
Abstract
Selective catalytic aromaticity-breaking epoxidation of arenes remains an ongoing research challenge, attributed to the stability of aromatic systems, the occurrence of over-oxidation, and the instability of epoxides. Epoxides, recognized as short-lived and highly reactive intermediates, are prone to undergo the “NIH-shift” rearrangement to yield the hydroxylated products. Herein, we report the synthesis and characterization of several electron-deficient naphthalene epoxides that disrupt the aromaticity of naphthalene. Their formation is highly dependent on the substrate, especially on the specific substitution pattern. This achievement is enabled by using mild biomimetic manganese porphyrins as catalysts and iodosylbenzene as an oxidant. The strategic employment of electron-withdrawing substitution on naphthalenes enables a chemoselective switch from hydroxylation to epoxidation. Experimental studies and theoretical calculations elucidate the possible underlying mechanisms of epoxidation and hydroxylation: epoxidation is likely mediated by a radical mechanism, whereas hydroxylation predominantly follows a zwitterionic pathway.
Similar content being viewed by others


Introduction
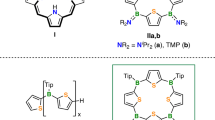
Oxidative functionalization of hydrocarbons represents a cornerstone in biological and chemical transformations, playing a vital role in the synthesis of oxygen-containing organic molecules1,2,3,4,5,6,7,8,9,10. Aromatic systems are inherently thermodynamically and kinetically inert, and the occurrence of over-oxidation during oxidation further complicates the process, rendering selective oxyfunctionalization a challenging yet significant area of research11,12,13. Despite significant advancements in biocatalysis and biomimetic hydroxylation14,15,16, the epoxidation reaction remains challenging, primarily due to over-oxidation, aromaticity-breaking, and instability of epoxides. The epoxides are short-lived and highly reactive intermediates in reactions, prone to undergo the “NIH-shift”17,18,19 rearrangement to yield the corresponding hydroxylated products (Fig. 1a). This propensity further exacerbates the challenges associated with the synthesis of epoxides, which is particularly regrettable given their potential as valuable building blocks in chemical transformations and drug metabolism20,21,22,23,24,25. Over the past decade, only a few cases of epoxidation of naphthalene, anthracene, tetracene and acridine have been reported26,27,28. These reactions predominantly result in the formation of diepoxides or peroxides rather than desired monoepoxides. Sarlah et al. have recently reported a general arenophile-based strategy for the oxidative dearomatization of benzenes and pyridines, primarily yields electron-rich arene monoepoxides. (Fig. 1c)29,30. However, this strategy, which requires a two-step process and involves the use of N-methyl-1,2,4-triazoline-3,5-dione (MTAD) in combination with potentially explosive peroxyacetic acid. The work of Hollmann group demonstrates that naphthalene epoxide can be directly generated by fungal peroxygenases31. Unfortunately, the monoepoxidation of electron-deficient naphthalenes remains largely unexplored. Consequently, the development of more straightforward, mild, and highly selective catalytic epoxidation methods for electron-deficient arenes is in high demand.

a Oxidative functionalization of arenes. b Mn-catalyzed two-step epoxidation of benzenes and pyridines. c This work: biomimetic catalysis enabled one-step epoxidation of electron-deficient naphthalene. TPFPP: 5,10,15,20-tetrakis(pentafluorophenyl) porphyrin.
Biomimetic metalloporphyrins have been pivotal in oxidative functionalization due to their facile structural modification and low catalyst loading32,33,34. We previously employed the highly fluorinated manganese porphyrin Mn(TPFPP)Cl as a catalyst for the oxidation of electron-rich 2,7-dimethoxynaphthalene, with iodosylbenzene (PhIO) as the oxidant. This process yielded hydroxylated products, which were then further oxidized to the observed quinones35. Encouragingly, our present work demonstrates that diethyl-2,6-naphthalenedicarboxylate, featuring a distinct substitution pattern undergoes epoxidation under mild catalytic conditions, enabling the dearomative synthesis of the corresponding epoxide with high chemoselectivity (Fig. 1c). By adjusting the substituents, we accomplished a selective shift from hydroxylation to epoxidation. Notably, while revising our manuscript, Costas and co-workers reported a breakthrough manganese-catalyzed enantioselective diepoxidation of a broad range of arenes36. Their success arises from the synergistic combination of an electron-rich, sterically demanding manganese complex and amino acid co-ligands containing a tert-butyl-leucine moiety. In contrast, our study employs a highly fluorinated Mn(TPFPP)Cl catalyst operating without any co-catalyst, enabling selective mono- and diepoxidation of electron-deficient naphthalenes. It is particularly intriguing that these distinct catalytic systems afford analogous epoxidation products. Moreover, our work complements this recent advance by providing detailed mechanistic insights into the competition between epoxidation and hydroxylation pathways, thereby elucidating the origin of substituent-controlled selectivity reversal.
Results
Experimental studies
The initial attempts have unveiled the potential of electron-deficient diethyl-2,6-naphthalenedicarboxylate 1a in epoxidation under the catalysis of Mn(TPFPP)Cl and with PhIO as the oxidant. Subsequently, the influence of oxidant and catalyst loadings, reaction time and temperature on the reactivity and selectivity has been comprehensively evaluated. The representative outcomes are presented in Table 1, while the others are provided in the Supporting Information (SI) (Tables S1–S4). To our delight, epoxide 3a was formed with no hydroxylation product 5a’ observed (Table 1, entry 1), and the structure of this epoxide has been confirmed by X-ray diffraction (See Table S5 for details). It was observed that the amounts of oxidant PhIO and catalyst Mn(TPFPP)Cl significantly influence the reaction activity and the selectivity (Table 1, entries 2–4 and 7–10). The optimal amounts for PhIO and Mn(TPFPP)Cl are 1.6 equiv and 1.0 mol%, respectively. Further observations reveal that extending the reaction time leads to a decrease in yield of 3a and an increase for 5a’ (Table 1, entries 5–7). The yield of 3a is maximized when the reaction duration is controlled at one hour. This epoxide is inherently stable, and its rearrangement is catalytically driven (see SI Fig. S1 for details). Additionally, an assessment of the effect of temperature on the reaction shows that at 40 °C, the conversion yield is the highest, reaching 65% (Table 1, entry 11). However, when the temperature is raised to 80 °C, the selectivity for epoxide formation is optimal (Table 1, entry 9). Considering both conversion yield and selectivity, the optimal reaction conditions are 1.6 equiv of PhIO, 1.0 mol% Mn(TPFPP)Cl, 50 °C, and 1 h (Table 1, entry 12). Notably, arene oxidation is inherently complex and often generates a variety of side products beyond the reported epoxides and phenols—including dihydroxylated compounds, diepoxides, and quinones. We made efforts to isolate and identify these possible by-products. However, due to the complexity of the reaction mixtures, the presence of isomeric species, and their closely related polarities, effective separation and characterization were not achievable under our current conditions. We believe that these uncharacterized by-products are responsible for the observed mass imbalance.
With optimal conditions established, we systematically investigated the reactivity of various naphthalene derivatives, focusing on both substitution pattern and electronic effects (Fig. 2). Our prior work had already indicated that electron-rich naphthalenes preferentially yield quinone products under similar conditions35. Consistent with this, when simple naphthalene was subjected to the current reaction conditions, quinones were obtained as the major products (see Fig. S2 for details), indicating that electron-donating gruops disfavor epoxide formation. We therefore turned our attention to electron-deficient substrates (Fig. 2). Among di-substituted naphthalenes, 2,6- or 2,7-diester-substituted derivatives afforded mono-epoxides 3a, 3b and 3c in moderate yields. In contrast, methyl 6-bromo-2-naphthoate afforded 3d in low yields. Cyano-substituted derivatives such as 2,3- and 1,4-cyano-substituted naphthalenes predominantly formed diepoxides (4e and 4 f), while methyl 5-bromo-2-naphthoate yielded a mixture of epoxide 3g and quinone 5g. For mono-substituted naphthalenes, only sulfonate-substituted substrates (-SO3Ph or -SO3Me derivatives) produced mono-epoxides (3k, 3l), along with diepoxides (4k, 4l). Other electron-withdrawing groups such as -CO2Me, -CN, and NO2 primarily gave diepoxides or mixtures of diepoxides and quinones (4h/5h, 4i/5i, 4j/5j, 4m). Mono-substituted quinoline derivatives bearing -CO2Me or -CN mainly afforded dioxetanes 4n and 4o. The X-ray crystal structures of 4e and 4j confirmed the anti-configuration of the diepoxides (See Tables S6, 7 for details). Notably, no epoxides were detected for naphthalene-2,6-dicarbonitrile 1p, 2-(trifluoromethyl) naphthalene 1q, or 1-(trifluoromethyl) naphthalene 1r. Experimental results demonstrate that the type and substitution pattern of substituents on the naphthalene ring effectively modulate the selectivity between mono- and diepoxides formation. Substrates 1a–1d and 1g, bearing electron-withdrawing groups at the 2,5-, 2,6-, and 2,7-positions, afforded monoepoxides exclusively without diepoxidation, a behavior ascribed to the combined effects of enhanced electrophilicity and sterically constrained environment at the substituted ring. In contrast, substrates 1e, 1f, 1h–1j, and 1m–1o underwent selective diepoxidation on the unsubstituted ring, highlighting the influence of steric accessibility. Interestingly, the sulfonate-substituted substrates 1k and 1l yielded mixtures of mono- and diepoxides: monoepoxidation occurred preferentially on the substituted ring, likely directed by the sulfonate group, while diepoxidation the unsubstituted ring due to steric accessibility.

aThe yields were based on the conversion yields. bThe yields were based on isolated yields.
Mechanistic consideration
Despite extensive studies on the diverse and intricate mechanisms of enzymatic hydroxylation of arenes, the debate over the competitive pathways between hydroxylation and epoxidation persists37,38,39,40,41,42. It is widely accepted that aromatic epoxide is an obligatory intermediate for the formation of hydroxylated product37. These studies indicate that the Mn-oxo porphyrin intermediate B attacks the π-system of A through two distinct oxygen atom attack mechanisms: radical and electrophilic pathways. This results in the formation of radical intermediate C and zwitterionic intermediate D, respectively (Fig. 3a). It is proposed that both intermediate C and D can undergo a ring closure process (path a) to form the epoxide-coordinated intermediate E. Alternatively, they can undergo hydroxylation via either an “NIH shift” (path b) or a “proton shuttle” mechanism (path c). The NIH shift involves the intramolecular migration of a hydrogen atom (highlighted in green) to an adjacent carbon atom, yielding the ketone intermediate F, which subsequently undergoes keto-enol tautomerization. The proton shuttle mechanism involves a proton transfer mediated by the porphyrin ligand, whereby the proton first migrates to a nitrogen atom on the porphyrin ring to form the N-protonated intermediate G, and is then shuttled to the oxygen atom of the Mn-oxo species. After obtaining the epoxide H, it may further undergo the NIH shift to form the hydroxylated I (path d). It should be noted that path a is defined as either the radical or zwitterionic form, depending on whether it originates from C or D. Similarly, path b and c also exist in both radical and zwitterionic forms.

a Proposed mechanism. b Mechanistic experiments.
The experimental results indicate that the chemoselectivity between hydroxylation and epoxidation can be reversed through the modification of substituents (Fig. 3b). It should be noted that the 2,6- and 2,7-substitution patterns of 1a and 1s have no significant influence on the chemoselectivity (see Fig. S3 for details). Despite the successful isolation of the epoxide without rearrangement to naphthol, the underlying mechanism of its formation and the origin of the reversal of chemoselectivity are yet to be elucidated. To gain more insights into the mechanism of this reaction, a series of experiments and DFT calculations were conducted. First, we monitored the reaction of 2,7-dimethoxynaphthalene 1s over time. Thin-layer chromatography and mass spectrometry showed that quinone product 5s formed within 1–10 minutes, with no detectable intermediate spots between 1s and 5s including the epoxide 3s (see Fig. S4 for details). After extended monitoring (10–60 min), no trace of 3s was detected. (Fig. 3b, left). Additionally, we calculated the relative thermodynamic stability of the epoxide 3 and hydroxylation product 5’ (Fig. 3b, right). For substrate 1a and 1s, the hydroxylation products 5a’ and 5s’ were more stable than the corresponding epoxide 3a and 3s by 37.1 and 36.0 kcal/mol, respectively. This suggests that both electron-rich and electron-poor epoxides are similarly less favorable thermodynamically. Moreover, kinetic isotope effect (KIE) experiments on 1s and [D]-1s indicate that C − H bond cleavage is unlikely to be the rate-determining step (see Fig. S5 for details).
Next, reaction (1), which incorporates diethyl-2,6-naphthalenedicarboxylate 1a, was first employed as a model to investigate the mechanism for epoxide formation. DFT-calculated free energy profile of oxidative functionalization of 1a is given in Fig. 4. We have considered the possible spin states, including singlet (closed- and open-shell), triplet and quintet. Only the most favorable spin states are presented in Fig. 4, while other spin states and their corresponding minimum energy crossing points (MECP) are detailed in the SI (see Figs. S6–S8 for details). The reaction initiates with the coordination of the quintet Mn(TPFPP)Cl 2 with the oxidant PhIO to afford the intermediate 5INT1. Subsequently, a high-valent Mn−O porphyrin intermediate 5INT2, with radical character localized on the oxygen atom, is formed via the oxidation transition state 5TS1 (see Fig. S9 for natural population analysis (NPA) spin population data). Thereafter, this oxygen atom initially reacts with 1a, and then attacks the C4 atom of π-system via the radical or electrophilic transition states 3TS2 and 3TS3, leading to the radical intermediate 5INT3 and zwitterionic intermediate 3INT4, respectively. It should be noted that for this C − O bond formation, there are four possibilities, depending on which carbon atom of arene is attacked by the oxyl group. Only the lowest-energy pathway is presented in the main text, and the other possibilities are detailed in Figs. S10, S11. The hydrogen atom abstraction (HAA)43 between 1a and 5INT2 was also considered (see Fig. S12 for details). Geometries and NPA charges of 3TS2 and 3TS3 are detailed in Fig. S13. It was found that the radical intermediate 5INT3 tends to undergo epoxidation through the ring closure transition state 5TS4, delivering epoxide 3a (Radical path a). Compared to undergoing epoxidation via the ring closure transition state 3TS5 (Zwitterionic path a), the zwitterionic intermediate 3INT4 is more inclined to experience hydroxylation through the NIH shift transition state 3TS6 (Zwitterionic path b) to form the INT5, which then proceeds through the keto-enol tautomerization to give the hydroxylated product 5a’. The proton shuttle mechanism mediated by the porphyrin was calculated to be less favorable compared with 3TS6 (see the SI Fig. S14 for details). Computations indicate that the oxygen atom attack is the rate-determining step of the reaction, with the energy barrier of 18.5 kcal/mol relative to 5INT1. More importantly, the energies for forming epoxide 3a via the radical pathway through 3TS2 and 5TS4 are lower than those for the hydroxylated product 5a’ via the zwitterionic pathway through 3TS3 and 3TS6, respectively. This is consistent with the chemoselectivity observed in our experiments.

Calculations were performed at the level of B3LYP-D3/def2-TZVP-SMD(Acetonitrile)//B3LYP-D3/6-31 G (d)-LANL2DZ.
To ascertain the nature of the intermediates 5INT3 and 3INT4, NPA charges (Q) and spin densities (ρ) analysis were performed (Fig. 5a). Naphthalene and Mn−O porphyrin are represented in purple and green, respectively. The results reveal that 5INT3 exhibits single-electron distributions in the naphthalene and the Mn−O porphyrin, with the spin density of 0.918 and 3.082, respectively. It also shows minimal charge distributions for the naphthalene (Q = 0.323) and the Mn−O porphyrin (Q = − 0.323), thereby displaying radical characteristics of 5INT3. Conversely, 3INT4 has a substantial charge distribution between the naphthalene and the Mn−O porphyrin, with charges of 0.917 and −0.917, respectively, and shows an absence of single electron distribution in the naphthalene (ρ = 0.011), thus presenting zwitterionic traits. Further analysis of 5INT3 and 3INT4 indicate that the differences in geometries affect their distinct reactivity profiles. As shown in Fig. 5a, the C4 − O bond in 5INT3 is perpendicular to the plane of naphthalene ring, with an OC4C10C9 dihedral angle of 89°. In the case of 3INT4, the conjugative effect of the oxygen atom aligns the C4 − O bond in the same plane as the naphthalene, resulting in the OC4C10C9 dihedral angle of 168°. This conjugation replaces the position of the hydrogen atom at the C4 − H bond with an oxygen atom of the C4 − O bond, positioning the H perpendicular to the naphthalene and placing it in an agostic state. This change facilitates the NIH shift or proton shuttle. Additionally, compared to 5INT3, the C4 − O bond in 3INT4 is shortened (1.34 vs. 1.44 Å), while the Mn−O bond (1.91 vs. 1.80 Å) and C4 − H bond (1.19 vs. 1.09 Å) are elongated. These alterations bring the hydrogen atom closer to the N atom within the porphyrin and the C3 atom in naphthalene, with distance of 1.94 and 1.89 Å, respectively. This further confirms the tendency of 3INT4 to undergo NIH shift and proton shuttle. In contrast, the extended C3···H (2.17 Å) and N···H (2.61 Å) distances in 5INT3 are less conducive to the proton transfer, and instead, they are more likely to promote epoxidation. Moreover, the electron-shift diagrams44,45,46,47 for the formation of 5INT3 and 3INT4 from arene 1a and Mn−O porphyrin intermediate 5INT2 are presented in Fig. 5b. The process of forming 5INT3 involves transfer of a single electron from the ϕ orbital of 1a (ϕarene1a) to the πyz orbital in 5INT2. In contrast, the formation of 3INT4 requires simultaneous or sequential transfer of two electrons from the ϕarene1a to the δxy and π*yz orbitals of 5INT2, respectively. An empty orbital on the naphthalene gives electron-deficient cationic characteristics, which enhances the conjugation with the oxygen’s lone pair and significantly alters the structural configuration of 3INT4. Notably, the cationic properties of naphthalene in 3INT4, exacerbated by the electron-withdrawing ester group, reduces its stability relative to 5INT3, consequently enhancing the radical pathway.

a Geometries, NPA charges and spin densities. b Electron-shift diagrams for the formation of 5INT3 and 3INT4. Hydrogen atoms omitted for clarity. The energies and distances are shown in kcal/mol and Å, respectively.
Ultimately, we employed 2,7-dimethoxynaphthalene 1s and 2,6-dimethoxynaphthalene 1t as model substrates to explore the origin of substituent-controlled reversal of chemoselectivity. The corresponding DFT-calculated free energy profile were presented in Fig. S15-S16. The proton shuttle mediated by the porphyrin was also considered (see Fig. S17 for details). Calculations reveal that 1s and 1t follow the same reaction mechanism and both behave similar to the results observed with 1a, the hydroxylation preferentially proceeds via a zwitterionic mechanism, whereas epoxidation occurs through a radical pathway. However, a reversal in reaction selectivity was observed, with hydroxylation now becoming the predominant reaction. Notably, replacing the electron-withdrawing -CO2Et with the electron-donating -OMe reverses the stability of the radical and zwitterionic intermediates, resulting in the zwitterionic intermediate 3INT13 being 5.3 kcal/mol more stable than the radical intermediate 5INT12 (Fig. 6a). This trend is consistent with the behavior observed for 5INT8 and 3INT9 (see Fig. S18 for details). This stability reversal can be ascribed to the electron-donating effect, which significantly enhances the stability of 3INT13 and narrows the energy gap between the ϕ orbital of 1t (ϕarene1t) and the π*yz orbital of 3INT2. The increased orbital energy of the ϕarene1t at −5.14 eV, is 1.04 eV higher than that of the ϕarene1a (−6.18 eV), shrinking the energy gap from 3.13 to 2.09 eV (Fig. 6b). The smaller gap facilitates electron transfer from the ϕarene1t to the π*yz orbital of 3INT2, promoting formation of 3INT13 and thereby reversing the observed chemoselectivity.

a Key intermediates. b Electron transfer from ϕ orbital of arene to π*yz orbital of INT2.
Discussion
In this work, we have developed a Mn(III) porphyrin-catalyzed method for the selective epoxidation of electron-deficient naphthalenes that is highly substrate-dependent, enabling the dearomative synthesis of naphthalene epoxides. Leveraging the electron-withdrawing substituents was crucial for precisely controlling the selectivity reversal from hydroxylation to epoxidation. Theoretical studies show that after oxidation and oxygen atom attack, both of zwitterionic and radical intermediates can be formed. Mechanistic experiments and theoretical calculations indicate that diethyl-2,6-naphthalenedicarboxylate and 2,6-dimethoxynaphthalene may follow two distinct reaction mechanisms. Electron-withdrawing -CO2Et group on the naphthalene ring favor the formation of the radical intermediate, thereby promoting the ring closure for epoxidation. In contrast, electron-donating -OMe group stabilize the zwitterionic intermediate, which subsequently undergoes an NIH shift followed by keto–enol tautomerization to complete hydroxylation. The reversal in selectivity is mainly ascribed to the energy gap between the ϕ orbital in the arene and the π*yz orbital of the high-valent Mn−O porphyrin. Our studies have significantly advanced the field of Mn(III) porphyrin-catalyzed epoxidation of electron-deficient naphthalene derivatives, particularly in synthetic methodologies and mechanistic studies. Currently, our laboratory is devoted to employing multivariate linear regression (MVLR)48 analysis to unravel quantitative structure-activity relationships governing substituent effects in epoxidation and hydroxylation, thereby facilitating the synthesis of a broader array of epoxides.
Methods
Synthesis of TPFPP
Under a nitrogen atmosphere and protected from light, the following reagents were added sequentially to a 2 L round-bottom flask equipped with a magnetic stir bar: dichloromethane (1.5 L), freshly distilled pyrrole (1.0 g, 15 mmol, 1.0 equiv.), and pentafluorobenzaldehyde (5.88 g, 30 mmol, 2.0 equiv.). After 15 min of reaction, boron trifluoride diethyl etherate (0.71 g, 5.0 mmol, 0.33 equiv.) was added. The mixture was stirred for 3 h, followed by the addition of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (6.81 g, 30 mmol, 2.0 equiv.) at room temperature. After an additional 3 h of stirring, triethylamine (5.0 mL) was added, and the mixture was stirred for another 30 minutes. The crude product was purified by silica gel column chromatography (eluent: petroleum ether/dichloromethane = 2/1, v/v) to afford TPTPP as a purple solid (443 mg, 12% yield).
Synthesis of Mn(TPFPP)Cl
In a 100 mL round-bottom flask equipped with a stir bar and under a nitrogen atmosphere, TPFPP (974 mg, 1 mmol, 10 equiv.) and manganese(II) acetate (1.76 g, 10 mmol, 10 equiv.) were charged. Dimethylformamide (50 mL) was added to the flask, and the reaction mixture was heated at 140 °C for 24 h. After completion, the mixture was cooled to room temperature and treated with 10 mL of concentrated hydrochloric acid (36%). The resulting precipitate was collected by filtration and washed with deionized water until the filtrate became nearly colorless. The product was dried overnight under reduced pressure prior to use, affording 560 mg of a dark red solid (53% yield).
General procedure for oxidation reaction
A 38 mL sealed tube (with a Teflon-lined cap) equipped with a magnetic stir bar was charged with substrate (1.0 mmol, 1.0 equiv.), Mn(TPFPP)Cl (0.01 mmol, 1.0 mol%), PhIO (1.6 mmol, 1.6 equiv.), and CH₃CN (5.0 mL). The tube was then sealed and immersed in a preheated oil bath at 50 °C. The reaction mixture was stirred for 1 h at this temperature before being cooled to room temperature. The solvent was removed under reduced pressure to afford the crude product, which was subsequently purified by flash chromatography on silica gel using petroleum ether/ethyl acetate as the eluent to yield the target product.
Computational methods
All DFT calculations are performed using the Gaussian16 package49. Geometric optimizations are performed at (U)B3LYP-D350,51,52,53 level of theory. The LANL2DZ basis set54,55,56 with ECP was used for Mn and I atoms, and the 6-31 G (d) basis set57,58,59 was used for other atoms. Frequency analyses were also performed at the same level of theory as geometry optimization to confirm whether optimized stationary points were either local minimum or transition state, as well as to evaluate zero-point vibrational energies and thermal corrections for enthalpies and free energies at 298.15 K. At the theoretical level of (U)B3LYP-D3/def2-TZVP60,61, the single point solvation energy is calculated using the SMD62 solvation model (solvent = acetonitrile) on the gas phase optimized geometry. Stability tests were performed on all structures to ensure that the correct unrestricted wave functions were obtained. The CYLview was used to visualize the optimized structure63. Orbital analyses were performed with Multiwfn64 and VMD65 software package. The minimum energy crossing point (MECP) was calculated using the sobMECP program66,67, which interfaces with Gaussian16. The NPA charges (Q) and spin densities (ρ) were computed at the (U)B3LYP-D3/6-31 G (d)-LANL2DZ level.
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2404381 (3a), 2482279 (4e) and 2485413 (4j). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. The experimental data generated in this study are provided in the Supplementary Information. The computational source data generated in this study have been deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.28130495)68. All data are available from the corresponding author upon request.
References
Zhang, L. & Ritter, T. A perspective on late-stage aromatic C-H bond functionalization. J. Am. Chem. Soc. 144, 2399–2414 (2022).
Chen, L. et al. Selective, catalytic oxidations of C-H bonds in polyethylenes produce functional materials with enhanced adhesion. Chem. 7, 137–145 (2021).
Litman, Z. C., Sharma, A. & Hartwig, J. F. Oxidation of hindered allylic C-H bonds with applications to the functionalization of complex molecules. ACS Catal. 7, 1998–2001 (2017).
Kawamata, Y. et al. Scalable, electrochemical oxidation of unactivated C-H bonds. J. Am. Chem. Soc. 139, 7448–7451 (2017).
Zhang, Q. et al. Oxidative upcycling of polyethylene to long chain diacid over Co-MCM-41 catalyst. Angew. Chem. Int. Ed. 63, e202407510 (2024).
Borgel, J., Tanwar, L., Berger, F. & Ritter, T. Late-stage aromatic C-H oxygenation. J. Am. Chem. Soc. 140, 16026–16031 (2018).
Genovino, J., Sames, D., Hamann, L. G. & Toure, B. B. Accessing drug metabolites via transition-metal catalyzed C-H oxidation: the liver as synthetic inspiration. Angew. Chem. Int. Ed. 55, 14218–14238 (2016).
Musolino, S. F. & Wulff, J. E. Catalytic C-H oxidation enhances polyethylene bonding. Chem. 7, 7–9 (2021).
Feng, X. et al. Rational construction of an artificial binuclear copper monooxygenase in a metal–organic framework. J. Am. Chem. Soc. 143, 1107–1118 (2021).
Guo, M., Corona, T., Ray, K. & Nam, W. Heme and nonheme high-valent iron and manganese oxo cores in biological and abiological oxidation reactions. ACS Cent. Sci. 5, 13–28 (2019).
Y. R. Luo. Handbook of bond dissociation energies in organic compounds (1st ed.). CRC Press. https://doi.org/10.1201/9781420039863 (2002).
Alonso, D. A., Najera, C., Pastor, I. M. & Yus, M. Transition-metal-catalyzed synthesis of hydroxylated arenes. Chem. Eur. J. 16, 5274–5284 (2010).
Ottenbacher, R. V., Talsi, E. P. & Bryliakov, K. P. Recent progress in catalytic oxygenation of aromatic C–H groups with the environmentally benign oxidants H2O2 and O2. Appl. Organomet. Chem. 34, e5900 (2020).
Zhou, H. et al. Chemo- and regioselective dihydroxylation of benzene to hydroquinone enabled by engineered cytochrome P450 monooxygenase. Angew. Chem. Int. Ed. 58, 764–768 (2019).
Baker Dockrey, S. A., Lukowski, A. L., Becker, M. R. & Narayan, A. R. H. Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem. 10, 119–125 (2018).
Cheng, L. et al. Iron-catalyzed arene C–H hydroxylation. Science. 374, 77–81 (2021).
Boyd, D. R. et al. Isolation of a stable benzene oxide from a fungal biotransformation and evidence for an ‘NIH shift’ of the carbomethoxy group during hydroxylation of methyl benzoates. Chem. Commun. 16, 1481–1482 (2000).
Kasperek, G. J., Bruice, T. C., Yagi, H., Kaubisch, N. & Jerina, D. M. Solvolytic chemistry of 1,4-dimethylbenzene oxide. A new and novel mechanism for the NIH shift. J. Am. Chem. Soc. 94, 7876–7882 (1972).
Daly, J. W., Jerina, D. M. & Witkop, B. Arene oxides and the NIH shift: the metabolism, toxicity and carcinogenicity of aromatic compounds. Experientia. 28, 1129–1149 (1972).
Jerina, D. M. & Daly, J. W. Arene oxides: a new aspect of drug metabolism. Science. 185, 573–582 (1974).
McManus, M. J., Berchtold, G. A. & Jerina, D. M. Nucleophilic addition of azide ion to benzene oxide: a reinvestigation. J. Am. Chem. Soc. 107, 2977–2978 (1985).
Jeffrey, A. M. et al. Stereochemical course in reactions between nucleophiles and arene oxides. J. Am. Chem. Soc. 96, 6929–6937 (1974).
Bertozzi, F. et al. Unprecedented catalytic enantioselective trapping of arene oxides with dialkylzinc reagents. Chem. Commun. 2001, 2606–2607 (2001).
Rastetter, W. H., Chancellor, T. & Richard, T. J. Biogenesis of epidithiadioxopiperazines. Nucleophilic additions to benzene oxide and sym-oxepin oxide. J. Org. Chem. 47, 1509–1512 (1982).
Vogel, E. & Gunther, H. Benzene oxide-oxepin valence tautomerism. Angew. Chem. Int. Ed. 6, 385–401 (1967).
Rebelo, S. L. H., Simoes, M. M. Q., Neves, M. G. P. M. S., Silva, A. M. S. & Cavaleiro, J. A. S. An efficient approach for aromatic epoxidation using hydrogen peroxide and Mn(III) porphyrins. Chem. Commun. 2004, 608–609 (2004).
Costa, P., Linhares, M., Rebelo, S. L. H., Neves, M. G. P. M. S. & Freire, C. Direct access to polycyclic peripheral diepoxy-meso-quinone derivatives from acene catalytic oxidation. RSC Adv. 3, 5350–5353 (2013).
Linhares, M., Rebelo, S. L. H., Biernacki, K., Magalhaes, A. L. & Freire, C. Biomimetic one-pot route to acridine epoxides. J. Org. Chem. 80, 281–289 (2015).
Siddiqi, Z., Wertjes, W. C. & Sarlah, D. Chemical equivalent of arene monooxygenases: dearomative synthesis of arene oxides and oxepines. J. Am. Chem. Soc. 142, 10125–10131 (2020).
Siddiqi, Z. et al. Oxidative dearomatization of pyridines. J. Am. Chem. Soc. 146, 2358–2363 (2024).
Zhang, W. et al. Biocatalytic aromaticity-breaking epoxidation of naphthalene and nucleophilic ring-opening reactions. ACS Catal. 11, 2644–2649 (2021).
Pereira, M. M., Dias, L. D. & Calvete, M. J. F. Metalloporphyrins: bioinspired oxidation catalysts. ACS Catal. 8, 10784–10808 (2018).
Wang, L. et al. A green process for oxidation of p-nitrotoluene catalyzed by metalloporphyrins under mild conditions. Org. Process Res. Dev. 10, 757–761 (2006).
Hu, B., Zhou, W., Ma, D. & Liu, Z. Metallo-deuteroporphyrins as catalysts for the oxidation of cyclohexane with air in the absence of additives and solvents. Catal. Commun. 10, 83–85 (2008).
Ge, Y., Su, X., Li, G., Yang, Y.-F. & She, Y. Oxidative functionalization of naphthalene derivatives catalyzed by Mn(III)-porphyrins. Tetrahedron Lett. 123, 154535 (2023).
Afailal, N. C., Chan, S.-C. & Costas, M. Manganese-catalyzed enantioselective dearomative epoxidation of naphthalenes with aqueous hydrogen peroxide. Angew. Chem. Int. Ed. 64, e202504356 (2025).
Gan, Z. et al. Diverse mechanisms for the aromatic hydroxylation: insights into the mechanisms of the coumarin hydroxylation by CYP2A6. ACS Catal. 14, 16277–16286 (2024).
Wanna, W. H. et al. Selective oxidation of simple aromatics catalyzed by nano-biomimetic metal oxide catalysts: a mini review. Front Chem. 8, 589178 (2020).
de Visser, S. P. & Shaik, S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J. Am. Chem. Soc. 125, 7413–7424 (2003).
Coleman, T. et al. Different geometric requirements for cytochrome P450-catalyzed aliphatic versus aromatic hydroxylation results in chemoselective oxidation. ACS Catal. 12, 1258–1267 (2022).
Bathelt, C. M., Ridder, L., Mulholland, A. J. & Harvey, J. N. Aromatic hydroxylation by cytochrome P450: model calculations of mechanism and substituent effects. J. Am. Chem. Soc. 125, 15004–15005 (2003).
Bathelt, C. M., Mulholland, A. J. & Harvey, J. N. QM/MM modeling of benzene hydroxylation in human cytochrome P450 2C9. J. Phys. Chem. A. 112, 13149–13156 (2008).
Chen, X., Cao, C., Yang, Y.-F. & She, Y.-B. Computational insights into different regioselectivities in the Ir-porphyrin-catalyzed C–H insertion reaction of quinoid carbene. Org. Chem. Front. 9, 1143–1151 (2022).
Liu, N., Chen, X., Jin, L., Yang, Y.-F. & She, Y.-B. A mechanistic study of the manganese porphyrin-catalyzed C–H isocyanation reaction. Org. Chem Front. 8, 1858–1866 (2021).
Jin, L. et al. Computational studies on the mechanism and origin of the different regioselectivities of manganese porphyrin-catalyzed C − H bond hydroxylation and amidation of equilenin acetate. J. Org. Chem. 85, 14879–14889 (2020).
Chen, X. et al. Mechanism and stereoselectivity of benzylic C − H hydroxylation by Ru-porphyrin: a computational study. Org. Biomol. Chem. 18, 346–352 (2020).
Larson, V. A., Battistella, B., Ray, K., Lehnert, N. & Nam, W. Iron and manganese oxo complexes, oxo wall and beyond. Nat. Rev. Chem. 4, 404–419 (2020).
Qian, G. et al. CCS Chem. 7, 1797–1811(2024).
Frisch, M. J. et al. Gaussian 16 Rev. C.01, Wallingford, CT, (2019).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 19 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Yang, T. H. et al. Singlet versus triplet reactivity in an Mn(V)-oxo species: testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 138, 12375–12386 (2016).
Witte, J., Mardirossian, N., Neaton, J. B. & Head-Gordon, M. Assessing DFT-D3 damping functions across widely used density functionals: can we do better? J. Chem. Theory Comput. 13, 2043–2052 (2017).
Wadt, W. R. & Hay, P. J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 82, 284–298 (1985).
Ehlers, A. W. et al. A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au. Chem. Phys. Lett. 208, 111–114 (1993).
Roy, L. E., Hay, P. J. & Martin, R. L. Revised basis sets for the LANL effective core potentials. J. Chem. Theory Comput. 4, 1029–1031 (2008).
Becke, A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377 (1993).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. C. 98, 11623–11627 (1994).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Ricciardi, G., Baerends, E. J. & Rosa, A. Charge effects on the reactivity of oxoiron(iv) porphyrin species: a DFT analysis of methane hydroxylation by polycationic compound I and compound II mimics. ACS Catal. 6, 568–579 (2016).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 113, 6378–6396 (2009).
Legault, C. Y. CYLview, 1.0b; Université de Sherbrooke: Canada. https://www.cylview.org (2009).
Lu, T. & Chen, F. W. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graphics. 14, 33–38 (1996).
Lu, T. sobMECP program. http://sobereva.com/286.
Harvey, J. N., Aschi, M., Schwarz, H. & Koch, W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc. 99, 95–99 (1998).
Wu, H. et al. Biomimetic Mn(III) porphyrins-catalyzed aromaticity-breaking epoxidation of electron-deficient naphthalene. Figshare. Dataset. https://doi.org/10.6084/m9.figshare.28130495 (2025).
Acknowledgements
We thank the National Natural Science Foundation of China (22138011, Y.-B. She; 22371256, Y.-F. Yang), the Zhejiang Provincial Natural Science Foundation of China (LR25B020002, Y.-F. Yang), the Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-C2022006, Y.-F. Yang) for financial support. We thank Jiyong Liu from Zhejiang University for the help in the single crystal measurement and analysis. We are grateful to Jiren Liu for his helpful discussion on this article.
Author information
Authors and Affiliations
Contributions
H. Wu and Y.-F. Yang analyzed the data and wrote the manuscript; J. Xu performed experiments; J. Tai performed the DFT calculations; Y. Ge and J. Gao analyzed the data; G. Li provided the experimental guidance; Y.-B. She and Y.-F. Yang conceived the idea and supervised this project. All authors discussed the results and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rong-Zhen Liao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, H., Xu, J., Tai, J. et al. Biomimetic Mn(III) porphyrin-catalyzed aromaticity-breaking epoxidation of electron-deficient naphthalene. Nat Commun 16, 11552 (2025). https://doi.org/10.1038/s41467-025-67300-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67300-8
