
Abstract
AC699 is a novel, orally bioavailable chimeric estrogen receptor-α (ERα) degrader that induces proteasome-dependent ERα degradation via cereblon E3 ligase recruitment. Here we report findings from a first-in-human, phase 1, dose-escalation study of once-daily AC699 (100–600 mg) in patients with heavily-pretreated, locally-advanced/metastatic ER-positive/HER2-negative breast cancer (ClinicalTrials.gov Identifier: NCT05654532). Primary objectives are to assess dose-limiting toxicities and treatment-emergent adverse events (TEAEs). Secondary objectives are to evaluate pharmacokinetics, objective response rate (ORR), clinical benefit rate (CBR, including stable disease ≥24 weeks), duration of response (DOR), and progression-free survival (PFS). Among 37 treated patients, TEAEs occurred in 78% of patients, most commonly nausea (19%), fatigue (16%), and neutropenia (16%). All treatment-related adverse events were Grade 1/2, with no dose reductions/discontinuations; the maximum tolerated dose was not reached. Of 26 efficacy-evaluable patients, 4 (15%) achieved partial responses and CBR was 23%. In exploratory analysis of patients with ESR1 mutations, ORR and CBR were 40% and 45%, respectively. Median DOR and PFS were 6.5 and 3.6 months overall, and 6.5 and 7.4 months in ESR1-mutant patients. AC699 steady-state exposure increased approximately dose-proportionally between 100–400 mg, plateauing at 600 mg. AC699 demonstrated favorable safety, predictable PK, and encouraging antitumor activity, particularly in ESR1-mutant disease.
Similar content being viewed by others

Introduction
Breast cancer is the most frequently diagnosed cancer worldwide, representing 12% of all cancers1 accounting for 31% of new cancer cases, and responsible for 15% of cancer deaths in the United States in 20232 Approximately 70% of patients with breast cancer have hormone receptor (HR)+/human epidermal growth factor receptor 2-negative (HER2−) disease3 and endocrine therapy is the mainstay of treatment4 However, primary and acquired resistance remain substantial challenges5 Acquired resistance is frequently caused by mutations in the estrogen receptor 1 (ESR1) gene, which encodes estrogen receptor-alpha (ERα), resulting in constitutive ER activity5 ESR1 mutations are common, occurring in 20−40% of patients with ER+/HER2− breast cancer undergoing endocrine therapy in the metastatic setting6
The selective estrogen receptor degrader (SERD) fulvestrant is approved for patients with previously untreated HR+/HER2− advanced breast cancer and for patients who have progressed following endocrine therapy7 However, it is limited by low oral bioavailability and is administered via intramuscular injection8 The SERD elacestrant is approved for patients with ER+/HER2−, ESR1-mutated breast cancer following at least one line of endocrine therapy; however, progression-free survival (PFS) remains low at 3.8 months9 Other novel SERDs are currently being investigated in second and later lines of treatment, but PFS consistently remains poor, particularly as single agents, highlighting an unmet need for new treatments in this difficult-to-treat patient population.
AC699, a novel, orally bioavailable chimeric ERα degrader, binds, ubiquitinates, and degrades ERα by engaging cereblon E3 ligase. Here, we report safety, pharmacokinetics, and preliminary efficacy of AC699 in patients with locally advanced/metastatic ER+/HER2− breast cancer, including patients with ESR1 mutations.
Results
Patient disposition and demographics
Between January 10, 2023, and August 8, 2024, 37 patients were treated at five sites in the United States (Supplementary Fig. 1). The median age of patients was 60 years. Patients were heavily pretreated with a median of five prior lines of systemic therapy in all settings and a median of three prior lines of therapy in the advanced/metastatic setting. All patients had received prior treatment with CDK4/6 inhibitors and prior endocrine therapy. In addition, 12 patients had tumors with somatic ESR1 mutations at a variant allele frequency (VAF) of >1% (Table 1 and Supplementary Table 1).
Safety
Primary objectives are to assess dose-limiting toxicities (DLTs) and treatment-emergent adverse events (TEAEs). The median duration of treatment was 84 days (range, 12−336). There were no DLTs, and the maximum-tolerated dose was not reached. Overall, 78% of patients had TEAEs, the most common being nausea (19%), fatigue (16%), and neutrophil count decreased (16%) (Table 2). Forty-one percent of patients had treatment-related adverse events (TRAEs). All treatment-related adverse events were Grade 1 or 2, including nausea (14%), hot flush (14%), and neutrophil count decreased (11%) (Supplementary Data Table 2). There were no Grade ≥3 TRAEs. In addition, there were no dose reductions, discontinuations, or interruptions of AC699 due to TRAEs. Eight patients had treatment-emergent serious adverse events, with pneumonia in two patients and six unique events in one patient each; none were considered by the investigator to be related to AC699. Two patients died due to disease progression, and one patient died due to an adverse event of seizure followed by septic shock; no deaths were related to AC699.
Pharmacokinetics
Pharmacokinetics evaluation is one of the secondary objectives of this study. Preliminary steady-state pharmacokinetics data for AC699 were available for 34 patients on Cycle 1, Day 15. The mean plasma concentration‒time profiles and area under the curve from time 0–24 h (AUC0–24) values at different doses are shown in Fig. 1a, b, respectively. AUC0–24 of AC699 increased in an approximately dose-proportional manner within the dose range of 100‒400 mg once daily (QD) with mean values of 19,522 ng h/mL (100 mg), 26,017 ng h/mL (200 mg), 41,778 ng h/mL (300 mg), and 45,889 ng h/mL (400 mg). An exposure plateau was observed at the 600 mg QD dose with an AUC0–24 value of 48,580 ng h/mL.

a AC699 concentration-time profile after multiple-dose administration at steady state (C1D15) mean ± SD is shown. b Dose-exposure (AUC0–24) at steady state (C1D15) showed that AC699 exposure increased approximately dose-proportionally between 100 and 400 mg, with a plateau at 600 mg. (total n = 34 patients for both). n number of PK evaluable patients at each dose level, h hours, QD once daily, AUC area under the concentration-time curve, AUC0–24 area under the concentration-time curve from time 0 to 24 h; the symbol dots show the arithmetic mean at the indicated dose, and error bars denote standard deviation.
Efficacy
Secondary objectives also included evaluation of objective response rate (ORR), clinical benefit rate (CBR), duration of response (DOR), and PFS. Among 26 evaluable patients, four patients (15%) achieved a confirmed partial response overall. In a predefined exploratory subgroup efficacy analysis for patients with ESR1 mutations, a confirmed partial response was achieved by 40% (4/10) of patients with ESR1 mutations (VAF > 1%) (Fig. 2). The depth of responses and sustainable responses are demonstrated in Fig. 2c, d. In addition, there was a general trend towards a decrease in lesion size that was not great enough to be classified as a partial response in non-responders. All four responders had previously progressed on an ER-targeted agent. Three of these were previously treated with fulvestrant. The fourth patient did not receive prior fulvestrant but underwent four prior lines of endocrine therapy, including aromatase inhibitors and selective estrogen receptor covalent antagonists (SERCAs), as well as one line of chemotherapy in the metastatic setting. In the 10 patients with ESR1 mutations, seven (70%) had tumor shrinkage. Tumor shrinkage of liver lesions in a patient harboring an ESR1 mutation following treatment with AC699 is shown in Fig. 2e. We observed a substantial reduction in overall tumor burden on AC699 treatment. The median DOR was 6.5 months, both overall and in patients with ESR1 mutations. Median PFS was higher in patients with ESR1 mutations (7.4 months in patients with ESR1 mutations vs 3.6 months in all patients) (Fig. 3). The CBR (including stable disease ≥24 weeks) was 23% overall and 45% in patients with ESR1 mutations (Table 3). The longest duration of treatment was 48 weeks.

Waterfall plot of best percentage change in tumor size in a all evaluable patients (n = 26) and b patients with ESR1 mutations (n = 10), and spider plot of change in tumor size during treatment overtime in c all evaluable patients (n = 26) and d patients with ESR1 mutations; e baseline contrast-enhanced axial CT image of liver lesions at baseline and 6 months after treatment with AC699 200 mg QD in a patient harboring an ESR1 mutation. Red arrows indicate the location of the tumor. Mut mutated, PD progressive disease, PR partial response, pt patient, SD stable disease; *, The patient had a 23% increase in the size of a target node which was an increase of <5 mm, thus SD); #, patients had no decrease in the size target lesions and a response of SD; °, patients <20% who had PD, due to developed new lesions. Waterfall plots (a, b) represent patients with target lesions at baseline and at least one adequate post-baseline assessment. The largest decrease or smallest increase represents the best response to treatment. Spider plots (c, d) represent patients with target lesions at baseline and at least one adequate post-baseline assessment. Source data are provided as a Source data file.

Kaplan–Meier curve of PFS in a all patients (n = 37) and b patients with ESR1 mutations (n = 12). Solid lines represent Kaplan–Meier estimates, with shaded areas indicate 95% confidence intervals. Numbers below the x-axis indicate patients remaining at risk at each time point. Source data are provided as a Source data file.
Biomarkers
In a predefined exploratory analysis of 31 patients who completed both a baseline sample at Cycle 1, Day 1 and at least one post-baseline sample at up to Cycle 4, eight had a baseline circulating tumor cell (CTC) count of 0, while 23 had a baseline CTC count greater than 0 (Supplementary Fig. 2A). Among those 23 patients, 20 patients (87.0%) had a reduction in CTC counts after treatment. In addition, all 10 patients with ESR1 mutations had a reduction in CTC counts after treatment (Supplementary Fig. 2B). The vast majority of patients had an initial response to AC699 therapy as demonstrated by a decrease in CTC count, even in those who experienced disease progression shortly thereafter.
Discussion
This first-in-human phase 1 trial evaluated the safety, pharmacokinetics, and preliminary antitumor efficacy of orally, once daily (QD) AC699 in 28-day cycles in patients with advanced/metastatic ER2+/HER2− breast cancer, including patients with ESR1 mutations. There were no DLTs, and the maximum-tolerated dose was not reached. AC699 was safe and well-tolerated, with all TRAEs being mild or moderate. The most frequent TRAEs were Grade 1/2 nausea, hot flush, and neutrophil count decrease. Notably, there were no Grade 3 or higher TRAEs. In contrast, Grade 3/4 TRAEs occurred in 7.2% of patients in a phase 3 trial of elacestrant9 and in 6% of patients in a phase 2 trial of vepdegestrant in patients with ER+/HER2− advanced breast cancer10 In addition, pharmacokinetic findings showed that AC699 has favorable pharmacokinetic properties with a plateau above 400 mg, justifying doses of 400 mg or less for future clinical trials.
The patient population in this trial was heavily pretreated, with a median of three prior lines of systemic therapy in the advanced/metastatic setting, and all patients had received prior CDK4/6 inhibitors. They had also received a median of two lines of prior endocrine therapy (fulvestrant, other novel SERDs, chimeric ER degraders, or aromatase inhibitors) in the metastatic setting. Despite this poor prognostic setting, AC699 demonstrated promising antitumor activity with an ORR of 15%, a CBR of 23%, and a median PFS of 3.6 months in the overall study population. These results are comparable to those reported in studies in similar study populations of patients with ER+/HER2− advanced breast cancer treated with elacestrant and other novel SERDs (camizestrant, imlunestrant, giredestrant, amcenestrant) and the chimeric ER degrader vepdegestrant10,11,12,13,14 Notably, in patients with ESR1 mutations in the current study, the ORR was 40%, the CBR was 45%, and the median PFS was 7.4 months. These results compare favorably to those seen in a phase 2 trial of patients treated with the chimeric ER degrader vepdegestrant, which reported a median PFS of 5.7 months (ORR was not reported) and a CBR of 47.4%10. Results from the current study also compare favorably to those seen in phase 2 and 3 trials of elacestrant, imlunestrant, and camizestrant. A phase 3 trial of elacestrant in patients with ER+/HER2− advanced breast cancer reported a median PFS of 3.8 months, ORR of 7%, and CBR of 24% at 24 weeks in patients with ESR1 mutations and a median PFS of 2.8 months in all patients9 A phase 3 trial comparing imlunestrant, standard endocrine monotherapy, and imlunestrant plus abemaciclib in patients with ER+/HER2− advanced breast cancer, reported a median PFS of 5.5 months in patients with ESR1 mutations and 5.6 months in all patients in the imlunestrant group15 A phase 2 trial (SERENA-2) comparing camizestrant versus fulvestrant in patients with ER+/HER− advanced breast cancer reported a PFS of 7.2 months and 7.7 months in all patients in the 75 mg and 150 mg camizestrant groups, respectively, and 6.3 months and 9.3 months, respectively, in patients with ESR1 mutations. However, results from the SERENA-2 study are not directly comparable to those of the AC699 study due to notable differences in patient populations11 For example, in SERENA-2, one-third of patients had de novo advanced breast cancer; all had received 0 or 1 line of ET therapy across all settings; and about 50% had not been treated with CDK4/6 inhibitors. In contrast, the AC699 study involved patients who had received at least 2 lines of ET therapy or 1 line of ET if combined with CDK4/6 inhibitors, with a median of 5 lines of therapy in the metastatic setting. Thus, indicating a more heavily pretreated cohort in the AC699 study, in which all patients had received CDK4/6 inhibitors. These differences affect the comparability of PFS results between the two studies. Overall, we note that cross-trial comparisons should be performed with caution, given there are differences in dose levels used, patient eligibility criteria, and small patient numbers in the phase 1 and 2 studies discussed here. However, they are presented here to provide context for the results of our study.
The measurement of CTCs using peripheral blood samples is non-invasive and convenient. It may also provide a more comprehensive representation of the characteristics of metastatic cells, as these cells originate from various metastatic lesions in patients. In recent years, advancements in CTC technology have led to widespread exploration of CTC analysis for assessing cancer prognosis and monitoring therapeutic responses16 In this current study of AC699, a reduction in CTC counts was observed in nearly all patients after just one cycle of treatment, suggesting that AC699 effectively removes cancer cells from the bloodstream or reduces the release of CTCs from tumors. While a substantial reduction in CTC counts appears to be associated with tumor regression, additional patient data are required to establish a clear correlation between changes in CTC counts and Response Evaluation Criteria in Solid Tumors (RECIST) clinical responses. However, these results should be interpreted with caution, given that there is not a comparator arm in the study.
Several limitations of this study should be noted, including the small sample size, single-arm design, and lack of direct comparison with other treatment options.
In conclusion, findings from this phase 1 trial of AC699 in patients with ER+/HER2− advanced/metastatic breast cancer show promising safety, tolerability, and antitumor activity, at doses up to 600 mg orally QD.
Methods
Trial oversight
This phase 1 study is registered with ClinicalTrials.gov (NCT05654532).
The study design was conducted in accordance with the protocol, the Declaration of Helsinki, the International Council for Harmonisation for Guidelines on Good Clinical Practice, and the US Food and Drug Administration Code of Federal Regulation. At enrollment, all patients signed informed consent forms approved by the local institutional review board or independent ethics committee, which also approved the protocol and all amendments. The following institutional review board or independent ethics committee provided approval of the study: Western-Copernicus Group (WCG) Institutional Review Board (IRB000000533), MD Anderson Institutional Review Board (IRB00000121), and US Oncology, Inc. Institutional Review Board (IRB00001130)
Trial design
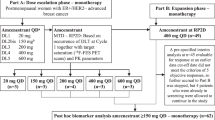
This was a phase 1, first-in-human, open-label dose-escalation trial in which AC699 monotherapy was administered orally, once daily (QD) in 28-day cycles. Primary endpoints are DLTs and TEAEs. Secondary endpoints include ORR, CBR (CR + PR + SD ≥ 24 weeks), DOR, PFS, and PK. The starting dose was 100 mg, and the dose was increased using a 3 + 3 dose-escalation design with the modification that four patients could be enrolled initially, following approval from the Safety Review Committee which determined the next dose level and/or schedule until the initially planned highest dose of 600 mg was reached. Pending the totality of clinical data, selected dose levels that had been cleared could be backfilled to expand to a maximum of 20 patients per dose level, if additional pharmacokinetic data were required.
Patients
Patients were eligible if they were ≥18 years of age; had an Eastern Cooperative Oncology Group (ECOG) performance status score of 0 or 1; had a confirmed diagnosis of advanced, unresectable, and/or metastatic breast cancer following disease progression on standard treatment, or for whom no therapy of proven efficacy existed, or who were not amenable to standard therapies; had a histologically and/or cytologically confirmed diagnosis of estrogen receptor positive (ER+) breast cancer; and had human epidermal growth factor 2-negative (HER2−) breast cancer as defined by American Society of Clinical Oncologists/College of American Pathologists guidelines17 Patients also had to have received at least two prior endocrine regimens in any setting (i.e., neoadjuvant, adjuvant, or advanced/metastatic) or at least one prior line of endocrine therapy if combined with a CDK4/6 inhibitor. Prior chemotherapy was not required, but up to three prior regimens of cytotoxic chemotherapy were allowed in the locally advanced/metastatic setting. In addition, patients had to have at least one measurable lesion according to RECIST Version 1.1 or at least one predominantly lytic bone lesion in the absence of measurable disease. Patients also had to have acceptable organ function (creatinine clearance of ≥60 mL/min by the Cockcroft-Gault equation or equivalent; total bilirubin ≤1.5 × upper limit of normal [ULN; ≤5 × ULN for patients with known Gilbert’s syndrome]; aspartate aminotransferase [AST] ≤2.5 × ULN or ≤5 × ULN in the presence of liver metastases; alanine aminotransferase [ALT] ≤2.5 × ULN or ≤5 × ULN in the presence of liver metastases; international normalized ratio ≤2), and acceptable hematologic function (hemoglobin ≥9 g/dL; absolute neutrophil count ≥1000 cells/mm3; platelet count ≥75,000 cells/mm3).
Premenopausal or perimenopausal patients had to be receiving concurrent treatment with a luteinizing hormone-releasing hormone (LHRH) agonist at least 4 weeks before the start of trial therapy and agree to continue the LHRH agonist throughout the duration of study treatment, have a negative serum pregnancy test within 7 days of initiating treatment, and agree to follow guidelines for use of highly effective contraception during the study and for 90 days following the last dose of study drug. Postmenopausal women had to meet one of the following requirements: age ≥60 years, spontaneous amenorrhea (i.e., in the absence of chemotherapy, tamoxifen, toremifene, or ovarian suppression) for ≥12 months following cessation of all exogenous hormonal treatment, 6 months of spontaneous amenorrhea with serum follicle-stimulating hormone levels and an estradiol value in the postmenopausal range per institutional standards, or prior bilateral oophorectomy performed at least 6 weeks before screening, with or without hysterectomy.
Male patients with female partners of childbearing potential were required to use contraception during the study, and life expectancy had to be greater than 12 weeks. Backfill patients also had to have available historical testing to confirm ESR1 mutations.
Exclusion criteria included: prior treatment with cytotoxic chemotherapy, investigational agents, or other anticancer drugs for the treatment of locally advanced or metastatic breast cancer within 14 days before the first administration of AC699; more than three prior chemotherapy regimens for locally advanced or metastatic breast cancer; radiation therapy within 14 days prior to the first study drug administration that did not resolve to tolerable toxicity, or prior irradiation to more than 25% of bone marrow (prior palliative radiotherapy to metastatic lesions was permitted, provided it had been completed 7 days prior to study enrollment and no clinically significant toxicities were expected, such as mucositis or esophagitis); major surgery within 21 days prior to the first study drug administration (with the exception that patients could enroll if fully recovered or without intolerable or clinically significant adverse effects, but at least 14 days had to have elapsed between major surgery and the first study drug administration); use of prophylactic growth factors and blood transfusions within 14 days prior to the first study drug administration was excluded; and use of proton pump inhibitors within at least 48 h prior to Cycle 1, Day 1. Additional exclusion criteria are detailed in the study protocol.
The sex of patients enrolled in the trial was self-reported, and data for gender were not collected. No analyses by sex or disaggregated data are presented. Classification terms for race and ethnicity were provided by the FDA. Racial and ethnic identity was self-reported.
Objectives
Primary objectives are to evaluate the safety and tolerability of AC699. Secondary objectives are to evaluate the preliminary antitumor activity of AC699 and to characterize the pharmacokinetic profile after a single dose and multiple doses of AC699. Exploratory objectives include evaluation of the relationship between circulating tumor DNA levels, CTCs, and post hoc analysis of ESR1 mutational status with antitumor activity of AC699.
Assessments
Safety
Safety assessments included analysis of reported incidence of TEAEs, treatment-related adverse events, and DLTs, all of which were graded by National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTCAE Version 5.0). Additional assessments included laboratory results, vital signs, and electrocardiogram findings.
DLTs were assessed during the DLT period (first 28 days following the first dose of AC699). The definition of DLTs is described in the study protocol and in the Supplementary Note. The study aimed to identify a maximum-tolerated dose if possible and the total data collected could be used to suggest a recommended phase 2 dose.
Pharmacokinetics
Blood samples for pharmacokinetic analyses were taken before dosing and post dosing at 1, 2 and 4 h (±5 min for each), 6, and 8 h (±10 min), and 24 h (±1 h) of Cycle 1, Day 1 and Cycle 1, Day 15, as well as before Day 1 dosing of Cycles 2, 3, 4 and 6.
Efficacy
Response and progression were evaluated using RECIST (Version 1.1) and included the ORR (defined as the percentage of patients with measurable disease at baseline and at least one post-baseline evaluation) with confirmed complete response or partial response; the CBR (defined as the percentage of patients with complete response, partial response, or stable disease, where stable disease is maintained for 24 weeks or more), the DOR (defined as the time between the first documentation of a complete response or partial response and the first evidence of progressive disease, or death due to any cause) and PFS (defined as the time from the first day of study drug administration [Day 1] until disease progression, or death on study, whichever occurred first). Patients who were alive and free from disease progression were censored at the date of the last tumor assessment. Response assessments were performed after 2 cycles and every 2 cycles/8 weeks (±5 days) for the first year of treatment, and then every 4 cycles/16 weeks (±5 days) thereafter.
Biomarkers
Blood samples were collected to assess CTC counts on Day 1 of Cycles 1, 2, and 4, and at all even-numbered cycles. CTC counts were analyzed using the CELLSEARCH® CTC18 assays. Plasma samples were collected to assess ESR1 mutation status on the Cycle 1, Day 1 visit. cfDNAs purified from plasma samples were sequenced using Guardant360 gene (74 gene-panel based; Guardant Health Inc.) assays to determine gene mutation status19.
Statistical analyses
The actual number of dose levels to be explored in this study depended on the determination of the non-tolerable dose based on DLTs. The maximum-tolerated dose was defined based on DLTs. If one patient experienced a DLT at a given dose level, then that dose level was expanded to 6 patients. Evaluation of a cohort of at least 3 patients completing 1 cycle of treatment (28 days) was required before proceeding to the next dose level. In addition, selected dose levels could be expanded to approximately 20 patients (per dose level). Qualitative variables were summarized as frequencies and percentages, and continuous variables were summarized with the number of non-missing values, mean, standard deviation, median, minimum, and maximum values. Time-to-event variables (DOR, PFS) were summarized using Kaplan–Meier analyses with medians and 95% confidence intervals (CIs). The ORR and CBR were summarized with frequency and percentage with 95% CIs using the Clopper-Pearson method.
The safety analysis set included all patients who had received at least one dose of study treatment. The efficacy-evaluable set included all patients who received any dose of study treatment. The pharmacokinetic analysis set included all patients who had received at least one dose of study treatment and had at least one sample collection of blood with a measurable concentration of study drug in plasma.
All data were analyzed using SAS (v9.4, SAS Institute, Cary, NC), GraphPad (San Diego, CA), and R software (v4.1.2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Health information from study participants is protected under applicable privacy laws and may only be shared with entities directly involved in the conduct and oversight of the trial, or as otherwise permitted upon receipt of the subject’s explicit authorization. Researchers or parties interested in collaboration for non-commercial use are encouraged to contact the corresponding author with a formal request. Applications should clearly specify the data being requested, the intended use, and ensure that all proposed uses comply with the country or region-specific regulations. Upon request, subject to review and execution of a formal data-sharing agreement with Accutar, deidentified participant data and target sequencing data supporting the findings of this study could be provided. The clinical trial protocol may also be made available upon request. All shared data will be available beginning immediately and ending 24 months following the publication of this article. Data from the data presented in graphs within the figures in this manuscript are provided with the paper. Supplementary Information or Source data files are provided with this paper. Source data are provided with this paper.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Giaquinto, A. N. et al. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 72, 524–541 (2022).
Cardoso, F. et al. 6th and 7th International consensus guidelines for the management of advanced breast cancer (ABC guidelines 6 and 7). Breast 76, 103756 (2024).
Hartkopf, A. D., Grischke, E. M. & Brucker, S. Y. Endocrine-resistant breast cancer: mechanisms and treatment. Breast Care 15, 347–354 (2020).
Brett, J. O., Spring, L. M., Bardia, A. & Wander, S. A. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 23, 85 (2021).
AstraZeneca. FASLODEX (fulvestrant) prescribing information. https://medicalinformation.astrazeneca-us.com/home/prescribing-information/faslodex.html. Accessed 1 November 2024 (2024).
Robertson, J. F. R. et al. A randomized, open-label, presurgical, window-of-opportunity study comparing the pharmacodynamic effects of the novel oral SERD AZD9496 with fulvestrant in patients with newly diagnosed ER(+) HER2(-) primary breast cancer. Clin. Cancer Res. 26, 4242–4249 (2020).
Bidard, F. C. et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from the randomized Phase III EMERALD Trial. J. Clin. Oncol. 40, 3246–3256 (2022).
Hurvitz, S. A. et al. Abstract PO3-05-08: Updated results from VERITAC evaluating vepdegestrant, a PROteolysis TArgeting Chimera (PROTAC) estrogen receptor (ER) degrader, in ER–positive/human epidermal growth factor receptor 2 (HER2)–negative advanced breast cancer. Cancer Res. 84, PO3-05-08 (2024).
Oliveira, M. et al. Camizestrant, a next-generation oral SERD, versus fulvestrant in post-menopausal women with oestrogen receptor-positive, HER2-negative advanced breast cancer (SERENA-2): a multi-dose, open-label, randomised, phase 2 trial. Lancet Oncol. 25, 1424–1439 (2024).
Bardia, A. et al. Phase I study of elacestrant (RAD1901), a novel selective estrogen receptor degrader, in ER-Positive, HER2-negative advanced breast cancer. J. Clin. Oncol. 39, 1360–1370 (2021).
Jhaveri, K. L. et al. Imlunestrant, an oral selective estrogen receptor degrader, as monotherapy and in combination with targeted therapy in estrogen receptor–positive, human epidermal growth factor receptor 2–negative advanced breast cancer: Phase Ia/Ib EMBER Study. J. Clin. Oncol. 0, JCO.23–02733 (2024).
Martín, M. et al. Giredestrant for estrogen receptor-positive, HER2-negative, previously treated advanced breast cancer: results from the randomized, phase ii acelera breast cancer study. J. Clin. Oncol. 42, 2149–2160 (2024).
Jhaveri, K. L. et al. Imlunestrant with or without abemaciclib in advanced breast cancer. N. Engl. J. Med. 392, 1189–1202 (2025).
Lin, D. et al. Circulating tumor cells: biology and clinical significance. Signal Transduct. Target. Ther. 6, 404 (2021).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. Arch. Pathol. Lab Med. 142, 1364–1382 (2018).
Galardi, F. et al. Circulating tumor cells and palbociclib treatment in patients with ER-positive, HER2-negative advanced breast cancer: results from a translational sub-study of the TREnd trial. Breast Cancer Res. 23, 38 (2021).
Mukohara, T. et al. Inhibition of lysine acetyltransferase KAT6 in ER(+)HER2(-) metastatic breast cancer: a phase 1 trial. Nat. Med. 30, 2242–2250 (2024).
Acknowledgements
The sponsor was involved in study design, data collection, and analysis, and in manuscript writing. Medical writing support, funded by Accutar Biotechnology Inc., was provided by Mark English, Ph.D., and Tricia Newell, Ph.D of Bellbird Medical Communications Ltd. This study has been presented at the American Society for Clinical Oncology Annual Meeting, Chicago, IL, 2024: Manish R. Patel et al., Preliminary results from a phase 1 study of AC699, an orally bioavailable chimeric estrogen receptor degrader, in patients with advanced or metastatic breast cancer. JCO 42, 3074-3074(2024). https://doi.org/10.1200/JCO.2024.42.16_suppl.3074. This study has also been presented at the European Society for Medical Oncology meeting, Barcelona, Spain, 2024: Erika Hamilton et al., – AC699, a novel chimeric estrogen receptor degrader, in a phase I study in breast cancer. This study was funded and sponsored by Accutar Biotechnology Inc.
Author information
Authors and Affiliations
Contributions
E.H., S.Y.K., and J.F. provided overall study leadership. S.Y.K. and H.Z. designed the study and coordinated data analysis. E.H., R.M.L., D.C., M.D., and M.R.P. enrolled patients. W.H. designed and conducted the biomarker analysis. H.Z. provided data management. E.H., H.Z., W.H., S.Y.K., and J.F. wrote the first draft. All authors contributed to data interpretation, reviewed the manuscript, and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests. D.C. and M.P. have no relevant disclosures to declare. H.Z., W.H., S.Y.K., and J.F. are employees of Accutar Biotechnology Inc. R.L. has received research support from Accutar Biotechnology, Eli Lilly, Puma, Celcuity, Arvinas, Biotheryx, and Boundless Bio, participated in advisory boards for Eli Lilly, Novartis, Celcuity, Gilead, and Biotheryx, and received honoraria from Pfizer. M.D. has worked in consulting or advisory roles for Novartis, Pfizer, Immunomedics, Seattle Genetics, Agendia, AstraZeneca, bioTheranostics, Genentech, Gilead Sciences, Lilly, Stemline Therapeutics, and Puma Biotechnology. E.H. has received research funding from Abbvie, Accutar Biotechnology, Artios, Arvinas, AstraZeneca, AtlasMedx, BeiGene, Bicycle Therapeutics, Biohaven Pharmaceuticals, BioNTech, Compugen, Cullinan, Daiichi Sankyo, Dantari, Day One Biopharmaceuticals, Duality Biologics, Ellipses Pharma, Elucida Oncology, Exelixix, FujiFilm, Genmab, Gilead Sciences, H3 Biomedicine, Iambic Therapeutics, Immunogen, Inspirna, InventisBio, Jacobio, Jazz Pharmaceuticals, K-Group Beta, Kind Pharmaceuticals, Lilly, Loxo Oncology, Mabspace Biosciences, Mabwell Bioscience, Marengo Therapeutics, MediLink Therapeutics, Merck, Mersana, Novartis, Olema, Orinove, Orum Therapeutics, Pfizer, Pionyr Immunotherapeutics, Prelude Therapeutics, Profound Bio, Regeneron, Relay Therapeutics, Rgenix, Roche/Genentech, SeaGen, Shattuck Labs, Simcha Therapeutics, Stemline Therapeutics, Sutro, Systimmune, Taiho, Tesaro, TheRas, Treadwell Therapeutics, Verastem, Xadcera Biopharmaceutical, Zymeworks, and has had consulting/advisory roles with Arvinas, AstraZeneca, BeOne Medicines, Boehringer Ingelheim, Boundless Bio, Bristol-Myers Squibb, Circle Pharma, Daiichi Sankyo, Gilead Sciences, Halda Therapeutics, Incyclix Bio, IQVIA, Janssen, Jazz Pharmaceuticals, Jefferies LLC, Johnson and Johnson, Lilly, Mersana Therapeutics, Novartis, Pfizer, Precede Biosciences, Pyxis Oncology, Roche/Genentech, Samsung Bioepis, Shorla Pharma, Stemline Therapeutics, and Tempus Labs.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hamilton, E., Layman, R.M., Cosgrove, D. et al. ER degradation for ER+/HER2– advanced or metastatic breast cancer: a phase 1 trial. Nat Commun 17, 796 (2026). https://doi.org/10.1038/s41467-025-67485-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67485-y
