
Abstract
The stereoselective construction of chiral vicinal diamines bearing multiple contiguous stereocenters remains a formidable challenge in modern organic synthesis. Herein, we report an Ir-catalyzed sequential dynamic kinetic resolution of 2,3-diamino-1,4-diketones that furnishes acyclic vicinal diamines containing four contiguous stereocenters in high yields with excellent diastereo- and enantioselectivity. The protocol exhibits broad substrate generality and high catalytic efficiency, enabling streamlined access to structurally diverse, functionally enriched chiral vicinal diamines. A gram-scale reaction proceeds smoothly with only 0.1 mol% catalyst loading, and versatile downstream derivatizations further highlights the synthetic utility of the method. Mechanistic investigations support a stepwise dynamic kinetic resolution pathway operative in this transformation.
Similar content being viewed by others

Introduction
The simultaneous construction of multiple contiguous stereocenters in an acyclic molecule is an important goal of modern organic synthesis1. Considerable efforts have been devoted to this pursuit, leading to the development of numerous approaches for synthesizing acyclic molecules with two consecutive chiral centers2,3,4,5,6. However, methods for synthesizing chiral acyclic molecules that bear three or more contiguous stereogenic centers have been rarely reported due to the free rotation of the acyclic chain and the formidable challenges in controlling stereoselectivity7,8,9,10. Thus, the development of new methods to efficiently prepare acyclic molecules with multiple continuous stereocenters at once is highly desirable and of great significance for improving synthesis efficiency.
Chiral α-hydroxy substituted vicinal diamines are a family of unique molecules that contain both vicinal diamine and vicinal aminoalcohol subunits, widely occurring in many natural products, pharmaceuticals, and exhibited important bioactivities11,12,13,14,15,16,17,18,19. In particular, chiral 2,3-diamino-1,4-diols and its analogues are known to be medicinally important motifs with a wide range of biological activities (Fig. 1a)20,21,22,23. For example, Oseltamivir phosphate and Zanamivir are common drugs for the treatment of influenza20,21, 1,2-diaminocyclitols are glucoceresidase activators and potential therapeutics for Gaucher disease22, DACH-PtCl2 has antitumor activity against P388 leukemia23.

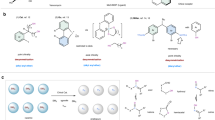
a Represent bioactive molecules containing α-hydroxyl substituted chiral vicinal diamines. b The state of art for asymmetric synthesis of chiral vicinal amines. c The synthesis of chiral acyclic 2,3-diamino-1,4-diols from chiral starting materials. d Our strategy for asymmetric synthesis of chiral acyclic 2,3-diamino-1,4-diols.
As a result, the synthesis of chiral 2,3-diamino-1,4-diols has received considerable attention. Nonetheless, efficient catalytic methods for constructing functionalized vicinal diamines remain scarce (Fig. 1b). Consequently, current syntheses of chiral 2,3-diamino-1,4-diols typically rely on multistep sequences that employ valuable chiral starting materials (Fig. 1c), resulting in low overall efficiency and limited substrate scope24,25. To the best of our knowledge, no catalytic method has yet been developed for the asymmetric synthesis of 2,3-diamino-1,4-diols containing four contiguous stereocenters. Accordingly, the development of new catalytic strategies to address this challenge is highly desirable.
Dynamic kinetic resolution (DKR), which enables the simultaneous establishment of multiple stereogenic centers from racemic starting materials, has emerged as a powerful strategy for synthesizing chiral molecules26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. In this context, the DKR of α-amino substituted ketones has been extensively explored44,45,46,47,48, offering an efficient pathway for the synthesis of vicinal aminoalcohols49,50,51,52,53,54,55,56,57,58. However, the sequential DKR of 2,3-diamino-1,4-diketones, which could provide an ideal route to acyclic vicinal diamines bearing four stereogenic centers remains unachieved. The possible reasons are as follows: (1) Racemization of stereocenters is greatly influenced by the equilibrium of multiple isomers, making the DKR process very complicated; (2) Selective production of a single double-reduction product from the 32 possible reduction products is extremely difficult due to the complex stereoselectivity inherent in the double DKR system, which includes 16 mono-reduction stereoisomers and 16 double-reduction stereoisomers. (3) Presence of multiple contiguous polar functional groups in the target product may attenuate the catalyst’s activity to some extent, further complicating the reaction. As a result, achieving the double DKR of 2,3-diamino-1,4-diketones is exceedingly challenging.
As our ongoing interest in synthesis of valuable chiral amines59,60,61,62,63,64,65, we aim to develop a new catalytic system to achieve the double DKR of 2,3-diamino-1,4-diols, thereby providing efficient access to functionalized vicinal diamines. Encouraged by our recent work on ferrocene-based multidentate ligands-mediated double reduction of enones66, we believe that the double DKR process can also be accomplished through rational substrate design and the selection of an appropriate catalytic system, thereby enabling the formation of vicinal diamines with four contiguous stereocenters in a single transformation. Herein, we report the sequential DKR of 2,3-diamino-1,4-diketones to stereospecifically afford chiral 2,3-diamino-1,4-diols in high yields (Fig. 1d).
Results and Discussion
Reaction optimization
Our initial studies commenced with the optimization of reaction conditions by choosing the Ir-catalyzed asymmetric hydrogenation of the mixture isomers of N-Boc protected 2,3-diamino-1,4-diphenylbutane-1,4-dione 1a as a model reaction. The tridentate chiral ligands f-Ampha L1 and f-Amphol L2 which have excellent performance in the AH of ketones were evaluated67,68, to our depression, both of them gave a very complicated mixture of mono-reduction product, double-reduction product and their isomers (Table 1, entries 1-2). Then the f-PNNO type tetradentate ligands which were developed by our group69 and Prof. Zhang group were employed70,71, and the results revealed that the f-PNNO type ligands are very efficient for this transformation, afford the target product with high yields and excellent stereoselectivities (90-99% yield and 93-99% ee, Table 1, entries 1-7). Among them, L6 had the best performance in this transformation, affording 2a with 99% yield and more than 99% ee (Table 1, entry 6), thus it was chosen as the best ligand for further optimization. Subsequently, the solvent effect was investigated, and the results disclosed that solvents have only a slight effect on the enantioselectivity of this reaction but a significant impact on the yield. When EtOAc, n-hexane, and i-PrOH were used as solvents, only moderate yields were obtained albeit the enantioselectivity remained very high (Table 1, entries 8-10). The reaction was inhibited when the reaction was conducted in MeOH or 1,4-dioxane (Table 1, entries 11-12). Next, a series of bases were evaluated to examine their effect on the DKR process. The results revealed that the base exerted a significant influence on this transformation, with Cs₂CO₃ emerging as the most efficient (Table 1, entries 13–18). Its superior performance is likely due to the synergistic effects of its optimal basicity and the favorable contribution of the cesium cation.

Substrate scope
With the optimal conditions in hand, the substrate scope of this transformation was investigated and the results were summarized in Fig. 2. Gratifyingly, the reaction has a broad substrate scope and exhibits good tolerance to a variety of functional groups, such as alkyl (Me, Et, 2 d, 2e), alkoxyl (MeO, 2k), aryl (Ph, 2 f), halides (F, Cl, Br, 2g-2i), trifluoromethyl (2j), and amino group (2ab). Moreover, the reaction was not affected by the electronic properties and the position of substituents on the benzene ring (para, meta and ortho), furnishing target products with almost quantitative yields and excellent enantioselectivities (98-99% yield and 96-99% ee). Installing multiple substituents on the benzene ring of the substrate didn’t cause any change in reactivity and enantioselectivity (2 s, 2t). Substrates containing other aromatic fragments, such as 2-naphthyl, and 2-thienyl, were also successfully compatible in this transformation (2 u, 2 v). To our delight, the sequential DKR of unsymmetrical 2,3-diamino-1,4-diketones, which possess four isomers as the starting material, also performed very well, delivering acyclic vicinal diamines with four contiguous stereocenters in high yields with excellent enantioselectivities (2w-2ah). Alkyl substituted substrate was also compatible in this reaction, affording target product 2ai in high yields with excellent enantioselectivities, which demonstrates the good compatibility of this catalytic system(See Supplementary Table 2 for unsuitable substrates). Notably, the diastereoselectivity of this reaction was excellent, and only a single isomer was detected in this transformation, indicating that the discrimination of chiral ketones was stereospecific and only the matched ketones could be hydrogenated during the second DKR process. The absolute configuration of 2 h was unambiguously determined as (1 R, 2S, 3S, 4 R) by X-ray crystallography.

Unless otherwise mentioned, all reactions were performed on a 0.2 mmol scale at room temperature in 1 mL THF with 0.5 mol% [Ir(COD)Cl]2, 1.1 mol% ligand, 50 bar H2, and a reaction time of 24 h. All yields are reported as isolated yields unless otherwise noted. The ee and dr were determined by chiral HPLC and ¹H NMR analysis, respectively. All products were obtained with > 20:1 dr.
To demonstrate the utility of the current methodology, the gram-scale reaction was conducted with 0.1 mol% catalyst loading, and the reaction proceeded very smoothly to afford the desired product 2a without any loss in yield and enantioselectivity (98% yield, 99% ee, Fig. 3a), which indicated that this methodology has potential practical uses. Subsequently, the applications of 2a in organic synthesis were investigated. The Boc protecting group of 2a can be easily removed in the presence of TFA, affording free α-hydroxy vicinal diamines 3 in quantitative yield (Fig. 3b-1). The Dess-Martin regent enabled oxidation of 2a proceeded smoothly at room temperature, delivering valuable chiral (S)-1a in quantitative yield without any erosion in enantioselectivity (Fig. 3b–2). In the presence of sodium hydride, 2a can be easily transformed into a novel dioxazolidinone 4 in 90% yield (Fig. 3b–3). Treatment of substrate 2a with LiAlH4 leads to the formation of product 5 through reduction (Fig. 3b–4). In addition, compound 7, a bisoxazoline ligand with a novel structure, can be efficiently prepared by treating 2c with DAST at -78 °C (Fig. 3b–5).

a Gram scale DKR of 2,3-diamino-1,4-diketones 1a with 0.1 mol% catalyst loading. b The transformation of chiral 2,3-diamino-1,4-diols.

a The possible reduction products. b Yield as a function of time for the hydrogenation of 1a. c ee as a function of time for the hydrogenation of 1a.
Mechanistic investigations
To shed light on the reaction mechanism, a series of control experiments were conducted. Initially, the effect of the base on the distribution of the stereoisomers of starting material was investigated (See Supplementary Table 3 and Fig. 1 for details). It was observed that the ratio of meso-1a to dl-1a was gradually increased in the presence of 5 mol% Cs2CO3 at room temperature, reaching an equilibrium when the ratio of meso-1a to dl-1a was 17:83. Subsequently, the change of product distribution with reaction time was investigated. As shown in Fig. 4, the ratio of meso-1a increased slightly in the first three hours before gradually decreasing. Concurrently, (S,S)-1a was gradually decreased along with the reaction time, while there was almost no consumption for (R,R)-1a in the first three hours, which indicates that (S,S)-1a, as the matched substrate, was preferentially hydrogenated in this transformation. The mono-reduction products 6 increased gradually until their consumption exceeded their production. Interestingly, the kinetic resolution of dl-1a was detected in the mono-reduction process, and the ee value of 1a gradually increased with reaction time, and the highest ee value of 1a was obtained after 7 h of reaction. It’s worth noting that the double DKR of 1a is a stepwise process, and the the second DKR process was completed after 12 h, affording the single chiral product 2a.
In conclusion, we have developed Ir/f-PNNO complex enabled asymmetric hydrogenation of the mixture of racemic 2,3-diamino-1,4-diketones, affording chiral 2,3-diamino-1,4-diols in high yields and excellent stereoselectivities. The mechanism studies revealed that a stepwise dynamic kinetic resolution was involved in this transformation. We anticipate that this facile, effective, and practical synthetic method will not only significantly facilitate the synthesis of functionalized vicinal diamines, but also provides a general strategy for the construction of challenging acyclic chiral molecules with four adjacent stereocenters. The application of the double DKR strategy in synthesis of complicated molecules with multiple stereocenters is undergoing in our lab.
Methods
General procedure of asymmetric hydrogenation of 2,3-diamino-1,4-diketones
To a 4.0 mL vial was added the catalyst precursor [Ir(COD)Cl]2 (3.3 mg, 5 × 10-3 mmol, 1.0 eq.), (SC, SC, RFC)-L6 (6.3 mg, 1.1 × 10-2mmol, 2.2 eq.) and anhydrous i-PrOH (1.0 mL) in the argon-filled glovebox. The mixture was stirred for 1.0 h at 25 °C. The resulting orange solution (50 μL) was transferred by syringe into a vial (5.0 mL) charged with substrate (0.05 mmol), Cs2CO3 (0.8 mg, 0.0025 mmol) and anhydrous THF (1.0 mL). The vial was transferred to an autoclave, which was then charged with of H2 (50 bar) and stirred at room temperature for 24 h. The hydrogen gas was released slowly in a well-ventilated hood and the solution was concentrated and purified by flash chromatography on silica gel (CH2Cl2/MeOH, 10:1) to afford the product.
Data availability
The data supporting the findings of this study are available in the paper and its Supplementary Information, further data are available from the corresponding author on request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers CCDC 2034549 (2 h). These data can be obtained free of charge from the Cambridge Crystallographic Data Center via (www.ccdc.cam.ac.uk/data_request/cif).
References
Eppe, G., Didier, D. & Marek, I. Stereocontrolled formation of several carbon–carbon bonds in acyclic systems. Chem. Rev. 115, 9175–9206 (2015).
Kraft, S., Ryan, K. & Kargbo, R. B. Recent advances in asymmetric hydrogenation of tetrasubstituted olefins. J. Am. Chem. Soc. 139, 11630–11641 (2017).
Holmes, M., Schwartz, L. A. & Krische, M. J. Intermolecular metal-catalyzed reductive coupling of dienes, allenes, and enynes with carbonyl compounds and Imines. Chem. Rev. 118, 6026–6052 (2018).
Hamada, Y. et al. Diastereo- and enantioselective anti-selective hydrogenation of α-amino-β-keto ester hydrochlorides and related compounds using transition-metal-chiral-bisphosphine catalysts. Chem. Rec. 14, 235–250 (2014).
Beletskaya, I. P., Nájera, C. & Yus, M. Stereodivergent catalysis. Chem. Rev. 118, 5080–5200 (2018).
Wei, L., Fu, C., Wang, Z.-F., Tao, H.-Y. & Wang, C.-J. Synergistic dual catalysis in stereodivergent synthesis. ACS Catal. 14, 3812–3844 (2024).
Watson, C. G. et al. Construction of multiple, contiguous quaternary stereocenters in acyclic molecules by lithiation-borylation. J. Am. Chem. Soc. 136, 17370–17373 (2014).
Anderson, J. C. et al. Enantioselective conjugate addition nitro-mannich reactions: solvent controlled synthesis of acyclic anti- and syn-β-nitroamines with three contiguous stereocenters. J. Org. Chem. 76, 1961–1971 (2011).
Wang, J. et al. Facile synthesis of enantiopure sugar alcohols: asymmetric hydrogenation and dynamic kinetic resolution combined. Angew. Chem. Int. Ed. 59, 18166–18171 (2020).
Chen, T. et al. Dynamic kinetic resolution of β-substituted α-diketones via asymmetric transfer hydrogenation. J. Am. Chem. Soc. 145, 585–599 (2023).
Paul, B. J. et al. Synthesis, structure, and biological evaluation of novel N- and O-Linked Diinositols. J. Am. Chem. Soc. 124, 10416–10426 (2002).
Coverdale, J. P. C. et al. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 10, 347–354 (2018).
Vu, B. et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 (2013).
Bakhonsky, V. V. et al. Synthesis and antiproliferative activity of hindered, chiral 1,2-diaminodiamantane platinum(II) complexes. Dalton Trans. 49, 14009–14016 (2020).
Foubelo, F., Nájera, C., Retamosa, M. G., Sansano, J. M. & Yus, M. Catalytic asymmetric synthesis of 1,2-diamines. Chem. Soc. Rev. 53, 7983–8085 (2024).
Gan, X.-C., Zhang, C.-Y., Zhong, F., Tian, P. & Yin, L. Synthesis of chiral anti-1,2-diamine derivatives through copper(I)-catalyzed asymmetric α-addition of ketimines to aldimines. Nat. Commun. 11, 4473 (2020).
Zhou, M. et al. Enantioselective reductive coupling of imines templated by chiral diboron. J. Am. Chem. Soc. 142, 10337–10342 (2020).
Tao, Z., Gilbert, B. B. & Denmark, S. E. Catalytic, enantioselective syn-diamination of alkenes. J. Am. Chem. Soc. 141, 19161–19170 (2019).
Chen, Y., Pan, Y., He, Y.-M. & Fan, Q.-H. Consecutive intermolecular reductive amination/asymmetric hydrogenation: facile access to sterically tunable chiral vicinal diamines and N-heterocyclic carbenes. Angew. Chem. Int. Ed. 58, 16831–16834 (2019).
von Itzstein, M. et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363, 418–423 (1993).
Magano, J. Recent synthetic approaches to oseltamivir phosphate (TamifluTM) for the treatment of influenza. Tetrahedron 67, 7875–7899 (2011).
Trapero, A. & Llebaria, A. The myo-1,2-Diaminocyclitol Scaffold Defines Potent Glucocerebrosidase Activators and Promising Pharmacological Chaperones for Gaucher Disease. ACS Med. Chem. Lett. 2, 614–619 (2011).
Hanessian, S. & Wang, J. Hydrophilic analogs of (R, R)-diaminocyclohexane dichloroplatinum (DACH) and the influence of relative stereochemistry on antitumor activity. Can. J. Chem. 71, 2102–2108 (1993).
Hanessian, S., Gauthier, J.-Y., Okamoto, K., Beauchamp, A. L. & Theophanides, T. Synthesis of diaminodideoxyalditol analogs of cisplatin as antitumor agents. Can. J. Chem. 71, 880–885 (1993).
Fiorelli, C., Maini, L., Martelli, G., Savoia, D. & Zazzetta, C. Study of the regioselectivity and diastereoselectivity in the addition of 3-substituted-2-propenylmetal reagents to N, N′-di[1(S)-phenylethyl]ethanediimine. Tetrahedron 58, 8679–8688 (2002).
Xie, Q.-X., Liu, L.-X., Zhu, Z.-H., Yu, C.-B. & Zhou, Y.-G. Asymmetric transfer hydrogenation of 2,3-disubstituted flavanones through dynamic kinetic resolution enabled by retro-oxa-Michael addition: construction of three contiguous stereogenic centers. J. Org. Chem. 87, 7521–7530 (2022).
Li, J. et al. RuPHOX–Ru catalyzed asymmetric hydrogenation of α-substituted tetralones via a dynamic kinetic resolution. Chem. Commun. 58, 4905–4908 (2022).
Vyas, V. K., Clarkson, G. J. & Wills, M. Sulfone group as a versatile and removable directing group for asymmetric transfer hydrogenation of ketones. Angew. Chem. Int Ed. 59, 14265–14269 (2020).
Zhang, L., Wang, Z., Han, Z. & Ding, K. Manganese-catalyzed anti-selective asymmetric hydrogenation of α-substituted β-ketoamides. Angew. Chem. Int. Ed. 59, 15565–15569 (2020).
Touge, T. et al. Multiple absolute stereocontrol in cascade lactone formation via dynamic kinetic resolution driven by the asymmetric transfer hydrogenation of keto acids with oxo-tethered ruthenium catalysts. J. Am. Chem. Soc. 141, 16354–16361 (2019).
Zhou, A., Amer, M. M. K. & Yin, Q. Recent advances on asymmetric reduction via dynamic kinetic resolution. Chin. Chem. Lett. https://doi.org/10.1016/j.cclet.2025.111929 (2025).
Liu, J., Krajangsri, S., Yang, J., Li, J.-Q. & Andersson, P. G. Iridium-catalysed asymmetric hydrogenation of allylic alcohols via dynamic kinetic resolution. Nat. Catal. 1, 438–443 (2018).
Cotman, A. E., Cahard, D. & Mohar, B. Stereoarrayed CF3-substituted 1,3-diols by dynamic kinetic resolution: ruthenium(ii)-catalyzed asymmetric transfer hydrogenation. Angew. Chem. Int. Ed. 55, 5294–5298 (2016).
Chen, M.-W., Cai, X.-F., Chen, Z.-P., Shi, L. & Zhou, Y.-G. Facile construction of three contiguous stereogenic centers via dynamic kinetic resolution in asymmetric transfer hydrogenation of quinolines. Chem. Commun. 50, 12526–12529 (2014).
Corbett, M. T. & Johnson, J. S. Diametric stereocontrol in dynamic catalytic reduction of racemic acyl phosphonates: divergence from α-keto ester congeners. J. Am. Chem. Soc. 135, 594–597 (2013).
Steward, K. M., Gentry, E. C. & Johnson, J. S. Dynamic kinetic resolution of α-keto esters via asymmetric transfer hydrogenation. J. Am. Chem. Soc. 134, 7329–7332 (2012).
Xie, J.-H. et al. Highly enantioselective and diastereoselective synthesis of chiral amino alcohols by ruthenium-catalyzed asymmetric hydrogenation of α-amino aliphatic ketones. J. Am. Chem. Soc. 131, 4222–4223 (2009).
Xie, J.-H., Zhou, Z.-T., Kong, W.-L. & Zhou, Q.-L. Ru-catalyzed asymmetric hydrogenation of racemic aldehydes via dynamic kinetic resolution: efficient synthesis of optically active primary alcohols. J. Am. Chem. Soc. 129, 1868–1869 (2007).
Noyori, R., Tokunaga, M. & Kitamura, M. Stereoselective organic synthesis via dynamic kinetic resolution. Bull. Chem. Soc. Jpn. 68, 36–55 (1995).
Noyori, R. et al. Stereoselective hydrogenation via dynamic kinetic resolution. J. Am. Chem. Soc. 111, 9134–9135 (1989).
Liu, W. et al. Regio- and stereoselective transfer hydrogenation of aryloxy group-substituted unsymmetrical 1,2-diketones: synthetic applications and mechanistic studies. J. Am. Chem. Soc. 146, 20092–20106 (2024).
Liang, M.-R. et al. Dynamic kinetic resolution-based asymmetric transfer hydrogenation of racemic 2-substituted quinolines. J. Am. Chem. Soc. 147, 4239–4248 (2025).
Rong, N., Zhou, A., Liang, M., Wang, S.-G. & Yin, Q. Asymmetric hydrogenation of racemic 2-substituted indoles via dynamic kinetic resolution: an easy access to chiral indolines bearing vicinal stereogenic centers. J. Am. Chem. Soc. 146, 5081–5087 (2024).
Huerta, F. F., Minidis, A. B. E. & Bäckvall, J.-E. Racemisation in asymmetric synthesis. Dynamic kinetic resolution and related processes in enzyme and metal catalysis. Chem. Soc. Rev. 30, 321–331 (2001).
Pellissier, H. et al. Chirality from Dynamic Kinetic Resolution. The Royal Society of Chemistry, 49–143 (2011).
Echeverria, P.-G., Ayad, T., Phansavath, P. & Ratovelomanana-Vidal, V. Recent developments in asymmetric hydrogenation and transfer hydrogenation of ketones and imines through dynamic kinetic resolution. Synthesis 48, 2523–2539 (2016).
Bhat, V., Welin, E. R., Guo, X. & Stoltz, B. M. Advances in stereoconvergent catalysis from 2005 to 2015: transition-metal-mediated stereoablative reactions, dynamic kinetic resolutions, and dynamic kinetic asymmetric transformations. Chem. Rev. 117, 4528–4561 (2017).
Betancourt, R. M., Echeverria, P.-G., Ayad, T., Phansavath, P. & Ratovelomanana-Vidal, V. Recent progress and applications of transition-metal-catalyzed asymmetric hydrogenation and transfer hydrogenation of ketones and imines through dynamic kinetic resolution. Synthesis 53, 30–50 (2021).
Ohkuma, T., Ishii, D., Takeno, H. & Noyori, R. Asymmetric hydrogenation of amino ketones using chiral RuCl2(diphophine)(1,2-diamine) complexes. J. Am. Chem. Soc. 122, 6510–6511 (2000).
Liu, S., Xie, J.-H., Wang, L.-X. & Zhou, Q.-L. Dynamic kinetic resolution allows a highly enantioselective synthesis of cis-α-aminocycloalkanols by ruthenium-catalyzed asymmetric hydrogenation. Angew. Chem. Int. Ed. 46, 7506–7508 (2007).
Seashore-Ludlow, B., Saint-Dizier, F. & Somfai, P. Asymmetric transfer hydrogenation coupled with dynamic kinetic resolution in water: synthesis of anti-β-hydroxy-α-amino acid derivatives. Org. Lett. 14, 6334–6337 (2012).
Rasu, L. et al. Highly enantioselective hydrogenation of amides via dynamic kinetic resolution under low pressure and room temperature. J. Am. Chem. Soc. 139, 3065–3071 (2017).
Lu, B. et al. Highly diastereo- and enantioselective access to syn-α-amido β-hydroxy esters via ruthenium-catalyzed dynamic kinetic resolution-asymmetric hydrogenation. J. Org. Chem. 84, 3201–3213 (2019).
Cao, J. & Hyster, T. K. Pyridoxal-catalyzed racemization of α-aminoketones enables the stereodivergent synthesis of 1,2-amino alcohols using ketoreductases. ACS Catal. 10, 6171–6175 (2020).
Gediya, S. K., Clarkson, G. J. & Wills, M. Asymmetric transfer hydrogenation: dynamic kinetic resolution of α-amino ketones. J. Org. Chem. 85, 11309–11330 (2020).
Bin, H.-Y. et al. Asymmetric hydrogenation of exocyclic γ,δ-unsaturated β-ketoesters to functionalized chiral allylic alcohols via dynamic kinetic resolution. Chem. Sci. 12, 7793–7799 (2021).
Xu, Y. et al. Rh-catalyzed sequential asymmetric hydrogenations of 3-amino-4-chromones via an unusual dynamic kinetic resolution process. J. Am. Chem. Soc. 144, 20078–20089 (2022).
Ishikawa, H. et al. Asymmetric hydrogenation of α-amino esters into optically active β-amino alcohols through dynamic kinetic resolution catalyzed by ruthenabicyclic complexes. Org. Lett. 25, 2355–2360 (2023).
Wang, Q. et al. Rhodium-catalyzed enantioselective hydrogenation of tetrasubstituted α-acetoxy β-enamido esters: a new approach to chiral α-hydroxyl-β-amino acid derivatives. J. Am. Chem. Soc. 136, 16120–16123 (2014).
Gao, M., Meng, J., Lv, H. & Zhang, X. Highly regio- and enantioselective synthesis of γ,δ-unsaturated amido esters by catalytic hydrogenation of conjugated enamides. Angew. Chem. Int. Ed. 54, 1885–1887 (2015).
Gao, W. et al. Nickel-catalyzed asymmetric hydrogenation of β-acylamino nitroolefins: an efficient approach to chiral amines. Chem. Sci. 8, 6419–6422 (2017).
Guan, Y.-Q. et al. A cheap metal for a challenging task: nickel-catalyzed highly diastereo- and enantioselective hydrogenation of tetrasubstituted fluorinated enamides. Chem. Sci. 10, 252–256 (2018).
Zhao, X., Zhang, F., Liu, K., Zhang, X. & Lv, H. Nickel-catalyzed chemoselective asymmetric hydrogenation of α,β-unsaturated ketoimines: an efficient approach to chiral allylic amines. Org. Lett. 21, 8966–8969 (2019).
Ma, Y., Liu, K., He, L. & Lv, H. Divergent synthesis of chiral amines via Ni-catalyzed chemo- and enantioselective hydrogenation of alkynone imines. Sci. China Chem. 66, 3186–3192 (2023).
Jiao, B., Wang, F. & Lv, H. Rhodium-catalyzed asymmetric hydrogenation of tetrasubstituted α,β-unsaturated amides: efficient access to chiral β-amino amides. Chin. J. Chem. 42, 2641–2646 (2024).
Li, W. et al. Ir/f-Ampha complex catalyzed asymmetric sequential hydrogenation of enones: a general access to chiral alcohols with two contiguous chiral centers. Chem. Sci. 13, 1808–1814 (2022).
Yu, J. et al. Iridium-catalyzed asymmetric hydrogenation of ketones with accessible and modular ferrocene-based amino-phosphine acid (f-Ampha) ligands. Org. Lett. 19, 690–693 (2017).
Yu, J. et al. Readily accessible and highly efficient ferrocene-based amino-phosphine-alcohol (f-Amphol) ligands for iridium-catalyzed asymmetric hydrogenation of simple ketones. Chem. Eur. J. 23, 970–975 (2017).
Ma, J., Li, W., He, L. & Lv, H. Iridium-catalyzed chemoselective asymmetric hydrogenation of conjugated enones with ferrocene-based multidentate phosphine ligands. Chem. Commun. 58, 5841–5844 (2022).
Yin, C. et al. A 13-million turnover-number anionic Ir-catalyst for a selective industrial route to chiral nicotine. Nat. Commun. 14, 3718 (2023).
Yu, J. et al. Discovery and development of ferrocene-based tetradentate ligands for Ir-catalysed asymmetric hydrogenation of ketone. Green. Synth. Catal. 3, 175–178 (2022).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant Nos. 22371217, 22071188), Hubei Provincial Natural Science Foundation of China (2023AFA011).
Author information
Authors and Affiliations
Contributions
H.L. directed the project. H.L. contributed to the concept and design of the experiments. J.Y. gave valuable advice for this project. J.M. performed the experiments and data analysis. J.M. wrote the manuscript with feedback and guidance from H.L. and J.Y. All authors discussed the experimental results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, J., Yuan, J. & Lv, H. Efficient synthesis of chiral vicinal diamines with four contiguous stereocenters via sequential dynamic kinetic resolution of 2,3-diamino-1,4-diketones. Nat Commun 17, 821 (2026). https://doi.org/10.1038/s41467-025-67526-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67526-6
