
Abstract
This report presents the primary analysis of the endometrial cancer (EMC) cohort of FRUSICA-1 (ClinicalTrials.gov identifier, NCT03903705), a multicenter, single-arm, phase Ib/II study evaluating fruquintinib plus sintilimab. The cohort included Chinese patients with inoperable or advanced mismatch-repair proficient (pMMR) EMC who had progressed on or could not tolerate up to two prior platinum-based therapies, and comprised exploratory and pivotal phases. Patients received fruquintinib (5 mg orally once daily on a 2 weeks on/1 week off schedule) plus sintilimab (200 mg intravenously once every 3 weeks). The primary endpoint was objective response rate (ORR) assessed by an independent review committee (IRC). Secondary endpoints included ORR as assessed by the investigator, disease control rate, time to response, duration of response, progression-free survival (PFS) and tumor shrinkage as assessed by both the IRC and investigator, overall survival, and safety. By May 15, 2024, 98 patients with pMMR EMC were enrolled and treated. IRC-assessed ORR was 32.7% (95% confidence interval [CI] 23.5–42.9) for the total pMMR population (n = 98) and 31.6% (95% CI 21.4–43.3) for the pivotal population (n = 76). Median PFS was 8.6 months (95% CI 5.5–16.6) for the total population and 7.1 months (95% CI 5.4–16.6) for the pivotal population. The most common grade ≥3 treatment-related adverse event was hypertension (17.3%). In conclusion, fruquintinib plus sintilimab showed promising efficacy and tolerable safety in previously treated, advanced pMMR EMC.
Similar content being viewed by others

Introduction
Endometrial cancer (EMC) is the sixth most common cancer among women worldwide1, with 420,368 new cases reported globally2. Globally, there were 97,723 deaths caused by EMC in 20222, and the 5-year survival rate associated with advanced/metastatic EMC is about 18%3. In China, EMC represented ~2% of all reported cancer cases in 2022 and was responsible for 13,511 deaths4.
Among the current standard regimens, platinum-based chemotherapy remains the backbone of first-line treatment for advanced EMC5,6,7, although some immune checkpoint inhibitors alone or plus chemotherapy have recently shown promising efficacy for the first-line treatment of patients with advanced EMC8,9,10,11.
Patients with EMC and proficient mismatch repair (pMMR) status account for approximately 70% of the population12. For the treatment options after failure of first-line chemotherapy, such as doxorubicin or paclitaxel chemotherapy, the objective response rate (ORR), median progression-free survival (PFS), and median overall survival (OS) of previously treated patients with advanced pMMR EMC were 15.1%, 3.8 months, and 12.2 months, respectively13. Other treatment options have similarly limited efficacy for those with pMMR status. For example, in the phase II PHAEDRA study of durvalumab (a programmed death-ligand 1 [PD-L1] inhibitor) in previously treated and untreated patients with pMMR, the ORR, median PFS, and median OS were only 3%, 1.8 months, and 12.1 months, respectively14.
However, efficacy outcomes have improved with the introduction of targeted therapies. Based on KEYNOTE-146 and KEYNOTE-77513,15, lenvatinib combined with pembrolizumab was fully approved by both the United States Food and Drug Administration and European Medicines Agency in 2021 for the second-line treatment of patients with advanced EMC that is not microsatellite instability (MSI)-high or mismatch-repair deficient, who have progressive disease (PD) following prior systemic therapy in any setting and are not candidates for curative surgery or radiation16. But this combination regimen is not yet approved in China, highlighting an unmet medical need for this patient population.
Fruquintinib is a potent, selective small-molecule inhibitor of vascular endothelial growth factor (VEGF) receptors 1, 2, and 3 that is approved globally for patients with metastatic colorectal cancer17,18,19,20, and sintilimab is an anti-programmed cell death protein-1 (PD-1) monoclonal antibody that is approved in China for multiple indications21. Combination therapy with fruquintinib plus sintilimab has been evaluated as a treatment for patients with previously treated EMC in the phase Ib/II FRUSICA-1 study. Based on the primary results of that study in the cohort of patients with advanced EMC and pMMR status treated in the second- or later-line setting, this therapeutic combination has been granted conditional approval as the first antiangiogenesis inhibitor combined with anti–PD-1 therapy in this disease setting in China.
Here, we report the results of the primary analysis in previously treated patients with advanced pMMR EMC in FRUSICA-1.
Results
Patient disposition
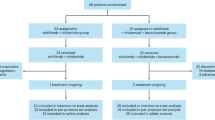
A total of 98 patients with previously treated, advanced EMC and centrally confirmed pMMR (i.e., target patients) were enrolled between October 9, 2020, and April 24, 2023, including 22 in the exploratory phase and 76 in the pivotal phase.
At data cut-off (May 15, 2024), the last enrolled target patient was followed up for more than 12 months, which met the prespecified timepoint for the primary analysis. The median follow-up duration was 22.0 months (95% confidence interval [CI] 20.5–23.7). Eleven (11.2%) of 98 patients were still receiving study treatment (fruquintinib and/or sintilimab) and 87 (88.8%) patients discontinued both study drugs, the primary reason for discontinuation being PD (n = 56; 57.1%) (Fig. 1).

AE adverse event, FAS full analysis set, PD progressive disease, pMMR proficient mismatch repair.
Patient baseline characteristics
Baseline characteristics of patients are presented in Table 1. Among 98 target patients (total pMMR population), the median (range) age was 57.8 years (29.5–75.5), all patients were Asian, and the proportions of patients with endometrioid adenocarcinoma and serous carcinoma were 64.3% and 27.6%, respectively. Twenty-six (26.5%) patients had a PD-L1 expression combined positive score (CPS) of ≥1. All patients had previously received at least first-line platinum plus taxane combined systemic therapy, and 22.4% of patients had received bevacizumab (VEGF receptor inhibitor) therapy. A total of 33 (33.7%) patients had received prior pelvic radiotherapy. The baseline characteristics of patients in the pivotal stage (n = 76) were comparable, as presented in Table 1.
Efficacy
Tumor response is summarized in Table 2 and Fig. 2. In the full analysis set (FAS), the primary endpoint of ORR per independent review committee (IRC) assessment was 32.7% (95% CI 23.5–42.9) in the total pMMR population (n = 98) and 31.6% (95% CI 21.4–43.3) in the pivotal population (n = 76). The lower bound of the 95% CI in the pivotal population exceeded the prespecified efficacy boundary of 16%, which indicates that fruquintinib plus sintilimab treatment was effective in the target population. The secondary endpoint of disease control rate (DCR) was 83.7% (95% CI 74.8–90.4) and 82.9% (95% CI 72.5–90.6), respectively. Median duration of response (DoR) reached 20.5 months (95% CI 11.1–not estimable [NE]) in the total pMMR population and 17.9 months (95% CI 11.1–NE) in the pivotal population, and median time to response (TTR) was 1.9 months (95% CI 1.4–4.0) and 1.9 months (95% CI 1.4–4.2), respectively. Tumor responses evaluated by investigators were comparable with those assessed by IRC (Table 2, Fig. 2; Supplementary Table 1). The efficacy findings for the tumor response-evaluable set (TRES) were similar to those in the FAS (Supplementary Table 2).

a IRC assessment and b investigator assessment in the total pMMR population, and by c IRC assessment and d investigator assessment in the pivotal population–TRES. CPS combined positive score, CR complete response, IRC independent review committee, PD progressive disease, pMMR proficient mismatch repair, PR partial response, SD stable disease, TRES tumor response-evaluable set. Source data are provided as a Source data file.
As of the data cut-off date, the PFS maturity per IRC assessment was 53.1% and 55.3% in the total pMMR population and the pivotal population. Median PFS was 8.6 months (95% CI 5.5–16.6) and 7.1 months (95% CI 5.4–16.6), respectively, as shown in Fig. 3a, c. The median investigator-assessed PFS was 7.2 months (95% CI 5.6–9.5) and 6.9 months (95% CI 5.5–9.5), respectively (Fig. 3b, d).

a IRC assessment and b investigator assessment in the total pMMR population, and by c IRC assessment and d investigator assessment in the pivotal population–FAS. CI confidence interval, FAS full analysis set, IRC independent review committee, PFS progression-free survival, pMMR proficient mismatch repair. Source data are provided as a Source data file.
At a median OS follow-up of 22.0 months in the total pMMR population and 21.7 months in the pivotal population, 45 (45.9%) and 32 (42.1%) patients, respectively, had died. The median OS was 21.8 months (95% CI 17.3–NE) in both the total pMMR and pivotal populations (Fig. 4a, b). The OS rate at 24 months was estimated to be 45.6% and 41.2%, respectively.

a The total pMMR population and b the pivotal population–FAS. CI confidence interval, FAS full analysis set, NE not evaluable, OS overall survival, pMMR proficient mismatch repair. Source data are provided as a Source data file.
Subgroup analysis (post-hoc analysis)
In subgroups of PD-L1 expression CPS < 1 or ≥1, patients with or without prior pelvic radiotherapy, and patients with or without prior bevacizumab therapy, efficacy outcomes were generally consistent between the groups, except for ORR, which was slightly higher for patients with PD-L1 expression CPS ≥ 1, patients with prior pelvic radiotherapy, and patients with prior bevacizumab therapy (Supplementary Tables 3–5 and Supplementary Figs. 1–3).
Safety
Among 98 target patients, median (range) duration of combination treatment was 6.2 months (0.7–41.9), and the median (range) number of cycles was 9.0 (1–61). During the study, 96 (98.0%) patients had at least one treatment-related treatment-emergent adverse event (TRAE, related to fruquintinib and/or sintilimab) (Table 3). Fifty-nine (60.2%) patients had grade ≥3 TRAEs. Grade ≥3 TRAEs with an incidence of ≥10% included hypertension (17.3%), palmar-plantar erythrodysesthesia syndrome (11.2%), and hypertriglyceridemia (10.2%).
TRAEs leading to permanent discontinuation of fruquintinib and/or sintilimab occurred in 15 (15.3%) out of 98 patients. These were most commonly gastrointestinal disorders, respiratory, thoracic, and mediastinal disorders, and reproductive system and breast disorders. TRAEs leading to interruption of fruquintinib occurred in 51 (52.0%) patients, and TRAEs leading to dose reduction of fruquintinib occurred in 40 (40.8%) patients. TRAEs leading to dose interruption/delay of sintilimab occurred in 37 (37.8%) patients. A TRAE leading to death (sintilimab-related) occurred in one patient due to immune-mediated lung disease; it was considered that multi-organ failure caused by an aggravated immune condition was the cause of the patient’s death.
Immune-related treatment-emergent adverse events (TEAEs) occurred in 74 (75.5%) patients; the most frequent included hypothyroidism (45.9%), hyperthyroidism (25.5%), blood thyroid-stimulating hormone increased (24.5%), and hyperglycemia (15.3%) (Supplementary Table 6).
TRAEs and immune-related TEAEs observed in the pivotal population (N = 76) are summarized in Table 3 and Supplementary Table 6.
Discussion
Antiangiogenic therapy plus anti–PD-(L)1 therapy has demonstrated clinically meaningful outcomes in previously treated pMMR EMC. In the KEYNOTE-775 study, pembrolizumab plus lenvatinib conferred an ORR (by blinded independent central review [BICR]) of 32.4%, and median PFS (also by BICR) and OS durations of 6.7 months and 18.0 months, respectively, in this patient population13,22; however, that regimen has not yet been approved in China. Based on the results of the present study, combination therapy with fruquintinib plus sintilimab recently received conditional approval in China for previously treated pMMR EMC, and is the first antiangiogenesis inhibitor plus anti–PD-1 combination regimen for this disease setting in China.
The ORR (primary objective) was reached in the present pivotal population, with the lower limit of the 95% CI exceeding 16% (ORR 31.6%, 95% CI 21.4–43.3). The results of our study thus showed a potentially improved response with this new combination therapy over chemotherapy or immunotherapy alone, for which the reported ORRs were 15.1% and 3.1%, respectively13,14. In addition, the PFS and OS were also numerically longer with fruquintinib plus sintilimab compared with either chemotherapy or immunotherapy. The efficacy findings of the present study in the Chinese population are aligned with those reported for pembrolizumab plus lenvatinib in patients with previously treated advanced EMC13,15.
Bevacizumab in combination with chemotherapy has been prescribed in some patients for the first-line treatment of EMC, or as monotherapy in the second- and subsequent-line setting in clinical practice23. Post-hoc analysis in the present study showed that patients have the potential to benefit from treatment with fruquintinib plus sintilimab in subsequent lines. Patients who have received prior pelvic radiotherapy may also benefit from this combination therapy. Moreover, the level of PD-L1 expression (CPS) appeared to have limited impact on the efficacy of fruquintinib plus sintilimab. It should be noted, however, that these post-hoc analyses were exploratory and limited by low patient numbers, and the findings should therefore be interpreted with caution.
In this study, fruquintinib plus sintilimab was tolerable in patients with pMMR EMC. The overall TRAE incidence (98.0%) was comparable with the previously reported incidence for fruquintinib or sintilimab monotherapies and combination therapy21,24,25, and no new safety issues were identified. The safety profile of fruquintinib plus sintilimab was also comparable with those of other antiangiogenesis plus immune checkpoint inhibitor combinations in patients with EMC, such as lenvatinib plus pembrolizumab13.
The most frequently reported TRAE in the present study (with fruquintinib plus sintilimab) was hypothyroidism (all grade 1–2), but this was reported less often with fruquintinib monotherapy25. Rates of other frequently reported TRAEs, such as proteinuria, hypertension, and palmar-plantar erythrodysesthesia syndrome, were consistent between fruquintinib plus sintilimab combination therapy and fruquintinib monotherapy24,25.
A limitation of this study was the non-randomized design and the lack of a comparator arm. The ongoing randomized phase III study of fruquintinib plus sintilimab versus paclitaxel or doxorubicin in patients with advanced EMC (NCT06584032) will allow further understanding of the baseline patient characteristics that predict better outcomes with this combination. Similarly, a larger patient population will be required to evaluate potential biomarkers related to the efficacy of fruquintinib plus sintilimab. Another limitation is that enrollment was exclusively in China, and the collected biomarker information was restricted to only MMR status and PD-L1 expression levels.
In conclusion, fruquintinib plus sintilimab showed promising antitumor activity in Chinese patients with previously treated advanced EMC with pMMR, and was tolerable in this patient population.
Methods
Study design and eligibility
FRUSICA-1 is an open-label, multi-center, single-arm, phase Ib/II study in China (ClinicalTrials.gov identifier: NCT03903705) evaluating fruquintinib plus sintilimab as a treatment for patients with solid tumors. The study comprised a dose-escalation phase and dose-expansion phase. A basket design was applied in the dose-expansion phase. The recommended phase II dose of fruquintinib plus sintilimab was determined based on the findings from the dose-escalation phase and the colorectal cancer cohort in the dose-expansion phase24. Here, we report the results of patients with confirmed pMMR status who had received prior systemic platinum-based therapy from the EMC cohort in the dose-expansion phase.
Patients were required to have histologically or cytologically confirmed inoperable or advanced EMC and had progressed on, or were unable to tolerate, no more than two prior lines of systemic platinum-based doublet therapy. Eligible patients were aged between 18 and 75 years (inclusive), with a body mass index of ≥18.5 kg/m2. Patients had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1, at least one measurable lesion as per Response Evaluation Criteria in Solid Tumors, version 1.1 (RECIST v1.1), and adequate organ function. Patients were required to provide a tumor tissue sample for the determination of PD-L1 expression and/or MSI/mismatch repair status. Patients previously treated with fruquintinib or any immune checkpoint inhibitor were excluded from this study. Full inclusion and exclusion criteria are provided in the Clinical Study Protocol (see Supplementary Information, Clinical Study Protocol).
This study was approved by the ethics committees and conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and applicable regulations and guidelines. All patients provided written informed consent to participate.
Treatments and procedures
Patients who were enrolled received fruquintinib, 5 mg orally once daily for 2 weeks on/1 week off, plus sintilimab, 200 mg intravenously once every 3-week cycle (the recommended phase II dose)24. Study treatment was continued until PD, death, intolerable toxicity, withdrawal, poor compliance, pregnancy, investigator’s decision, loss to follow-up, or the start of new antitumor therapy. The maximum duration of sintilimab treatment was 24 months.
Tumor evaluation was performed by enhanced computed tomography/magnetic resonance imaging per RECIST v1.1. During the study, tumor assessments were performed at baseline, every 6 weeks (±7 days) from the first dose, and every 12 weeks (±7 days) after 48 weeks. Objective response (complete response [CR] or partial response [PR]) was confirmed at least 4 weeks after the first documented response.
Tumor tissues were collected for the detection of PD-L1 expression, which was assessed via an immunohistochemical assay using a monoclonal Mouse Anti-Human PD-L1 Clone 22 C3 (Dako, Carpinteria, CA). The pMMR phenotype was confirmed by central laboratory testing. Mouse and rabbit antibodies against MMR proteins were used to perform automated immunohistochemistry staining and chromogenic labeling of the MLH1, MSH2, MSH6, and PMS2 proteins on the Ventana Benchmark Ultra (Roche Diagnostics, Tucson, AZ). The MLH1 (clone M1, mouse monoclonal, Ventana, Cat# 790–5091), PMS2 (clone A16-4, mouse monoclonal, Ventana, Cat# 790–5094), MSH2 (clone G219-1129, mouse monoclonal, Ventana, Cat# 790–5093) and MSH6 (clone SP93, rabbit monoclonal, Ventana, Cat# 790–5092) antibodies were used to perform immunohistochemistry staining. MMR status was determined by pathologist evaluation.
Safety evaluation was based on grade and severity of TEAEs, laboratory assessments, electrocardiogram, ECOG PS, vital signs, and physical examination. A TEAE was defined as an adverse event that occurred either on or after the first dose of the study drug, or within 90 days after the final dose of the study drug, or prior to the initiation of a new antitumor treatment, whichever occurred first. Only serious adverse events reported 90 days after the last dose or the start of new antitumor therapy were considered causally related to the study drug. TRAEs were TEAEs that were considered related to fruquintinib or sintilimab or both.
Endpoints
The primary endpoint was ORR assessed by an IRC, defined as the proportion of patients with the best overall response (BOR) of confirmed CR or PR.
Secondary endpoints included the ORR as assessed by the investigator, and the following assessments by both the IRC and investigator: DCR, defined as the proportion of patients with a BOR of confirmed CR, PR, stable disease, or non-CR/non-PD; TTR, defined as the time from the first dose of study drug to first response (CR or PR) in patients with confirmed objective response; DoR, defined as the time from initial response (CR or PR) to PD or death from any cause, whichever occurred first, in patients with confirmed objective response; PFS, defined as the time from the first dose of study drug to imaging-confirmed PD or death from any cause, whichever occurred first; and tumor shrinkage, defined as the minimum of the percentage change (including negative value) per patient from baseline to all post-baseline visits on or before PD and prior to the initiation of new antitumor therapy. OS was defined as the time from the first dose of study drug to death from any cause. Safety was also assessed as a secondary endpoint.
Post-hoc analyses
Post-hoc analyses for ORR, DCR, DoR, TTR, PFS, and OS were conducted in the following subgroups: patients with a PD-L1 expression CPS of <1 or ≥1; patients with or without prior pelvic radiotherapy; and patients with or without prior bevacizumab therapy.
Sample size determination
The EMC cohort comprised an exploratory phase and a pivotal phase for the target population of patients with advanced EMC and centrally confirmed pMMR who experienced PD after previous systemic therapy and were ineligible for surgery or radiotherapy. The planned sample size for the target patients in the EMC cohort (hereafter the “total pMMR population”) was 95 patients, including 20 in the exploratory phase and 75 in the pivotal phase (hereafter the “pivotal population”). The sample size for the exploratory phase population was calculated based on feasibility. Further expansion to the pivotal phase was based on the results of the exploratory phase (a confirmed ORR rate of approximately 35%). The sample size of 75 target patients in the pivotal population was calculated per the following hypotheses: the null hypothesis was an ORR of ≤16% and the alternative hypothesis was an ORR of >16%. Assuming an ORR of 35% with 75 target patients would provide approximately 95% power to reject the null hypothesis (ORR ≤ 16%) using an exact probability test at a one-sided 0.025 level. Statistical hypothesis testing was performed on the basis of a two-sided exact 95% CI. The null hypothesis was rejected if the lower bound of the two-sided exact 95% CI exceeded 16%.
Data analysis sets
The FAS, for the analysis of efficacy and safety data, included patients who had received at least one dose of the study drug. Additionally, patients in the FAS who had measurable disease at baseline and at least one post-baseline tumor-imaging evaluation were included in the TRES for supportive analysis of ORR, DCR, DoR, and TTR outcomes.
Statistical analysis
95% CIs for ORR and DCR were calculated using the Clopper–Pearson method. TTR, DoR, PFS, and OS were estimated by the Kaplan–Meier method. SAS version 9.4 (SAS Institute, Cary, NC) was used for all statistical analyses. Further details of the statistical analyses are available in the Statistical Analysis Plan (see Supplementary Information, Statistical Analysis Plan).
Ethics approval and consent to participate
This study was approved by the ethics committees and conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and applicable regulations and guidelines. All patients provided written informed consent. The trial protocols and any amendments were reviewed by independent ethics committees or institutional review boards (see Supplementary Table 7).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The clinical study protocol and statistical analysis plan are available in Supplementary Information. De-identified source data used in the figures are provided with this paper. The clinical study report can be obtained by contacting the corresponding author or S.F. (songhuaf@hutch-med.com) upon reasonable request. Requests for the clinical study report will be processed within 4 weeks, and access will be granted for a month. The remaining raw data is not publicly available due to patient privacy protection laws. Requests to access the de-identified data for further scientific use can be sent to X.W. (wu.xh@fudan.edu.cn) and S.F. (songhuaf@hutch-med.com) and will be considered on a case-by-case basis in a timely manner beginning 3 months and ending 5 years after publication of this article. Source data are provided with this paper.
References
World Health Organization. Global Cancer Observatory. Absolute numbers, Incidence, Females, in 2022. https://gco.iarc.who.int/today/en/dataviz/pie?mode=cancer&types=0&sexes=2&populations=900 (2022).
World Health Organization. Global Cancer Observatory. Corpus Uteri. https://gco.iarc.who.int/media/globocan/factsheets/cancers/24-corpus-uteri-fact-sheet.pdf (2022).
American Cancer Society. Survival Rates for Endometrial Cancer. https://www.cancer.org/cancer/types/endometrial-cancer/detection-diagnosis-staging/survival-rates.html (2024).
World Health Organization. Global Cancer Observatory. China. https://gco.iarc.who.int/media/globocan/factsheets/populations/160-china-fact-sheet.pdf (2022).
Coleman, R. L., Garside, J., Hurteau, J., Nguyen, J. & Kobayashi, M. Treatment patterns and outcomes among patients with advanced or recurrent endometrial cancer initiating first-line therapy in the United States. J. Health Econ. Outcomes Res. 10, 82–90 (2023).
Kalampokas, E. et al. Current approaches to the management of patients with endometrial cancer. Cancers 14, 4500 (2022).
PDQ Adult Treatment Editorial Board. https://www.cancer.gov/types/uterine/hp/endometrial-treatment-pdq (2024).
Oaknin, A. et al. Safety, efficacy, and biomarker analyses of dostarlimab in patients with endometrial cancer: interim results of the phase I GARNET study. Clin. Cancer Res. 29, 4564–4574 (2023).
ICI-chemo new standard for endometrial cancer. Cancer Discov. 13, 1030–1031 (2023).
Eskander, R. N. et al. Pembrolizumab plus chemotherapy in advanced endometrial cancer. N. Engl. J. Med. 388, 2159–2170 (2023).
Mirza, M. R. et al. Dostarlimab for primary advanced or recurrent endometrial cancer. N. Engl. J. Med. 388, 2145–2158 (2023).
Kommoss, S. et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 29, 1180–1188 (2018).
Makker, V. et al. Lenvatinib plus pembrolizumab in previously treated advanced endometrial cancer: updated efficacy and safety from the randomized phase III study 309/KEYNOTE-775. J. Clin. Oncol. 41, 2904–2910 (2023).
Antill, Y. et al. Clinical activity of durvalumab for patients with advanced mismatch repair-deficient and repair-proficient endometrial cancer. A nonrandomized phase 2 clinical trial. J. Immunother. Cancer 9, e002255 (2021).
Lee, C. H. et al. Lenvatinib plus pembrolizumab in patients with either treatment-naive or previously treated metastatic renal cell carcinoma (Study 111/KEYNOTE-146): a phase 1b/2 study. Lancet Oncol. 22, 946–958 (2021).
US Food and Drug Administration. FDA grants regular approval to pembrolizumab and lenvatinib for advanced endometrial carcinoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-pembrolizumab-and-lenvatinib-advanced-endometrial-carcinoma#:~:text=On%20July%2021%2C%202021%2C%20the%20Food%20and%20Drug,are%20not%20candidates%20for%20curative%20surgery%20or%20radiation. (2022).
US Food and Drug Administration. FDA D.I.S.C.O. Burst Edition: FDA approval of Fruzaqla (fruquintinib) for adult patients with metastatic colorectal cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approval-fruzaqla-fruquintinib-adult-patients-metastatic-colorectal (2023).
HUTCHMED. HUTCHMED Announces Japan Approval for FRUZAQLA® (fruquintinib) Received by Takeda. https://www.hutch-med.com/japan-approval-for-fruzaqla-fruquintinib/ (2024).
European Medicines Agency. Fruzaqla (fruquintinib). https://www.ema.europa.eu/en/documents/overview/fruzaqla-epar-medicine-overview_en.pdf (2024).
Shirley, M. Fruquintinib: first global approval. Drugs 78, 1757–1761 (2018).
Sintilimab Package Insert. Innovent Biologics (Suzhou) Co., Ltd. https://img.innoventbio.com/ppdf/new1.pdf.
Yonemori, K. et al. Analysis of East Asia subgroup in Study 309/KEYNOTE-775: lenvatinib plus pembrolizumab versus treatment of physician’s choice chemotherapy in patients with previously treated advanced or recurrent endometrial cancer. J. Gynecol. Oncol. 35, e40 (2024).
Restaino, S. et al. Management of patients diagnosed with endometrial cancer: comparison of guidelines. Cancers 15, 1091 (2023).
Guo, Y. et al. Phase 1b/2 trial of fruquintinib plus sintilimab in treating advanced solid tumours: the dose-escalation and metastatic colorectal cancer cohort in the dose-expansion phases. Eur. J. Cancer 181, 26–37 (2023).
Li, J. et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: the FRESCO randomized clinical trial. JAMA 319, 2486–2496 (2018).
Acknowledgements
This study was funded by HUTCHMED and Innovent Biologics, Inc. HUTCHMED was involved in the study design, providing fruquintinib, data collection, data analysis, data interpretation, writing of the report, and the decision to submit for publication. Innovent Biologics, Inc. provided sintilimab. Medical writing and editorial support were provided by Ian J Phillips and Catherine Bowe of Parexel, funded by HUTCHMED.
Author information
Authors and Affiliations
Contributions
X.W., J.W., D.W., and G.L. contributed equally to this work; X.W. and the Sponsor HUTCHMED designed the study; X.W., J.W., D.W., G.L., J.Z., H.C., H.Y., Q.Z., Ke.W., Yumei.W., T.Y., J.L., Y.H., Y.B., Keming.W., K.J., H.L., R.A., X.L., Y.P., Yue.W., W.T., K.S., Y.K., Y.L., Wenyu.S., A.L., Y.Y., and Yijun.W. enrolled patients and collected the data. X.W., J.W., D.W., G.L., J.Z., H.C., H.Y., Q.Z., Ke.W., Yumei.W., T.Y., J.L., Y.H., Y.B., Keming.W., K.J., H.L., R.A., X.L., Y.P., Yue.W., W.T., K.S., Y.K., Y.L., Wenyu.S., A.L., Y.Y., Yijun.W., K.C., H.S., P.L., P.T., S.F., M.S., and Weiguo.S. contributed to the interpretation of data, participated in writing the manuscript and critically reviewed and revised the manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication. All authors have directly accessed and verified the underlying data.
Corresponding author
Ethics declarations
Competing interests
K.C., H.S., P.L., P.T., S.F., M.S., and Weiguo.S. are employees of HUTCHMED. X.W., J.W., D.W., G.L., J.Z., H.C., H.Y., Q.Z., Ke.W., Yumei.W., T.Y., J.L., Y.H., Y.B., Keming.W., K.J., H.L., R.A., X.L., Y.P., Yue.W., W.T., K.S., Y.K., Y.L., Wenyu.S., A.L., Y.Y., and Yijun.W. have no conflicts of interests to declare.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, X., Wang, J., Wang, D. et al. Fruquintinib plus sintilimab in patients with advanced endometrial cancer with mismatch-repair proficient status: a multicenter, single-arm, phase Ib/II trial. Nat Commun 17, 658 (2026). https://doi.org/10.1038/s41467-025-67375-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67375-3
