
Abstract
The surface ocean is the largest sunlit environment on Earth where marine microalgae are known as the main drivers of global productivity. However, rhodopsin phototrophs are actually the most abundant metabolic group, suggesting a major role in the biogeochemical cycles. While previous studies have shown that rhodopsin-containing bacterioplankton thrive in the most severely nutrient-depleted environments, growing evidence suggest that this type of phototrophy may also be relevant in nutrient-rich environments. To examine its role in productive waters, we investigated the monthly rhodopsin dynamics in the upwelling system of the Southern California Bight by measuring retinal–the photoreactive chromophore essential for rhodopsin function–in seawater. Unlike oligotrophic regions, rhodopsin levels peaked during the highly productive spring phytoplankton bloom, coinciding with the highest chlorophyll concentrations. Heterotrophic bacterial abundances, particularly within the order Flavobacteriales, correlated strongly with rhodopsin concentrations, allowing us to build linear models to predict rhodopsin distributions in a productive environment. Metagenomic data further showed that Flavobacteriales also dominated the rhodopsin gene pool when the highest rhodopsin levels were recorded, underscoring their key contribution to light-driven energy capture. Overall, our findings reveal that rhodopsin phototrophy plays a substantial role in productive marine systems, broadening its recognized importance far beyond oligotrophic oceans.
Similar content being viewed by others

Introduction
Sunlight is the ultimate energy source fueling almost all of the Earth’s biosphere1. Yet, life can only channel this energy through two fundamentally distinct transducing mechanisms–those based on chlorophyll or retinal chromophores2,3,4. While chlorophyll photoautotrophy (i.e., photosynthesis) relies on a complex light-harvesting apparatus that drives organic matter synthesis, rhodopsin-based photoheterotrophy consists of simple light-driven ion pumps that do not generate reducing power and therefore support bacteria in the inverse process: organic matter utilization. Rhodopsins’ structural simplicity–requiring only six genes for their synthesis5–may be the key to their high incidence in surface ocean microbes6. Although various microbial rhodopsins perform distinct functions, ranging from light-driven ion pumps to light sensors7, light-driven proton pumps are the most widespread in marine systems8, potentially providing critical amounts of previously unaccounted-for energy entering the ocean’s biosphere9,10. However, their actual concentrations in distinct marine regions and the broader ecosystem-wide implications of the incident light energy they transduce remain largely unknown.
Because of their low biosynthesis costs, rhodopsin photosystems play a substantial role in nutrient-depleted environments where primary productivity is low8,11,12. Multiple omics studies observed that the relative abundance of rhodopsin genes, transcripts, or proteins is often inversely related to chlorophyll concentrations in different marine systems11,13,14. For instance, in the Atlantic Ocean, rhodopsin gene distributions were negatively correlated to chlorophyll, nitrate, ammonium, silicate, and phosphate concentrations15. Similarly, a study from the P-limited Eastern Mediterranean showed that most bacterioplankton genomes (> 80%) contained rhodopsin genes16. Notably, the fraction of rhodopsin-bearing microbes in the most oligotrophic stations was the highest ever reported–highlighting that understanding these trends along productivity gradients is central to elucidating rhodopsin phototrophy’s role in microbial niche adaptation.
Metagenomic studies across numerous marine environments indicate that the SAR11 cluster and Flavobacteria are two of the primary rhodopsin-containing bacterial clades in the ocean8,17. These photoheterotrophic taxa, however, exhibit very contrasting lifestyles: SAR11 are typically oligotrophic, whereas the Flavobacteria are generally copiotrophic, thriving mainly during phytoplankton blooms18. Laboratory studies on the Candidatus Pelagibacter ubique strain HTCC1062 (a member of the SAR11 clade) showed that rhodopsin phototrophy may partially replace aerobic carbon respiration as a source of ATP under starvation19. Consistent with this, in situ SAR11 rhodopsin gene abundances were inversely related to the trophic richness in oligotrophic areas within the Atlantic Ocean and Mediterranean Sea15. However, in more productive regions such as the California upwelling regime, SAR11 rhodopsin genes showed no significant correlation with chlorophyll levels20. While rhodopsins may confer survival advantages under extreme oligotrophic conditions in SAR1121, studies on the Flavobacterium Dokdonia sp. MED134 showed that rhodopsin phototrophy enhances substrate uptake22 and yields higher cell densities in natural seawater under light conditions compared to darkness23. These findings in Flavobacteria suggest that rhodopsin phototrophy may play important roles beyond oligotrophic environments, though its function in more productive oceanic regions remains poorly characterized.
Most existing studies of rhodopsin distributions rely on molecular data, offering only semi-quantitative estimates of their presence based on genes, transcripts, and proteins within microbial communities6. However, because each rhodopsin photosystem contains a single retinal molecule, retinal concentration can serve as a quantitative proxy of rhodopsin abundance in microbial biomass9. Despite the promise of this direct approach to examine the importance of rhodopsin phototrophy in the ocean, field data remain scarce. To date, only three studies have used this direct quantification approach and showed that (i) the highest rhodopsin levels are found in ultraoligotrophic regions of the eastern Mediterranean Sea9, (ii) iron limitation appears to favor rhodopsin phototrophy over photosynthesis in the high-nutrient low chlorophyll areas of the Southern Ocean24, and (iii) rhodopsin phototrophy is widespread in polar environments25. These studies, however, represent isolated snapshots, and none have addressed rhodopsin distribution in coastal upwelling regions or its temporal dynamics. Furthermore, no study has yet integrated quantitative rhodopsin data with microbial community analyses, an essential step for identifying the major rhodopsin-bearing microbial taxa and understanding how this light-capturing function is regulated in situ. Ultimately defining the factors controlling the two major phototrophic mechanisms in surface waters remains essential not only for vast oceanic regions but also for productive environments, which collectively comprise 44% of the annual net primary productivity of the oceans26.
Some of the largest phytoplankton blooms occur in the world’s four main coastal upwelling regions (California, Humboldt, Canary, and Benguela ecosystems) when chlorophyll-a and inorganic nutrients reach their annual maximum concentrations27. Within the California upwelling regime, the Southern California Bight (SCB) exhibits oligotrophic conditions throughout most of the year, with seasonal wind-driven upwelling in the spring that brings nutrients to surface waters, resulting in increased productivity during those months28,29,30. A study in the SCB regime showed that rhodopsin genes and transcripts were continuously present year-round, with >90% of organisms smaller than 1 μm containing these genes, even during the upwelling season20. Thus, the importance of marine microbial rhodopsins may extend beyond solely oligotrophic regimes. However, to date, there are no data on the actual abundances of rhodopsin photosystems in microbial communities of any of the world’s upwelling regimes.
In this study, we investigated the temporal distribution of microbial rhodopsins in a dynamic upwelling system at the San Pedro Ocean Time Series in the Eastern North Pacific coast (Fig. 1). We found that, in contrast to observations in oligotrophic systems, rhodopsin concentrations peaked during the high productivity months. These trends were largely driven by bacterioplankton abundance and the incidence of Flavobacteriales–a microbial group typically associated with phytoplankton blooms and the degradation of complex organic matter–suggesting a link between rhodopsin light capture and complex substrate utilization.

Circle and star symbols denote SPOT (33.55°N, 118.40°W) and Catalina (33.44 N, 118.49 W), respectively.
Results
Monthly samplings over 15 months at SPOT revealed shallow rhodopsin maxima located at or above the deep chlorophyll maxima (Fig. 2A). Below the photic zone (60–250 m depth), however, rhodopsin levels were undetectable, suggesting a strictly phototrophic role in this coastal ocean system. Contrary to previous observations linking rhodopsin distributions with oligotrophy9,15,16, the highest rhodopsin abundances coincided with the spring phytoplankton blooms (Fig. 2). Peak annual levels of both rhodopsin and chlorophyll occurred in May, approximately one month after the onset of the 5-month-long upwelling season (Supplementary Fig. S1).

A Distribution of total microbial rhodopsin obtained through retinal and B chlorophyll-a quantifications. Black dots indicate the depths of sampling. Total rhodopsin and chlorophyll-a concentrations were calculated by adding the three microbial size fractions of >10 µm, 3–10 µm, and 0.2–3 µm. Months in boldface (March, April, May, June, and July) denote upwelling season. Datapoints inside black open circles indicate the months and depths used for metagenome analysis. Source data are provided as a Source Data file.
Although the concentrations of both chromophores were not significantly correlated (Fig. 3A), rhodopsin and chlorophyll-a presented their highest concentrations during the upwelling months (March through July, Fig. 2). This apparent synchronization between the increase of photoheterotrophs and the phytoplankton bloom suggests that microbial rhodopsins are not merely restricted to oligotrophic systems but may also play important roles in productive settings. While their overall temporal variation was significantly less pronounced than that of chlorophyll-a (from 490 to 6500 pM; 13-fold change; Supplementary Fig. S2 and Supplementary Data 1), total rhodopsin concentrations still varied substantially across the different seasons, with peak concentrations differing by as much as ninefold (from 13 to 120 pM; Fig. 2A and Supplementary Data 1). In contrast to these monthly patterns, daily and hourly rhodopsin concentrations measured at the nearby coastal station off Catalina Island (Fig. 1 and Supplementary Fig. S3A) revealed smoother temporal variation at these narrower scales (Supplementary Fig. S3B). Diel measurements performed over 60-h long periods (4-h sampling intervals) showed that total rhodopsin concentrations varied only by 2.8-fold in spring (between 18 and 51 pM) and by 2.2-fold in summer (10–22 pM) (Supplementary Fig. S3). Consistent with the findings from SPOT, rhodopsin concentrations at Catalina Island were higher in the mid-upwelling season (June 2018; 33 ± 9 pM, n = 15) compared to later in July-August, when the upwelling event started to relax (August 2018; 14 ± 4 pM, n = 16), Supplementary Figs. S1 and S3). Thus, hourly changes in light availability did not significantly alter rhodopsin abundances when compared to longer monthly variations.

A Spearman correlation matrix. The color scale on the right represents the Spearman correlation factor (rho). Asterisks indicate statistically significant correlations p < 0.05. “LNA” and “HNA bacteria” denote Low and High Nucleic Acid bacterial cell counts, respectively. Specific rho (ρ) and p values for the correlations are also shown in Supplementary Data 2. B Linear model of surface rhodopsin concentrations in picoplankton (≤45 m depth, 0.2–3.0 µm microbial size fraction, n = 48) shown as the regression of rhodopsin molecules per mL (left y-axis) and pico moles per liter (right y-axis) against the total heterotrophic bacteria abundances measured with flow cytometry. Rhodopsin molecules per milliliter were calculated applying the Avogadro number conversion (1 mol equals 6.022 × 1023 molecules). Grey shadings show 95% confidence intervals of the linear model. The green color gradient of the filled circles indicates the total concentration of chlorophyll-a and serves as a visual measure of the water’s productivity for each data point. Source data are provided as a Source Data file.
Of all seawater samples collected in the photic zone at SPOT, 80% of the microbial rhodopsin content was found in the picoplankton size fraction (0.2–3.0 µm, Supplementary Data 1 and Supplementary Figs. S4 and S5), whereas most of the chlorophyll content (57%) was detected in size fractions >3 µm (Figs. S4 and S6). These results align with previous studies showing that rhodopsins in the global ocean are predominantly a prokaryotic light-capturing feature of free-living bacteria9,24,31. Nevertheless, 35% of the total rhodopsin signal was in the >10 µm size-fraction during the spring phytoplankton bloom peak (Supplementary Data 1), suggesting that rhodopsins were also present in some particle-associated bacteria24,31,32 or even in eukaryotic phytoplankton25,26,27,28,29,30,31,32,33. These observations support the notion that rhodopsin photoheterotrophy may boost multiple and diverse organic matter utilization processes8, including extracellular hydrolytic enzymatic activities to break down large particles34, substrate uptake across a wide molecular size range, and organic matter catabolism itself, which also requires ATP at initial catabolic steps22,35.
To further examine the environmental variables underlying these temporal patterns, we evaluated potential drivers of rhodopsin distribution. While nitrogen levels appeared to be unrelated to rhodopsin concentrations (Fig. 3A, Supplementary Data 2, and Supplementary Fig. S7), some biotic parameters, such as the abundance of total heterotrophic and high nucleic acid (HNA) bacteria36, showed significant and strong correlations (Fig. 3A and Supplementary Data 2). Total rhodopsin distributions also correlated weakly but significantly with photosynthetic active radiation (PAR), water temperature, and low nucleic acid (LNA) bacterial abundance. Although phytoplankton biomass (as chlorophyll-a) and rhodopsin concentrations showed synchronized peak dynamics during the spring season, the correlation was not statistically significant (ρ = 0.04, p = 0.8; Fig. 3A and Supplementary Data 2). Similarly, small picophytoplankton groups potentially present in the 0.2–3.0 µm size fraction (e.g., Synechococcus, Prochlorococcus, and picoeukaryotes) did not covary with rhodopsin levels. Rhodopsin concentrations in the picoplankton fraction and total heterotrophic bacteria showed the strongest correlation among all biological indicators (ρ = 0.96, p < 0.01, Fig. 3A and Supplementary Data 2), suggesting that rhodopsins are persistent among most marine bacteria in this marine environment year-round.
To explore these temporal trends, we built a linear model to estimate the environmental concentration of rhodopsins found in picoplankton (i.e., all cells between 0.2 and 3.0 µm) as a function of heterotrophic bacterial abundance. The slope of this linear regression (Fig. 3B) represents the number of rhodopsin molecules per heterotrophic bacterial cell in the photic zone at SPOT, providing a cellular quota of ~ 10,000 rhodopsins per cell–consistent with previous culture- and field-based estimates9,10,37,38. Furthermore, using the regression intercept (5.3 × 105 heterotrophic bacterial cells ml−1 at zero rhodopsin) and the 15-month average (1.78 × 106 heterotrophic bacterial cells ml−1), we infer that, on average, ~70% of heterotrophic bacteria contained rhodopsins. Overall, the association between rhodopsin and bacterial abundance suggests that rhodopsins are widespread among fast-growing bacteria that are stimulated during phytoplankton blooms.
Next, we analyzed the microbial communities present at SPOT to identify potential rhodopsin-containing bacteria, in particular the taxa associated with the periods of high chlorophyll and high rhodopsin levels (Figs. 2 and 4A). 16S rRNA sequencing of picoprokaryotes revealed that the major heterotrophic groups were SAR11 (relative abundance of 15–44%) and Flavobacteriales (relative abundance of 14–42%, Fig. 4A), consistent with prior studies at this location28,29 (Supplementary Fig. S8). Rhodopsin concentrations were independent of SAR11 or SAR86 clade abundance (Supplementary Fig. S9) but showed a strong positive covariance trend with Flavobacteriales (r2 = 0.75, p = <0.01; Fig. 4B). This relationship (Fig. 4B) suggests a higher rhodopsin content per cell (~ 30,000) within Flavobacteriales compared to the community average (~ 10,000; Fig. 3B). The larger cell size of Flavobacteriales relative to other bacterioplankton39 may provide additional membrane area to house more photosystems.

A Relative abundance of prokaryotic picoplankton (0.2–3.0 µm size-fraction) at 5 m and Deep Chlorophyll Maximum (DCM) depths based on 16S rDNA gene. Rhodopsin and cell concentrations are shown for reference. DCM depths are noted with grey italic letters under the sampling month. Relevant known rhodopsin-containing bacteria (Flavobacteriales and SAR11 clade) and their estimated total abundances are noted in boldface. Taxa abbreviations: Other CFB (Other Bacteroidota), Other Proteob. (Other Proteobacteria), Other bact. (other bacteria). Asterisks under the columns denote the samples subsequently used for metagenome analyses. B Linear model of surface rhodopsin concentrations in picoplankton (0.2–3.0 µm) against the estimated Flavobacteriales concentrations n = 12, with 95% confidence intervals (grey shading). Green filling on the datapoints indicates total chlorophyll-a concentrations as a visual measure of the water’s productivity. The datapoints chosen for metagenomic analysis (with asterisks) and two additional ones outside the confidence range are labeled with date and depth. Source data are provided as a Source Data file.
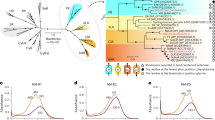
To better understand the taxon-specific trend, we further explored the taxonomic affiliation of rhodopsin genes in four metagenomes from the spring bloom (May 2017, surface and the Deep Chlorophyll maximum, DCM) and oligotrophic season (November 2017, surface and DCM) (Fig. 5). Rhodopsin gene composition revealed that SAR11 and Flavobacteriales dominated the rhodopsin gene pool in all four metagenomes (Fig. 5A). SAR11 rhodopsin genes were most abundant overall (25–42%), except in May-DCM, when Flavobacteriales genes predominated (with 26% of all rhodopsin sequences), coinciding with the highest rhodopsin concentrations. Furthermore, the relative abundance of Flavobacterial rhodopsin was significantly correlated with retinal levels (Supplementary Fig. S10), whereas no clear patterns were observed for SAR11, SAR86, or any other group. Eukaryotic and viral rhodopsin genes were also detected but comprised less than 3% of the total sequences (Supplementary Data 4). Together, these results reinforce our conclusion that the order Flavobacteriales drove the temporal variations in rhodopsin levels.

A Relative abundance of rhodopsin genes by taxa in metagenomes within the 0.2–3.0 µm size fraction collected in bloom (May of 2017; 5 m and DCM at 20 m depth) and oligotrophic season (November of 2017, 5 m and DCM at 31 m depth). Taxon abbreviations: Flavobac. (Flavobacteriales), Rhodobac. (Rhodobacterales), Actinom. (Actinomycetales). B Relative abundance of green (L, M) and blue (Q) rhodopsin variants according to their spectral tuning residues (105 amino acid position). C Relative abundance of rhodopsin gene motifs. D Summary of the recovered metagenome-assembled genomes (MAGs), together with their rhodopsin gene incidence by taxa, and their combined relative abundances expressed as RPKM (metagenome Reads recruited Per Kilobase of MAG per Million mapped reads). Source data are provided as a Source Data file.
Other relevant features of the rhodopsin gene pool include their spectral tuning and functional motifs. As previously observed in samples of this coastal region20, most of the rhodopsin genes encoded the amino acids leucine or methionine at position 105, characteristic of green-tuned rhodopsins (Fig. 5B). Interestingly, though, the proportion of green-tuned rhodopsin genes was significantly lower in November (63% ±0.5) than during the May bloom (76% ±3). This difference between seasons likely reflects a stronger attenuation of blue and UV light by chlorophyll and dissolved organic matter (DOM) during phytoplankton blooms. Nearly all recovered rhodopsin sequences (> 97%) contained the DTE, DTK, and DTV motifs, characteristic of light-driven proton pumping rhodopsins7,40,41,42 (Fig. 5C), suggesting energy-harvesting functions.
Metagenome Assembled Genome (MAG) reconstruction yielded 74 high-quality genomes (completeness ≥70% and contamination ≤5%). The best-represented taxa in the MAG collection belonged to Bacteroidota (n = 26), 84% of which contained rhodopsin genes (Fig. 5D and Supplementary Data 5). The next best-represented taxa were classified within Gammaproteobacteria (n = 17) and Alphaproteobacteria (n = 15), both with a 53% incidence of rhodopsin genes. MAGs within the Bacteroidota, a phylum that contains the order Flavobacteriales, recruited the most reads during the spring bloom of May 2017, particularly at the DCM (82 and 124 RPKM at surface and DCM, respectively; Fig. 5D), when rhodopsin chromophore levels were also highest (Fig. 4). In contrast, Alphaproteobacteria and Gammaproteobacteria MAGs were most abundant in samples with lower rhodopsin levels, such as May at 5 m (122 RPKM for Alphaproteobacteria) and November 5 m (77 RPKM for Gammaproteobacteria, Fig. 5D). Another interesting observation was that, regardless of taxonomic affiliation, the combined read sequences of MAGs containing rhodopsin genes were highest in May at the DCM, while MAGs without rhodopsin genes presented the lowest number of reads in that particular sample (Fig. 5D).
Discussion
The importance of understanding rhodopsin spatial and temporal dynamics
Chlorophyll quantifications have traditionally been used to study phytoplankton dynamics, a crucial parameter for the ocean’s carbon cycle43. As a result, such measurements are routinely collected in all the oceanographic cruises aimed at understanding the biogeochemical cycles that control ecosystem functioning and, ultimately, our climate. Yet, equivalent data regarding rhodopsins remain rare, limiting our ability to identify the rules that control their distribution in the ocean4. Given that rhodopsins are the gates to significant additional amounts of solar energy entering the marine ecosystems9, their temporal and spatial dynamics are likely to influence carbon biogeochemistry as well. In this study, our goal was to quantify rhodopsin photosystems in a dynamic coastal upwelling system and identify the environmental factors potentially controlling their seasonality.
Rhodopsins are widespread and most abundant during the upwelling months in the California system
A defining feature of the California upwelling regime is the annual phytoplankton bloom, which we identified by the increased chlorophyll levels (up to 13-fold) in the spring season. During this period, wind-driven upwelling transports macronutrients from deep waters to the photic zone where primary productivity is mostly limited by low nitrate concentrations (Supplementary Fig. S7). In our study, the upwelling season began in March, and the annual chlorophyll maxima occurred between April and June, with the highest peak occurring in May. In contrast to previous observations from oligotrophic systems where rhodopsins have an inverse distribution pattern to chlorophyll9,15,24, the annual rhodopsin maximum co-occurred with the spring phytoplankton bloom when both chromophores reached their highest concentrations over the 15-month sampling period. This apparent synchronization to phytoplankton biomass indicates that microbial rhodopsins are regulated differently in nutrient-poor versus rich environments. Thus, rhodopsins are not merely restricted to open ocean oligotrophic systems. This, in turn, suggests that rhodopsin phototrophy plays a relevant role in energizing organic matter processing in productive settings, broadening the known range of retinal-based photoheterotrophy to include coastal environments.
Rhodopsin levels can be explained by bacterioplankton abundance
Rhodopsin dynamics were directly associated with total heterotrophic bacterioplankton abundance. Thus, the greater the number of bacterioplankton cells in the water column, the more rhodopsin molecules can be found. This relationship aligns with genomic evidence showing that most bacterioplankton contain rhodopsin genes in this dynamic coastal environment20. Measuring of rhodopsin concentrations in natural bacterioplankton communities provides a quantitative view of their role in the coastal environment–something that gene or transcript measurements cannot achieve. Among the correlations we found, the strongest was between rhodopsin concentrations and bacterial abundance. If this relationship is consistent across other environments, rhodopsin distributions could then be estimated from bacterial abundance, offering a more complete picture of sunlight utilization in the marine ecosystem.
Rhodopsins are related to non-oligotrophic fast-growing bacteria in productive marine systems
The positive trend showing that rhodopsin levels increase with bacterial abundance as phytoplankton biomass develops suggests that rhodopsins are widespread among fast-growing bacteria that are seasonally stimulated during the bloom seasons. Notably, rhodopsin concentrations were also correlated with the abundance of high HNA cells, which typically represent bacteria with larger genomes and non-oligotrophic lifestyles. Thus, rhodopsin utilization is not limited to low-nutrient-adapted photoheterotrophs acting on reduced levels of DOM. Although some eukaryotic phytoplankton species at SPOT contained rhodopsin genes, as previously reported in other marine systems and cultures25,33,44,45, our data indicate that rhodopsin-containing bacteria contribute the most to the total rhodopsin phototrophy in this upwelling environment.
Flavobacterial rhodopsins are the dominant types at SPOT, suggesting the use of light for complex organic matter utilization
The dominant heterotrophic taxa in the SCB during our study were SAR11 and Flavobacteriales, two of the primary rhodopsin-bearing bacterial clades in the ocean8,17. SAR11 abundance varied only by fourfold between upwelling and non-upwelling months (range: 2.5 × 105–1.0 × 106 cells.ml−1) and showed no clear relationship with the ambient rhodopsin levels. In contrast, Flavobacteria abundance (range: 1.0 × 105–1.5 × 106 cells.ml−1) increased by 15-fold during upwelling and correlated positively with measured rhodopsin concentrations in picoplankton. These observations suggest a baseline rhodopsin pool year-round, likely dominated by SAR11 bacteria, with a sharp increase during upwelling driven by Flavobacteriales. Cellular rhodopsin estimates for the bacterioplankton community support a seasonal shift in the rhodopsin-containing bacterial groups. The average rhodopsin content per cell at SPOT was in the order of 10,000 molecules per cell, which corresponds to levels reported for the SAR11 clade17. Yet, the amount of rhodopsin per cell increases by a factor of three during upwelling associated with the increase in Flavobacteriales. Thus, the rhodopsin temporal dynamics can be explained by the contrasting lifestyles of these two bacterial clades, as we discuss below.
Members of the SAR11 clade are streamlined oligotrophs that constitutively express many of their genes, including proteorhodopsin19,37. Using this strategy, they optimize cell and genome size, minimizing the energy dedicated to replicating genomes with few gene regulatory elements21. In contrast, Flavobacteria are versatile heterotrophs capable of using organic matter of diverse complexities and broader concentration ranges46. Consistent with our results, Flavobacteriales are found to be the major contributors to in situ community rhodopsin transcripts during the annual chlorophyll maximum in other coastal waters (e.g., northeast Atlantic; Arandia‐Gorostidi et al.47). Thus, among other features of Flavobacteriales, the additional light energy harvested by rhodopsins may help them to establish their ecological niche and outcompete other bacteria lacking this light-transducing mechanism.
Previous studies have shown that some Flavobacteriales grow better in the light because they become more efficient at resource acquisition through rhodopsin photoheterotrophy. For instance, Dokdonia sp. MED134 achieves higher cell yields in the light under low organic matter concentrations23, partly due to light-induced uptake of vitamin B122, an essential coenzyme for organic matter degradation. Similarly, Polaribacter sp. MED152, can take up substrates at higher rates in the light, even during growth in rich conditions48. Our microbial community structure and metagenomics data show that both Flavobacteriales and their rhodopsin genes, dramatically increase with rhodopsin concentrations during the spring phytoplankton bloom. Furthermore, although MAGs represent only a fraction of the genomes present in the microbial community, most Flavobacteriales MAGs had rhodopsin genes and exhibited their highest abundance in May. Other in situ data indicate that photoheterotrophs may use sunlight to outcompete other bacteria at organic matter utilization in oligotrophic systems49,50. Yet, the extent of rhodopsin-enhanced substrate uptake and the potential competitive advantage to bacterioplankton at high organic matter concentrations remains unknown.
Traditionally, heterotrophic bacterial dynamics have been viewed as tightly coupled to phytoplankton production, which serves as their primary organic carbon source. However, the concentrations and composition of this organic matter fluctuate throughout the seasons and over the course of a bloom event18,46, especially as algal cells decay. The presence of rhodopsins in Flavobacteria suggests that light may be a complementary mechanism to boost the utilization of a wide range of organic molecules (including complex ones) in the surface marine environment, with still unknown implications for the carbon cycle.
Overall, our findings shows that rhodopsin distribution patterns in upwelling systems can be shaped by nutrient availability and community composition–particularly the abundance of Flavobacteriales. Combined with prior studies revealing the importance of rhodopsin phototrophy in oligotrophic regimes, these results highlight the central role of microbial rhodopsins across a broad range of nutrients regimes, validating their global importance in solar energy and organic carbon acquisition in the surface ocean.
Methods
Seawater sampling
Samples were collected at the San Pedro Ocean Time Series (SPOT) station (33°33’N, 118°24’W) (Fig. 1A) monthly with Niskin bottles attached to a rosette equipped with a CTD or in Go-Flo’s (12 L capacity) when the rosette was unavailable. Seawater was generally collected at 5, 20, 40, and 60 m depth (Supplementary Data 1). However, to prioritize the deep chlorophyll maximum (DCM) sampling, the depth closest to the DCM was replaced by the actual DCM, which was determined using the in situ fluorescence reading of the CTD. On the same day, 3–7 L of seawater were serially filtered using a peristaltic pump9, through 10, 3, and 0.22 μm pore-size filters at a rate of 75 mL/min. Filters were immediately stored at −80 °C until analysis. Two liters of seawater were separately filtered for microbial community analysis.
Chromophore extraction and quantification
Chromophores were extracted from filters, one replicate for each depth, in methanol9,24. Chlorophyll-a was quantified fluorometrically using the non-acidification method51, and rhodopsin was measured using the retinal chromophore as a proxy by means of a previously established liquid chromatography tandem-mass spectrometry (LC-MS/MS) protocol9,24 using triple injections for each extract. This rhodopsin extraction and quantification method has been previously used in bacterioplankton and phytoplankton cultures9,25, natural microbial plankton samples9,24,25,34. Chromophore distribution plots and the map shown in Fig. 1 were generated using Ocean Data View52.
Ancillary environmental parameters
10 mL aliquots of seawater were fixed with 200 μL of formalin (37% formaldehyde, BDH® histology/cytology grade) and stored at −80 °C for flow cytometry cell count analysis, using a Becton–Dickinson FACSCalibur36. Specifically, heterotrophic bacteria, picoeukaryotes, and Prochlorococcus sp. and Synechococcus sp. type of cyanobacteria, were enumerated using a FACScalibur (Becton and Dickinson) flow cytometer36. Subsamples (400 µL) were stained with SYBR Green I, Molecular Probes Inc. (final concentration 1000× dilution of the commercial product) for heterotrophic bacterial counts. High nucleic-acid content (HNA) bacteria and low nucleic-acid content (LNA) bacteria were identified in bivariate scatter plots of side scatter versus green fluorescence. Cyanobacteria and eukaryotes were discriminated in plots of orange fluorescence versus red fluorescence. Additionally, 50–100 mL of 0.22 μm filtered seawater was stored at −20 °C for dissolved nitrite and nitrate analyses using a standard colorimetric protocol53.
DNA extraction and microbial community structure analysis
Filters for genomic DNA extraction were removed from the cryovial tubes with sterile forceps, and in the case of the SterivexM (GV) cartridges, were broken to retrieve the filter54. All extractions were performed using the QIAGEN® “DNeasy PowerWater” kit. DNA concentration was estimated spectrophotometrically with the Nanodrop 2000 equipment (Thermo Scientific®).
Libraries of the V4-V5 region of the 16S rRNA gene were constructed using the primers 515FY/926R55. The PCR products were cleaned and normalized with the SequalPrep™ Normalization Plate kit (Invitrogen). A second PCR reaction attached the Illumina dual indices and sequence adapters. The PCR products were then purified, and their concentrations normalized using the SequalPrep™ Normalization Plate kit (Invitrogen). dsDNA concentrations were determined using a Qubit® fluorometer (Thermo Fisher Scientific) and the labeling HS detection kit (Thermo Fisher Scientific). PhiX was used as an internal control library. Sequencing was performed using paired-end strategy on the Illumina® MiseqTM platform at CICESE (Ensenada, BC, Mexico).
Microbial community structure analyses based on amplicons were carried out with DADA2 in R to assess read quality, filter, pair read, remove chimeras, and make taxonomic assignments, using the Silva 138 database (2020 prokaryote update) as a reference. The Phyloseq library was used to combine the resulting Amplicon Sequence Variant and create the microbial diversity and taxonomy graphs.
Rhodopsin gene presence in Metagenomes
Four environmental samples (Nov 2017, 5 m; Nov 2017 DCM; May 2017 5 m; May 2017, DCM) were sequenced on the Illumina PE150 platform (Novogene Bioinformatics Technology Co., Ltd.), with quality assessment performed using FastQC v0.11.956 retrieving 52,803,862 to 86,313,956 150-bp paired-end reads per sample after quality control. Raw reads were trimmed using Trimmomatic v0.3657 and assembled using MEGAHIT v1.2.958. Assembly settings included a minimum contig length of 2000 bp and were optimized for meta-sensitive analyses, all conducted on the KBase platform59. Contigs produced by MEGAHIT were analyzed with EukRep v0.6.560 to classify the contigs into Eukaryotes and Prokaryotes, generating one fasta file for the contigs of each group. Each file was processed independently.
For prokaryotic gene prediction, MetaGeneMark v3.3861 and Prodigal v2.6.362 were used on prokaryotic contigs from individual samples. Predicted genes (ORFs) shorter than 250 bp were excluded before concatenating all predictions from different samples. Redundancies were removed by clustering using linclust from MMseqs2 v.15c776263 at 99% identity and 80% coverage. This generated the prokaryotic gene catalog. Predicted genes were then translated into amino acids using transeq from EMBOSS v6.6.064, and rhodopsin-like genes were identified and classified using diamond v2.0.14 blastp65 and the UniRef90 database (version 202402)66, using a percentage of identity >90%.
For eukaryotic gene prediction, MetaEuk v1-ea903e567 was used on the eukaryotic contigs from the different samples. Predicted genes shorter than 250 bp were excluded before concatenating all genes from the different samples. Redundancy was removed by clustering genes at 99% identity with 80% coverage using linclust from MMseqs2 v.15c776263. This resulted in the Eukaryotic Gene Catalog. Predicted genes from the eukaryotic gene catalog were translated into amino acids using transeq from EMBOSS v6.6.064 and searched against a UniRef90 database (version 202402)66 of rhodopsins genes using diamond v2.0.14 blastp65 and a percentage of identity >90% to recover all rhodopsin-like genes.
Taxonomy was assigned to genes from both prokaryotic and eukaryotic catalogs using MMseqs2 v.15c776263, with the MarFERReT v1 database68 for eukaryotic and GTDB release 8969 for prokaryotic taxonomy. Gene abundance was estimated by mapping clean reads against the catalogs using bwa-mem2 v2.2.170. Samtools v1.871 was used to generate the filtered BAM files. Fasta files were converted to GFF formats for counting hits with HTSeq v2.0.472. Finally, abundance tables including raw counts from HTSeq were generated, providing detailed insights into gene distribution across samples.
The rhodopsin gene functional classification was guided by a motif analysis to identify key structural features associated with specific rhodopsin families. All sequences were screened for the presence of the retinal-binding lysine in helix G73 and characteristic motifs in helix C, critical for functional differentiation74,75. DTK and DTE motifs were indicative of proteorhodopsins, xanthorhodopsins, and actinorhodopsins41. In addition, TAT domains were used to identify pH-dependent light sensors42, the ETX motif for sensory eukaryotic rhodopsins or heliorhodopsins76, and the DTV motif for viral rhodopsins77. Finally, sequences containing leucine or methionine in helix C (105 amino acid position) were classified as green-absorbing, whereas those with glutamine were classified as blue-absorbing rhodopsins, based on known color-tuning properties23,38,78. Following this motif-based classification, a comprehensive phylogenetic analysis was performed to refine and validate the sequence grouping. Multiple sequence alignment was conducted using MAFFT79, trimmed with Gblocks80, and used to build a Hidden Markov Model (HMM) with HMMER 3.481. A maximum likelihood (ML) phylogenetic tree was inferred using RAxML 8.2.1282, applying the WAG substitution matrix and a Gamma model of rate heterogeneity to account for among-site rate variation.
Metagenome-assembled genome (MAG) reconstruction and annotation
Four metagenomes were co-assembled using Megahit. Contig abundances across samples were calculated by mapping reads from individual metagenomes to the co-assembly using BWA-MEM2 version 2.2.170. The resulting alignments were sorted with SAMtools version 1.1971. Contig coverage profiles across all samples were calculated using jgi_summarize_bam_contig_depths. Binning was performed using MetaBAT2 v 2.1583, CONCOCT v 1.1.084, and SemiBin285. A minimum contig length of 2500 base pairs was applied in all cases. Bins were first refined and merged using the bin-refinement module of MetaWRAP v1.386. This resulted in 174 metagenome-assembled genomes (MAGs) with completeness > 50% and contamination <10% and an N50 ≥ 10,000 bp. Dereplication was conducted with dRep version 3.5.087 using identity thresholds of 90 and 99 percent to define primary and secondary clusters, respectively, retaining the highest-quality genome per group. A final set of 74 High-Quality MAGs (99% dereplicated) were used in downstream analyses. Gene prediction was performed using Prokka version 1.14.688, and functional annotation was carried out with eggNOG-mapper version 2.1.989. Taxonomic classification was assigned with GTDB-Tk version 2.4.069. Relative abundance of each dereplicated MAG across the four samples was estimated using CoverM v 0.7.090 in genome mode, with alignments performed using BWA-MEM270. Only read alignments with a minimum of 95% identity and 80% read coverage were considered. Abundance metrics included read counts, RPKM, CPM, and covered bases. Finally, a stricter filtering step was applied to retain only high-quality MAGs with a threshold of ≥ 70% completeness, ≤ 5% contamination and an N50 ≥ 10,000 bp.
Statistical analyses
Significant correlations between environmental variables (shown in Fig. 3A and Supplementary Data 2) were determined with Spearman’s correlations (p-values < 0.05) using data from ≤45 meters depth, after testing for data distribution normality with the Shapiro-Wilk test. Preliminary analyses, including data at the edge of the euphotic zone (60 meters depth, ≤1% PAR; Supplementary Data 1), unrealistically magnified correlations reflecting a drop in multiple parameters associated directly or indirectly to phototrophy. This could be explained by the bottom of the euphotic zone being a critical depth limit for phototrophic processes and all naturally associated parameters, such as chlorophyll levels, bacterioplankton and phytoplankton cell abundances. Since the extreme values biased the correlation patterns, we limited the analysis to depths ≤45 m to better observe the temporal interactions between parameters measured during the different months of the year. A likelihood ratio statistical test was used to compare variances among groups of samples shown in Supplementary Fig. S2 (n = 48), while a Welch’s t-test was used to compare average values of smaller sample sizes (n < 20) shown in Supplementary Fig. S3. Linear regression analyses were used to further examine the co-variance of different parameters against rhodopsin concentrations, shown in Figs. 3B and 4B and Supplementary Figs. S9 and S10.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. 16S rDNA amplicon and shotgun sequencing data are available on Genbank (https://www.ncbi.nlm.nih.gov/genbank/) under the Bioproject PRJNA1040444. Metagenome Assembled Genomes (MAGs) are available on Figshare https://doi.org/10.6084/m9.figshare.2985686691 Source data are provided with this paper.
References
Falkowski, P. G. The role of phytoplankton photosynthesis in global biogeochemical cycles. Photosynth Res. 39, 235–258 (1994).
Larkum, A., Ritchie, R. & Raven, J. J. P. Living off the sun: chlorophylls, bacteriochlorophylls and rhodopsins. Photosynthetica 56, 11–43 (2018).
Karl, D. M. Solar energy capture and transformation in the sea. Elementa 2, 000021 (2014).
Béjà, O. et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906 (2000).
Martinez, A., Bradley, A. S., Waldbauer, J. R., Summons, R. E. & DeLong, E. F. Proteorhodopsin photosystem gene expression enables photophosphorylation in a heterologous host. Proc. Natl. Acad. Sci. USA 104, 5590–5595 (2007).
Finkel, O. M., Béjà, O. & Belkin, S. Global abundance of microbial rhodopsins. ISME J. 7, 448–451 (2013).
Kandori, H. Ion-pumping microbial rhodopsins. Front. Mol. Biosci. 2, 52 (2015).
Pinhassi, J., DeLong, E. F., Béjà, O., Gonzalez, J. M. & Pedrós-Alió, C. Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol. Mol. Biol. Rev. 80, 929–954 (2016).
Gómez-Consarnau, L. et al. Microbial rhodopsins are major contributors to the solar energy captured in the sea. Sci. Adv. 5, eaaw8855 (2019).
Kirchman, D. L. & Hanson, T. E. Bioenergetics of photoheterotrophic bacteria in the oceans. Environ. Microbiol. Rep. 5, 188–199 (2013).
Morris, R. M. et al. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 4, 673–685 (2010).
Martínez-García, S. & Pinhassi, J. Adaptations of microorganisms to low nutrient environments: managing life in the oligotrophic ocean. In Encyclopedia of Microbiology 4th edn, (ed. Schmidt, T. M.) (Academic Press, Oxford, 2019).
Brindefalk, B. et al. Distribution and expression of microbial rhodopsins in the Baltic Sea and adjacent waters. Environ. Microbiol. 18, 4442–4455 (2016).
Nguyen, D. et al. Winter diversity and expression of proteorhodopsin genes in a polar ocean. ISME J. 9, 1835–1845 (2015).
Campbell, B. J., Waidner, L. A., Cottrell, M. T. & Kirchman, D. L. Abundant proteorhodopsin genes in the North Atlantic Ocean. Environ. Microbiol. 10, 99–109 (2008).
Dubinsky, V. et al. Metagenomic analysis reveals unusually high incidence of proteorhodopsin genes in the ultraoligotrophic Eastern Mediterranean Sea. Environ. Microbiol. 19, 1077–1090 (2017).
Lami, R., Cottrell, M. T., Campbell, B. J. & Kirchman, D. L. Light-dependent growth and proteorhodopsin expression by Flavobacteria and SAR11 in experiments with Delaware coastal waters. Environ. Microbiol. 11, 3201–3209 (2009).
Teeling, H. et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611 (2012).
Steindler, L., Schwalbach, M. S., Smith, D. P., Chan, F. & Giovannoni, S. J. Energy starved Candidatus Pelagibacter ubique substitutes light-mediated ATP production for endogenous carbon respiration. PLoS ONE 6, e19725 (2011).
Sieradzki, E. T., Fuhrman, J. A., Rivero-Calle, S. & Gómez-Consarnau, L. Proteorhodopsins dominate the expression of phototrophic mechanisms in seasonal and dynamic marine picoplankton communities. PeerJ 6, e5798 (2018).
Giovannoni, S. J. SAR11 bacteria: the most abundant plankton in the oceans. Ann. Rev. Mar. Sci. 9, 231–255 (2017).
Gómez-Consarnau, L. et al. Proteorhodopsin light-enhanced growth linked to vitamin-B1 acquisition in marine Flavobacteria. ISME J. 10, 1102–1112 (2016).
Gómez-Consarnau, L. et al. Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature 445, 210–213 (2007).
Hassanzadeh, B. et al. Microbial rhodopsins are increasingly favoured over chlorophyll in high nutrient low chlorophyll waters. Environ. Microbiol. Rep. 13, 401–406 (2021).
Andrew, S. M. et al. Widespread use of proton-pumping rhodopsin in Antarctic phytoplankton. Proc. Natl. Acad. Sci. USA 120, e2307638120 (2023).
Longhurst, A., Sathyendranath, S., Platt, T. & Caverhill, C. An estimate of global primary production in the ocean from satellite radiometer data. J. Plankton Res. 17, 1245–1271 (1995).
Capone, D. G. & Hutchins, D. A. Microbial biogeochemistry of coastal upwelling regimes in a changing ocean. Nat. Geosci. 6, 711–717 (2013).
Chow, C.-E. T. et al. Temporal variability and coherence of euphotic zone bacterial communities over a decade in the Southern California Bight. ISME J. 7, 2259 (2013).
Cram, J. A. et al. Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. ISME J. 9, 563 (2015).
Countway, P. D., Vigil, P. D., Schnetzer, A., Moorthi, S. D. & Caron, D. A. Seasonal analysis of protistan community structure and diversity at the USC Microbial Observatory (San Pedro Channel, North Pacific Ocean). Limnol. Oceanogr. 55, 2381–2396 (2010).
Maresca, J. A., Miller, K. J., Keffer, J. L., Sabanayagam, C. R. & Campbell, B. J. Distribution and diversity of rhodopsin-producing microbes in the Chesapeake Bay. Appl. Environ. Microbiol. 84, 00137–00118 (2018).
Gómez-Consarnau, L., Needham, D. M., Weber, P. K., Fuhrman, J. A. & Mayali, X. Influence of Light on particulate organic matter utilization by attached and free-living marine bacteria. Front. Microbiol. 10, 1204 (2019).
Strauss, J. et al. Plastid-localized xanthorhodopsin increases diatom biomass and ecosystem productivity in iron-limited surface oceans. Nat. Microbiol. 8, 2050–2066 (2023).
Bar-Shalom, R. et al. Rhodopsin-mediated nutrient uptake by cultivated photoheterotrophic Verrucomicrobiota. ISME J. 17, 1063–1074 (2023).
Bergauer, K. et al. Organic matter processing by microbial communities throughout the Atlantic water column as revealed by metaproteomics. Proc. Natl. Acad. Sci. USA 115, E400–E408 (2018).
Arístegui, J. et al. Variability in plankton community structure, metabolism, and vertical carbon fluxes along an upwelling filament (Cape Juby, NW Africa). Prog. Oceanogr. 62, 95–114 (2004).
Giovannoni, S. J. et al. Proteorhodopsin in the ubiquitous marine bacterium SAR11. Nature 438, 82–85 (2005).
Béjà, O., Spudich, E. N., Spudich, J. L., Leclerc, M. & DeLong, E. F. Proteorhodopsin phototrophy in the ocean. Nature 411, 786–789 (2001).
Lauro, F. M. et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 106, 15527–15533 (2009).
Olson, D. K. et al. Proteorhodopsin variability and distribution in the North Pacific Subtropical Gyre. ISME J. 12, 1047–1060 (2018).
Rozenberg, A., Inoue, K., Kandori, H. & Béjà, O. Microbial rhodopsins: the last two decades. Annu. Rev. Microbiol. 75, 427–447 (2021).
Mannen, K. et al. Multiple roles of a conserved glutamate residue for unique biophysical properties in a new group of microbial rhodopsins homologous to TAT rhodopsin. J. Mol. Biol. 436, 168331 (2024).
Kolber, Z. Energy cycle in the ocean: powering the microbial world. Oceanography 20, 79–88 (2007).
Vader, A., Laughinghouse, H. D., Griffiths, C., Jakobsen, K. S. & Gabrielsen, T. M. Proton-pumping rhodopsins are abundantly expressed by microbial eukaryotes in a high-Arctic fjord. Environ. Microbiol. 20, 890–902 (2018).
Marchetti, A. et al. Marine diatom proteorhodopsins and their potential role in coping with low iron availability. ISME J. 9, 2745–2748 (2015).
Fernández-Gomez, B. et al. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037 (2013).
Arandia‐Gorostidi, N. et al. Light supports cell‐integrity and growth rates of taxonomically diverse coastal photoheterotrophs. Environ. Microbiol. 22, 3823–3837 (2020).
González, J. M. et al. Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria). Proc. Natl. Acad. Sci. USA 105, 8724–8729 (2008).
Mary, I. et al. Light enhanced amino acid uptake by dominant bacterioplankton groups in surface waters of the Atlantic Ocean. FEMS Microbiol. Ecol. 63, 36–45 (2008).
Gómez-Pereira, P. R. et al. Comparable light stimulation of organic nutrient uptake by SAR11 and Prochlorococcus in the North Atlantic subtropical gyre. ISME J. 7, 603–614 (2013).
Knap, A., Michaels, A., Close, A., Ducklow, H. & Dickson, A. Protocols for the Joint Global Ocean Flux Study (JGOFS) Core Measurements. IOC Manuals and Guides No. 29, UNESCO, Paris (1994).
Schlitzer, R. Ocean Data View. https://odv.awi.de (2018).
Strickland, J. D. & Parsons, T. R. A Practical Handbook of Seawater Analysis. 2nd ed. Bulletin No. 167. (Ottawa: Fisheries Research Board of Canada, 1972).
Cruaud, P., Rasplus, J.-Y., Rodriguez, L. J. & Cruaud, A. High-throughput sequencing of multiple amplicons for barcoding and integrative taxonomy. Sci. Rep. 7, 1–12 (2017).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, D. et al. MEGAHIT v1. 0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016).
Arkin, A. P. et al. KBase: the United States department of energy systems biology knowledgebase. Nat. Biotechnol. 36, 566–569 (2018).
West, P. T., Probst, A. J., Grigoriev, I. V., Thomas, B. C. & Banfield, J. F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 28, 569–580 (2018).
Zhu, W., Lomsadze, A. & Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, e132 (2010).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 1–11 (2010).
Mirdita, M., Steinegger, M., Breitwieser, F., Söding, J. & Levy Karin, E. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031 (2021).
Li, W. et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Suzek, B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Levy Karin, E., Mirdita, M. & Söding, J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 8, 1–15 (2020).
Groussman, R. D., Blaskowski, S., Coesel, S. N. & Armbrust, E. V. MarFERReT, an open-source, version-controlled reference library of marine microbial eukaryote functional genes. Sci. Data 10, 926 (2023).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 6, 1925–1927 (2020).
Vasimuddin, M., Misra, S., Li, H. & Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) 314–324 (IEEE, Rio de Janeiro, Brazil, 2019). https://doi.org/10.1109/IPDPS.2019.00041.
Li, H. The sequence alignment/map (SAM) format and SAMtools 1000 Genome Project data processing subgroup. Bioinformatics 25, 1 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Ernst, O. P. et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 8, 126–163 (2014).
Nagata, T. & Inoue, K. Rhodopsins at a glance. J. Cell Sci. 134, jcs258989 (2021).
Yamauchi, Y. et al. Molecular properties of a DTD channelrhodopsin from Guillardia theta. Biophys. Physicobiol. 14, 57–66 (2017).
Bulzu, P., Kavagutti, V. S., Andrei, A. & Ghai, R. The evolutionary kaleidoscope of rhodopsins. mSystems 7, e00405–22 (2022).
Needham et al. A distinct lineage of giant viruses brings a rhodopsin photosystem to unicellular marine predators. Proc. Natl. Acad. Sci. USA 116, 20574–20583 (2019).
Man, D. et al. Diversification and spectral tuning in marine proteorhodopsins. EMBO J. 15, 725–731 (2003).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinf. 11, 431 (2010).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Pan, S., Zhao, X.-M. & Coelho, L. P. SemiBin2: self-supervised contrastive learning leads to better MAGs via deep learning. Bioinformatics 39, i21–i29 (2023).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Bio. Evol. 38, 5825–5829 (2021).
Aroney, S. T. N. et al. CoverM: read alignment statistics for metagenomics. Bioinformatics 41, btaf147 (2025).
Cuevas-Cruz, M. et al. Annotated metagenome-assembled genomes (MAGs) from the Southern California Bight. Data sets. Figshare https://doi.org/10.6084/m9.figshare.29856866.
Acknowledgements
We thank Yamne Ortega Saad, Hiram Zayola and Lidia Montiel for their assistance in DNA extractions, statistical and bioinformatics analyses, and the crew on board the R/V Yellowfin for sample collection. This project was partially funded by the United States National Science Foundation grant (NSF, OCE1924464 and OCE-2220546), the Ministry of Economy and Competitiveness - Spanish Agencia Estatal de Investigación PID2023-152792NB-I00 and the United States-Israel Binational Science Foundation (BSF, No. 2019612).
Author information
Authors and Affiliations
Contributions
L.G.-C., B.H., and S.A.S.-W. designed research, L.G.-C., B.H., and S.A.S.-W. collected and processed samples, L.G.-C., B.H., E.V., M.C.-C., J.A., R.L., F.L., A.L.-L., L.S. and S.A.S.-W. performed research and analyzed data; L.G.-C., B.H., and S.A.S.-W. wrote the paper with input from the other coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shiqiang Gao, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gómez-Consarnau, L., Hassanzadeh, B., Villarreal, E. et al. Unexpected microbial rhodopsin dynamics in sync with phytoplankton blooms. Nat Commun 17, 790 (2026). https://doi.org/10.1038/s41467-025-67474-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67474-1
