
Abstract
Kidney fibrosis is a key pathological feature of chronic kidney disease (CKD), characterized by the activation and accumulation of αSMA-positive myofibroblasts. The cellular origin of myofibroblasts in kidney fibrosis has been a subject of extensive research and debate. Previous studies suggested that myofibroblasts might arise from various cellular sources through mesenchymal transition processes, including epithelial-mesenchymal transition (EMT) of tubular epithelial cells (TECs), endothelial-mesenchymal transition (EndoMT), and macrophage-mesenchymal transition (MMT) of bone marrow-derived cells. In this study, we systematically investigated the origin of myofibroblasts using renal cell lineage tracing tools during kidney fibrosis. Our findings indicated the absence of EMT, EndoMT, and MMT contributing to myofibroblasts, whereas resident fibroblasts were the primary source. Next, we developed and employed specialized dual recombinase-mediated lineage tracing tools, including EMTracer, EndoMTracer, and MMTracer, which revealed reversible partial mesenchymal transition occurring predominantly in the renal urothelial cells (UroCs) and parietal epithelial cells (PECs), minimally in endothelial cells, and not in TECs or macrophages. Our work elucidated the contribution of both complete and partial mesenchymal transition in various renal cell lineages and revealed the origin of myofibroblasts during kidney fibrosis, which may provide important insights into the diagnosis and treatment of CKD.
Similar content being viewed by others

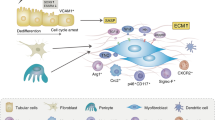

Introduction
CKD is a widespread condition that affects over 10% of the global population1. A hallmark of advanced CKD is tissue fibrosis, marked by the excessive accumulation of extracellular matrix (ECM)2. This process leads to scar formation and a progressive decline in kidney function. Myofibroblasts, characterized by αSMA expression, are usually rare in normal kidneys but become significantly more abundant during kidney fibrosis, serving as the primary ECM-producing cells and playing a crucial role in the fibrotic process2,3. Understanding the mechanisms behind myofibroblast generation is essential for the development of effective CKD treatment strategies. Despite extensive research, the origin of kidney myofibroblasts remains a subject of debate2,3,4. Previous studies have suggested that interstitial mesenchymal cells (resident fibroblasts/pericytes)5,6,7,8,9, TECs10,11, endothelial cells11,12,13,14,15, and bone marrow-derived cells11,16,17,18,19,20,21 could serve as the cellular sources of myofibroblasts. While the role of resident fibroblasts as a source of myofibroblasts is generally accepted, the other proposed sources remain controversial2,4,22.
Among the non-fibroblast origins, TECs were first proposed to undergo EMT to transform into myofibroblasts. Previous studies primarily focused on proximal TECs, utilizing genetic lineage tracing tools such as γGT-Cre10,11, Six2-GFPCre6, Ksp-Cre23, Slc34a1-CreER24, with varying results indicating major, minor, or no contribution to myofibroblast formation. However, these genetic tools possess certain limitations, in that constitutive Cre-based lineage tracing may lead to nonspecific labeling, since Cre is continuously expressed throughout development and following injury, potentially causing false positives if transiently expressed in non-target cells. In contrast, tamoxifen-induced, temporally controlled CreER-based lineage tracing offers more precise labeling. Additionally, previous studies using Ksp-Cre showed low labeling efficiency (21%) in proximal TECs, and Slc34a1-CreER only labeled S1/S2 segments, excluding S3, of the proximal TECs. Furthermore, aside from TECs, other renal epithelial cells, such as the renal urothelium––a monolayered epithelium that covers the renal pelvis25, and PECs, which line the outer membrane of Bowman’s capsule26, are often overlooked in kidney EMT research.
EndoMT-related studies predominantly relied on Tie-Cre12,13,14 and Cdh5-Cre11,15. Both Tie2 and CDH5 are expressed in most hematopoietic cells during the embryonic stage27,28, with Tie2 also found in perivascular cells29,30. MMT studies used bone marrow transfer or Lyz2-Cre-based lineage tracing11,16,17,18,19,20,21,24, yielding controversial conclusions and lacking macrophage-specific inducible genetic tools for reliable analysis. These studies often relied on immunostaining to analyze αSMA colocalization with genetically labeled cells. However, due to the abundance of endothelial cells and macrophages in the renal interstitium and the fact that they are in proximity to myofibroblasts, distinguishing true colocalization from mere proximity is technically challenging.
The concept of “partial EMT” in TECs has been proposed to play a significant role in kidney fibrosis31,32,33,34. This process involves epithelial cells activating αSMA without fully converting into myofibroblasts, allowing them to remain within the renal tubules. Despite its potential importance, the existence of partial EMT has not been conclusively established in the past decade. One of the primary challenges in studying partial EMT is the low detection resolution in studies that examine αSMA colocalization with TECs, which has limited our understanding of this phenomenon. Furthermore, the existence and potential roles of other partial mesenchymal transitions, such as “partial EndoMT” and “partial MMT” in the context of kidney fibrosis, remain unexplored. These processes could similarly involve the activation of mesenchymal markers without complete transdifferentiation, potentially contributing to the fibrotic environment in the kidney.
To address these challenges, we developed inducible genetic lineage tracing tools for lineage analysis of renal epithelial cells, endothelial cells, and macrophages. For epithelial cells, our focus extends beyond the commonly studied proximal TECs to non-proximal TECs, glomerular PECs, and renal UroCs. To overcome the limitations of traditional lineage tracing tools, which often depend on colocalization analysis of genetically labeled cells with myofibroblast marker αSMA to detect the mesenchymal transition events, we developed and employed specialized dual recombinase-mediated lineage tracing tools, XMTracer (including EMTracer, EndoMTracer, and MMTracer). These specially designed genetic tools facilitated the capture and direct visualization of both complete and partial XMT events, enabling genetic labeling of transformed cells without relying on colocalization analysis, thus effectively distinguishing them from untransformed cells. Moreover, XMTracer is particularly effective for detecting rare XMT events and transient αSMA activation, both posing significant challenges for traditional lineage tracing methods.
Results
Lineage tracing of proximal TECs by Hnf4a-CreER in kidney fibrosis
Given that previous studies on EMT have primarily focused on proximal TECs, we began by re-evaluating EMT in proximal TECs. HNF4a has been reported to be expressed in proximal TECs35 (Fig. 1a). To trace proximal TECs specifically, we developed an inducible genetic lineage tracing mouse model, Hnf4a-CreER (Fig. 1b). By crossing this mouse model with NR1 reporter mouse line36, we generated Hnf4a-CreER; NR1 mice, in which, cells expressing HNF4a and their progeny were labeled with green fluorescent protein ZsGreen following tamoxifen-induced Cre-loxP recombination (Fig. 1c). Seven days post-tamoxifen induction, ZsGreen+ cells were detected in the renal cortex (Fig. 1d, e). Immunostaining for ZsGreen alongside the proximal TEC marker HNF4a and the non-proximal TEC marker EpCAM confirmed that all ZsGreen+ cells were HNF4a+ proximal TECs, whereas no ZsGreen was observed in EpCAM+ non-proximal TEC cells (Fig. 1f, g). Further immunostaining with other proximal TEC markers, such as CD1337 and Lotus tetragonolobus lectin (LTL)38, demonstrated the efficient and specific labeling of proximal TECs by the Hnf4a-CreER line (Supplementary Fig. 1a–c). We also evaluated ZsGreen expression in the liver, where HNF4a is a known hepatocyte marker36. Immunostaining for HNF4a and the biliary epithelial cell (BEC) marker CK19 indicated that ZsGreen was specifically expressed in HNF4a+ hepatocytes but not in CK19+ BECs (Supplementary Fig. 1d–f). Overall, our results demonstrated that the HNF4a-CreER mouse line is an effective tool for specifically labeling HNF4a+ cells, which provides a robust genetic method for tracing proximal TECs in the kidney.

a Schematic diagram of renal nephron structure and HNF4a+ proximal TECs. b Strategy for generating Hnf4a-CreER knock-in mouse line. c Lineage tracing strategy for proximal TECs. d Experimental design for labeling proximal TECs. e Whole-mount bright-field and fluorescent images of the kidney (left) and immunostaining for ZsGreen on kidney sections (right). f Immunostaining for ZsGreen, HNF4a, and EpCAM on kidney sections. g Quantification of the percentage of ZsGreen+ cells in HNF4a+ and EpCAM+ cells respectively. Data are the mean ± SEM, n = 5. h Experimental design for tracing proximal TECs during kidney fibrosis. i Immunostaining for ZsGreen, HNF4a, and αSMA on kidney sections. j Quantification of percentage of ZsGreen+ cells in HNF4a+ cells and αSMA+ cells. Data are the mean ± SEM, n = 5. k Cartoon image showing proximal TECs do not undergo EMT during kidney fibrosis. PT proximal tubule, DT distal tubule, LH loop of Henle, CD collecting duct. Scale bars: white 100 µm; yellow 1 mm. Each figure is representative of five individual biological samples.
We then used the Hnf4a-CreER; NR1 mice to trace the fate of proximal TECs during kidney fibrosis. Following tamoxifen treatment, mice were subjected to unilateral ureteral obstruction (UUO) to induce kidney fibrosis, and kidneys were harvested at 7 and 14 days post-UUO injury (Fig. 1h). Immunostaining for αSMA, HNF4a, and ZsGreen, along with z-stack imaging, showed that ZsGreen was exclusively expressed in HNF4a+ proximal TECs, with no overlap between ZsGreen and αSMA after UUO injury (Fig. 1i). Quantitative analysis revealed that 100% of ZsGreen+ cells remained as proximal TECs, with no αSMA+ myofibroblasts detected in either sham or UUO injury groups (Fig. 1j). Additionally, immunostaining for ZsGreen and PDGFRa, a universal fibroblast marker, confirmed that ZsGreen+ cells did not differentiate into PDGFRa+ fibroblasts or myofibroblasts in kidney fibrosis (Supplementary Fig. 1g). Together, these findings suggest that proximal TECs do not undergo EMT to become myofibroblasts during kidney fibrosis, instead, they retain their epithelial cell identity, regardless of the fibrosis stage (Fig. 1k).
Absence of αSMA activation in proximal TECs during kidney fibrosis revealed by proximal TEC EMTracer
EMT might be a dynamic and reversible process, with αSMA potentially being expressed only transiently. Consequently, our inability to detect EMT using traditional lineage tracing methods could be due to an improper time window of tissue analysis. Additionally, the detection of EMT could be challenging because the occurrence of EMT events might be rare, leading to a low probability of observing EMT at any given time point. To effectively capture the transient or rare expression of αSMA, we developed a novel lineage tracing system utilizing a dual recombinase-mediated genetic approach, termed XMTracer. This system consists of X-CreER, αSMA-loxP-Stop-loxP-Dre (αSMA-LSL-Dre)39, and NR1 reporter (Fig. 2a). In this context, “XMT” can refer to EMT, EndoMT, or MMT, depending on the gene promoter X in X-CreER is specific to epithelial, endothelial, or macrophage cells. In this XMTracer system, after tamoxifen induction, CreER enters the nucleus to excise the “Stop” sequence between the two loxP sites via Cre-loxP recombination in both αSMA-LSL-Dre and the NR1 reporter, converting αSMA-LSL-Dre into αSMA-Dre specifically in X cells, meanwhile, labeling these cells with ZsGreen. If αSMA is activated at any time in X cells or their progeny after tamoxifen induction, Dre will target the rox sites in NR1, removing the “Stop” and “ZsGreen” sequence to activate tdTomato expression, thereby labeling the X cells and their progeny with tdTomato. The labeling is permanent due to the irreversibility of Dre-rox recombination, even if αSMA subsequently turns off (Fig. 2a, b). Thus, with XMTracer, ZsGreen+ cells indicate the absence of XMT, tdTomato+ cells that maintain the X cell identity indicate partial XMT, and tdTomato+ cells that became myofibroblasts indicate XMT (Fig. 2b). Compared with conventional lineage tracing methods, XMTracer allows efficient detection of both complete and partial XMT, even with transient and rare αSMA activation (Fig. 2c).

a XMTracer Strategy for capturing transient αSMA activation. b Schematic diagram showing the readout and corresponding cell fate of XMTracer. c Schematic diagram showing the capture window of αSMA activation using XMTracer compared with conventional methods. d Experimental design for capture αSMA activation in PT epithelial cells by proximal TEC EMTracer. e Immunostaining for ZsGreen, tdTomato, and HNF4a on kidney sections. f Immunostaining for ZsGreen, LTL, and CD13 on kidney sections. g Quantification of the percentage of tdTomato+ cells. Data are the mean ± SEM, n = 5. h PCR analysis of genomic DNA from ZsGreen+ and ZsGreen– cells. The image corresponds to three biological replicates, which are DNA samples extracted respectively from sorted cells of three individual mice. i Cartoon image showing proximal TECs do not undergo XMT and partial EMT during kidney fibrosis. Scale bars: white 100 µm; yellow 1 mm. Each figure is representative of five individual biological samples.
To investigate complete and partial EMT in proximal TECs, we crossed Hnf4a-CreER with αSMA-LSL-Dre and NR1 mice to obtain the proximal TEC EMTracer. After tamoxifen induction, we performed sham or UUO injury on these mice and harvested the kidney 7 days and 14 days post-surgery for analysis (Fig. 2d). Immunostaining showed that all the ZsGreen+ cells express proximal TEC marker HNF4a, LTL, and CD13 (Fig. 2e, f), while no tdTomato+ cells were observed in either the sham or UUO injury groups (Fig. 2e, g). As a technical control, we examined the excision efficiency of the “Stop” sequence in αSMA-LSL-Dre by Hnf4a-CreER to ensure that αSMA activation in proximal TECs could be labeled by tdTomato. Genomic DNA from ZsGreen+ and ZsGreen− cells isolated from proximal TEC EMTracer mice was analyzed by PCR, the results showed that nearly 100% of ZsGreen+ PT cells had excised the Stop cassette, indicating that αSMA-LSL-Dre has been converted to αSMA-Dre in all ZsGreen+ proximal TECs, but not in ZsGreen– cells (Fig. 2h). The data suggested that the proximal TEC EMTracer can faithfully and efficiently capture αSMA activation in proximal TECs. In summary, proximal TEC EMTracer did not capture any αSMA activation in proximal TECs, indicating the absence of EMT or partial EMT in proximal TECs during kidney fibrosis (Fig. 2i).
Lineage tracing of non-proximal TECs by Cdh1-CreER in kidney fibrosis
In light of the absence of EMT and partial EMT in proximal TECs, we evaluated whether EMT or partial EMT occurs in non-proximal TECs. Given that CDH1 is expressed in all non-proximal TECs40 (Fig. 3a), we generated a Cdh1-CreER mouse line and crossed it with NR1 mice to produce the Cdh1-CreER; NR1 double knock-in mice for tracing non-proximal TECs (Fig. 3b, c). Due to the low recombination efficiency of Cdh1-CreER; NR1, we administered five doses of tamoxifen to enhance labeling efficiency (Fig. 3d). Following tamoxifen induction, ZsGreen+ cells were observed in both the cortex and medulla, predominantly in the medulla (Fig. 3e). Immunostaining and quantitative analyses revealed the ZsGreen+ cells labeled by Cdh1-CreER; NR1 are EpCAM+ non-proximal TECs, with no HNF4a+ proximal TECs detected (Fig. 3f, g). These results demonstrate Cdh1-CreER; NR1 serves as an effective genetic lineage tracing tool for non-proximal TECs. We next assessed the contribution of CDH1+ non-proximal TECs to the myofibroblast formation following UUO-induced kidney fibrosis (Fig. 3h). Remarkably, we did not observe any ZsGreen signals in the αSMA+ or PDGFRa+ myofibroblasts/fibroblasts at either 7 days or 14 days after obstruction; all ZsGreen+ cells retained their epithelial identity and expressed EpCAM (Fig. 3i–k). These results indicated that CDH1+ non-proximal TECs did not contribute to myofibroblast formation via EMT in kidney fibrosis (Fig. 3l).

a Schematic diagram of renal nephron structure and CDH1+ non-proximal TECs. b Strategy for generating Cdh1-CreER knock-in mouse line. c Lineage tracing strategy for CDH1+ EpCs. d Experimental design for labeling CDH1+ EpCs. e Whole-mount bright-field and fluorescent images of the kidney (left) and immunostaining for ZsGreen on kidney sections (right). f Immunostaining for ZsGreen, HNF4a, and EpCAM on kidney sections. g Quantification of the percentage of ZsGreen+ cells in HNF4a+ cells and EpCAM+ cells, respectively. Each data point represents the average of statistics from three fields of view per biological sample, with a total of five biological replicates in each group. Data are the mean ± SEM, n = 5. h Experimental design for tracing CDH1+ EpCs during kidney fibrosis. i Immunostaining for ZsGreen, EpCAM, and αSMA on kidney sections. j Immunostaining for ZsGreen and PDGFRa on kidney sections. k Quantification of the percentage of ZsGreen+ cells in HNF4a+, αSMA+, and PDGFRa+ cells. Each data point represents the average of statistics from three fields of view per biological sample, with a total of five biological replicates in each group. Data are the mean ± SEM, n = 5. l Cartoon image showing CDH1+ EpCs do not undergo EMT during kidney fibrosis. Scale bars: white 100 µm; yellow 1 mm. Each figure is representative of five individual biological samples.
Recognizing the limitations of conventional lineage tracing with Cdh1-CreER; NR1 in detecting transient or rare αSMA expression within CDH1 cell lineage, we generated a non-proximal TEC EMTracer by crossing Cdh1-CreER with αSMA-LSL-Dre and NR1 reporter. Similar to proximal TEC EMTracer (Fig. 2), this tool allows detection of EMT and partial EMT in non-proximal TECs, even with rare or transient αSMA activation (Fig. 4a). We activated the system by tamoxifen administration, following UUO injury to induce kidney fibrosis (Fig. 4b), tdTomato signals were difficult to observed in sham and 7-day post-UUO groups. Surprisingly, tdTomato+ cells appeared in the 14-day post-UUO group, located in the renal urothelium, but not in the TECs (Fig. 4c). Immunostaining for EpCAM and PDGFRa showed that tdTomato+ cells were EpCAM+ epithelial cells, but not PDGFRa+ fibroblast (Fig. 4c–f), which indicated αSMA expression in UroCs without transdifferentiation into myofibroblast/fibroblasts. To verify whether tdTomato+ cells in UroCs were due to Cre-rox crosstalk in Cdh1-CreER; NR1, causing tdTomato leakiness, we examined tdTomato signals on UUO injured Cdh1-CreER; NR1 mice, and observed no tdTomato+ cells, which suggested the tdTomato signals resulted from αSMA activation in UroCs 14 days post-UUO injury (Fig. 4h, i). We also evaluated the “Stop” sequence excision efficiency in αSMA-LSL-Dre by Cdh1-CreER. PCR analysis of genomic DNA revealed the conversion of αSMA-LSL-Dre to αSMA-Dre in most ZsGreen+ cells (Fig. 4j, k). Overall, our findings suggested that CDH1+ UroCs, but not CDH1+ TECs, underwent partial EMT during kidney fibrosis (Fig. 4g).

a Schematic diagram of EMT and Partial EMT revealed by Cdh1-CreER-driven EMTracer. MFCs, myofibroblasts. b Experimental design for capturing αSMA activation in CDH1+ EpCs. c, d Immunostaining for ZsGreen, tdTomato, and EpCAM on kidney sections. e Immunostaining for ZsGreen, tdTomato (tdT), and PDGFRa on kidney sections. f Quantification of the percentage of tdTomato+ cells in EpCAM+ cells and in PDGFRa+ cells. Each data point represents the average of statistics from three fields of view per biological sample, with a total of five biological replicates in each group. Data are the mean ± SEM, n = 5, *P < 0.05. **P < 0.01.). g Cartoon image showing CDH1+ UroCs undergo partial EMT but not full EMT during kidney fibrosis, while CDH1+ TECs do not undergo EMT and Partial EMT during kidney fibrosis. h Experimental design for tracing CDH1+ EpCs 14 days after UUO injury. i Immunostaining for ZsGreen and tdTomato on kidney sections. j Experimental design for cell isolation for PCR analysis. k PCR analysis of genomic DNA from ZsGreen+ and ZsGreen– cells. Scale bars: yellow 1 mm; white 100 µm; red 20 µm. Each figure is representative of five individual biological samples.
Lineage tracing of TECs, PECs, and UroCs by Cdh16-CreER in kidney fibrosis
To examine EMT in all TECs, PECs, and UroCs during kidney fibrosis, we generated a Cdh16-CreER mouse model (Supplementary Fig. 2a). CDH16, also known as kidney-specific (ksp) cadherin, is expressed in all these cell types (Fig. 5a). We crossed Cdh16-CreER mice with NR1 reporter, which enables ZsGreen labeling of all TECs, PECs, and UroCs following tamoxifen induction (Fig. 5b). To characterize this mouse model, we harvested kidney tissues at 7 days post-tamoxifen induction (Fig. 5c). ZsGreen+ cells were observed throughout the renal cortex and medulla, including both the tubular and glomerular area (Fig. 5d, e). To further identify the cell types labeled by the Cdh16-CreER; NR1 mouse model, we performed staining with PEC marker WT1, proximal TEC marker HNF4a, and non-proximal TEC and UroC marker EpCAM. Although WT1 is also expressed in podocytes, we could distinguish PECs from podocytes based on their location. Similarly, UroCs can be distinguished from TECs by their location. Our results indicated that Cdh16-CreER; NR1 efficiently labeled TECs, PECs, and UroCs at 100% (Fig. 5f), demonstrating it is a powerful tool for the lineage tracing of renal TECs, PECs, and UroCs.

a Schematic diagram of renal nephron structure and CDH16+ TECs and PECs. b Lineage tracing strategy for CDH16+ epithelial cells (EpCs). c Experimental design for labeling CDH16+ EpCs. d Whole-mount bright-field and fluorescent images of the kidney. e Immunostaining for ZsGreen on kidney sections. f Immunostaining for ZsGreen, HNF4a, and EpCAM on kidney sections from the sham group and quantification of the percentage of ZsGreen+(ZsG+) cells in WT1+ PECs, HNF4a+ proximal TECs, and EpCAM+ non-proximal TECs and UroCs respectively. Data are the mean ± SEM, n = 5. g Immunostaining for ZsGreen, HNF4a, EpCAM, and αSMA on kidney sections from the mice after UUO injury at 7 days and 14 days (left) and quantification of the percentage of ZsGreen+ cells in HNF4a+/EpCAM+ TECs and αSMA+ cells (right). Data are the mean ± SEM, n = 5. h Immunostaining for ZsGreen, αSMA, and PDGFRa on kidney sections from the mice after UUO injury at 7 days (left) and quantification of the percentage of ZsGreen+ PECs and UroCs in αSMA+ or PDGFRa+ cells (right). Data are the mean ± SEM, n = 5. i Cartoon image showing CDH16-CreER labeled TECs, PECs, and UroCs do not undergo EMT during kidney fibrosis. Scale bars: yellow 1 mm; white 100 µm; red 50 µm. Each figure is representative of five individual biological samples.
We next assessed the contribution of renal TECs, PECs, and UroCs to myofibroblast formation in kidney fibrosis using this genetic tool. Consistent with the results from the Hnf4a-CreER- and Cdh1-CreER-based lineage tracing, Cdh16-CreER; NR1 labeled ZsGreen+ proximal and non-proximal TECs still maintain their epithelial cell identity, with no ZsGreen signals co-localizing with αSMA or PDGFRa at 7 days and 14 days post-UUO injury (Fig. 5g and Supplementary Fig. 2b, c). In the glomerular area and urothelium, αSMA+ and PDGFRa+ cells surrounded the ZsGreen+ PECs and UroCs, respectively, with no expression of αSMA or PDGFRa in ZsGreen+ PECs and UroCs following UUO injury (Fig. 5h and Supplementary Fig. 2d–f). These results indicated that CDH16+ renal TECs, PECs, and UroCs did not give rise to myofibroblasts during kidney fibrosis (Fig. 5i).
Partial EMT in PECs and UroCs instead of TECs during kidney fibrosis revealed by Cdh16-CreER-driven EMTracer
We next generated Cdh16-CreER-driven EMTracer by crossing Cdh16-CreER with αSMA-LSL-Dre and NR1, for the detection of transient or rare αSMA+ activation in TECs, PECs, and UroCs (Fig. 6a). This model was induced with tamoxifen, followed by either sham or UUO surgery, and kidneys were collected at indicated times for analysis (Fig. 6b). As expected, no tdTomato+ cells were detected in the sham group. However, tdTomato signals were observed in PECs and UroCs 7 days post-UUO injury, becoming more pronounced at 14 days post-UUO. At this time point, 49.48 ± 15.25% of UroCs and 47.50 ± 6.65% of PECs showed tdTomato expression, while no tdTomato+ TECs were detected (Fig. 6c, d). We further examined the identity of tdTomato+ cells in UroCs and PECs using immunostaining with specific markers. We found that all tdTomato+ cells lacked PDGFRa but expressed EpCAM in the UroCs and WT1 in PECs (Fig. 6e, f), indicating αSMA activation in epithelial cells without transdifferentiation into myofibroblasts. In addition, αSMA antibody staining showed no expression of αSMA in the UroCs and PECs, both at 7 days and 14 days post-injury, indicating αSMA activation in the UroCs and PECs is transient and reversible (Fig. 5h and Supplementary Fig. 2d–f).

a Schematic diagram of EMT and Partial EMT revealed by Cdh16-CreER-driven EMTracer. MFCs, myofibroblasts. b Experimental design for capture αSMA activation in CDH16+ EpCs. c Immunostaining for ZsGreen and tdTomato on kidney sections (left), and quantification of the percentage of tdTomato+ cells in UroCs, PECs, and TECs, respectively (right). Each data point represents the average of statistics from three fields of view per biological sample, with a total of five biological replicates in each group. Data are the mean ± SEM, n = 5. d Immunostaining for ZsGreen, tdTomato, and EpCAM on kidney sections. Immunostaining for ZsGreen, tdTomato, PDGFRa, and EpCAM (e) or WT1 (f) on kidney sections. Yellow arrowheads indicate tdTomato co-localizes with EpCAM (e) or WT1(f), while white arrowheads indicate tdTomato doesn’t co-localize PDGFRa. Cartoon image showing PECs and UroCs, but not TECs, undergo partial EMT (g), and none of the undergo EMT during kidney fibrosis (h). Scale bars: yellow 1 mm; white 100 µm; red 20 µm. Each figure is representative of five individual biological samples.
To ensure that tdTomato expression was due to αSMA activation rather than Cre-rox crosstalk in Cdh16-CreER; NR1, we analyzed tdTomato expression in kidney sections from Cdh16-CreER; NR1 mice 14 days post-UUO injury and observed no tdTomato+ cells (Supplementary Fig. 2g, h). This indicates that Cdh16-CreER-driven EMTracer faithfully captures αSMA activation. Compared with the Cdh1-CreER-driven EMTracer, more tdTomato+ cells were detected in UroCs using Cdh16-CreER-driven EMTracer, for the higher efficiency of Cdh16-CreER mouse model relative to the Cdh1-CreER mouse model. PCR analysis of genomic DNA confirmed the high excision efficiency of the “Stop” sequence in αSMA-LSL-Dre by Cdh16-CreER (Supplementary Fig. 2i, j).
Our results showed that partial EMT occurs in vivo, not in the previously reported TECs, but rather in UroCs and PECs (Fig. 6g). Despite the presence of partial EMT, these cells ultimately retain their epithelial identity and do not contribute to the formation of myofibroblasts (Fig. 6g).
Minimal partial EndoMT in kidney fibrosis revealed by EndoMTracer
In addition to epithelial cells, endothelial cells have also been reported as a significant source of myofibroblasts in kidney fibrosis through EndoMT11,12,13,14,15. We next investigated the contribution of endothelial cells to myofibroblast formation. CDH5 is a common endothelial marker (Fig. 7a), and we used a Cdh5-CreER41 mouse line for endothelial cell lineage tracing during kidney fibrosis (Fig. 7b). By crossing Cdh5-CreER with NR1, we generated Cdh5-CreER; NR1 mice and performed sham or UUO surgery on this double knock-in mice following tamoxifen administration (Fig. 7c). Immunostaining for ZsGreen and endothelial cell marker CD31 confirmed that ZsGreen was efficiently and specifically expressed in endothelial cells in the sham group (Fig. 7d). In the UUO group, we examined the co-localization of ZsGreen with CD31, αSMA, and PDGFRa using immunostaining (Fig. 7e and Supplementary Fig. 3a). Due to the proximity of endothelial cells to fibroblasts, determining overlap with fibroblast markers can be challenging. Therefore, we performed z-stack imaging to analyze the co-localization of ZsGreen with CD31 and αSMA/PDGFRa in the same sections. Z-stack images showed that ZsGreen signals were near αSMA/PDGFRa signals rather than completely overlapping, instead merging with CD31 (Fig. 7e, f and Supplementary Fig. 3a). These results indicated that endothelial cells did not transdifferentiate into myofibroblasts during kidney fibrosis (Fig. 7g).

a Schematic diagram of CDH5+ renal endothelial cells (EnCs). b Lineage tracing strategy for renal EnCs. c Experimental design for tracing renal EnCs in sham and UUO-injured mice. d Immunostaining for ZsGreen and CD31 on kidney sections from the sham group. e Immunostaining for ZsGreen, CD31, and αSMA on kidney sections 7 and 14 days after UUO injury. f Quantification of the percentage of ZsGreen+ cells in CD31+ EnCs and αSMA+ cells. Each data point represents the average of statistics from three fields of view per biological sample, with a total of five biological replicates in each group. Data are the mean ± SEM, n = 5. g Cartoon image showing renal EnCs do not undergo EndoMT during kidney fibrosis. h Schematic diagram of EndoMT and partial EndoMT revealed by EndoMTracer. i Experimental design for capture αSMA activation in renal EnCs. j, k Immunostaining for ZsGreen, tdTomato (tdT), and CD31 on kidney sections. Yellow arrowheads indicate tdTomato co-localizes with CD31. l Immunostaining for ZsGreen, tdTomato, and PDGFRa on kidney sections. White arrowheads indicate tdTomato doesn’t co-localize with PDGFRa. m Quantification of the percentage of tdTomato+ cells in PDGFRa+ cells and CD31+ cells. Data are the mean ± SEM, n = 5. n Cartoon image showing renal EnCs undergo minimal partial EndoMT during kidney fibrosis. Scale bars: yellow 1 mm; white 100 µm; red 20 µm. Each figure is representative of five individual biological samples.
Cdh5-CreER; NR1-based conventional lineage tracing method requires analyzing the co-localization of genetically labeled cells with myofibroblast marker. However, due to their proximity with each other, the results are not always reliable. Besides, if only a few endothelial cells transform into myofibroblasts, these rare EndoMT events can easily be missed. Therefore, direct labeling of the transformed endothelial cells, distinguishing them from non-transformed endothelial cells and non-endothelial cell-derived myofibroblasts, would enable more effective detection of EndoMT. To efficiently capture EndoMT, we developed EndoMTracer by crossing Cdh5-CreER with αSMA-LSL-Dre and NR1, treated these mice with tamoxifen, and performed sham or UUO surgery. Compared with the sham group, we observed tdTomato+ cells after UUO injury, but at a very low percentage (0.05 ± 0.04%) (Fig. 7j, m). Further immunostaining revealed that these rare tdTomato+ cells were CD31+ endothelial cells rather than PDGFRa+ fibroblasts/myofibroblasts, indicating αSMA activation in endothelial cells without conversion to myofibroblasts, which suggested partial EndoMT (Fig. 7j–l).
To exclude the possibility that tdTomato+ cells resulted from Cre-rox crosstalk in Cdh5-CreER; NR1, we assessed tdTomato expression in Cdh5-CreER; NR1 mice 14 days after subjected to UUO, and no tdTomato+ cells were observed (Supplementary Fig. 3b, c), which suggested that αSMA was activated in some endothelial cells after kidney fibrosis. We also examined the “Stop” sequence excision efficiency in αSMA-LSL-Dre by Cdh5-CreER to determine if the rare tdTomato detected in EndoMTracer was due to low excision efficiency. PCR analysis of genomic DNA revealed complete conversion of αSMA-LSL-Dre to αSMA-Dre in ZsGreen+ cells (Supplementary Fig. 3d, e), indicating that EndoMTracer can efficiently capture EndoMT and partial EndoMT events.
Taken together, EndoMTracer revealed that during kidney fibrosis, endothelial cells did not undergo EndoMT to contribute to myofibroblast, and only minimal endothelial cells activated αSMA expression, termed partial EndoMT, which indicates a negligible contribution to kidney fibrosis (Fig. 7n).
Absence of MMT during kidney fibrosis revealed by MMTracer
During kidney fibrosis, a substantial of macrophages infiltrate the renal interstitium. These macrophages typically originate from resident macrophages or are derived from bone marrow-derived monocytes that are recruited and subsequently differentiate into macrophages. It has been suggested that macrophages can transform into myofibroblasts through a process known as MMT. To assess the MMT events during kidney fibrosis, we employed Lyz2-CreER42 and Cx3cr1-CreER43 to trace recruited and resident macrophages, respectively (Fig. 8a, b). Immunostaining for ZsGreen and F4/80 showed that Lyz2-CreER and Cx3cr1-CreER collectively labeled more than 90% renal macrophages in both sham and UUO groups (Supplementary Fig. 4a, b), confirming that our tools effectively captured the vast majority of renal macrophages. Given the proximity of macrophages to fibroblasts/myofibroblasts, we performed immunostaining for ZsGreen alongside αSMA/PDGFRa and macrophage marker F4/80 on the same tissue sections, employing z-stack imaging to visualize colocalization of ZsGreen signals with F4/80 and αSMA/PDGFRa. No ZsGreen signals were detected in fibroblasts or myofibroblasts after UUO surgery in either Lyz2-CreER; NR1 or Cx3cr1-CreER; NR1 mice, which suggested that neither recruited nor resident macrophages transdifferentiated into myofibroblasts during kidney fibrosis. (Fig. 8c, d, and Supplementary Fig. 4c–f).

a Schematic diagram of recruited macrophages (MCs) and resident MCs in the kidney. b Experimental design for tracing macrophages in sham and UUO-injured mice. Immunostaining for ZsGreen, F4/80, and CD31 on kidney sections from Lyz2-CreER; NR1 (c) or Cx3cr1-CreER; NR1 (d) mice after UUO injury, and quantification of the percentage of ZsGreen+ cells in F4/80+ cells and αSMA+ cells, respectively. Each data point represents the average of statistics from three fields of view per biological sample, with a total of five biological replicates in each group. Data are the mean ± SEM, n = 5. Cartoon image showing recruited MCs and resident MCs didn’t undergo MMT during kidney fibrosis. e Schematic diagram of MMT and partial MMT revealed by MMTracer. f Experimental design for tracing recruited MCs and resident MCs in sham and UUO-injured mice. g Immunostaining for ZsGreen and tdTomato on kidney sections from MMTracer. h Quantification of the percentage of tdTomato+ cells in MMTracer mice. Data are the mean ± SEM, n = 5. i Cartoon image showing MCs do not undergo partial MMT and MMT during kidney fibrosis. Scale bars: white 100 µm; yellow 1 mm. Each figure is representative of five individual biological samples.
To directly visualize macrophages undergoing the MMT process, if any, and differentiate them from non-transformed macrophages and non-macrophage-derived myofibroblasts, we generated Lyz2-CreER driven and Cx3cr1-CreER driven MMTracer models (Fig. 8e). Following tamoxifen induction, UUO surgery was performed on these MMTracer mice, and kidneys were analyzed 7 days and 14 days post-surgery (Fig. 8f). Immunostaining and quantitative assessments showed an absence of tdTomato+ cells in the kidneys of either MMTracer mice after UUO surgery (Fig. 8g, h), suggesting the absence of αSMA activation in macrophages and their progenies.
To confirm whether the lack of tdTomato signal detection was due to low excision efficiency of the “Stop” sequence in αSMA-LSL-Dre by Lyz2-CreER or Cx3cr1-CreER, we assessed the excision efficiency through PCR analysis of genomic DNA from these MMTracer mice. PCR analysis revealed the complete conversion of αSMA-LSL-Dre to αSMA-Dre in ZsGreen+ cells (Supplementary Fig. 4g, h).
Overall, the MMTracer demonstrated that during kidney fibrosis, both recruited and resident macrophages did not undergo MMT to form myofibroblasts, and there was no αSMA activation in macrophages, indicating that partial MMT did not occur (Fig. 8i).
Myofibroblasts primarily originate from fibroblasts in kidney fibrosis
Fibroblasts are considered the major source of myofibroblasts during fibrosis in various organs, including the kidney22,39. To reassess the contribution of fibroblasts to myofibroblast formation, we employed the Col1a2-CreER44 mouse model (Fig. 9a, b). Using Col1a2-CreER; NR1-based genetic lineage tracing, we showed that PDGFRa+ fibroblasts are efficiently labeled with ZsGreen in this model, with 91.8 ± 1.08% of fibroblasts being labeled (Supplementary Fig. 5a, b). Both sham and UUO surgeries were conducted on Col1a2-CreER; NR1 mice (Fig. 9c). Compared to the sham group, an increase in ZsGreen signals was observed in the fibrotic kidney following UUO surgery, (Supplementary Fig. 5c). Immunostaining analysis showed that ZsGreen+ cells in the injured area expressed PDGFRa and αSMA, indicating that fibroblasts contribute to myofibroblasts during kidney fibrosis (Fig. 9d). To confirm that myofibroblasts primarily arise from fibroblast activation, we quantified and compared the proportion of ZsGreen+ fibroblasts/myofibroblasts in sham and injured kidneys (Supplementary Fig. 5d). If non-fibroblast cells significantly contributed to myofibroblasts after injury, a notable number of ZsGreen–myofibroblasts derived from these non-fibroblast cells would be expected, which would lead to a dilution of the of ZsGreen+ fibroblasts/myofibroblasts proportion in injured kidneys. However, if the proportion of ZsGreen+ fibroblasts/myofibroblasts remains similar between sham and injured kidneys, this suggests minimal contribution from non-fibroblast cells to myofibroblast formation. Quantitative analysis revealed no significant dilution in the percentage of ZsGreen+/PDGFRa+ after surgery, and the percentage of ZsGreen+/αSMA+ cells was similar to that of ZsGreen+/PDGFRa+ cells, suggesting that non-fibroblast cells did not significantly contribute to myofibroblasts (Supplementary Fig. 5d). Overall, these results indicated that myofibroblasts primarily derived from fibroblasts during kidney fibrosis, rather than from other cell sources (Fig. 9e).

a Schematic diagram of Col1a2+ renal fibroblasts. b Lineage tracing strategy for renal fibroblasts. c Experimental design for tracing renal fibroblasts in sham and UUO-injured mice. d Immunostaining for ZsGreen, PDGFRa, and αSMA on kidney sections. e Fibroblasts give rise to myofibroblasts during kidney fibrosis. f Schematic diagram of capture αSMA activation in fibroblasts by Col1a2-CreER; αSMA-LSL-Dre; NR1. g Experimental design for capturing αSMA activation in fibroblasts in sham and UUO-injured mice. h Immunostaining for ZsGreen and tdTomato on kidney sections. i Quantification of the percentage of ZsGreen+ and tdTomato+ fibroblasts in all ZsGreen+/tdTomato+ fibroblasts. Data are the mean ± SEM, n = 5. Immunostaining for tdTomato with αSMA (j) or PDGFRa (k) on kidney sections from UUO-injured mice after 14 days. Scale bars: white 100 µm; yellow 1 mm. Each figure is representative of five individual biological samples.
To directly visualize αSMA activation in fibroblasts and assess their contribution to myofibroblasts, we generated Col1a2-CreER; αSMA-LSL-Dre; NR1 by crossing Col1a2-CreER with αSMA-LSL-Dre and NR1 (Fig. 9f). PCR analysis of genomic DNA confirmed complete removal of “Stop” sequence in ZsGreen+, converting αSMA-LSL-Dre to αSMA-Dre for efficient capturing αSMA activation in fibroblasts (Supplementary Fig. 5e, f). Subsequently, we performed UUO and sham surgeries on these triple knock-in mice and analyzed kidneys at 7 days and 14 days post-injury (Fig. 9g). In the sham group, rare tdTomato+ cells were detected in the kidneys, whereas a substantial number were observed after injury. At 7 days, 41.39 ± 5.65% of fibroblasts activated αSMA and converted the cell tagging from ZsGreen to tdTomato, and by 14 days post-injury, 90.56 ± 1.86% of fibroblasts had activated αSMA(Fig. 9h, i). The tdTomato+ cells expressed myofibroblast marker αSMA and PDGFRa (Fig. 9j, k), indicating the activation of αSMA in fibroblasts and their transformation into myofibroblasts. FACS analysis of all XMTracer models (EMTracer, MMTracer, EndoMTracer, and Fibroblast-to-Myofibroblast Tracer) at 14 days post-injury/sham validated immunostaining results: only fibroblast-derived cells showed tdTomato+ labeling (Supplementary Fig. 6). Notably, Cdh16-CreER-driven EMTracer showed no detectable tdTomato+ cells by FACS, likely due to the low frequency of labeled cells in live populations.
We found that tdTomato+ cells are primarily located in the cortex and outer medulla, with few in the inner medulla (Supplementary Fig. 5g). Nearly all fibroblasts in the cortex and more than half in the outer medulla transitioned into tdTomato+ myofibroblasts (Supplementary Fig. 5g). αSMA antibody staining also confirmed a scarcity of myofibroblasts in the inner medulla (Supplementary Fig. 5h).
Our results indicated that during kidney fibrosis, fibroblasts became activated to transform into myofibroblasts. As fibrosis progresses, more fibroblasts are labeled by tdTomato in the cortex and outer medulla, while rare in the inner medulla, with the majority of the fibroblasts eventually transforming into myofibroblasts at the later stage of kidney fibrosis.
Discussion
Over the past two decades, the origin of kidney myofibroblasts has been a central question in kidney fibrosis research, particularly concerning the transformation of non-fibroblast cells through mesenchymal transition processes, such as EMT, into myofibroblasts. This topic has been extensively studied and debated. In this study, we developed and employed multiple genetic tools targeting renal epithelial cells, endothelial cells, macrophages, and fibroblasts to systematically assess the contribution of each of these cell lineages to myofibroblast formation. By leveraging the Dre-rox and Cre-loxP dual recombinase system36, we introduced novel genetic techniques—EMTracer, EndoMTracer, and MMTracer—for directly capturing and visualizing both complete and partial EMT, EndoMT, and MMT. These methods eliminate the need to analyze the colocalization of genetically labeled cells with myofibroblast markers, enabling more effective and precise detection of mesenchymal transition events. Our findings indicate that renal myofibroblasts primarily arise from resident fibroblasts, with no significant contribution from other cell lineages via mesenchymal transition. This conclusion carries critical biological and translational implications: by definitively ruling out epithelial, endothelial, and macrophage lineages as major sources of myofibroblasts, our study redirects fibrosis research toward fibroblast-specific mechanistic investigations. Importantly, these insights have substantial relevance for advancing research into human IRI. They suggest that therapeutic strategies to combat IRI-induced renal fibrosis may be more effective if they prioritize targeting fibroblast-specific pathways (such as fibroblast activation, proliferation, or extracellular matrix production), instead of focusing on interventions aimed at EMT, EndoMT, or MMT processes. Among these cell lineages, TECs were initially suggested to predominantly contribute to myofibroblast formation in kidney fibrosis through EMT10. However, debates on the existence of EMT in kidney fibrosis have persisted for over 20 years45,46. Previous studies employing different genetic lineage tracing tools yielded conflicting results, often due to inherent limitations in these methodologies. Some of these limitations include the use of constitutively active Cre recombinase, which can lead to unintended recombination events, low efficiency in labeling target cell populations, and incomplete labeling of tubular segments. In this study, we first developed a highly specific and efficient (100%) proximal TEC lineage tracing tool, Hnf4a-CreER, given the focus on proximal TECs in kidney EMT studies. We then explored EMT in non-proximal TECs (including the epithelial cells in loop of Henle, distal tubule, and collecting duct) by generating the Cdh1-CreER mouse line. Due to the insufficient labeling efficiency (approximately 60%) of the Cdh1-CreER line, we generated the Cdh16-CreER line, which efficiently labels both proximal and non-proximal TECs. Lineage tracing studies based on these three genetic tools showed no contribution of TECs in any tubule segments to myofibroblast formation.
In addition to EMT, “partial EMT” has been proposed in kidney fibrosis31,32,33,34. However, our genetic lineage tracing study using the aforementioned tools did not detect αSMA expression in TECs, indicating no partial EMT in kidney fibrosis, which is in conflict with previous studies. The lack of αSMA detection might be due to incorrect timing of tissue analysis or its transient expression in TECs during fibrosis, making it challenging to capture. Additionally, rare EMT or partial EMT events might be missed by commonly used colocalization analysis of αSMA with genetically labeled cells. To address these challenges, we developed EMTracer, which detects EMT events without relying on αSMA colocalization. Instead, only cells that undergo either complete or partial EMT were genetically labeled, distinguishing them from other untransformed TECs and the non-EMT-derived myofibroblasts, facilitating direct visualizing EMT events. This method improved the signal-to-noise ratio and resolution, effectively capturing complete and partial EMT events, particularly benefiting the detection of rare events. Furthermore, EMTracer permanently labels cells even with transient or reversible αSMA expression, allowing for the capture of reversible and transient EMT events. Moreover, EMTracer records all EMT events from their occurrence to the time of tissue analysis, providing comprehensive detection beyond mere snapshots, which is advantageous for identifying rare and reversible EMT events. EndoMTracer and MMTracer function similarly to capture EndoMT and MMT events, respectively.
Similar to our traditional lineage tracing, EMTracer revealed neither complete nor partial EMT in TECs contributing to myofibroblast formation in kidney fibrosis. Interestingly, we detected reversible partial EMT in PECs and UroCs using EMTracer. The underlying mechanisms driving reversible partial EMT in PECs and UroCs, and their potential physiological roles in kidney fibrosis, remain underexplored and merit further investigation.
We speculate that transient αSMA expression in PECs may reflect activation of pro-fibrotic signaling to coordinate early inflammatory responses and renal capsule remodeling. Conversely, transient αSMA expression in urothelial cells lining the renal pelvis could represent a mechanosensitive response to injury, modulating the microenvironment through cytokine secretion or extracellular matrix remodeling. The absence of partial EMT in tubular cells—contrary to previous reports—suggests non-tubular epithelial compartments employ distinct stress response pathways. This highlights the need for future studies to explore compartment-specific roles in fibrosis pathogenesis.
Regarding EndoMT, previous Tie2-Cre and Cdh5-Cre-based lineage tracing studies reported that 30–50%12,14 and 10%11,15 of myofibroblast in kidney fibrosis derived from endothelial cells through EndoMT, respectively. These two genetic tools are not specific for endothelial cells, with Tie2-Cre labeling hematopoietic cells27 and perivascular cells29,30, and Cdh5-Cre labeling hematopoietic cells28. In this study, we adopted an inducible genetic lineage tracing tool Cdh5-CreER, which we previously generated to specifically and efficiently label endothelial cells47, to study EndoMT in kidney fibrosis. Since endothelial cells are very thin and close to myofibroblasts, making it difficult to distinguish them, we generated EndoMTracer for direct visualization of both complete and partial EndoMT events. Both Cdh5-CreER-based traditional lineage tracing and EndoMTracer showed no contribution of endothelial cells to myofibroblast formation via EndoMT in kidney fibrosis. Although EndoMTracer revealed partial EndoMT, it was very rare (0.05 ± 0.04%) and negligible, suggesting it does not play a significant role in kidney fibrosis.
For MMT, the extent of macrophage to renal myofibroblast formation has also been debated for many years22,48. Some studies reported a high percentage (35–50%)11,16,20 of renal myofibroblasts originating from bone marrow-derived macrophages, while others suggested a limited contribution (10%)24. Most previous studies relied on BM transfer, parabiosis experiments, or Lyz2-Cre-based lineage tracing to analyze bone marrow-derived macrophages. After kidney injury, in addition to bone marrow-derived monocytes recruited to the injury area and transforming into macrophages, resident macrophages also increased through proliferation in the injury area. In this study, we assessed the contribution of both recruited and resident macrophages for renal myofibroblast formation using Lyz2-CreER and Cx3cr1-CreER, respectively. Similar with endothelial cells, macrophages are also very thin and close to myofibroblasts, making them difficult to distinguish. We also generated MMTracer for direct visualization of both complete and partial EndoMT events. Both traditional lineage tracing methods and MMTracer showed no contribution of bone marrow-derived and resident macrophages to myofibroblast formation via MMT in kidney fibrosis.
To verify that resident fibroblasts serve as the primary source of renal fibroblasts, we utilized a Col1a2-CreER mouse line to trace renal fibroblasts during fibrosis, observing that fibroblasts significantly contribute to the population of renal myofibroblasts. By combining Col1a2-CreER with the XMTracer key component αSMA-LSL-Dre, which enables the labeling and visualization of fibroblast-derived myofibroblasts, we demonstrated that renal myofibroblasts predominantly originate from resident fibroblasts. As fibrosis progresses, tdTomato-labeled fibroblasts increase in the cortex and outer medulla, while remaining rare in the inner medulla. This reduced fibroblast activation in the inner medulla may reflect regional injury responses, potentially influenced by the medulla’s unique microenvironment—characterized by high osmolarity and low oxygen tension—which could limit fibroblast proliferation or transdifferentiation.
Overall, our study has developed advanced genetic tools to resolve the origin of myofibroblasts in renal fibrosis, indicating that basic and clinical research on renal fibrosis should focus on fibroblasts rather than transdifferentiation of other cell types. Given the critical role of myofibroblasts in fibrosis across multiple organs, our findings provide valuable genetic tools and conceptual frameworks for investigating fibrosis in various tissues.
However, our study has limitations. Although we observed transient αSMA expression in parietal epithelial cells (PECs) and urothelial cells during renal fibrosis, we did not investigate whether this phenomenon plays a significant role in fibrosis or its specific functional mechanisms. Additionally, our EMT-related studies primarily utilized αSMA-based genetic tools. We did not construct tools targeting EMT transcription factors like Snail or Twist, as our research focus was on myofibroblast origin—with αSMA being the primary myofibroblast marker. Thus, for EMT studies in other contexts (especially cancer research), reliance solely on our αSMA-based tools may have limitations.
Methods
Mice
All mouse studies were conducted in accordance with the current guidelines of the Institutional Animal Care and Use Committee (IACUC) at Westlake University. Mouse lines NR136, αSMA-LSL-Dre39, Cdh5-CreER41, Cx3cr1-CreER43 (Jax020940), and Col1a2-CreER44 were reported previously. Lyz2-CreERT242 mice (Strain NO. T052789) were purchased from GemPharmatech (Nanjing, China). Two new knock-in mouse lines, Hnf4a-CreER and Cdh1-CreER, were generated by inserting 2A-CreER into the translational stop codon of the Hnf4a or Cdh1 gene, respectively. These two mouse lines were generated by the Shanghai Model Organisms Center Inc. (SMOC). The Cdh16-CreER mouse line was generated by inserting CreER into the translational start codon of the Cdh16 locus via homologous recombination using CRISPR/Cas9 technology, and it was generated by GemPharmatech (Nanjing, China). All mice used in this study were maintained on a C57BL/6J x ICR background, and all were housed in a specific pathogen-free environment at Westlake University under conditions of 25 °C, 50% humidity, and a 12 h light/dark cycle. Both male and female mice were randomly assigned to different groups. Tamoxifen (Sigma-Aldrich, T5648) was dissolved in corn oil (20 mg/ml) and administered by gavage at a dose of 0.2 mg per g of mouse body weight at the indicated time points. For the Cdh16-CreER and Hnf4a-CreER mouse lines, a lower dose of tamoxifen (0.1 mg per g mouse body weight) was administered. The primers used for all the mouse lines in this study are provided in Supplementary Table 1.
UUO surgery
UUO surgery was performed as previously described49. Briefly, mice were anesthetized with a continuous supply of 1–2% isoflurane. A lateral incision was made to expose the left ureter, which was then ligated at two points, typically in the mid-region between the renal pelvis and bladder, using a 4–0 silk suture. The ureter was then cut between the ligatures to ensure complete blockage of urine flow. Mice in the sham group underwent the same procedure but without ureter ligation.
Whole-mount fluorescence microscopy
Mouse kidneys were washed in cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (PFA) at 4 °C for 1 h, followed by PBS washing three times (for 15 min each time). Tissues were placed on an agar gel for whole-mount brightfield or fluorescence imaging using a Zeiss stereo microscope (AxioZoom V16).
Tissue embedding and immunostaining
Immunostaining was performed as previously described50. Tissues were collected in cold PBS and fixed in 4% PFA at 4 °C for 1 h. After three washes with PBS, tissues were placed in 30% sucrose/PBS overnight at 4 °C, then embedded in OCT compound (Sakura) and stored at −80 °C until sectioning. Cryosections, 10 µm thick, were collected on positively charged slides and stored at −20 °C until use. For immunostaining, tissue sections were blocked with PBSST (0.1% Triton X-100/2.5% normal donkey serum/PBS) containing DAPI (1:1000 dilution) for 30 min at room temperature, followed by overnight incubation with primary antibodies at 4 °C. The next day, sections were washed three times with PBS and incubated with fluorescence-conjugated secondary antibodies (Jackson ImmunoResearch) for 30 min at room temperature. After another three washes with PBS, sections were mounted with mounting medium without DAPI. The following antibodies were used: ZsGreen (Clontech, 632474, 1:2000), HNF4a (CST, 3113S, 1:1000), EpCAM (Abcam, ab92382, 1:2000), αSMA (Sigma, C6198, 1:500), RFP (Rockland, 200-101-379, 1:1000), WT1 (Abcam, ab89901, 1:100), PDGFRa (R&D, AF1062, 1:500), CD31 (BD-Pharmingen, 553370, 1:500), F4/80 (Abcam, ab6640, 1:200), CD13 (R&D, AF2335, 1:100), LTL (Vector Lab, FL-1321, 1:100). Images were acquired using a Nikon confocal microscope (Nikon A1R) and analyzed with ImageJ and Photoline software.
To acquire z-stack confocal images, a z-step size of 1 μm was used to capture serial optical sections through the tissue. Images were obtained using a 60x objective lens on a Nikon A1R confocal microscope, with 10–15 consecutive XY scans acquired along the z-axis. Image analysis was performed using ImageJ (NIH) software. Fluorescent channels were first merged using the “Color Merge Channels” function, followed by Z-projection with “Average Intensity” to generate composite stacks. Orthogonal views (XZ and YZ axes) were reconstructed from the 3D stacks to visualize cellular relationships in multiple planes. Merged and single-channel images were generated to resolve signals at the single-cell level, enabling clear delineation of labeled structures.
Kidney cell isolation and sorting
After euthanizing the mice, the kidneys were removed and promptly immersed in ice-cold PBS. The kidneys were then finely chopped into small pieces and transferred to 6-well plates for further processing. A digestion solution, comprising 1 ml of Type 1 A Collagenase (1.6 mg/ml) and DNase I (60 U/ml) in RPMI 1640 medium, was added to each well to facilitate tissue breakdown. To ensure thorough digestion, the tissues were incubated in this solution at 37 °C for 30 min with intermittent agitation. Following the incubation period, the tissue and digestate were filtered through a 100 µm cell strainer into a 50 ml centrifuge tube. The filtrate was then passed through a 40 µm cell strainer to achieve further dissociation of the tissue. The resulting cell suspension was centrifuged at 211 × g for 5 min at room temperature to pellet the cells. The supernatant was discarded, and the cell pellet was resuspended in 5 ml of ice-cold red blood cell (RBC) lysis buffer and incubated on ice for 3-4 min to remove erythrocytes. Subsequently, 5 ml of ice-cold PBS was added to the cells, and the suspension was centrifuged at 211 × g for 5 min at 4 °C. The cell pellet was resuspended with 10 ml PBS and then passed through a 40 µm cell strainer to ensure complete dissociation of the tissue. After centrifugation, the supernatant was discarded, and the cells were resuspended in RPMI 1640 medium. Nuclear staining was performed by adding DAPI (1:1000) to the cell suspension and incubating at 4 °C for 30 min. For flow cytometric analysis, cells were gated as depicted in the figures. Viable cells were identified as DAPI– and analyzed for tdTomato and ZsGreen fluorescence signals using a Beckman Cytoflex LX-6L. For flow cytometric sorting, DAPI–/ZsGreen– and DAPI–/ZsGreen+ cells were sorted by FACSAria Fusion (BD) and collected in RPMI 1640 supplemented with 10% FBS for subsequent experiments.
Genomic DNA extraction and PCR analysis
Mouse tails or sorted cells were lysed in lysis buffer (100 mM Tris HCl (pH 7.8), 5 mM EDTA, 0.2% SDS, 200 mM NaCl, and 100 mg/ml proteinase K) overnight at 55 °C, followed by centrifugation at for 20,000 × g for 5 min to obtain supernatants with genomic DNA. Genomic DNA was purified by adding isopropanol and washed with 70% ethanol. PCR primers spanning the genomic DNA and inserted cassette were designed to test for the correct genotype in all mouse lines, as shown in Supplementary Table 1. For examining the excision efficiency of the “Stop” sequence in αSMA-LSL-Dre, primers spanning the αSMA promoter and inserted Dre cassette were designed (Forward: CTGTGACACTCCCGCTCTTT, Reverse: CATCCAGCAGCTTCAGGTCA).
Statistical analysis
All data were analyzed following an assessment of normality and variance across data sets. A two-sided unpaired Student’s t test was used to compare the two groups’ differences. For comparisons involving three or more groups, one-way ANOVA was performed, with statistical significance defined as P < 0.05. Each figure legend specifies the analysis of five independent biological replicates, with results presented as mean values ± SEM. Mice were randomly assigned to different experimental groups to ensure unbiased results. Additionally, the researcher responsible for quantifying the immunostaining images was blinded to the group assignments to maintain objectivity.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All of the data generated or analyzed during this study are included in the Figs. 1–9, Supplementary Figs. 1–6, and Supplementary Table 1. The statistical raw data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Webster, A. C., Nagler, E. V., Morton, R. L. & Masson, P. Chronic kidney disease. Lancet 389, 1238–1252 (2017).
Yamashita, N. & Kramann, R. Mechanisms of kidney fibrosis and routes towards therapy. Trends Endocrinol. Metab. 35, 31–48 (2024).
Yuan, Q., Tan, R. J. & Liu, Y. Myofibroblast in kidney fibrosis: origin, activation, and regulation. Adv. Exp. Med Biol. 1165, 253–283 (2019).
Falke, L. L., Gholizadeh, S., Goldschmeding, R., Kok, R. J. & Nguyen, T. Q. Diverse origins of the myofibroblast—implications for kidney fibrosis. Nat. Rev. Nephrol. 11, 233–244 (2015).
Lin, S. L., Kisseleva, T., Brenner, D. A. & Duffield, J. S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 173, 1617–1627 (2008).
Humphreys, B. D. et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176, 85–97 (2010).
Asada, N. et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Invest 121, 3981–3990 (2011).
Kramann, R. et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16, 51–66 (2015).
Kuppe, C. et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 589, 281–286 (2021).
Iwano, M. et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 110, 341–350 (2002).
LeBleu, V. S. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med 19, 1047–1053 (2013).
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M. & Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19, 2282–2287 (2008).
Phua, Y. L., Martel, N., Pennisi, D. J., Little, M. H. & Wilkinson, L. Distinct sites of renal fibrosis in Crim1 mutant mice arise from multiple cellular origins. J. Pathol. 229, 685–696 (2013).
Xavier, S. et al. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 26, 817–829 (2015).
Lovisa, S. et al. Endothelial-to-mesenchymal transition compromises vascular integrity to induce Myc-mediated metabolic reprogramming in kidney fibrosis. Sci. Signal 13, eaaz2597 (2020).
Broekema, M. et al. Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J. Am. Soc. Nephrol. 18, 165–175 (2007).
Yang, J. et al. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J. Am. Soc. Nephrol. 24, 1644–1659 (2013).
Yan, J., Zhang, Z., Yang, J., Mitch, W. E. & Wang, Y. JAK3/STAT6 stimulates bone marrow-derived fibroblast activation in renal fibrosis. J. Am. Soc. Nephrol. 26, 3060–3071 (2015).
Reich, B. et al. Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int. 84, 78–89 (2013).
Wang, Y. Y. et al. Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J. Am. Soc. Nephrol. 28, 2053–2067 (2017).
Tang, P. M. et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 93, 173–187 (2018).
Tanaka, S., Portilla, D. & Okusa, M. D. Role of perivascular cells in kidney homeostasis, inflammation, repair and fibrosis. Nat. Rev. Nephrol. 19, 721–732 (2023).
Li, L., Zepeda-Orozco, D., Black, R. & Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 176, 1767–1778 (2010).
Kramann, R. et al. Parabiosis and single-cell RNA sequencing reveal a limited contribution of monocytes to myofibroblasts in kidney fibrosis. JCI Insight 3, e99561 (2018).
Girshovich, A. et al. Ureteral obstruction promotes proliferation and differentiation of the renal urothelium into a bladder-like phenotype. Kidney Int. 82, 428–435 (2012).
Liu, W. B. et al. Single-cell landscape of parietal epithelial cells in healthy and diseased states. Kidney Int. 104, 108–123 (2023).
Tang, Y., Harrington, A., Yang, X., Friesel, R. E. & Liaw, L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 48, 563–567 (2010).
Chen, M. J., Yokomizo, T., Zeigler, B. M., Dzierzak, E. & Speck, N. A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457, 887–891 (2009).
De Palma, M. et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8, 211–226 (2005).
Cai, J., Kehoe, O., Smith, G. M., Hykin, P. & Boulton, M. E. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 49, 2163–2171 (2008).
Lovisa, S. et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21, 998–1009 (2015).
Grande, M. T. et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997 (2015).
Lovisa, S., Zeisberg, M. & Kalluri, R. Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol. Metab. 27, 681–695 (2016).
Menon, M. C. & Ross, M. J. Epithelial-to-mesenchymal transition of tubular epithelial cells in renal fibrosis: a new twist on an old tale. Kidney Int. 89, 263–266 (2016).
Xiong, L. et al. Direct androgen receptor control of sexually dimorphic gene expression in the mammalian kidney. Dev. Cell 58, 2338–2358.e5 (2023).
He, L. et al. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat. Med. 23, 1488–1498 (2017).
Van der Hauwaert, C. et al. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling. PLoS ONE 8, e66750 (2013).
Wang, G. et al. Spatial dynamic metabolomics identifies metabolic cell fate trajectories in human kidney differentiation. Cell Stem Cell 29, 1580–1593.e7 (2022).
Zhang, S. et al. Seamless genetic recording of transiently activated mesenchymal gene expression in endothelial cells during cardiac fibrosis. Circulation 144, 2004–2020 (2021).
Wang, G. et al. Analyzing cell-type-specific dynamics of metabolism in kidney repair. Nat. Metab. 4, 1109–1118 (2022).
Liu, K. et al. Lineage tracing clarifies the cellular origin of tissue-resident macrophages in the developing heart. J. Cell Biol. 221, e202108093 (2022).
Wang, X. et al. Palmitic acid in type 2 diabetes mellitus promotes atherosclerotic plaque vulnerability via macrophage Dll4 signaling. Nat. Commun. 15, 1281 (2024).
Yona, S. et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013).
Tian, X. et al. Generation of a self-cleaved inducible Cre recombinase for efficient temporal genetic manipulation. EMBO J. 39, e102675 (2020).
Hadpech, S. & Thongboonkerd, V. Epithelial-mesenchymal plasticity in kidney fibrosis. Genesis 62, e23529 (2024).
Youssef, K. K. & Nieto, M. A. Epithelial-mesenchymal transition in tissue repair and degeneration. Nat. Rev. Mol. Cell Biol. 25, 720–739 (2024).
Liu, K. et al. Triple-cell lineage tracing by a dual reporter on a single allele. J. Biol. Chem. 295, 690–700 (2020).
Tang, P. M., Nikolic-Paterson, D. J. & Lan, H. Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 15, 144–158 (2019).
Chevalier, R. L., Forbes, M. S. & Thornhill, B. A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152 (2009).
He, S. et al. Spatial-temporal proliferation of hepatocytes during pregnancy revealed by genetic lineage tracing. Cell Stem Cell 30, 1549–1558 (2023).
Acknowledgements
We are grateful to the Laboratory Animal Resources Center of Westlake University for mouse husbandry. We thank the Biomedical Research Core Facilities and Supercomputer Center of Westlake University for providing technical support. This work was supported by the National Key Research & Development Program of China (2021YFA0805100, to L.H.), the National Science Foundation of China (323B2028 to S.H., 32370885 to L.H., 92368103 to L.H.), the Westlake Education Foundation, the Westlake Laboratory of Life Sciences and Biomedicine, and the Benyuan Charity Fund.
Author information
Authors and Affiliations
Contributions
S.H. and L.H. designed the study; S.H. performed experiments and analyzed the data; T.X., Z.G., X.S., H.W., H.H., K.J., and R.C. bred the mice and performed experiments; B.Z. provided important mouse lines; L.H. conceived and supervised the study and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ton Rabelink and Katalin Susztak for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, S., Xia, T., Guo, Z. et al. Tracing the origin of myofibroblasts in kidney fibrosis. Nat Commun 17, 653 (2026). https://doi.org/10.1038/s41467-025-67373-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67373-5
