had 1–49%, and none had a TPS ≥ 50%. At the data cut-off (March 7, 2025), after a median follow-up duration of 23.9 months, the study achieved its primary endpoint, with one patient (2.5%) remaining on treatment. Among the 39 patients who discontinued therapy, 36 experienced disease progression, two (5%) discontinued due to adverse events (AEs), and one patient (2.5%) completed the full two-year course of therapy (Fig. 1B).

A Flow diagram depicting the overall study design. Created with BioGDP.com (https://biogdp.com/)54. B Flowchart outlining patient screening and treatment process. NSCLC non-small cell lung cancer, ORR objective response rate, BOR best objective response, DCR disease control rate, DOR duration of response, OS overall survival, PFS progression-free survival, Q2W every 2 weeks, QD quaquedie, once daily, AEs adverse events, TME tumor microenvironment.
Efficacy
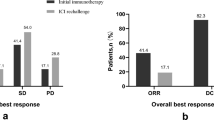
All enrolled patients received at least one cycle of investigational treatment. The 12-week ORR (primary endpoint) was determined to be 40.0% (16/40, 95% confidence intervals [CI]: 24.9–56.7) (Table 2). Specifically, 16 patients (40.0%) exhibited a PR, while 12 patients (30.0%) showed SD, and 12 patients (30.0%) had progressive disease (PD) (Fig. 2A, B). The best overall response rate (BOR) was 47.5% (19/40; 95% CI: 31.5–63.9) (Table 2 and Supplementary Fig. 1). The disease control rate (DCR) reached 90.0% (36/40; 95% CI: 76.3–97.2) (Table 2).

A Waterfall plot illustrating changes in tumor size relative to baseline, stratified by tumor response at approximately 12 weeks after treatment initiation (n = 39). Colors indicate the 12-week overall response. The hash symbol (#) indicates patients who experienced disease progression due to developing new lesions. One patient who died within 12 weeks after treatment initiation was not included in the plot. Dotted lines at +20% and −30% represent thresholds for progressive disease and partial response, respectively. B Bar plot summarizing the overall response rates at 12 weeks for the study cohort (n = 40). Source data are provided as a Source Data file.
The clinical characteristics and treatment outcomes of all 40 patients are presented in Fig. 3A. The median progression-free survival (PFS) was 6.9 months (95% CI: 4.8–9.5), with a 6-month PFS rate of 57.5% (95% CI: 40.8–71.0) (Fig. 3B). The median OS was 18.2 months (95% CI: 10.7–29.1; Fig. 3C). The median duration of response (DOR) was 8.5 months (95% CI: 4.3–10.8; Supplementary Fig. 2).

A Swimmer plot displaying the individual patient timelines, treatment duration, and clinical characteristics (n = 40). PR partial response, SD stable disease, PD disease progression, Colors denote clinical characteristics as indicated in the legends. Kaplan-Meier survival curves for progression-free survival (PFS) (B) and overall survival (OS) (C) in patients who received IBI318 plus lenvatinib as second-line treatment (n = 40). Shaded areas indicates the 95% confidence intervals (CI). Dotted lines mark the 6-month,12-month or 24-month survival milestones. Cross marks indicate censored data. The risk table below shows the number of patients included in the analysis per time point. Source data are provided as a Source Data file.
To assess factors associated with survival outcomes, univariate and multivariate Cox regression analyses were conducted, evaluating variables such as age, sex, smoking status, histology, ECOG performance status, PD-L1 TPS, and the presence of liver, brain, or bone metastases. Brain metastases emerged as a significant predictor of PFS. Specifically, in the multivariate analysis, patients with brain metastases demonstrated significantly worse PFS (adjusted hazard ratio [HR]: 4.962, 95% CI: 1.398–17.613) compared to those without brain metastases. In this cohort, no statistically significant associations were observed between survival outcomes and age, sex, histology, ECOG PS, or the presence of liver or bone metastases (Supplementary Fig. 3).
Safety
Treatment-related adverse events (TRAEs) were assessed in all 40 patients. Grade 3–4 TRAEs occurred in five patients (12.5%) (Table 3). Ten patients (25.0%) required temporary treatment interruption due to AEs (IBI318-related AE, n = 4 and lenvatinib-related AE, n = 6) and two patients (5%) permanently discontinued treatment.
The most frequently reported TRAEs were increased blood thyroid-stimulating hormone (n = 11, 27.5%), increased blood bilirubin (n = 8, 20.0%), infusion-related reaction (n = 8, 20.0%), and fatigue (n = 8, 20.0%). Additional TRAEs observed in ≥10% of the cohort included increased alanine aminotransferase (n = 7, 17.5%), increased aspartate aminotransferase (n = 5, 12.5%), rash (n = 5, 12.5%), decreased neutrophil count (n = 4, 10.0%), decreased blood thyroid-stimulating hormone (n = 4, 10.0%), hemoptysis (n = 4, 10%), and cough (n = 4, 10.0%). Grade ≥3 TRAEs included increased alanine aminotransferase, increased aspartate aminotransferase, headache, infusion-related reactions, and rash, each affecting one patient (2.5%).
scRNA-seq analysis identified key immune cell subsets associated with treatment response
As a post-hoc exploratory analysis to investigate immune correlates of response to IBI318 plus lenvatinib, scRNA-seq was performed on pre-treatment tumor biopsy samples from all 40 patients. After stringent quality control, a total of 581,212 cells were retained for further analysis. Eighteen distinct cell populations were identified based on canonical markers, including B cells (CD19, MS4A1), plasma cells (MZB1, CD79A), T cells and natural killer cells (NK cells, CD3D, CD3E, KLRF1), neutrophils (CSF3R, FCGR3B), macrophages (CD68, CD163), monocytes (LYZ), mast cells (TPSAB1, KIT), conventional type 1 dendritic cells (cDC1s, CLEC9A, XCR1), conventional type 2 dendritic cells (cDC2s, CD1C, FCER1A), plasmacytoid dendritic cells (pDCs, LILRA4, IL3RA), osteoclasts (CTSK, ACP5), endothelial cells (ECs, VWF, CLDN5), fibroblasts (COL1A1, LUM), mural cells (ACTA2, RGS5), alveolar epithelial cells (SFTPB, SFTPC), ciliated cells (PIFO, FOXJ1), cancer cells (EPCAM, KRT8), and club cells (SCGB3A1, SCGB1A1) (Fig. 4A and Supplementary Fig. 4A). Cancer cells were validated via copy number variation analysis using InferCNV, with numerous copy number gains and losses identified in cancer cells (Supplementary Fig. 4B).

A Uniform manifold approximation and projection (UMAP) plot showing the annotation of cell subsets from non-responders (NR, n = 24) and responders (R, n = 16). Different colors represent different cell types. B Stacked bar graph displaying the relative proportion of each cell type in patients from the NR and R groups. Colors represent different cell types. C Bubble plots illustrating the percentage of cells expressing PDCD1 and CD274 across different cell types. Bubble size indicates the proportion of cells expressing the gene, while color reflects the average expression level within each cell subset. UMAP plot illustrating the T cell/NK cell subsets (D) and myeloid cell subsets (E), each subset represented by a unique color. F Differential mRNA expression analysis of CD274 and PDCD1 across all annotated clusters. Clusters with adjusted p < 0.05 (Benjamini–Hochberg corrected Wilcoxon test) and average log₂ fold change > 0.2 were defined as high-expression clusters and are highlighted. Significantly upregulated clusters are shown in red. AECs alveolar epithelial cells, DC dendritic cell. Source data of are provided as a Source Data file.
Patients were stratified into responders (R group; patients with CR or PR) and non-responders (NR group, patients with SD or PD) based on their tumor response at approximately 12 weeks after initiating treatment. Responders exhibited a higher baseline proportion of T cells/NK cells and macrophages, and a lower proportion of epithelial cells (normal alveolar epithelial cells and cancer cells) compared with non-responders (Fig. 4B and Supplementary Fig. 4C). These findings suggest that TME composition may serve as a predictor of treatment response.
Given that IBI318 targets both PD-1 and PD-L1, gene expression of PDCD1 and CD274 was examined across cell types. PD-1 (PDCD1) was predominantly expressed in T cells/NK cells (19.2%), while PD-L1 (CD274) was mainly expressed in myeloid cells (8.8%) (Fig. 4C). Unsupervised clustering identified 23 T cell /NK cell subsets, including 8 subsets of CD4+ T cells (CD4_Naive/Tcm_SELL, CD4_Naive/Tcm_IL7R, CD4_Th1-like_CCL4, CD4_Tfh_CXCL13, CD4_Treg_FOXP3, CD4_Treg_TNFRSF4, CD4_IFI6, CD4_MKI67), 7 subsets of CD8+ T cells (CD8_Tem_GZMK, CD8_Tem_GZMA, CD8_Trm_ZNF683, CD8_Tex_CTLA4, CD8_Tex_TNFRSF9, CD8_IFIT2, CD8_MKI67), 6 NK cell subsets (NK_CX3CR1, NK_TNF, NK_XCL1, NKT_TRGC2, NK_FGFBP2, NK_KLRC1), and a subset of innate lymphoid cell (ILC3_IL4I1) (Fig. 4D and Supplementary Fig. 5A–C). Myeloid cells were divided into 13 cell subsets, including 2 subsets of neutrophils (Neutrophils_VEGFA, Neutrophils_CXCR2), 7 subsets of macrophages (Macrophages_SELENOP, Macrophages_CXCL1, Macrophages_SPP1, Macrophages_CHI3L1, Macrophages_FABP4, Macrophages_CXCL10, Macrophages_MKI67), 2 dendritic cell subsets (cDC1s, cDC2s), a monocyte subset (Monocytes_FCN1), and a subset of osteoclasts (Fig. 4E and Supplementary Fig. 5D). PD-1 (PDCD1) expression was enriched in CD4_Th1-like_CCL4, CD4_Tfh_CXCL13, CD8_Tex_CTLA4, and CD8_Tex_TNFRSF9 subsets of T cells/NK cells, while PD-L1 (CD274) expression was highest in Macrophages_CXCL10 in myeloid cells (Fig. 4F). These findings suggest that these immune cell populations may be primary targets of IBI318.
Subsequently, we developed a classification model, termed scPro-X (single‑cell Proportion‑based XGBoost model), to predict early treatment response in patients undergoing IBI318 plus lenvatinib therapy (Fig. 5A). This model utilizes the relative proportions of immune cell subsets identified from scRNA-seq data within tumor tissues as input features and outputs the objective response status at 12 weeks of treatment (responder vs. non-responder). Performance evaluation demonstrated that scPro-X achieved exceptional classification performance, with an area under the receiver operating characteristic curve (AUC) of 0.919, indicating high predictive accuracy (Fig. 5B). By comparison, a control model using the same algorithm with conventional PD-L1 TPS as the input feature yielded an AUC of only 0.534, which was markedly lower than that of scPro-X (Fig. 5B). These findings suggest that the immune cell composition derived from scRNA-seq data provides a substantially higher predictive accuracy compared to PD-L1 expression levels.

A Schematic overview of the single-cell proportion-based (scPro-X) model construction, which uses XGBoost to predict 12-week objective response rate (ORR) based on cluster proportion data derived from single-cell RNA sequencing (scRNA-seq). Model performance was compared to a conventional PD-L1 tumor proportion score (TPS)-based classifier. IHC immunohistochemistry, PD-L1 Programmed Cell Death Ligand 1, ROC Receiver Operating Characteristic, AUC Area Under the Curve. Created with BioGDP.com (https://biogdp.com/)54. B Receiver operating characteristic (ROC) curve comparing the predictive performance of scPro-X and PD-L1 TPS-based model (n = 40). C SHAP (SHapley Additive exPlanations) summary plot showing the top predictive cell clusters ranked by their impact on model output. Each dot represents a patient sample, colored by the relative abundance (feature value) of the corresponding cluster. Clusters with high PDCD1 or CD274 mRNA expression are color-coded accordingly (n = 40).
To further interpret the model’s decision-making process and identify key features driving its predictions, we employed SHAP (SHapley Additive exPlanations) analysis to assess feature importance within the scPro-X model (Fig. 5C). The analysis identified 11 cell subsets that significantly contributed to the model’s predictive power. Among these, VEGA highly expressed neutrophils (Neutrophils_VEGFA) and endothelial cells (Endothelial_cells), which are associated with angiogenesis, were notable contributors. Importantly, three immune cell subsets with high SHAP values and elevated expression of IBI318 target molecules—CD8_Tex_CTLA4, CD4_Tfh_CXCL13, and Macrophages_CXCL10—were identified as key predictive features. Further subgroup analysis of clinical responses revealed that the abundance of CD4_Tfh_CXCL13 and CD8_Tex_CTLA4 subsets was significantly higher in responders compared to non-responders (p < 0.05; Supplementary Fig. 6), suggesting their potential functional roles in mediating the response to IBI318 plus lenvatinib therapy. In summary, the scPro-X model demonstrates robust predictive performance while also identifying several immune cell subsets with potential mechanistic significance. These findings provide a theoretical basis for the discovery of potential biomarkers and offer valuable insights into the mechanisms underlying the efficacy of IBI318 combined with lenvatinib.
Multiplex immunofluorescence confirms differential TME cell composition between treatment responders and non-responders
To corroborate our results of scRNA-seq analysis, we conducted Multiplex Immunofluorescence (mIHC) on pre-treatment tumor samples within our cohort. As illustrated in Fig. 6A, B, mIHC revealed the presence of CD4_Tfh_CXCL13 (CD4 + CD3 + BCL6 + CXCL13+), CD8_Tex_CTLA4 (CD8 + CD3 + CTLA4+) in the tumor tissue of patients who responded to treatment. All these cell subsets expressed PD-1, consistent with our scRNA-seq analysis (Fig. 6A, B and Supplementary Figs. 7, 8). The quantitative analysis of mIHC revealed an elevated proportion of these cells within the pre-treatment TME of responders compared to non-responders (p < 0.05, Fig. 6C, D). Additionally, we also detected the presence of Macrophages_CXCL10 based on mIHC, which exhibited PD-L1 expression (Supplementary Fig. 9). These findings corroborate the results obtained from scRNA-seq analysis, suggesting that CD4_Tfh_CXCL13, CD8_Tex_CTLA4 may be associated with therapeutic response and could serve as potential predictive biomarkers.

Representative hematoxylin-eosin (HE)-stained (Scale bar = 500 μm and Scale bar = 800 μm) and multiplex immunofluorescence (mIF) micrographs of CD4_Tfh_CXCL13 and CD8_Tex_CTLA4 in pre-treatment tissue specimens from a patient who experienced partial response with IBI318 plus lenvatinib (A) and a typical non-responder (B). Dotted circles on HE-stained images (top panels) highlight regions of interest corresponding to mIF views shown in the lower panels (Scale bar = 20 μm). Red or white arrowheads indicate cells of the identified subsets. Insets in the top-right corner of mIF images provide magnified views of selected regions to enhance visualization of identified cells. Different colors denote different cell markers. C Box plots comparing median CD4_Tfh_CXCL13 cell counts across three ROIs per patient in pre-treatment tissues from responders (R, n = 5) and non-responders (NR, n = 4). D Box plots comparing median CD8_Tex_CTLA4 cell counts across three ROIs per patient in pre-treatment tissues from responders (R, n = 5) and non-responders (NR, n = 5). The “box” shows the interquartile range (IQR) between the 25th and 75th percentiles, and the “center line” marks the median. Whiskers extend to the minimum and maximum values. P-values were calculated using a two-sided Wilcoxon rank-sum test without adjustment. Source data are provided as a Source Data file.
Discussion
In patients with advanced NSCLC who developed acquired resistance following first-line PD-1/PD-L1 inhibitor therapy, current second-line treatment options offer limited clinical benefit. There remains a critical need for more effective therapeutic strategies to improve prognosis in this patient population8. In this prospective phase II clinical trial, we evaluated the efficacy and safety of IBI318 combined with lenvatinib as a second-line treatment for patients previously treated with ICIs. Our findings demonstrate that the combination of IBI318 and lenvatinib confers meaningful clinical benefits, including improved survival outcomes, with a favorable and manageable safety profile. The study met its primary endpoint, achieving a 12-week ORR of 40.0%, which notably exceeds the response rates reported in prior studies evaluating PD-1 or PD-L1 inhibitors in combination with anti-angiogenic agents in previously ICI-treated NSCLC (ORR: 11.3–15.3%)13,28,29. Furthermore, the PFS and OS outcomes observed in this study compare favorably to those reported in similar patient populations (mPFS: 2.5–4.6 months, mOS: 10.7–14.5 months)13,16,28,29. Differences in tumor response and survival outcomes between studies may reflect variability in drug mechanisms of action and differences in the patient population.
To the best of our knowledge, this is the first attempt to combine a PD-1/PD-L1 bispecific antibody with an anti-angiogenic agent to overcome acquired resistance in NSCLC. Ruan et al. reported an ORR of 45.5% with IBI318 monotherapy in treatment-naïve NSCLC. However, among ten patients who had progressed after prior ICI therapy, none achieved a PR, with only three patients experienced SD30. This contrasts with our findings and suggests that the combination with lenvatinib may contribute to improved efficacy in patients with ICI-resistant NSCLC. Notably, our study exclusively enrolled patients with resistance to first-line ICIs, whereas Ruan et al’s cohort included a higher proportion (60%) of patients with resistance to later-line ICIs, which may further account for differences in outcomes.
The safety profile of IBI318 plus lenvatinib was manageable, with no unexpected TRAEs observed. The incidence of TRAEs (82.5%) was lower than that reported in other studies involving lenvatinib combined with PD-1/PD-L1 inhibitors in other cancer types31,32,33. For comparison, any-grade TRAEs were observed in 96.8% of patients with endometrial cancer treated with lenvatinib plus pembrolizumab in the KEYNOTE-146 study33, 97.0% of patients with various solid tumors treated with lenvatinib plus pembrolizumab in another phase Ib/II, multicenter, open-label study31, and 96.1% of patients with melanoma treated with lenvatinib plus pembrolizumab in the LEAP-004 study34. This difference in AE rates may be partially attributed to the lower dose of lenvatinib (8 mg/day) used in our study compared to higher doses in other trials, and to potential synergistic effects between IBI318 plus lenvatinib. As expected, the most common TRAEs were generally consistent with the known safety profiles of PD-1/PD-L1 inhibitors and lenvatinib and were generally manageable30. However, a relatively higher proportion of patients in our cohort experienced elevations in aspartate aminotransferase and alanine aminotransferase levels, suggesting the potential for hepatic toxicity, which warrants careful monitoring during treatment.
The TME plays a pivotal role in modulating immune response to ICIs35,36. Using scRNA-seq, we explored the TME to identify cellular subsets associated with clinical response to the IBI318 and lenvatinib combination. Our analysis revealed that responders exhibited a higher proportion of T cells/NK cells and a lower proportion of epithelial cells within their TME at baseline. Anti-PD1/PD-L1 therapies function by disrupting PD-L1−PD-1 interactions on activated or exhausted cytotoxic T cells, thereby restoring T cell-mediated tumor killing37,38. Additionally, NK cells contribute to the antitumor effects of PD-1 blockade39. Increasing evidence supports the predictive value of specific immune cell subsets within the pre-treatment TME for responses to ICIs40,41. Our results indicate that elevated levels of CD4_Tfh_CXCL13 and CD8_Tex_CTLA4— subsets that express PD-1—are associated with favorable treatment outcomes. These findings are consistent with prior studies showing that increased infiltration of T follicular helper (Tfh) cells correlates with better responses to anti-PD-1 therapy42. Tfh cells are direct targets of immune checkpoint blockade, and upon activation, they promote anti-tumor immunity by enhancing the functions of B cells and cytotoxic T lymphocytes through IL-21 and IL-4 secretion43,44. Similarly, exhausted CD8+ T cell subsets have also been associated with ICI response across multiple cancer types41,45,46. For example, Chen et al. identified CD8_Tex_LAYN, a CD8+ T cell exhaustion subset, as a predictive biomarker of ICI response in colorectal cancer, noting its enrichment in responders’ pre-treatment TME and its reduction following treatment46. Although our study did not identify this specific CD8+ T cell subset, other exhausted CD8+ T cell populations were identified to be associated with treatment efficacy. These differences may stem from variations in treatment regimens and tumor types.
This study has several limitations. It was a single-arm trial with a relatively small sample size, which may constrain the generalizability of our findings. The sample size of 40 patients was pre-determined using PASS software, considering 80% statistical power, a one-sided alpha of 0.05, and a 15% dropout rate. While this sample size was sufficient for a phase II study designed to detect an initial efficacy signal, larger, randomized phase III trials with appropriate control arms (such as docetaxel or other standard second-line therapies) are necessary to validate these findings. Although both univariate and multivariate Cox regression analyses were performed to identify clinical factors associated with PFS and OS in patients treated with IBI318 plus lenvatinib, the limited sample size necessitates cautious interpretation, particularly for non-significant associations. These may reflect insufficient statistical power rather than a true absence of relationships. Although our single-cell analysis provided valuable insights by identifying several key immune and stromal cell subsets associated with treatment response, confirmatory evidence and mechanistic investigations to delineate the underlying biological mechanisms were not undertaken. Several factors known to influence ICI efficacy—such as disruptions in double-stranded (ds) RNA and/or dsDNA sensing, interferon signaling, PD-L1 expression, and tumor metabolic reprogramming—were outside the scope of this study47,48. Further research is needed to investigate these mechanisms and their roles in the response to IBI318 plus lenvatinib. Furthermore, given the potential of IBI318 to reshape immune–tumor cell interactions by altering their spatial organization, future studies incorporating spatial transcriptomics will be critical to more comprehensively elucidate its mechanism of action.
In conclusion, our findings support the clinical activity and safety of IBI318 in combination with lenvatinib as a second-line treatment for patients with NSCLC who have developed acquired resistance to first-line ICI therapy. The dual-targeting approach, complemented by insights from TME profiling, offers a promising path forward in addressing resistance mechanisms. While preliminary, these results justify further investigation in larger trials to validate efficacy, clarify the immunologic mechanisms at play, and refine patient selection through biomarker discovery.
Methods
Ethics statement
This study complies with all relevant ethical regulations regarding research involving human participants. The study protocol was approved by the Institutional Review Board of Hunan Cancer Hospital (Ethics Approval No: CXSL1800119). The study design and conduct were performed in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines. Written informed consent was obtained from all participants prior to enrollment.
Study design and interventions
This phase II, open-label, single-center, non-randomized prospective cohort study evaluated the efficacy and safety of IBI318 in combination with lenvatinib in patients with advanced NSCLC lacking actionable driver mutations and exhibiting acquired resistance to first-line PD-1/PD-L1 inhibitor therapy. The trial was registered on ClinicalTrials.gov on March 2, 2021 (NCT04777084, https://clinicaltrials.gov/search?cond=Lung%20Cancer&term=NCT04777084) and was conducted at Hunan Cancer Hospital. All patients provided written informed consent before study enrollment (Dec 21, 2021 to Jun 21, 2023), including consent for the publication of deidentified medical data. The full study protocol is available in the Supplementary Information.
Patients received IBI318 (300 mg) intravenously every 2 weeks (Q2W, day 1 of each cycle) and lenvatinib (8 mg) orally once daily until treatment discontinuation due to disease progression, death, intolerable toxicity, withdrawal of informed consent, initiation of a new antineoplastic therapy, or other protocol-specified reasons. The maximum treatment duration was 2 years. Tumor imaging assessments, based on Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, were performed before initiating treatment, every 6 weeks during the first 24 weeks of treatment, and every 8 weeks thereafter. If radiological progression was suspected, treatment was discontinued following a second assessment. If PD was not confirmed, study treatment continued per protocol-specified assessment intervals.
Participants and sample collection
Eligible patients were aged between 18 and 75 years, with histologically or cytologically confirmed locally advanced (stage IIIB-IIIC) or metastatic (stage IV) NSCLC. All patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0−1 and at least one measurable lesion per the RECIST version 1.1. Key inclusion criteria required documented acquired resistance following first-line ICI therapy, defined by all of the following: (1) Receipt of anti-PD-1 /PD-L1 monotherapy or combination therapy in the first-line setting, with no additional immunotherapies; (2) A best response of CR, partial response, or SD lasting ≥6 months with first-line ICI-based treatment; (3) Radiologically confirmed disease progression during or after the most recent treatment. Key exclusion criteria included a diagnosis of small cell lung cancer (SCLC), including mixed SCLC/ NSCLC histology. All patients with lung adenocarcinoma submitted biological samples for DNA-based next-generation sequencing (NGS) prior to enrollment, and only those who were not detected with actionable oncogenic alterations were included in the study. Detailed inclusion and exclusion criteria are provided in the study protocol (see Supplementary Information). The biological sex (female/male) of patients was recorded. Gender identity was not collected. Written informed consent for the publication of clinical information, including potentially identifiable data, was obtained from all patients. Measures were taken to safeguard confidentiality and minimize the risk of patient identification.
Pre-treatment tumor samples were collected via needle biopsy and stored in GEXSCOPE tissue preservation solution for scRNA-seq.
Endpoints and assessment
The primary and secondary endpoints were assessed by the investigators in accordance with RECIST version 1.1. The primary endpoint was the 12-week ORR, defined as the proportion of subjects achieving a CR or PR approximately 12 weeks after treatment initiation. This timing was selected based on typical response kinetics of immunotherapy. Secondary endpoints included BOR, PFS, OS, DCR, DOR, and safety. BOR was defined as the proportion of participants who achieved CR or PR as the best overall response during the study. PFS was defined as the time from treatment initiation to the first radiologically confirmed PD or death. OS was defined as the time from treatment initiation to death from any cause. DCR was defined as the percentage of patients whose best response was CR, PR, or SD. DOR was defined as the interval from the first documented response (CR or PR) to disease progression or death. Safety was assessed based on the incidence and severity of TRAEs, classified according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 5.0). The exploratory endpoint assessed the association between tumor response and TME characteristics. Patients were classified according to their objective response status at 12 weeks: CR or PR as responders, and SD or PD as non-responders.
Tissue dissociation and preparation
Human tumor tissue samples were procured from patients at Hunan Cancer Hospital. Fresh tissue samples were obtained via needle biopsy before IBI318 treatment initiation. All patients provided written informed consent for tissue sampling and research use. Fresh tumor samples were promptly stored in sCelLive™ Tissue Preservation Solution (Singleron) on ice within 30 min of biopsy. Samples were washed with Hank’s Balanced Salt Solution (HBSS), minced, and enzymatically digested with 3 mL of sCelLive™ Tissue Dissociation Solution (Singleron) using the Singleron PythoN™ Tissue Dissociation System at 37 °C for 15 min. The resulting cell suspension was filtered through a 40-micron sterile strainer. Red blood cells were lysed using GEXSCOPE® red blood cell lysis buffer (RCLB, Singleron) at a 1:2 ratio for 5–8 min at room temperature. The mixture was centrifuged at 300 × g for 5 min at 4 °C, the supernatant was discarded, and the cell pellet was gently resuspended in phosphate-buffered saline (PBS). Cell viability was assessed using trypan blue exclusion under a microscope. All tissue samples have been exhausted during single-cell sequencing and related experiments, and are therefore no longer available for sharing with other researchers.
scRNA-seq library preparation and sequencing
Single-cell suspensions were adjusted to 2 × 10⁵ cells/mL with PBS (HyClone) and loaded onto microwell chips using the Singleron Matrix® Single Cell Processing System. Barcoded beads captured messenger RNA for reverse transcription into complementary DNA, which was subsequently amplified by polymerase chain reaction, fragmented, and ligated with sequencing adapters. ScRNA-seq libraries were prepared using the GEXSCOPE® Single Cell RNA Library Kit (Singleron), following the manufacturer’s protocol49. The individual libraries were diluted to a final concentration of 4 nM, pooled, and sequenced on an Illumina NovaSeq 6000 platform with 150 bp paired-end reads.
Initial processing of raw sequencing data
Raw sequencing reads were processed using the CeleScope (version 1.14.0, Singleron Biotechnologies), following standard parameters. Barcodes and unique molecular identifiers were extracted and corrected from R1 reads. R2 reads were trimmed of adapter sequences and poly (A) tails and aligned to the human reference genome GRCh38 (hg38) transcriptome using STAR aligner (version 2.6.1a). Reads uniquely mapped to exons were quantified using FeatureCounts (version 2.0.1), and reads with matching cell barcode, UMI, and gene were aggregated to generate the gene expression matrix.
scRNA-seq data processing
Gene expression matrices were processed with Scanpy (version 1.8.1, https://github.com/scverse/scanpy) in Python (version 3.7, https://www.python.org/)50. Quality control excluded: (1) cells within the top 2% of UMI count; (2) cells with fewer than 200 genes or in the top 2% of gene count; (3) cells with mitochondrial gene content greater than 30%; and (4) genes expressed in fewer than 5 cells. The raw counts were normalized by total counts per cell and log-transformed to generate a normalized data matrix. Highly variable genes (top 2000) were identified using the “Seurat” method. Principal Component Analysis was performed on a scaled variable gene matrix, and the top 22 principal components were used for clustering and dimensionality reduction.
Cell clustering was performed using the Louvain algorithm with a resolution parameter set at 1.2. T cells/NK cells and myeloid cells were separately extracted and re-clustered, adhering to the same procedures and employing clustering resolutions of 1.8 and 1.2, respectively. Uniform Manifold Approximation and Projection was used for visualization. Harmony (version 1.0, https://github.com/immunogenomics/harmony) corrected batch effects51. Seurat (version 4.0.4, https://github.com/satijalab/seurat) was used for visualization45.
scRNA-seq-based copy number detection
To identify cancer cells among cell clusters, large-scale chromosomal copy number variations were inferred using R package InferCNV (version 1.2.1, https://github.com/broadinstitute/inferCNV). Transcriptional profiles of non-malignant cells (fibroblast, mural cells, endothelial cells, ciliated cells, alveolar epithelial cells, and club cells) served as references.
Construction and comparison of predictive models
scPro-X model construction
We developed a predictive model, termed Single‑cell Proportion‑based XGBoost model (scPro-X), to estimate the 12‑week tumor response to IBI318 plus lenvatinib therapy using baseline scRNA‑seq data. For each patient, the relative proportions of immune cell subsets were computed and used as input features for the model. An XGBoost binary classifier (logistic objective function) was trained with the following hyperparameters: a learning rate (η) of 0.05, a maximum tree depth of 3, a subsampling ratio of 0.7, and a column sampling ratio of 0.8. The training process included a total of 1000 boosting iterations, with an early stopping criterion applied when model performance failed to improve over 20 consecutive rounds. Threefold cross-validation was used to determine the optimal number of boosting rounds, after which the final model was retrained on the entire dataset using the identified optimal parameters. Model construction was carried out using the xgboost package (version 1.7.7.1, https://github.com/dmlc/xgboost) in R.
SHAP analysis
To enhance interpretability and quantify the contribution of individual immune cell clusters to model predictions, SHAP (SHapley Additive exPlanations) values were calculated. These values provided insights into the relative importance of each feature in determining predictive outcomes. The SHAP values were visualized using the SHAPforxgboost package (version 0.1.3, https://github.com/liuyanguu/SHAPforxgboost) in R.
Model comparison
The performance of the scPro-X model was evaluated by comparing it to a comparator model constructed using PD-L1 TPS as input feature. Both models employed the same XGBoost algorithm and training procedures for consistency. Receiver operating characteristic curves were generated for both models, and the AUC was calculated to measure predictive performance. All ROC analyses were conducted using the pROC package (version 1.18.0, https://github.com/cran/pROC) in R.
Multiplex immunofluorescence (mIF) staining and analysis
Formalin-fixed paraffin-embedded lung cancer tissues (3–5 μm) were processed using Opal Polaris™ 7-color Manual immunohistochemistry Kit (Akoya Biosciences) following the manufacturer’s recommendation. After deparaffinization, rehydration, and epitope retrieval in citrate buffer, slides were stained with antibodies. The mIF panel included DAPI (Abcam, ab104139, 1:1000), anti-CD4 (Abcam, ab133616, 1:300), anti-CD3 (Abcam, ab16669, 1:200), anti-PD-1(Abcam, ab237728, 1:200), anti-CD8 (CST, 70306S, 1:200), anti-BCL6 (MAB, 0746, ready-to-use working solution), anti-CXCL13 (Proteintech, 10927-1-AP, 1:200), anti-CD163 (Abcam, ab156769, 1:150), anti-CD68 (Abcam, ab192847, 1:200), anti-CXCL10 (AP, 10937, 1:400), anti-PD-L1 (Abcam, ab237726, 1:200) and anti-CTLA4 (CST, 5360T, 1:100). Secondary antibody incubation was performed at room temperature for 10 min.
Hematoxylin-eosin (HE)-stained slides were scanned with the Olympus VS200. mIF imaging was conducted using the Polaris imaging system (Akoya Vectra Polaris, Institute of Clinical Pathology, West China Hospital, Sichuan University) under appropriate fluorescent filters for a multispectral microscope. Three regions of interest (ROIs) of uniform size and shape were selected per tumor sample for cell quantification. The number of positive cells within each ROI was identified and counted using the QuPath (version 0.4.3)52. Median cell counts across the three ROIs per patient were computed for subsequent statistical comparisons. Cells were classified as follows: CD4_Tfh_CXCL13 (co-expression of CD4, CD3, BCL6, CXCL13, and PD-1), CD8_Tex_CTLA4 (co-expression of CD8, CD3, CTLA4, and PD-1) and Macrophage_CXCL10 (co-expression of CD68, CD163, CXCL10, and PD-L1).
Statistical analysis
The primary endpoint was 12-week ORR. The sample size determination for this study was based on the primary hypothesis that IBI318 plus lenvatinib would achieve a clinically meaningful improvement in the 12-week ORR compared to historical data from second-line chemotherapy, which reported an ORR of 12%53. The study was powered to detect a 30% ORR, an improvement considered both clinically significant and indicative of the efficacy of the combination therapy. Based on these parameters, the sample size was calculated to provide 80% power with a one-sided alpha of 0.05, while accounting for a potential 15% dropout rate, resulting in a requirement of 40 patients. Sample size was calculated using PASS software (version 15.0, NCSS LLC, Kaysville, Utah, USA).
Statistical analyses were conducted for scRNA-seq data, mIF and clinical trial outcomes employing conventional methodologies, as detailed below. Categorical variables were summarized as frequencies. Continuous variables were reported as mean ± standard deviation or median and range, depending on data distribution. To investigate differences in the TME between responders (CR and PR) and non-responders (SD and PD) to IBI318 plus lenvatinib, statistical comparisons of patient-level data were performed. Specifically, group comparisons for continuous variables were performed using Wilcoxon rank-sum tests or t-tests, as appropriate, while categorical variables were compared using chi-square or Fisher’s exact tests.
All efficacy analyses were conducted in the intention-to-treat population, defined as all patients who received at least one cycle of the study treatment. Kaplan-Meier was used to estimate the median PFS, OS, and DOR, along with corresponding 95% CI. The 95% CI for 12-week ORR, BOR, and DCR were calculated using the Clopper–Pearson method.
HRs and their 95% CI were calculated using Cox proportional hazards models to assess the association of treatment outcomes with relevant covariates. Both univariate and multivariate Cox proportional hazards models were employed to explore differences in PFS or OS among subgroups receiving IBI318 plus lenvatinib.
All statistical analyses and data visualizations were performed using R (version 4.0.3, http://www.R-project.org/), Python (version 3.7, https://www.python.org/), and GraphPad Prism (version 9.0.2, https://www.graphpad.com/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Patient data remains confidential in accordance with privacy protection protocols. The complete study protocol and statistical analysis plan are provided in the Supplementary Information. Researchers seeking access to anonymized participant datasets should direct their requests to the corresponding author, including a detailed justification. All requests will undergo evaluation by the principal investigator site and sponsoring organization to ensure adherence to confidentiality requirements, with responses provided within an 8-week timeframe. Before any data release, applicants must execute a formal data-sharing agreement with the study sponsor. The raw single-cell RNA-seq data generated in this study have been deposited in the Genome Sequence Archive (GSA) of the National Genomics Data Center (NGDC), Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, under accession code HRA014481. These data are available under controlled access due to privacy restrictions related to patient consent. Access is granted to principal investigators from non-profit research institutions for research purposes only. Access applications can be submitted via the GSA-human portal following their guidelines. Requests are typically reviewed and responded to within 1 month. Once access is granted, the data will be available for download for a period of 1 month. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
Code availability
This study utilized established computational methods without developing novel algorithms. All custom scripts employed for data processing, statistical analysis, and figure generation are deposited at https://github.com/ZhaohuiRuan/IBI318_len_clinical_trial.
References
Arbour, K. C. & Riely, G. J. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: a review. JAMA 322, 764–774 (2019).
Schoenfeld, A. J. & Hellmann, M. D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37, 443–455 (2020).
Memon, D. et al. Clinical and molecular features of acquired resistance to immunotherapy in non-small cell lung cancer. Cancer Cell 42, 209–224 e209 (2024).
Antonia, S. J. et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 20, 1395–1408 (2019).
Schoenfeld, A. J. et al. Clinical definition of acquired resistance to immunotherapy in patients with metastatic non-small-cell lung cancer. Ann. Oncol. 32, 1597–1607 (2021).
Auclin, E. et al. Second-line treatment outcomes after progression from first-line chemotherapy plus immunotherapy in patients with advanced non-small cell lung cancer. Lung Cancer 178, 116–122 (2023).
Auclin, E. et al. OA07.06 second line treatment outcomes after progression on immunotherapy plus chemotherapy (IO-CT) in advanced non-small cell lung cancer (aNSCLC). J. Thorac. Oncol. 17, S22 (2022).
Park, S. E. et al. Increased response rates to salvage chemotherapy administered after PD-1/PD-L1 inhibitors in patients with non-small cell lung cancer. J. Thorac. Oncol. 13, 106–111 (2018).
Lee, W. S., Yang, H., Chon, H. J. & Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 52, 1475–1485 (2020).
Zhao, S. et al. Low-dose apatinib optimizes tumor microenvironment and potentiates antitumor effect of PD-1/PD-L1 blockade in lung cancer. Cancer Immunol. Res 7, 630–643 (2019).
Shigeta, K. et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology 71, 1247–1261 (2020).
Zhang, X. et al. Anlotinib combined with PD-1 blockade for the treatment of lung cancer: a real-world retrospective study in China. Cancer Immunol. Immunother. 70, 2517–2528 (2021).
Neal, J. et al. CONTACT-01: a randomized phase III trial of atezolizumab + cabozantinib versus docetaxel for metastatic non-small cell lung cancer after a checkpoint inhibitor and chemotherapy. J. Clin. Oncol. 42, 2393–2403 (2024).
Borghaei, H. et al. SAPPHIRE: phase III study of sitravatinib plus nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. Ann. Oncol. 35, 66–76 (2024).
Leighl, N. B. et al. LEAP-008: lenvatinib plus pembrolizumab for metastatic NSCLC that has progressed after an anti PD-(L)1 plus platinum chemotherapy. J. Thorac. Oncol. 20, 1489–1504.
Reckamp, K. L. et al. Phase II randomized study of ramucirumab and pembrolizumab versus standard of care in advanced non-small-cell lung cancer previously treated with immunotherapy-lung-MAP S1800A. J. Clin. Oncol. 40, 2295–2306 (2022).
Xiong, A. et al. Ivonescimab versus pembrolizumab for PD-L1-positive non-small cell lung cancer (HARMONi-2): a randomised, double-blind, phase 3 study in China. Lancet 405, 839–849 (2025).
Li, T., Niu, M., Zhou, J., Wu, K. & Yi, M. The enhanced antitumor activity of bispecific antibody targeting PD-1/PD-L1 signaling. Cell Commun. Signal 22, 179 (2024).
Kotanides, H. et al. Bispecific targeting of PD-1 and PD-L1 enhances T-cell activation and antitumor immunity. Cancer Immunol. Res. 8, 1300–1310 (2020).
Huehls, A. M., Coupet, T. A. & Sentman, C. L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 93, 290–296 (2015).
Kontermann, R. E. Dual targeting strategies with bispecific antibodies. MAbs 4, 182–197 (2012).
Labrijn, A. F., Janmaat, M. L., Reichert, J. M. & Parren, P. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 18, 585–608 (2019).
Ruan, R. et al. Unleashing the potential of combining FGFR inhibitor and immune checkpoint blockade for FGF/FGFR signaling in tumor microenvironment. Mol. Cancer 22, 60 (2023).
Lee, C. H. et al. Lenvatinib plus pembrolizumab in patients with either treatment-naive or previously treated metastatic renal cell carcinoma (Study 111/KEYNOTE-146): a phase 1b/2 study. Lancet Oncol. 22, 946–958 (2021).
Butterfield, L. H. & Najjar, Y. G. Immunotherapy combination approaches: mechanisms, biomarkers and clinical observations. Nat. Rev. Immunol. 24, 399–416 (2024).
Zhang, Y. et al. VEGFR2 activity on myeloid cells mediates immune suppression in the tumor microenvironment. JCI Insight 6, e150735 (2021).
Geindreau, M., Ghiringhelli, F. & Bruchard, M. Vascular endothelial growth factor, a key modulator of the anti-tumor immune response. Int. J. Mol. Sci. 22, e150735 (2021).
Reck, M. et al. P1.15-05 nintedanib in combination with Nivolumab in pretreated patients with advanced adenocarcinoma of the lung (AIO-TRK-0117 Phase IB/II Trial). J. Thorac. Oncol. 17, S121 (2022).
He, K. et al. MRTX-500 phase 2 trial: sitravatinib with nivolumab in patients with nonsquamous NSCLC progressing on or after checkpoint inhibitor therapy or chemotherapy. J. Thorac. Oncol. 18, 907–921 (2023).
Ruan, D. Y. et al. The first-in-class bispecific antibody IBI318 (LY3434172) targeting PD-1 and PD-L1 in patients with advanced tumors: a phase Ia/Ib study. J. Hematol. Oncol. 17, 118 (2024).
Taylor, M. H. et al. Phase IB/II trial of lenvatinib plus pembrolizumab in patients with advanced renal cell carcinoma, endometrial cancer, and other selected advanced solid tumors. J. Clin. Oncol. 38, 1154–1163 (2020).
Pal, S. K. et al. Cabozantinib in combination with atezolizumab for advanced renal cell carcinoma: results from the COSMIC-021 study. J. Clin. Oncol. 39, 3725–3736 (2021).
Makker, V. et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. J. Clin. Oncol. 38, 2981–2992 (2020).
Arance, A. et al. Phase II LEAP-004 study of lenvatinib plus pembrolizumab for melanoma with confirmed progression on a programmed cell death protein-1 or programmed death ligand 1 inhibitor given as monotherapy or in combination. J. Clin. Oncol. 41, 75–85 (2023).
Bejarano, L., Jordao, M. J. C. & Joyce, J. A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11, 933–959 (2021).
Jin, M. Z. & Jin, W. L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target Ther. 5, 166 (2020).
Yi, M. et al. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol. Cancer 21, 28 (2022).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 128, 4654–4668 (2018).
Liu, B., Zhang, Y., Wang, D., Hu, X. & Zhang, Z. Single-cell meta-analyses reveal responses of tumor-reactive CXCL13(+) T cells to immune-checkpoint blockade. Nat. Cancer 3, 1123–1136 (2022).
Liu, B. et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 3, 108–121 (2022).
Baumjohann, D. & Brossart, P. T Follicular helper cells: linking cancer immunotherapy and immune-related adverse events. J. Immunother. Cancer9, e002588 (2021).
Hollern, D. P. et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell 179, 1191–1206 e1121 (2019).
Sánchez-Alonso, S., et al. A new role for circulating T follicular helper cells in humoral response to anti-PD-1 therapy. J. Immunother. Cancer 8, e001187 (2020).
Zhang, Y. et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593 e1578 (2021).
Chen, Y. et al. Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer. Cancer Cell 42, 1268–1285.e1267 (2024).
Pitt, J. M. et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and extrinsic factors. Immunity 44, 1255–1269 (2016).
Alsaafeen, B. H., Ali, B. R. & Elkord, E. Resistance mechanisms to immune checkpoint inhibitors: updated insights. Mol. Cancer 24, 20 (2025).
Dura, B. et al. scFTD-seq: freeze-thaw lysis-based, portable approach toward highly distributed single-cell 3’ mRNA profiling. Nucleic Acids Res. 47, e16 (2019).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Horn, L. et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, Phase III trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 35, 3924–3933 (2017).
Jiang, S. et al. Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 53, D1670–d1676 (2025).
Acknowledgements
We are deeply grateful to the patients and their families for their participation and trust in this study. The authors also acknowledge the generous support provided by Innovent Biologics, Inc. and Xian Sheng Biotechnology Co., Ltd. We thank Li Li, Fei Chen, and Chunjuan Bao (Institute of Clinical Pathology, West China Hospital, Sichuan University) for their assistance with histological staining. We are especially grateful to Qun Zeng and Christoph Scheiermann (University of Geneva, Switzerland) for their valuable insights and guidance on the study design. We also thank Dr Analyn Lizaso (Weber Shandwick HK) for her editing assistance. We acknowledge Bioinformatics Center, Furong Laboratory and Bioinformatics Center, Xiangya Hospital, Central South University for support. This work was financially supported by the Natural Science Foundation of China (grant numbers: 82222048 to Y.C.Z., 82173338 to Y.C.Z., 82102747 to Q.Q.X., 82303629 to W.J.J., and 82160489 to Q.Q.X.), and the Key Basic Research Project of Qinghai Provincial Department of Science and Technology (2022-ZJ-733 to Q.Q.X.). The funding agencies had no role in the study design, data collection, analysis, interpretation, manuscript writing, or the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
Y.C.Z., N.Y., and L.Z. were responsible for conceptualizing and designing the study. Y.C.Z., N.Y., L.Z., W.J.J., F.X.Z., L.L., Y.X., Z.W., J.C.D., Z.H., S.D.X., L.X.S., Y.Q.C., Y.D.Z., S.Y.W., S.M.C., J.D., X.Z., Y.Z.S., D.Y., C.H.Z., H.Y.Y., Y.Z.L., L.D., Q.Q.X., C.F., J.W., T.L., G.Z., H.Z. and X.C. contributed to patient recruitment, treatment, and the collection of clinical data related to efficacy and safety. L.Z., Z.H.R., H.Y., and H.Y.Q. conducted the analysis of clinical data and performed statistical evaluations. Z.Z. and C.Z. carried out the experiments and associated data analyses. Z.H.R., H.Y., and H.Y.Q performed Bioinformatic analyses. Y.C.Z., N.Y., and L.Z. oversaw the execution of the experiments. All authors contributed to data interpretation, had full access to the study data, participated in drafting or critically reviewing the manuscript, and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
H.Z. is employed by Innovent Biologics (Suzhou) Co., Ltd. The other authors report no conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Nikolaj Frost, Peng Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, L., Ruan, Z., Yan, H. et al. PD-1/ PD-L1 bispecific antibody IBI318 combined with lenvatinib in advanced non-small cell lung cancer with acquired resistance to immune checkpoint inhibitors: a phase II trial. Nat Commun 17, 567 (2026). https://doi.org/10.1038/s41467-025-67262-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67262-x
