
Abstract
CO2 hydrogenation to ethanol serves as a potential route for carbon neutrality and renewable energy utilization, while its practical application is severely limited by the activity-selectivity trade-off. This challenge primarily arises from the difficulty of C–C coupling and the occurrence of multiple side reactions. Herein, we design a NiFe2O4 spinel-modified Fe2O3 catalyst via a solid-state co-precipitation method, achieving a high CO2 conversion rate of 49.3% with an ethanol space–time yield of 883.7 mg·gcat.−1· h−1. Further mechanism investigation reveals that the incorporation of NiFe2O4 spinel benefits the formation of active Fe5C2 phase. Meanwhile, the interfacial sites between NiFe2O4 and Fe2O3 endow the catalyst with superior hydrogenation ability, which effectively inhibits excessive carbon chain growth and promotes the orientated synthesis of ethanol. This work proposes an inspiring NiFe2O4 spinel engineering method for the efficient production of multi-carbon oxygenates from CO2 hydrogenation.
Similar content being viewed by others

Introduction
Under the global initiative toward carbon neutrality, the efficient conversion of carbon dioxide (CO2) into high-value chemicals has become an essential component in building sustainable low-carbon energy systems1,2,3. Among the various possible hydrogenation products, ethanol has attracted particular attention due to its high energy density, excellent fuel properties, and broad industrial applicability4,5,6. Iron-based catalysts are widely employed in CO2 hydrogenation to ethanol, where CO2 is first reduced to CO via the reverse water-gas shift (RWGS) reaction and subsequently converted to ethanol over an active Fe5C2 phase through Fischer-Tropsch synthesis (FTS)2,7,8,9. Despite extensive research efforts devoted to improving Fe-based catalysts, simultaneously achieving a superior CO2 conversion rate and remarkable ethanol selectivity remains a formidable challenge. The inherent high thermal stability of CO2 and the substantial energy barrier to C-C coupling impose significant obstacles to enhancing CO2 activation and conversion. Furthermore, the stringent conditions for the formation of reactive iron carbide phases limit effective enrichment of FeCx in FTS10,11. Meanwhile, multiple concurrent side reactions are prone to occur owing to the difficulty in precisely modulating C–C extension process, and the selectivity of target ethanol is generally restricted by the Anderson–Schulz–Flory (ASF) distribution over Fe-based catalysts12,13,14. The development of catalysts for the hydrogenation of CO2 to ethanol with industrialization potential is urgently required.
In recent years, spinel ferrite materials have emerged prominently in CO2 hydrogenation due to their tunable compositions, defect-rich sites, and high thermal stability15. Generally, spinel ferrites are Fe-based oxides in which part of the trivalent Fe3+ ions in the lattice are substituted by divalent transition metal ions15. The unique structural compositions endow spinel ferrites with sufficient oxygen localization to form abundant oxygen vacancies (OVs) for the adsorption and interaction of CO216,17. Besides, the inclusion of various divalent metal additives leads to charge redistribution, which promotes the reducibility and carburization of Fe species for the formation of active iron carbides18. Moreover, the spinel structure with tetrahedral and octahedral sites provides multiple sites for different metals, favoring the construction of potentially reactive centers, including the interfacial domain and the dual-metal sites19. The integration of appropriate secondary metals can modulate the microenvironment of Fe species and regulate the C–C coupling process for the selective synthesis of target products. Nickel-based species are a promising candidate as the secondary metal in spinel ferrites for the oriented production of ethanol. Firstly, Ni-based species display high activity in C-O cleavage and rapid hydrogenation, which boosts the formation of alkyls (CHx*) intermediates and further facilitates the C–C coupling process3. Additionally, the efficient H2 activation and dissociation capability of Ni-based species can tune the hydrogenation kinetics and suppress the random carbon chain growth, benefiting for the preferential formation of ethanol.
Herein, drawing inspiration from the advantageous spinel structures and the potent hydrogenation activity of Ni-based species, we innovatively introduced the NiFe2O4 spinel into Fe2O3 (NiFe2O4/Fe2O3) by a solid-state co-precipitation method. The modification of NiFe2O4 spinel broke the selectivity limitation caused by ASF distribution and further facilitated the space-time yield (STY) of ethanol. The selectivity of ethanol was boosted to 33.0% at a high CO2 conversion rate of 49.3% under a weight hourly space velocity (WHSV) of 24000 mL·gcat.−1·h−1. More impressively, NiFe2O4/Fe2O3 achieved a high ethanol STY of 883.7 mg·gcat.−1· h−1. Further mechanism investigations revealed that the interfacial sites between NiFe2O4 and Fe2O3 of NiFe2O4/Fe2O3 favored the activation and dissociation of H2 with the generation of sufficient active hydrogen species, which assisted to achieve the dynamic transformation of the adsorbed CO2 and the high CO2 conversion. Moreover, the modification of NiFe2O4 promoted the reducibility of Fe2O3, contributing to the rapid carburization process and the formation of Fe5C2 phase for carbon chain extension. Theoretical calculations further confirmed that NiFe2O4 spinel reduced the energy barrier of C–C coupling between CH2* and CHO*. Meanwhile, the efficient hydrogenation capability of NiFe2O4/Fe2O3 endowed by the interfaces also suppressed the excessive carbon chain growth, which facilitated the precise C–C coupling process and the oriented production of ethanol. The NiFe2O4 spinel engineering successfully breaks the trade-off between activity and selectivity in CO2 hydrogenation to ethanol.
Results
Catalytic properties of spinel-modified Fe2O3 catalysts in CO2 hydrogenation
To begin with, a series of spinel-modified Fe2O3 catalysts was fabricated via the solid-state co-precipitation method20. Pure NiFe2O4 and Fe2O3 were also prepared for further comparison. Besides, traditional liquid-state co-precipitation method was applied to synthesize Fe2O3 catalyst decorated with NiOx species (NiOx/Fe2O3). All the catalysts were further loaded with 2.0 wt% of alkali metal potassium (K). The catalytic properties of the as-obtained catalysts were evaluated toward CO2 hydrogenation over a fixed-bed reactor at 330 °C and 5 MPa for 6 h on stream. The main products over pure NiFe2O4 were CO and CH4 with negligible generation of ethanol, while sole Fe2O3 primarily produced C5+ hydrocarbons during CO2 hydrogenation (Fig. 1a). Impressively, the incorporation of NiFe2O4 into Fe2O3 significantly boosted the CO2 conversion rate to the 49.3% and ethanol selectivity to 33.0% under a high WHSV of 24,000 mL·gcat.−1·h−1, while reducing the selectivity of C5+ hydrocarbons to 5.8% (Fig. 1a and Table S1). NiFe2O4/Fe2O3 achieved an unprecedented STY of 883.7 mg·gcat.−1· h−1 towards ethanol, which was 64.5 and 8.0 times higher than that for NiFe2O4 and Fe2O3, respectively (Fig. 1b and Table S1). The prominent improvement in the catalytic property of NiFe2O4/Fe2O3 relative to sole NiFe2O4 and Fe2O3 suggested the potential synergistic effect between these two components for the oriented production of ethanol. As for NiOx/Fe2O3, despite suppressing the production of C5+ hydrocarbons to some extent, NiOx species impaired the conversion of CO2 while accelerating the production of CO, leading to a lower STY of ethanol (Fig. 1a and Table S1). Besides, the modification of Fe2O3 with other spinel species was also conducted, including CoFe2O4, CuFe2O4, and ZnFe2O4, among which NiFe2O4 presented the most pronounced enhancement in the selectivity and STY of ethanol, as well as the most intense inhibition of C5+ hydrocarbons production during CO2 hydrogenation (Figs. 1a, 1b). The above results demonstrated that the NiFe2O4 spinel exhibited strong hydrogenation capability and could modulate the C–C coupling process to restrain the excessive carbon chain growth, which was further verified by the hydrocarbon distribution analysis. As shown in Fig. S1, all catalysts except NiFe2O4/Fe2O3 exhibited typical random chain growth behavior of conventional FTS and followed the ASF distribution of hydrocarbon products. NiFe2O4/Fe2O3 broke the traditional ASF distribution with the lowest probability of chain growth (α) of 0.46 among the obtained catalysts, revealing that the modification of NiFe2O4 potentially controlled the excessive formation of long-chain hydrocarbons and further facilitated the preferential generation of ethanol (Figure S1).

a Catalytic performance and b oxygenated product distribution and STY of EtOH over the prepared catalysts. c Catalytic performance, product distribution, and STY of EtOH over NiFe2O4/Fe2O3 with different WHSV. d Comparison of WHSV and STY of EtOH over NiFe2O4/Fe2O3 with the results reported in other literature. e Stability of NiFe2O4/Fe2O3 in CO2 hydrogenation to EtOH. Reaction conditions: 330 °C, 5.0 MPa, WHSV = 24000 mL·gcat.−1·h−1, H2/CO2/Ar = 72/24/4.
The effect of alkali metal K on CO2 hydrogenation to ethanol was assessed for NiFe2O4/Fe2O3. As displayed in Fig. S2 and Table S2, variation of K loading from 0 to 4 wt% had a negligible impact on CO2 conversion rate (48.6–49.3%) and selectivity of CO (1.9-3.0%), while the product distributions were fine-tuned via K component. With the increase of K content to 2 wt%, the selectivity of CH4 significantly decreased with the concomitant enhancement in the production of ethanol. The selectivity and STY of ethanol slightly declined when further increasing K content to 4 wt%, likely due to over-saturation of basic sites or blockage of active sites at surfaces. Thus, an optimal K loading of 2 wt% effectively suppressed the methanation process and further contributed to the formation of ethanol. The catalytic performance of NiFe2O4/Fe2O3 was further investigated under various reaction temperatures, pressures, and WHSV. As illustrated in Fig. S3 and Table S3, increasing the temperature from 300 to 360 °C only slightly elevated the CO2 conversion as well as the selectivity and STY of ethanol, indicating that NiFe2O4/Fe2O3 operated efficiently even at relatively moderate temperatures. The effect of pressure on catalytic performance over NiFe2O4/Fe2O3 was displayed in Fig. S4 and Table S4. With the increase of pressure, both CO2 conversion and selectivity of ethanol greatly enhanced, while the production of CO and CH4 was suppressed. Higher pressure was thermodynamically favorable for the formation of ethanol, and the STY of ethanol significantly promoted from 22.5 to 883.7 mg·gcat.−1·h−1 when elevating the pressure from 1 to 5 MPa. The enhancement could be attributed to the increased the partial pressure of CO2 and H2 at higher pressure, which facilitated the adsorption and activation of CO2 and the C–C coupling process. WHSV affected CO2 conversion and the detailed distribution of products, where the STY of ethanol displayed a volcano-type trend against the WHSV over NiFe2O4/Fe2O3 (Fig. 1c).
NiFe2O4/Fe2O3 exhibited the supreme catalytic performance for CO2 hydrogenation to ethanol in this work, which also displayed a significant performance leap across all key metrics compared to the vast majority of catalysts reported previously, especially for the STY of ethanol (Fig. 1d, S5 and Table S5). Additionally, five independent catalytic tests under the same reaction conditions were also conducted for NiFe2O4/Fe2O3, verifying the repeatability and reliability of its pioneering catalytic performance towards CO2 hydrogenation to ethanol (Fig. S6 and Table S6). Furthermore, the stability of NiFe2O4/Fe2O3 towards CO2 hydrogenation was evaluated by prolonging the reaction time. Long-term durability test over 120 h revealed that NiFe2O4/Fe2O3 maintained sa table CO2 conversion rate and STY of ethanol with minor fluctuations, demonstrating the remarkable structural robustness and resistance to deactivation (Fig. 1c and Table S7). Overall, the outstanding catalytic performance and the facile fabrication process of NiFe2O4/Fe2O3 highlighted its substantial promise for efficient and durable ethanol production via CO2 resourcization.
Morphological and electronic properties of NiFe2O4/Fe2O3
To elucidate the origin of the superior catalytic performance of NiFe2O4/Fe2O3, multiple structural characterizations were first conducted for the fresh catalysts. Fig. S7a, b shows the X-ray diffraction (XRD) profiles of the as-obtained catalysts. Pure NiFe2O4 showed diffraction peaks at 30.3°, 35.7°, 43.4°, and 63.0°, well assigned to (220), (311), (400), and (440) planes of NiFe2O4 spinel phase (JCPDS No. 74-2081), respectively (Fig. S7a). The XRD patterns for NiOx/Fe2O3 and Fe2O3 were mainly ascribed to hematite Fe2O3 phase (JCPDS No. 84-0308), while both Fe2O3 and NiFe2O4 phase were clearly observed for NiFe2O4/Fe2O3. The successful incorporation of the NiFe2O4 spinel was further confirmed by Raman spectroscopy. As shown in Fig. S7c, NiFe2O4/Fe2O3 displayed additional characteristic features attributed to the typical T2g and A1g vibrational modes of NiFe2O4 spinel21. XRD patterns for other spinel-modified Fe2O3 catalysts were illustrated in Fig. S7b. Similar to NiFe2O4/Fe2O3, additional peaks for corresponding spinels also appeared for CoFe2O4/Fe2O3, CuFe2O4/Fe2O3, and ZnFe2O4/Fe2O3 (Fig. S7b). Inductively coupled plasma-optical emission spectrometry (ICP-OES) tests were also carried out for the as-obtained catalysts. As listed in Table S8, the K content and the molar ratio between Fe and M (M = Ni, Co, Cu, and Zn) were consistent with the experimental design. Besides, the Brunauer–Emmett–Teller (BET) surface areas and crystallite size of the catalysts of the obtained catalysts were shown in Fig. S8 and Table S9. Figure 2b and S9 depict the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of NiFe2O4/Fe2O3, which was composed of numerous irregular nanoparticles. Further observations in the high-resolution transmission electron microscopy (HRTEM) showed two sets of fringes with interplanar spacings of 0.251 and 0.250 nm, related to the (311) plane of NiFe2O4 and (110) plane of Fe2O3, respectively (Fig. 2a). Interfacial sites between NiFe2O4 and Fe2O3 formed in NiFe2O4/Fe2O3, which served as potential reactive center during CO2 hydrogenation. The STEM energy dispersive X-ray (EDX) elemental mapping images were also collected, indicating a uniform distribution of K, Fe, Ni, and O elements in NiFe2O4/Fe2O3 (Fig. 2c). Morphology characterizations for NiOx/Fe2O3 were performed as well, which showed similar nanoparticle stacking structure and homogeneous element distribution (Fig. S10). (012) plane for Fe2O3 phase was also observed in the HRTEM image of NiOx/Fe2O3, while the lack of visible NiOx nanoparticles could be attributed to the high dispersion or amorphous nature of Ni species (Fig. S10b).

a HRTEM image, b HAADF-STEM image, and c EDS mapping of NiFe2O4/Fe2O3. d Normalized Ni K-edge XANES spectra of the NiFe2O4/Fe2O3 and NiOx/Fe2O3 in reference to the Ni foil, NiO, and Ni2O3. Wavelet transform analyses of the EXAFS spectra on Ni K-edge of e NiFe2O4/Fe2O3 and f NiOx/Fe2O3. g EXAFS spectra of NiFe2O4/Fe2O3 and NiOx/Fe2O3 in reference to the Ni foil, NiO, and Ni2O3. The dotted line and solid line were the experimental data and the corresponding fitting curve, respectively.
The X-ray absorption near-edge spectroscopy (XANES) and extended X-ray fine structure (EXAFS) were further employed to differentiate the electronic and local structures of the samples. Ni K-edge XANES profiles showed that the adsorption edge positions for both NiFe2O4/Fe2O3 and NiOx/Fe2O3 were between NiO and Ni2O3, implying the mixed oxidation state of Ni species ranged from +2 to +3 (Fig. 2d). Additionally, the Ni species in NiFe2O4/Fe2O3 was in a lower oxidation state than that in NiOx/Fe2O3 due to the formation of NiFe2O4 spinel, as evidenced by its lower adsorption edge energy (Fig. 2d)22. The local coordination configurations of Ni species for the samples were further identified by EXAFS. As displayed in the Ni K-edge wavelet transformed EXAFS (WT-EXAFS) spectra, the prominent peak at k ≈ 5.3 Å−1 and k ≈ 6.7 Å−1 were associated with Ni–O scattering path of the first shell and Ni–Ni/Fe scattering path of the second shell for both NiFe2O4/Fe2O3 and NiOx/Fe2O3, respectively (Fig. 2e, 2f). The precise Ni-Ni and Ni-Fe coordination numbers (CNs) were hard to determine, while the lower k of the second shell in NiFe2O4/Fe2O3 indicated the higher degree of Ni-Fe interaction originating from the spinel-type NiFe2O4 structure23,24. Figure 2g displays the Fourier-transformed EXAFS spectra in R space. The CNs of Ni-Ni/Fe for NiFe2O4/Fe2O3 and NiOx/Fe2O3 were calculated to be 11.0 and 5.1, respectively (Table S10). The substantial increase of Ni–Ni/Fe CNs in NiFe2O4/Fe2O3 stemmed from the well-ordered Ni–Fe coordination network in NiFe2O4 spinel. This spinel structure was also able to establish efficient electron transfer and buffering pathways, thus improving the redox flexibility of catalysts15,25.
Structural evolution of NiFe2O4/Fe2O3 during CO2 hydrogenation
The structural evolution of the catalysts was probed via in situ XRD measurements. Pure H2 was firstly introduced, and the temperature was gradually increased to 400 °C, during which NiFe2O4/Fe2O3 showed the transformation of Fe2O3 into Fe3O4 (JCPDS No. 88-0315) (Fig. 3a). The diffraction peak at 44.7° appeared at around 80 min corresponding to the formation of metallic Fe (JCPDS No. 89-4186). After further treatment of CO2/H2 mixed gas at 330 °C, the peak for metallic Fe gradually disappeared with the concomitant generation of Fe5C2 phase (JCPDS No. 89-2544) even under atmospheric pressure. By contrast, no obvious peak for Fe phase or Fe5C2 phase was detected for NiOx/Fe2O3 under reduction and reaction conditions (Fig. S11a). As for Fe2O3, weak metallic Fe peaks appeared after H2 reduction, while the signals for Fe5C2 remained feeble after exposure to the reaction gas (Fig. S11b). The above in situ XRD results suggested that the NiFe2O4 spinel held the potential to promote the reduction of Fe2O3 to Fe and further the formation of active Fe5C2 phase. Moreover, the evolution of Ni species was explored by quasi in situ XPS measurement. As indicated by the Ni 2p3/2 spectra, partial reduction of Ni3+/2+ to Ni0 was observed after H2 pretreatment (Fig. S12). Upon exposure to the CO2/H2 atmosphere, the peak for metallic Ni disappeared with the generation a large portion of Ni3+/2+ species, indicating that Ni0 species underwent oxidation under reaction atmosphere. The reduced Ni species was speculated to reform the NiFe2O4 spinel under reaction conditions due to the redox flexibility nature of the spinel structure, which was further proved via Raman spectroscopy (Figs. S12 and 3b). For the spent NiOx/Fe2O3, A1g, T2g, and Eg were attributed to the vibrational modes of Fe3O4, consistent with the XRD results. In addition to the Fe3O4 phase, A1g, T2g, and Eg vibrational modes for NiFe2O4 spinel still retained for the spent NiFe2O4/Fe2O3, manifesting that NiFe2O4 phase was still preserved after the CO2 hydrogenation process.

a In situ XRD patterns of NiFe2O4/Fe2O3. b Raman spectra of spent NiFe2O4/Fe2O3 and spent NiOx/Fe2O3. c Mössbauer spectra of spent NiFe2O4/Fe2O3 catalyst. (d) H2-TPR, e CO2-TPD, and f H2-TPD profiles of NiFe2O4/Fe2O3, NiOx/Fe2O3, NiFe2O4, and Fe2O3. (g and h) In situ DRIFTS of NiFe2O4/Fe2O3.
Furthermore, the formation process of the pivotal Fe5C2 phase during CO2 hydrogenation was elucidated in detail. Figure S13a displays the HRTEM images of NiFe2O4/Fe2O3 after 6-h catalytic test. The lattice parameters of 0.251 and 0.210 nm corresponded to the (311) facet of NiFe2O4 and the (021) facet of Fe5C2, respectively. The interface between NiFe2O4 and Fe5C2 was clearly identified. Besides, (021) facets of the Fe5C2 phase were also observed for the spent NiOx/Fe2O3 and Fe2O3. The evolution of the iron phase during the reaction at different time on stream (TOS) for NiFe2O4/Fe2O3 was also monitored. The CO2 conversion and CO selectivity trends over TOS were shown in Fig. S14a, which was divided into two stages, including the induction period and the stabilization period. The CO2 conversion rapidly increased from 3.5% to 49.8% with the gradual decrease of the CO selectivity from 0 to 1.5 h. The reaction then reached a steady state with stable CO2 conversion and CO selectivity after 1.5 h. Figure S14b presents the corresponding XRD patterns of the spent NiFe2O4/Fe2O3 at different TOS. The Fe5C2 content sharply increased to 65.35% at 1.5 h and remained almost unchanged up to 120 h, which revealed that NiFe2O4/Fe2O3 underwent a fast carburization process in the initial stage and then maintained a stable ratio of Fe5C2 to Fe3O4 (Table S10). The K content and different reaction conditions also influenced the formation of Fe5C2 over NiFe2O4/Fe2O3. The addition of K facilitated the generation of Fe5C2 to some extent, but excess K possessed a limited impact on the Fe3O4–Fe5C2 phase distribution (Fig. S15a and Table S12). NiFe2O4/Fe2O3 maintained a relatively stable phase composition across the tested temperature range of 300-360 °C (Fig. S15b and Table S13). By contrast, high reaction pressure markedly promoted the formation of Fe5C2 phase (Fig. S15c and Table S13).
Mössbauer spectroscopy and XRD characterizations were further carried out to quantify the contents of different phases for the spent catalysts after a 6-hour standard catalytic test. As listed in Table S14, the proportions of reactive Fe5C2 phase of the total iron phase were determined to be 69.41%, 37.27%, and 59.83% for the spent NiFe2O4/Fe2O3, NiOx/Fe2O3, and Fe2O3, indicating the introduction of NiFe2O4 spinel promoted the formation of active Fe5C2 phase relative to sole Fe2O3 (Fig. 3c and S16). Besides, NiFe2O4/Fe2O3 exhibited the most prominent Fe5C2 features among these three catalysts in terms of the XRD patterns (Fig. S17a). The quantitative analysis of XRD revealed that Fe5C2 phase in spent NiFe2O4/Fe2O3 occupied 65.35% of all crystalline components, approaching that (69.41%) obtained from the Mössbauer spectra (Table S15). The formation of Fe5C2 phase over other spinel-modified Fe2O3 catalysts was also investigated via XRD and Mössbauer spectra (Figs. S17b and S18). The incorporation of CoFe2O4 and ZnFe2O4 also promoted the carburization of Fe phases, while CuFe2O4 spinel suppressed the generation of Fe5C2, possibly due to the weaker ability of Cu species to facilitate CO activation and carbon diffusion (Table S16)26,27. Furthermore, the electronic structures of the spent catalysts were investigated via X-ray photoelectron spectroscopy (XPS) measurements. Based on the Fe 2p XPS spectra, all the obtained catalysts contained a mixture of Fe3+ and Fe2+ species before and after CO2 hydrogenation process (Figs. S19b, S20b, S21a, and S22b). Additional peaks for Fe-C at around 706.4 eV were clearly observed for NiFe2O4/Fe2O3, Fe2O3, and NiFe2O47. The Ni 2p3/2 XPS spectra indicated the existence of Ni2+ and Ni3+ species for the fresh NiFe2O4, NiFe2O4/Fe2O3 and NiOx/Fe2O3 (Figs S19a, S20a, and S22a). The position of Ni3+/2+ peaks shifted towards the higher binding energy direction after reaction, which probably resulted from the electronic changes during the reaction process, such as NiFe2O4 or NiOx donating electrons to the Fe2O3 phase or the adsorbed reactants28. The O 1 s spectra of the as-obtained catalysts were shown in Figs. S19c, S20c, S21b, and S22c. The peaks located below 531.0 eV corresponded to the different lattice oxygens (OL)29. The binding energies at approximately 531.3 and 533.1 eV were ascribed to the OVs and surface hydroxyl group (–OH), respectively30. The concentration of OVs in Fe2O3 skyrocketed to 78.9% after CO2 hydrogenation, reflecting its excessive reduction and structural instability, while the decline of OVs in the spent NiOx/Fe2O3 indicated an attenuation of activity31,32. In contrast, OVs in NiFe2O4/Fe2O3 and NiFe2O4 steadily increased to a moderate concentration of 60.1% and 69.7%, respectively, showing the ability to regulate OVs generation (Table S17). This was attributed to the inherent reversible electron transfer mechanism of Ni2+/Ni3+ versus Fe3+/Fe2+ in the spinel structure, which effectively inhibited the overreduction and enhanced the electronic structure regulation.
Identification of the active sites in NiFe2O4/Fe2O3 for CO2 hydrogenation to ethanol
The role of NiFe2O4 spinel and the active sites in NiFe2O4/Fe2O3 for CO2 hydrogenation to ethanol were further investigated in detail. Temperature-programmed reduction of H2 (H2-TPR) was conducted to probe the reduction behavior and the interactions at the interfacial sites of the samples at first (Fig. 3d). Peaks at 378 and 560 °C corresponded to the reduction of Ni2+ and Fe3+ species in pure NiFe2O4, respectively33. As for Fe2O3, bands at 400–700 °C were assigned to the three-step reduction process of the Fe2O3 phase, where peaks α, β, and γ corresponded to the transformation of Fe2O3 to Fe3O4, Fe3O4 to FeO, and FeO to metallic Fe, respectively. The reduction peaks for NiFe2O4 shifted to higher temperatures over NiFe2O4/Fe2O3 due to the interactions between NiFe2O4 and Fe2O334. Conversely, Fe2O3 was more easily reduced in NiFe2O4/Fe2O3 with the reduction peaks moving to lower temperatures, ascribing to the donation of electrons from NiFe2O4 to Fe2O3 based on the XPS results (Fig. S19). The adsorption and activation of reactants were also investigated by temperature-programmed desorption of CO2 (CO2-TPD) and H2 (H2-TPD). As displayed in the CO2-TPD profiles, NiFe2O4/Fe2O3 exhibited the strongest adsorption peak of CO2 at 50–200 °C compared with that for Fe2O3 and NiFe2O4, proving that both NiFe2O4 and Fe2O3 components in NiFe2O4/Fe2O3 contributed to the adsorption of CO2 (Fig. 3e). Besides, although no obvious chemisorption of CO2 was observed in CO2-TPD profiles, both physisorbed and chemisorbed CO2 species existed on NiFe2O4/Fe2O3 under reaction conditions in terms of the in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) results (Fig. S23). Figure 3f shows the H2-TPD profiles for the obtained catalysts. Peaks at around 100-200 °C were observed for all the catalysts, which were assigned to the weakly chemisorbed H235. Impressively, bands on the higher temperature range of 310-460 °C only arose for NiFe2O4/Fe2O3, associated with the active hydrogen species adsorbed on the interfaces between NiFe2O4 and Fe2O3. As a result, the modification of NiFe2O4 created reactive NiFe2O4-Fe2O3 interfaces that greatly promoted the adsorption and dissociation of H2 at reaction temperature. Temperature-programmed desorption of CO (CO-TPD) was also performed to account for the adsorption properties of CO. As illustrated in Fig. S24, the peaks ranging from 100–200 °C and 200–400 °C corresponded to the physisorption and chemisorption of CO, respectively. In addition to physisorbed CO, NiFe2O4/Fe2O3 and NiOx/Fe2O3 exhibited two predominant chemisorption peaks at around 230 and 394 °C, while NiFe2O4 and Fe2O3 only showed weak chemically adsorbed CO at approximately 313 °C. This result indicated that the interactions of NiFe2O4 and Fe2O3 enhanced the adsorption and activation of CO, which favored the cleavage of C–O bond and further provided a carbon source for the generation of Fe5C2.
Based on the above characterizations, the interfacial sites in NiFe2O4/Fe2O3 served as the main reactive centers for CO2 hydrogenation to ethanol. Both NiFe2O4 and Fe2O3 components contributed to the adsorption of CO2. More importantly, the interfacial sites were in favor of the activation and dissociation of H2, contributing to the generation of active hydrogen species at reaction temperature. Sufficient active hydrogen species of NiFe2O4/Fe2O3 assisted to achieve the dynamic adsorption–activation–transformation of CO2 and further the high CO2 conversion rate. What’s more, the interactions promoted the reducibility of Fe2O3 and the activation of CO, contributing to the rapid carburization process and the formation of Fe5C2 phase for carbon chain growth. Meanwhile, the efficient hydrogenation capability endowed by the interfaces suppressed the excessive carbon chain extension, which facilitated the precise C–C coupling process and the oriented production of ethanol. As for NiOx/Fe2O3, the NiOx species diminished the formation of Fe5C2 due to weak reducibility and hydrogenation ability, leading to a decreased CO2 conversion rate and low selectivity of ethanol. The inferior catalytic properties of CoFe2O4-, CuFe2O4-, and ZnFe2O4-modified Fe2O3 catalysts were probably ascribed to the poor carbonization of Fe species or the inadequate activation of CO2 or H2 (Figs. S18 and S25).
Reaction pathways of ethanol synthesis over NiFe2O4/Fe2O3
We further implemented in situ DRIFTS measurements to investigate the evolution of key intermediate and reaction pathways for the synthesis of ethanol. To simulate the CO2 hydrogenation process, the catalysts were treated with CO2/H2 mixed gas at 330 °C under 5 MPa. As depicted in in situ DRIFT spectra over NiFe2O4/Fe2O3, the band at 2210–2400 cm−1 was associated with the gas-phase CO2, accompanied by the peak at 2180 and 2104 cm−1 assigned to the stretching vibrations of gaseous CO36,37 (Figs. 3g and 3h). Peaks at 1645 and 1360 cm−1 were attributed to the asymmetric and symmetric stretching vibrations of surface formate species (HCOO*), respectively37,38. Moreover, linearly adsorbed CO* and bridge-adsorbed CO* simultaneously appeared at respective 2012 and 1915 cm−1, further implying the rapid transformation of CO2 to CO at the initial stage of reaction though the RWGS process39,40 (Figs. 3g and 3h). Besides, the higher intensity of bridge-adsorbed CO* relative to that of linearly adsorbed CO* declared that the CO* species tended to dissociate to form CHx* species, which served as key intermediates for C–C coupling to hydrocarbons or ethanol. With the increase of treatment time, a series of intense peaks were then clearly observed for CHx* (3100 cm−1) intermediates and CH4 (1301 and 3011 cm−1)41,42,43. Notably, bending vibration and stretching vibration of C–H for CH2* species was clearly observed at 1455 and 2920 cm−1, respectively, and stretching vibration for CH3* species was also observed at 2948 cm−1[ 44,45. Meanwhile, pronounced bands for surface CHO* species with C=O stretching vibration were also detected at 2881 cm−1, which coupled with CHx* species to form the key intermediate CHxCHO* towards the production of ethanol45. The C–C coupling between CHx* and CHO* was further confirmed by the C = O stretching vibration of CH2CHO* (1550 cm−1) and CH3CHO* (1692 and 1750 cm−1)40,46. CHxCHO* species underwent subsequent hydrogenation and finally converted to ethanol featured at 1045 cm−1 47. In situ DRIFT spectra for NiOx/Fe2O3 and Fe2O3 exhibited similar characteristic bands but vastly different intensity of peaks (Fig. S26). Thus, the evolution of the characteristic peak intensity as a function of reaction time was plotted in Fig. S27 for further clarification. Compared with NiFe2O4/Fe2O3 and NiOx/Fe2O3, Fe2O3 possessed a much slower response and conversion of CO2. The intensity of the key intermediates over NiFe2O4/Fe2O3 was much higher than that over NiOx/Fe2O3. Notably, the introduction of NiFe2O4 induced a distinctive decay of linearly adsorbed and bridge-adsorbed CO* species at around 16 min, ascribed to the fast carburization behavior at the initial stage of the reaction. More importantly, NiFe2O4/Fe2O3 showed continuous CH2* build-up along with sustained high intensities of CHO* species, which accelerated the C–C coupling between CHx* and CHO* for the formation of ethanol rather than the C–C coupling of CHx* for hydrocarbons. In contrast, NiOx/Fe2O3 exhibited a plateauing trend in CH2* species accumulation and low-intensity signals for bridge-adsorbed CO* and CHO* species over time.
To further substantiate the proposed reaction pathways and clarify the intrinsic catalytic differences among the samples, density functional theory (DFT) calculations were carried out to evaluate the energetics of key elementary steps during CO2 hydrogenation. Based on the characterizations of the spent catalysts, three models were constructed, including Fe5C2(021) with the Fe5C2 exposing (021) facet, NiFe2O4/Fe5C2(021) with the modification of NiFe2O4 on Fe5C2, and NiO/Fe5C2(021) with the modification of NiO on Fe5C2 (Fig. 4a–4c). The formation of ethanol occurred via the formate pathway with C–C coupling between CHO* and CH2* in terms of the in-situ DRIFTS results. Figure 4 illustrates the reaction pathways for CO2 hydrogenation to ethanol over NiFe2O4/Fe5C2(021), NiO/Fe5C2(021), and Fe5C2(021) models, including the activation of CO2, C–C coupling process, and the subsequent hydrogenation process. Among the elementary steps considered, the initial hydrogenation of CO2* to HCOO* was difficult for all three models. For this step, NiFe2O4/Fe5C2(021) exhibited the lowest energy barrier of 1.01 eV relative to NiO/Fe5C2(021) (1.24 eV) and Fe5C2(021) (1.28 eV), which was ascribed to the efficient H2 activation and dissociation ability induced by NiFe2O4 to provide sufficient active hydrogen species. As for the critical C–C coupling step between CH2* and CHO*, the values of energy barriers were calculated to be 0.80, 0.72, and 0.98 eV for NiFe2O4/Fe5C2(021), NiO/Fe5C2(021), and Fe5C2(021), respectively (Fig. 4d–f). Figure S28 displays the differential charge density diagrams of CH2CHO* over the three models, and the Bader charge of CH2CHO* was calculated to determine the number of electrons (QTM) of CH2CHO* gained from the surfaces. The number of accepting electrons for CH2CHO* were 0.70, 0.53, and 0.38 e over NiFe2O4/Fe5C2(021), NiO/Fe5C2(021), and Fe5C2(021), respectively. Thus, the modification of Ni-based species favored the electron transfer for the formation of key intermediate CH2CHO* and further reduced the energy barrier for C–C coupling. Moreover, NiFe2O4 spinel significantly promoted the following hydrogenation process after C–C coupling and accelerated the final formation of ethanol. The energy barrier for the hydrogenation of CH2CHO* to CH3CHO* over NiFe2O4/Fe5C2(021) was only 0.45 eV, which was much lower than that over Fe5C2(021) of 1.12 eV. The enhanced efficiency of the subsequent hydrogenation process trended to shift the reaction pathway toward ethanol formation rather than further carbon-chain growth to long-chain hydrocarbons over NiFe2O4/Fe5C2(021). As a result, the modification of NiFe2O4 spinel endowed the catalyst with strong H2 activation and dissociation ability, facilitating the transformation of adsorbed CO2 and hydrogenation of pivotal intermediates. Additionally, NiFe2O4 spinel also lowered the energy barrier of C–C coupling and further contributed to the high catalytic activity towards CO2 hydrogenation to ethanol. The evolution of various intermediate species during the reaction was visually depicted through the structural model on NiFe2O4/Fe5C2(021), NiO/Fe5C2(021), and Fe5C2(021) (Figs. S29–S34). The distances between the C atom and the H/C atom in carbon-containing species became closer from the initial state (IS) to the transition state (TS) and further decreased at the final state (FS) (Table S18). Moreover, the reaction pathways considering dispersion correction (vdW), GGA + U, and dipole correction were also calculated, and the overall trend remained consistent with the original calculations (Figs. S35–S38).

Optimized models of a NiFe2O4/Fe5C2(021) for NiFe2O4/Fe2O3, b Fe5C2(021) for Fe2O3, and c NiO/Fe5C2(021) for NiO/Fe2O3. Illustrations of the reaction pathways for CO2 hydrogenation to ethanol over d NiFe2O4/Fe5C2(021), e Fe5C2(021), and f NiO/Fe5C2(021). Violet, green, gray, and red balls represented Fe, Ni, C, and O atoms, respectively. TS represented the transition state.
Discussion
In summary, we developed a NiFe2O4-modified Fe2O3 catalyst via the solid-state co-precipitation method towards CO2 hydrogenation. The interfaces between NiFe2O4 spinel and Fe2O3 served as the reactive sites for the activation and dissociation of H2, which contributed to the generation of sufficient active hydrogen species and further the rapid activation and transformation of the adsorbed CO2 even under a high WHSV. Besides, the incorporation of NiFe2O4 spinel enhanced the reducibility of Fe2O3 and facilitated the in-situ formation of active Fe5C2 phase. Furthermore, the NiFe2O4 spinel substantially suppressed multiple side reactions via regulating the excessive carbon chain growth, steering the reaction route for ethanol production. NiFe2O4/Fe2O3 exhibited an extraordinary catalytic performance, achieving a high CO2 conversion rate (49.3%) and exceptional ethanol STY (883.7 mg·gcat.−1· h−1) under an identical WHSV (24000 mL·gcat.−1·h−1). This work provides creative insights into the structure–activity relationships of NiFe2O4/Fe2O3 and proposes an effective strategy for concurrently achieving high CO2 conversion rate and ethanol selectivity in the industrial catalytic conversion of CO2 to multi-carbon oxygenates.
Methods
Catalyst preparation
Synthesis of MFe2O4/Fe2O3 (M = Ni, Co, Cu, and Zn)
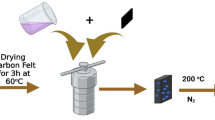
MFe2O4/Fe2O3 catalysts were obtained via a solid-state co-precipitation method20. Compared with the traditional liquid-phase co-precipitation method, the solid-state co-precipitation method creates a unique solvent-deficient environment, which exhibits abundant advantages in the aspects of the products, such as increased crystallinity, formation of porous structure, and stronger interactions between different components48,49. The presence of a small quantity of crystallization water favors the formation of a liquid layer around the precursors and causes the typically solid reagents to liquefy and bubble vigorously. This significantly accelerates the component diffusion and simultaneous nucleation of precursors, promoting the formation of heterometallic spinels. Take the synthesis of NiFe2O4/Fe2O3 as an example. To begin with, 0.06 mol of Fe(NO3)3·9H2O and 0.01 mol of Ni(NO3)2·6H2O were first mixed and ground in a mortar for 30 min. 0.6 mol of NaOH was then added, followed by grinding for another 60 min. Subsequently, the resulting mixture was washed with deionized water via the vacuum suction filter system until the filtrate reached a neutral pH. The filtered product was then dried overnight at 60 °C in an oven, which was further calcined in a muffle furnace at 600 °C for 4 h with a rate of 2 °C·min−1. 2 g of the product obtained from the above preparation was loaded with 2.0 wt% of K by ultrasonic impregnation. The final NiFe2O4/Fe2O3 catalyst was obtained by calcination in a muffle furnace at 550 °C for 4 h with a ramp rate of 2 °C·min−1. Pure NiFe2O4, Fe2O3, and other MFe2O4/Fe2O3 catalysts were synthesized via similar procedures, except adjusting the metallic precursors. NiFe2O4/Fe2O3 catalysts with different K content (0, 1 wt%, and 4 wt%) were also prepared via the above process, except adjusting the usage of KNO3 precursor. In this work, all the Fe2O3-based catalysts were impregnated with 2 wt% of K unless otherwise specified.
Synthesis of NiOx/Fe2O3
NiOx/Fe2O3 was synthesized via the traditional liquid-state co-precipitation method for further comparison. Initially, 0.06 mol of Fe(NO3)3·9H2O and 0.01 mol of Ni(NO3)2·6H2O were dissolved in deionized water and heated at 80 °C for 3 h. After stirring, the solution was allowed to cool to room temperature, and the pH was adjusted to 11 using NaOH. The solution was further stirred for 3 h, followed by filtration, washing with deionized water to neutrality, and drying at 60 °C overnight. The catalyst was then calcined at 600 °C in a muffle furnace for 4 h with a heating rate of 2 °C·min−1. 2.0 wt% of K was further loaded onto the product via ultrasonic impregnation, resulting in the final NiOx/Fe2O3.
Catalytic tests
Hydrogenation of CO2 over the obtained catalysts was conducted in a fixed-bed reactor with an inner diameter of 8 mm. In a typical catalytic test, 0.25 g of catalyst (20–40 meshes) diluted with 1 g of quartz sand (20–40 meshes) was packed in the reactor tube, with 1 g of glass beads placed above as a preheating layer of the gas. During the packing process, a thermocouple was inserted into the void space of the reactor tube to monitor the actual reaction temperature in real time. Prior to each reaction, the catalyst was reduced in H2 gas flow (40 mL·min−1) at 400 °C for 8 h under atmospheric pressure. After pre-reduction, the reactor was cooled to 160 °C to replace the reductive gas with reaction gas (H2/CO2/Ar = 72/24/4). The standard catalytic measurement was conducted at 330 °C under 5.0 MPa with a WHSV of 24000 mL·gcat.−1·h−1 for a total duration of 6 h. The reaction officially started after the reaction system reached the target temperature and pressure, which was defined as the “0 h” for the reaction. During the reaction, the effluent gas was detected by two online gas chromatographs. One was equipped with a thermal conductivity detector (TCD, Shimadzu, GC-2014) using an active charcoal column to analyze Ar, CO2, CO, and CH4. The other was equipped with a flame ionization detector (FID, Shimadzu, GC-2014) using a Porapak Q column to analyze C1-C6 hydrocarbons. An ice trap was installed between the reactor and the back pressure valve to collect the liquid-phase products, which were further analyzed via two offline gas chromatographs (GC-2014) equipped with FID. The DB-624 column and InerCap 5 column were used to analyze oxygenated compounds and C4+ hydrocarbons, respectively.
The CO2 conversion, CO selectivity, selectivity of product i (exclude CO), and STY were calculated through the following equations:
where CO2-in and CO2-out represented the mole fractions of CO2 in the intake and exit, respectively.
where CO-out symbolized the mole fraction of CO in the outlet.
where Ni and ni represented the mole percentage and carbon number of product i, respectively.
where M(C2H6O) represented the relative molecular mass of ethanol; nCO2 and nCO represented the amount of converted CO2 and produced CO in mol·g−1·h−1, respectively50.
The carbon balance data of all catalysts were calculated between 93.7% and 105.7% based on the following formula:
where nProd., nCO2, and nCO represented the amounts of hydrocarbons and oxygenates, converted CO2, and produced CO in mol·g−1·h−1, respectively.
DFT calculations
All the spin-polarized DFT calculations were performed by the Vienna Ab initio Simulation Package (VASP 5.4.4)51 with the projector augmented wave (PAW) method52. The exchange functional was treated using the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof (PBE)53 functional. The energy cutoff for the plane wave basis expansion was set to 450 eV. Partial occupancies of the Kohn−Sham orbitals were allowed using the Gaussian smearing method and a width of 0.2 eV. The bulk structure of Fe5C2 was calculated first towards the optimization of the lattice parameter, where the k-point of 5×5×4 was used. The Fe5C2 (021) surface was then built with three Fe layers and five C layers (60 Fe atoms and 20 C atoms in the slab) with parameters of a = 10.95, b = 12.42 and c = 20.71 Å. Cluster structures of NiFe2O4 with 32 atoms and NiO with 26 atoms loaded on the (021) surface of Fe5C2 were established, named as NiFe2O4/Fe5C2(021) and NiO/Fe5C2(021), respectively. A vacuum of 15 Å was adopted to prevent the interaction between two adjacent layers. The Monkhorst k-point of 2 × 3 × 1 was used during the calculations. The self-consistent calculations apply a convergence energy threshold of 10−5 eV, and the force convergence was set to 0.02 eV/Å for either structure optimizations or transition state calculations. The transition state calculations were performed using the climbing image nudged elastic band (CI-NEB) method54. The adsorption Gibbs free energy (ΔG) was considered at the temperature of 603 K, following:
where ΔE was the DFT calculated energy change, ZPE was zero-point energy and was calculated by ZPE = Σi\(\frac{1}{2}\)hvi, i was the frequency number, and vi was the frequency with unit cm−1. ΔZPE represented the difference of zero-point energy between the adsorbed and un-adsorbed states. Besides, ΔGU was the correction from inner energy, which was calculated based on the following equation55:
where R = 8.314 J·mol−1·K−1, T = 603 K, h = 6.63 ×10−34 J·s, and kB = 1.38 × 10−23 J·K−1.
For T∆S, T was 603 K and ∆S was the correction from entropy. For the entropy of gas-phase, it was obtained from the NIST database (T = 603 K, PCO2: PH2 = 1.25: 3.75 MPa)55,56. For the entropy of adsorbed species, the standard molar entropy (Sv) was calculated as follows:
where R = 8.314 J·mol−1·K−1, T = 603 K, h = 6.63 ×10−34 J·s, and kB = 1.38 × 10−23 J· K−1 55.
The adsorption Gibbs free energies of adsorbed species were calculated as follows:
where G* and Gadsorbed-species are the free energies without and with adsorbed species on the slab, respectively. GH2O(g), GCO2(g), and GH2(g) are the free energy of gas-phase H2O, CO2, and H2, respectively. All values of Gibbs free energy (G) intermediates were summarized in Table S21.
The values of detailed ΔZPE, ΔGU, and TΔS for different intermediates over NiFe2O4/Fe5C2(021), NiO/Fe5C2(021), and Fe5C2(021) were listed in Table S19. Besides, vibrational frequencies of corresponding carbon species over NiFe2O4/Fe5C2(021), NiO/Fe5C2(021), and Fe5C2(021) were provided as Table S20.
To ensure transparency and reproducibility, the atomic coordinates for all calculated structures were provided in the Source Data file. The total energies (E) for all calculated structures were also listed in Table S22. The above DFT calculations only considered dispersion correction (vdW)57. We also calculated the reaction pathways considering dispersion correction (vdW), GGA+U (U-J = 3 eV), and dipole correction, and all the relevant diagrams were shown in Figures S35-S3858.
Data availability
The source data generated in this study are provided in the Source Data file. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Ding, L. et al. CO2 hydrogenation to ethanol over Cu@Na-Beta. Chem 6, 2673–2689 (2020).
Ye, R. et al. Design of catalysts for selective CO2 hydrogenation. Nat. Synth. 4, 288–302 (2025).
Simons, J. F. M. et al. Structure sensitivity of CO2 hydrogenation on Ni revisited. J. Am. Chem. Soc. 145, 20289–20301 (2023).
Wang, X. et al. Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat. Energy 5, 478–486 (2020).
Wang, X. et al. Cesium-induced active sites for C–C coupling and ethanol synthesis from CO2 hydrogenation on Cu/ZnO(0001̅) surfaces. J. Am. Chem. Soc. 143, 13103–13112 (2021).
Xu, D. et al. Advances in higher alcohol synthesis from CO2 hydrogenation. Chem 7, 849–881 (2021).
Xiang, W. et al. Potassium-driven pathway modulation in CO2 hydrogenation: Tuning ethanol and liquid fuels synthesis over FeCuAl catalysts. Appl. Catal. B 369, 125157 (2025).
Wang, H. et al. Synergistic interactions of neighboring platinum and iron atoms enhance reverse water–gas shift reaction performance. J. Am. Chem. Soc. 145, 2264–2270 (2023).
Wu, W. et al. Facet sensitivity of iron carbides in Fischer-Tropsch synthesis. Nat. Commun. 15, 6108 (2024).
Wang, P. et al. Efficient conversion of syngas to linear α-olefins by phase-pure χ-Fe5C2. Nature 635, 102–107 (2024).
Han, J. et al. Low-temperature CO2 Hydrogenation to Olefins on Anorthic NaCoFe Alloy Carbides. Angew. Chem. Int. Ed. 64, e202420621 (2025).
Hong, X. et al. Visualizing Phase Evolution of Co2C for Efficient Fischer–Tropsch to Olefins. Adv. Mater. 36, 2404046 (2024).
Yang, H. et al. Tuning the selectivity of CO2 hydrogenation to alcohols by crystal structure engineering. Chem 10, 2245–2265 (2024).
Liu, T. et al. Selectivity control by zeolites during methanol-mediated CO2 hydrogenation processes. Chem. Soc. Rev. 54, 2726–2761 (2025).
Orege, J. I. et al. Emerging spinel ferrite catalysts for driving CO2 hydrogenation to high-value chemicals. Matter 6, 1404–1434 (2023).
Wang, M. et al. Spinel Nanostructures for the Hydrogenation of CO2 to Methanol and Hydrocarbon Chemicals. J. Am. Chem. Soc. 146, 14528–14538 (2024).
Zhang, J. et al. Selective formation of light olefins from CO2 hydrogenation over Fe–Zn–K catalysts. J. CO2 Util. 12, 95–100 (2015).
Yang, Q. et al. Understanding Mn-modulated restructuring of Fe-based catalysts for controlling selectivity in CO2 hydrogenation to olefins. Nat. Catal. 8, 595–606 (2025).
Li, C. et al. Catalytic Tandem CO2 Hydrogenation and Hydroformylation for High-Yield Synthesis of C2+ Alcohols. ACS Catal. 15, 11440–11451 (2025).
Smith, S. J. et al. Synthesis of metal oxide nanoparticles via a robust “solvent-deficient” method. Nanoscale 7, 144–156 (2015).
Zeng, X. et al. Ni modulates the coordination environment of cations in Fe3O4 to efficiently catalyze lignin depolymerization. Chem. Eng. Sci. 288, 119798 (2024).
Huang, Z. et al. Investigation on reactivity of iron nickel oxides in chemical looping dry reforming. Energy 116, 53–63 (2016).
Shah, S. et al. Dynamic Tracking of NiFe Smart Catalysts using In Situ X-Ray Absorption Spectroscopy for the Dry Methane Reforming Reaction. ACS Catal. 13, 3990–4002 (2023).
Qin, T. et al. Layered Porous Ring-Like Carbon Network Protected FeNi Metal Atomic Pairs for Bifunctional Oxygen Electrocatalysis and Rechargeable Zn-Air Batteries. Small 21, 2402762 (2025).
Yu, A. et al. Structural transformation between rutile and spinel crystal lattices in Ru–Co binary oxide nanotubes: enhanced electron transfer kinetics for the oxygen evolution reaction. Nanoscale 13, 13776–13785 (2021).
Xu, D. et al. Selective C2+ alcohol synthesis from direct CO2 hydrogenation over a Cs-promoted Cu-Fe-Zn catalyst. ACS Catal. 10, 5250–5260 (2020).
Xu, Y. et al. Enhancement of toluene oxidation performance over Cu–Mn composite oxides by regulating oxygen vacancy. Appl. Surf. Sci. 560, 149983 (2021).
Liao, H. et al. Unveiling Role of Sulfate Ion in Nickel-Iron (oxy)Hydroxide with Enhanced Oxygen-Evolving Performance. Adv. Funct. Mater. 31, 2102772 (2021).
Zou, H. et al. Enhanced degradation of tetracycline via Visible-light-assisted peroxymonosulfate activation over oxygen vacancy rich Fe2O3-CoFe2O4 heterostructures. Sep. Purif. Technol. 314, 123586 (2023).
An, C. et al. Porous NiCo2O4 nanostructures for high performance supercapacitors via a microemulsion technique. Nano Energy 10, 125–134 (2014).
Wu, L. et al. Boosting oxygen evolution reaction of (Fe,Ni)OOH via defect engineering for anion exchange membrane water electrolysis under industrial conditions. Adv. Mater. 35, 2306097 (2023).
Li, C.-F. et al. Fe doping and oxygen vacancy modulated Fe-Ni5P4/NiFeOH nanosheets as bifunctional electrocatalysts for efficient overall water splitting. Appl. Catal. B 291, 119987 (2021).
Vajglová, Z. et al. Interactions between Iron and Nickel in Fe–Ni Nanoparticles on Y Zeolite for Co-Processing of Fossil Feedstock with Lignin-Derived Isoeugenol. ACS Appl. Nano Mater. 6, 10064–10077 (2023).
Wei, G. et al. Chemical-looping reforming of methane using iron based oxygen carrier modified with low content nickel. Chin. J. Chem. 32, 1271–1280 (2014).
Sun, Y. et al. Plasma-catalytic CO2 hydrogenation over a Pd/ZnO catalyst: in situ probing of gas-phase and surface reactions. JACS Au 2, 1800–1810 (2022).
Wang, J. et al. In-situ-formed potassium-modified nickel-zinc carbide boosts production of higher alcohols beyond CH4 in CO2 hydrogenation. Angew. Chem. Int. Ed. 62, e202311335 (2023).
Wang, C. et al. CO2 hydrogenation to linear α-olefins on FeCx/ZnO catalysts: Effects of surface oxygen vacancies. Appl. Surf. Sci. 641, 158543 (2023).
Li, L. et al. CO2 hydrogenation selectivity shift over In-Co binary oxides catalysts: Catalytic mechanism and structure-property relationship. Chin. J. Catal. 43, 862–876 (2022).
Galhardo, T. S. et al. Optimizing active sites for high CO selectivity during CO2 hydrogenation over supported nickel catalysts. J. Am. Chem. Soc. 143, 4268–4280 (2021).
He, K. et al. Enhanced ethanol synthesis via CO2 hydrogenation using La-Doped CuFeOx catalysts. Chem. Eng. J. 501, 157608 (2024).
Wang, Y. et al. Direct conversion of CO2 to ethanol boosted by intimacy-sensitive multifunctional catalysts. ACS Catal. 11, 11742–11753 (2021).
Zhu, J. et al. Deconvolution of the particle size effect on CO2 hydrogenation over iron-based catalysts. ACS Catal. 10, 7424–7433 (2020).
Khan, M. K. et al. Selective conversion of carbon dioxide into liquid hydrocarbons and long-chain α-Olefins over Fe-amorphous AlOx bifunctional catalysts. ACS Catal. 10, 10325–10338 (2020).
Yang, Y. et al. Light-Induced Redox Looping of a Rhodium/CeWO3 photocatalyst for highly active and robust dry reforming of methane. Angew. Chem. Int. Ed. 61, e202200567 (2022).
Zhu, P. et al. Prebiotic synthesis of microdroplets from formate over a bimetallic Cobalt–Nickel Nanomotif. J. Am. Chem. Soc. 146, 25005–25015 (2024).
Xu, H. et al. Engineering Cr−O−Ti structure towards boosted vinyl chloride destruction: Oxygen species evolution and reaction mechanism. Appl. Catal. B 365, 124914 (2025).
Xu, B. et al. Pd–Sb Rhombohedra with an Unconventional Rhombohedral Phase as a Trifunctional Electrocatalyst. Adv. Mater. 34, 2206528 (2022).
De Bellis, J. et al. Direct Dry Synthesis of Supported Bimetallic Catalysts: A Study on Comminution and Alloying of Metal Nanoparticles. Angew. Chem. Int. Ed. 61, e202208016 (2022).
Carneiro, J. S. A. et al. Embracing the complexity of catalytic structures: a viewpoint on the synthesis of nonstoichiometric mixed metal oxides for catalysis. ACS Catal. 10, 516–527 (2020).
Tran, C.-C. & Kaliaguine, S. Rhodium-doped iron oxides promoted by sodium for highly selective hydrogenation of CO2 to ethanol and C2+ hydrocarbons. Chem. Eng. J. 496, 153636 (2024).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P. et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671 (1992).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Cao, X.-M. et al. An understanding of chemoselective hydrogenation on crotonaldehyde over Pt(111) in the free energy landscape: The microkinetics study based on first-principles calculations. Catal. Today 165, 71–79 (2011).
Banerjee, A. & Mushrif, S. H. Reaction Pathways for the Deoxygenation of Biomass-Pyrolysis-Derived Bio-oil on Ru: A DFT Study using Furfural as a Model Compound. ChemCatChem 9, 2828–2838 (2017).
Fritsch, D. & Ederer, C. Epitaxial strain effects in the spinel ferrites CoFe2O4 and NiFe2O4 from first principles. Phys. Rev. B 82, 104117 (2010).
Bhola, K. et al. Influence of Hubbard U Parameter in Simulating Adsorption and Reactivity on CuO: Combined Theoretical and Experimental Study. J. Phys. Chem. C. 121, 21343–21353 (2017).
Acknowledgements
This work was supported by the Grant-in-Aid from Japan Society for the Promotion of Science (JSPS) (22K14540) (S.Y.), (25K01806) (S.Y.), and JST SPRING (JPMJSP2145) of Japan (W.X.).
Author information
Authors and Affiliations
Contributions
W.X. conceived and conducted the catalyst evaluation experiments, performed data visualization, and wrote the initial draft of the manuscript. S.Y. carried out catalyst characterizations and contributed to manuscript revision. L.Z. and J.F. analyzed the in situ XRD data. K.T. synthesized the catalysts. Y.X. assisted in data analysis and manuscript revision. N.T. supervised the project and provided guidance on experimental design and manuscript revision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jian Liu, Chalachew Mebrahtu, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiang, W., Yasuda, S., Zhang, L. et al. NiFe2O4 spinel engineering for transcending the dilemma of activity–selectivity in CO2 hydrogenation to ethanol. Nat Commun 17, 572 (2026). https://doi.org/10.1038/s41467-025-67269-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67269-4
