
Abstract
Recent studies have indicated that traps play a crucial role in determining characteristics of organic persistent luminescence, yet constructing traps with controllable depth remains highly challenging. Herein, we propose a host energy-level engineering strategy, which enables precise control of trap depth. Using the Randall-Wilkins method, the trap depth can be tuned from 0.38 to 0.72 eV without altering emission wavelength and the result aligns well with density functional theory calculations. Specifically, a host-guest material namely CPND@DPEPO, which has a deep trap of ~ 0.72 eV, exhibits deep-blue persistent luminescence lasting for 27 hours and efficient energy storage over 14 days at room temperature. By utilizing these trap-containing materials as the emission layer in organic light-emitting diodes, we develop a pixel-programmable information storage device. This work establishes a fundamental principle for designing organic materials with controllable trap depth, potentially expanding their applications in night tracing, military communication and biological imaging.
Similar content being viewed by others

Introduction
Organic long-lived luminescent materials possess excellent biocompatibility, broad structural/functional designability, and high signal-to-noise ratio in sensing. These remarkable characteristics make them highly promising in applications of optoelectronic devices, biological imaging, ultrathin flexible glass, and optical information technologies1,2,3,4,5,6,7,8. Room-temperature phosphorescence (RTP) and thermally activated delayed fluorescence (TADF) are primary ways to achieve long-lived luminescence in organic systems9,10,11,12,13. Since the mid-20th century, researchers had observed phosphorescence in molecules at low temperatures, and it was soon attributed to the spin-forbidden transitions of triplet to singlet states14,15. A variety of strategies have been explored to enhance the lifetime of RTP, driving substantial interest in developing efficient organic light-emitting devices (OLEDs)16,17,18,19. The concept of TADF has been known from the early 1960s20,21,22. TADF materials can, in principle, utilize 100% of excitons by transforming triplet excitons into emissive singlet states via the reverse intersystem crossing mechanism (RISC)23,24,25. Both RTP and TADF have greatly advanced the design and understanding of organic long-lived luminescent materials26,27,28,29. However, due to the inherent spin-forbidden nature of triplet-to-singlet transitions, the emission from both RTP and TADF follows an exponential decay law, thereby restricting the emission durations to typical milliseconds or seconds30.
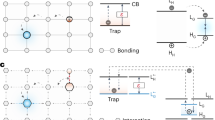
In 2017, Kabe and Adachi reported organic persistent luminescence (PersL) that lasted for hours, marking a new form of organic long-lived luminescence distinct from RTP and TADF31,32. The organic PersL was then explained with a charge separation-recombination model, where the electrons or holes in the charge-separated states need to overcome an energy barrier for recombination, resulting in a power-law decay of emission33. In subsequent research, the concept of traps was incorporated into the charge separation-recombination model by several groups, which facilitates the understanding of energy barrier34,35,36. Building on these pioneering studies, we recently proposed a mechanism for the trap-induced PersL in host-guest molecule systems (Fig. 1a)37. The mechanism includes four steps: (i) excitation, (ii) charge separation and trapping, (iii) detrapping, and (iv) recombination. Specifically, under irradiation, charge carriers from guest molecules are excited (i), then delocalize and transfer to other guest molecules via the hosts, generating metastable trap states as radical cations/anions (ii). This route avoid direct and rapid radiative transitions of the excited charge carriers. The trapped charge carriers are detrapped gradually under external stimulation (iii) and subsequently recombine with the original guest molecule (luminescent center) give trap-induced PersL (iv). The detrapping step is essentially a thermally activated process, with the detrapping energy (i.e., trap depth) playing a key role in shaping the luminescence dynamics. Our analyses suggest that the detrapping step may occur between the lowest unoccupied molecular orbitals (LUMO) of the host and guest radical anions (\({{{{\rm{H}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) and \({{{{\rm{G}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\)). Consequently, by changing the species of guest molecules, we are able to regulate the trap depth (Fig. 1b). However, since the guests also serve as recombination centers in the host-guest systems, this strategy inevitably leads to variation in emission wavelength, and thus a trade-off between trap depth and emission wavelength becomes necessary in applications38,39,40.

a Energy level diagram and electron transition route for trap-induced PersL. b Trap depth regulation by guest energy-level engineering. c Trap depth regulation by host energy-level engineering. d Chemical structure of TADF molecules as guest (left) and host molecules (right). One guest and five host molecules were studied, including 2,7-bis(4-(9H-carbazol-9-yl)phenyl)−1,8-naphthyridine (CPND), 3,3’-(5’-(3-(pyridin-3-yl)phenyl)-[1,1’:3’,1”-terphenyl]−3,3”-diyl)dipyridine (TMPyPB), 2,4,6-tri([1,1’-biphenyl]−3-yl)−1,3,5-triazine (T2T), dibenzo[b,d]furan-2,8-diylbis(diphenylphosphine oxide) (PPF), diphenyl(4-(triphenylsilyl)phenyl)phosphine oxide (TSPO1) and bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO). Abbreviations and labels: host (H), guest (G), recombination center (R), trap (T), electrons (black balls), LUMO of the guest (GL), HOMO of the guest (GH), LUMO of the host radical anion (\({{{{\rm{H}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\)), LUMO of the guest radical anion (\({{{{\rm{G}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\)), and trap depth (ε).
In this work, we introduce a strategy, namely host energy-level engineering, to regulate the trap depth in organic PersL materials. The guest molecule remains unchanged, but different host materials are selected based on their LUMO energy and molecular structure (Fig. 1c). Through this approach, we achieve precise control of trap depth in the range of 0.38 to 0.72 eV, while have almost the same emission wavelength in the blue-light region. The construction of traps with controllable depth in organic systems enables ultralong organic PersL lasting for 27 h and efficient energy storage for 14 days at room temperature (RT). Moreover, by incorporating these trap-containing host-guest materials into OLEDs, we develop a pixel-programmable array device with excellent energy storage capabilities, highlighting its potential in information storage applications. The trap depth regulation strategy by host energy-level engineering may pave the way for the development of high-performance organic light-emitting materials and devices, fostering advancements in next-generation optoelectronics.
Results
Host energy-level engineering
The CPND molecule, a TADF-type emitter with a donor-phenyl-acceptor-phenyl-donor (D-Ph-A-Ph-D) framework, was prepared as the guest molecule (Supplementary Figs. 1–3). Incorporating Ph-bridges into the intramolecular-charge-transfer molecules increases the spatial separation between the donor and acceptor units, which in turn narrows the energy difference between the S1 and T1 states (ΔEST)41. Low-temperature (77 K) fluorescence and phosphorescence measurements indicate that CPND exhibits a small ΔEST of 0.28 eV, supporting the occurrence of efficient RISC at room temperature (Supplementary Figs. 14a, b). Moreover, the delayed fluorescence intensity grows steadily with increasing temperature from 200 to 325 K, further confirming that CPND displays characteristic TADF behavior (Supplementary Fig. 14c). In addition, we calculated the reverse intersystem crossing rate (kRISC) of CPND (Supplementary Table 1) as high as 4.4 × 105 s−1 in the family of TADF materials42,43,44.
A series of electron-transporting materials, including TMPyPB, T2T, PPF, TSPO1, and DPEPO were selected as hosts, and their energy levels were investigated through density functional theory (DFT) calculations and verified by cyclic voltammetry measurements (Fig. 1d and Supplementary Table 3, Supplementary Data 1 and Supplementary Fig. 15). They were blended with CPND to prepare host-guest composites by using a melt-quenching method (Methods Section)45,46. The optimal guest/host ratio of these composites was determined as 1 wt% based on PersL intensity measurements (Supplementary Figs. 16, 17). As expected, all the samples exhibited intense long-lived luminescence that lasted for hours after ultra-violet (UV) irradiation (Fig. 2a, Supplementary Fig. 18, and Supplementary Videos 1–5). The emission intensity followed a power-law decay over time, and the intensity and decay rates varied in the five samples. These results indicate that the long-lived luminescence in the host-guest materials could be attributed to an energy barrier-dominated process (i.e., trap-induced PersL).

a PersL decay curves of the samples after UV irradiation (365 nm) for 3 min at RT. b TL glow curves of CPND@DPEPO with different heating rates of 2, 5, 10, 20, and 50 K min–1. The sample was irradiated by UV light for 3 min at 100 K prior to the TL measurements. c, Estimation of trap depths with the Randall-Wilkins model. The trap depth (ε) was derived by plotting ln(Tm2/β) against 1/(kB·Tm). d PersL spectra. The spectra were acquired during TL measurements at the peak temperature. e Absorption (Abs., dotted curves) and fluorescence spectra (Flu., solid curves). f Photographs of PersL in the dark (sample size: 2 × 2 × 0.1 cm³). The CPND@PPF, CPND@TSPO1, and CPND@DPEPO were irradiated with UV light at RT, kept for 10 min, and then heated to 350 K. The temperature curve is given on the top of the panel. Note that the color of the photographs with high PersL brightness may deviate from the original blue color due to overexposure of the camera sensor. Camera exposure settings: f/1.2, 5 s, ISO 80000. g Orbital configurations and energy levels of the radical anions CPND•−, TMPyPB•−, T2T•−, PPF•−, TSPO1•−, and DPEPO•−. The frontier molecular orbitals (FMO) energies were obtained from DFT calculations. The LUMO of the radical anions exhibited two sub-orbitals (α and β) due to two electron spin orientations in an open-shell system. h Comparison of the trap depths obtained from the TL measurements (top) and the energy gaps between \({{{{\rm{G}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) and \({{{{\rm{H}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) derived from the DFT calculations (bottom).
Intensity-time-wavelength thermoluminescence (TL) technology was used to evaluate the contribution of the possible traps in the host-guest composites. All samples showed TL glow curves with peaks in the range of 250 to 400 K (Supplementary Figs. 19–23). Using CPND@DPEPO as a representative case, we found that the maximum intensity temperature (Tm) of the TL glow curves increased from 310 to 350 K when the heating rate (β) was raised from 2 to 50 K min⁻¹ (Fig. 2b). The trap depth (ε) was estimated using the Randall-Wilkins model47:
where kB is the Boltzmann constant and s (s–1) is the frequency factor. The trap depth, derived by plotting ln(Tm2/β) against 1/(kB·Tm), was calculated to be 0.38 ± 0.01 eV for CPND@TMPyPB, 0.41 ± 0.01 eV for CPND@T2T, 0.54 ± 0.01 eV for CPND@PPF, 0.70 ± 0.01 eV for CPND@TSPO1, and 0.72 ± 0.02 eV for CPND@DPEPO, respectively (Fig. 2c and Supplementary Figs. 19-23). Conversely, all five samples displayed nearly identical PersL spectra, characterized by deep-blue emission centered at around 460 nm (Fig. 2d, Supplementary Fig. 24 and Supplementary Table 2). These observations indicate that adjusting the host matrix allows modulation of the trap depth in the organic systems, while the emission color remains almost unchanged.
The fluorescence spectra of the host-guest composites closely resembled their PersL spectra (Fig. 2e). Since the fluorescence spectra (also PersL spectra) were independent of host materials, the deep-blue emission could be reasonably attributed to the guest molecule CPND. The deep-blue emission of the host-guest composites at approximately 460 nm fell between the 424 nm emission observed from CPND monomers dispersed in toluene and the 486 nm emission from CPND aggregates in a pure film (Supplementary Figs. 25, 26). This suggests that the guest molecules might exist as multi-molecule aggregates within the composites (1 wt%), leading to emission wavelength shift relative to the monomers.
Upon determining the trap depth, we can understand the differences in the PersL properties. As the trap depth increased from CPND@TMPyPB to CPND@DPEPO, more electrons were stored after UV irradiation at RT, resulting in increase of PersL intensity and deceleration of emission decay. Interestingly, when we irradiated the samples at RT and subsequently heated them to 350 K (Fig. 2f), the TL intensity of CPND@PPF (with shallower traps) increased more rapidly and surpassed that of CPND@DPEPO (with deeper traps) in the range of 300–330 K. This occurrs because the electrons captured in the shallow traps of CPND@PPF are rapidly released under intensified thermal stimulation, thus causing a notable increase in intensity.
The orbital configurations and energy levels of the guest and host molecules were analyzed using DFT calculations at the B3LYP/def2-SVP level of theory. Detailed data regarding neutral molecules, radical cations, and radical anions are presented in Supplementary Table 3. In addition, we employed time-dependent density functional theory (TD-DFT) calculations to identify the possible interaction regions between the host and guest molecules (Supplementary Fig. 27 and Supplementary Data 1). Given that the electron capture and release likely occur between guest and host molecules, the energy barrier (i.e., trap depth) may be associated with the energy differences of LUMO between the guest and host radical anions (neutral molecules that acquire an electron). As shown in Fig. 2g, the LUMO of the guest radical anion (\({{{{\rm{G}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\)) exhibits two sub-orbitals (α and β) due to two electron spin orientations in an open-shell system, located at 0.96 and 0.96 eV, respectively. The LUMO of the host radical anion (\({{{{\rm{H}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\)) was situated at higher energy levels. From TMPyPB•− to T2T•−, PPF•−, TSPO1•−, and DPEPO•−, the energy gap between \({{{{\rm{G}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) and \({{{{\rm{H}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) (based on the average energies of the α and β sub-orbitals) gradually increased from 0.39 to 0.71 eV (Supplementary Table 4). Notably, the energy gap between \({{{{\rm{G}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) and \({{{{\rm{H}}}}}_{{{{\rm{L}}}}}^{{{{\rm{\bullet }}}}-}\) derived from the DFT calculations shows a high degree of consistency with the trap depths obtained from the TL measurements, with a deviation of approximately 10% (Fig. 2h and Supplementary Table 4). These results provide molecular orbital energy evidence that the selection of different hosts may offer an effective way to tailor the trap depth of organic PersL materials. The findings may promote the discovery of high-performance organic PersL materials with specific trap depths for advanced optoelectronic applications.
Photophysical properties
We focused on the photophysical properties of the CPND@DPEPO, as it possessed the largest trap depth (~0.72 eV) among the host-guest composites studied. Remarkably, the CPND@DPEPO showed ultralong PersL after UV irradiation, which lasted for over 27 h at RT, surpassing most previously reported organic long-lived luminescent materials (Fig. 3a). Meanwhile, we measured the PersL spectra from the beginning up to 16 h (Fig. 3b and Supplementary Fig. 28). It reveals that the PersL spectra recorded at RT remained unchanged throughout the entire period, consistently showing a blue emission peaked at ~460 nm. This indicates that during the long-term PersL, photon emissions always came from the same recombination center. In addition, TL spectra, recorded from 100 to 375 K, were initially dominated with an emission maximum of 515 nm (due to the T1-S0 transition) at 200 K, and they progressively shifted to 460 nm (S1-S0 transition) at higher temperatures (Fig. 3c and Supplementary Fig. 29). This result supports that CPDN with the TADF property was the only recombination center for PersL, which determines the final pathway of the de-trapped electrons returning to the S0 state.

a PersL decay curve at RT. The sample was excited by UV light (365 nm) for 3 min before measurement. The noise signal is given for comparison (grey dots). b Time-resolved PersL spectra (i, delayed time from 10 min to 16 h; ii, delayed time at 60 min). The PersL spectra were measured at RT after UV irradiation for 3 min. c Temperature-dependent TL spectra (i, temperature from 100 to 375 K; ii, temperature at 200 and 350 K). The heating rate was 50 K min−1. d ESR spectra recorded after UV irradiation at 110 and 300 K (top) and after 365 nm excitation at different storage time (bottom). e PersL decay curves under NIR photo-stimulation (980 nm) in continuous and pulsed output modes (the latter repeating on and off every 30 s). f PersL under thermal stimulation. The CPND@DPEPO was excited with UV light, kept at RT for 10 min, and heated to 300, 325, 350 and 375 K with a heating rate of 50 K min−1. g Photographs of the CPND@DPEPO showing PersL decay (top) and thermal-stimulated process (bottom, TL). Sample size: 2 × 2 × 0.1 cm³. Top: the photographs from left to right were taken with a delay time from 10 s to 10 h. Bottom: the photographs show the sample heated from RT to 350 K, and kept at 350 K for 20 min. Note that the color of the photographs with high PersL brightness may deviate from the original blue color due to overexposure of the camera sensor. Camera exposure settings: f/1.2, 1/100 s, ISO 40 for UV on; f/1.2, 5 s, ISO 80000 for PersL decay and TL.
Electron spin resonance (ESR) spectra of the CPND@DPEPO were examined. As depicted in Fig. 3d, a distinct ESR signal at g-factor of ~2.0031 emerged after UV irradiation. The ESR intensity measured at 110 K was greater than that at 300 K. Moreover, the intensity gradually weakened over time at RT, but it remained detectable even after 37 days. The ESR signals indicate the existence of species with an unpaired electron, thereby verifying the formation of radical anions or radical cations upon UV irradiation. At a lower temperature, more radical anions were stored, resulting in higher ESR signals. The long-term retention of ESR signals was probably attributed to the deep traps in CPND@DPEPO.
Furthermore, the PersL property of CPND@DPEPO under photo-stimulation or thermal stimulation was investigated. As illustrated in Fig. 3e, following its natural decay at room temperature, the sample shows a pronounced increase in PersL intensity (by ~48 times at the onset) when irradiated with near-infrared light (NIR, 980 nm). Meanwhile, the PersL decay process becomes noticeably accelerated. Furthermore, by employing a pulsed NIR source that alternates between on and off states every 30 s, the PersL output can be periodically and reversibly modulated. These results indicate that the electrons stored in the deep traps of CPND@DPEPO could be accelerated to release through photo-stimulation, which enables controllable photon emissions. In addition, we recorded the PersL at RT (291 K) and then heated the sample to higher temperatures. The PersL intensity increased by 1.5, 6.2, 14.3, and 16.5 times as the temperature was raised to 300, 325, 350, and 375 K, respectively (Fig. 3f). In addition, photographic images of the CPND@DPEPO showing PersL and thermal-stimulated emission were acquired (Fig. 3g and Supplementary Video 1) and the spectral signals retained within CPND@DPEPO film remained detectable via TL measurements even after a storage period of 14 days (Supplementary Fig. 30). Obviously, owing to the deep traps, the CPND@DPEPO demonstrates ultralong PersL and energy storage capability, presenting new opportunities for semiconductor applications.
To verify the universality of the host energy-level engineering strategy, we prepared another guest molecule 2,7-bis(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)−1,8-naphthyridine (tCPND) by incorporating tert-butyl functional groups into the carbazole segments of CPND (Supplementary Figs. 4–12). The basic photophysical properties of tCPND are given in Supplementary Fig. 31. The host-guest composites using tCPND as guest molecule exhibited blue PersL lasting for several hours with a consistent emission peak at ~475 nm (Supplementary Fig. 32). Changing the host material from PPF to TSPO1 and DPEPO, the peak temperature of the TL glow curves shifted to higher temperatures and the estimated trap depths increased from 0.62 ± 0.02 eV (tCPND@PPF) to 0.74 ± 0.01 eV (tCPND@TSPO1) and 0.76 ± 0.01 eV (tCPND@DPEPO, Supplementary Figs. 33–35). Benefited from the deep traps, tCPND@DPEPO showed excellent PersL and energy storage capability after UV irradiation (Supplementary Fig. 36 and Supplementary Video 6).
Application of information storage device
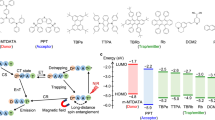
Considering the excellent electron storage capability of the host-guest composites, we applied them to fabricate an information storage device. The storage device has a typical OLED configuration (Fig. 4a), which consists of ITO/ 1,4,5,8,9,11-hexaazatriphenylene hexacarbonitrile (HAT-CN, 10 nm, hole injecting layer)/ 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC, 40 nm, hole transporting layer)/ tris(4-carbazoyl-9-ylphenyl)amine (TCTA, 10 nm, electron blocking layer)/ 5–15 wt% CPND@DPEPO (25 nm, emission layer (EML))/ TMPyPB (40 nm, electron transporting layer)/ Liq (1 nm, electron injecting layer)/ Al (120 nm). The electroluminescence (EL) performances of the device using different CPND@DPEPO as the EML were fully examined (Supplementary Fig. 37 and Supplementary Table 5). The device with 15 wt% CPND@DPEPO achieved a maximum external quantum efficiency (EQE) of 14.26% with deep-blue EL peaked at ~448 nm. The EQE value of the 15 wt% CPND@DPEPO sits at an intermediate level relative to previously reported organic PersL devices (Supplementary Table 6 and Supplementary Fig. 38). Importantly, the device contained 4 × 4 active array, and was controlled by a multi-channel switch and driven by a direct current (DC) power supply. This enables us to independently manipulate (e.g., to charge) each millimeter-scale pixel of the array (Fig. 4b).

a Configuration of a typical 4 × 4 array device. The size of each pixel is 2 mm × 2 mm. The components of multilayers from top to bottom are given. b Schematic diagram of a multiplexing controller to selectively charge the array. c PersL decay curves of the storage device (15 wt% CPND@DPEPO): EML thickness, from 50 to 250 nm; temperature, RT; driving voltage, 25 V. d PersL decay curves: EML thickness, 250 nm; temperature, RT; driving voltage, from 10 to 30 V. e PersL decay curves: EML thickness of 250 nm; temperature, from 100 K to RT; driving voltage, 25 V. f PersL decay curves with thermal stimulation: EML thickness, 250 nm; start temperature, 200 K; end temperature, 200, 250, 300 or 350 K; heating rate, 50 K min−1; driving voltage, 25 V. The applied temperature curve is given on the top of the panel. g PersL decay curves under repeated electric charging and decay: EML thickness, 250 nm; temperature, RT; driving voltage, 25 V; charging and decay time, 1 and 3 min in each cycle. The applied voltage curve is given on the top of the panel. h Photographs of various devices with different EML materials (noted at the top) after being charged: EML thickness, 250 nm; temperature, RT; driving voltage, 25 V. The devices were charged through a programmable multiplexing controller, exhibiting different display information on the 4 × 4 array devices. Camera exposure settings: f/1.2, 5 s, ISO 80000.
The device showed deep-blue PersL after being charged. The PersL intensity and duration increased as the EML thickness expanded. Impressively, when the EML thickness reached 250 nm, the PersL of the device lasted for ~10 min at RT before the intensity decayed to the noise ratio of the photodetector (Fig. 4c). The EL performance of this device is provided in Supplementary Fig. 39. The PersL properties of the device could be controlled by varying charging voltages. For example, as the driving voltage rose, the PersL intensity increased and it became saturated when the voltage exceeded 25 V (Fig. 4d). Moreover, the PersL could be further improved as the working temperature decreased from RT to a lower temperature. At 100 K, the PersL lasted for more than 30 min (Fig. 4e). This indicates that the PersL in the electrically charged device was temperature-dependent, which was similar to that of the CPND@DPEPO after light irradiation (Supplementary Fig. 40).
To explore the electron storage capacity of the device, we further studied the effect of thermal stimulation on the PersL intensity. After being charged at 200 K, the device was kept at the same temperature and subsequently heated to 250, 300, or 350 K (Fig. 4f). Notably, the PersL showed an clear response to the heat treatment, which reflected that the thermal stimulation promoted the release of the stored electrons. In addition, the repeatability of electron charging and release of the device was evaluated. As shown in Fig. 4g and Supplementary Video 7, when the PersL intensity decayed to a low value, it could be restored to the initial level by electric recharging. The excellent reproducibility indicates its potential in the information storage applications.
In addition to the aforementioned CPND@DPEPO-based device that required a vacuum deposition process to prepare the EML, we could alternatively employ the conventional spin-coating method to fabricate the device. This is due to the fact that the EML materials (e.g. tCPND@DPEPO) exhibit excellent solubility in N,N-dimethylformamide (DMF). A typical device configuration includes ITO/ poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS, 40 nm, hole injection layer)/ 1-5 mg ml-1 tCPND@DPEPO (10−30 nm)/ TmPyPB (40 nm)/ Liq (1 nm)/ Al (120 nm) (Supplementary Fig. 41a, b). The PersL property and electron storage capability, together with the effects of EML thickness, driving voltage, and working temperature on them, were analogous to those in the CPND@DPEPO-based device (Supplementary Fig. 41c–e). Also, the thermal stimulation effect and the reproducibility of electron charging were verified (Supplementary Fig. 41f, g).
Finally, we demonstrated the selective charging (information write-in) of the pixels on the 4 × 4 array device through a programmable multiplexing controller. As an example, when 9 out of 16 pixels were charged, some electrons were stored in the array, and the information corresponding to the letter “P” could be continuously read out through blue light emission (Fig. 4h). Broadly, we could select various host-guest composites as the EML materials of the devices to control the emission color. As we modified the guest molecule from CPND to 10-(4-(4,6-diphenyl−1,3,5-triazin-2-yl)phenyl)-9,9-dimethyl-9,10-dihydroacridine (DT), 4,4”-Di-10H-phenoxazin-10-yl[1,1’:2’,1”-terphenyl]-4’,5’-dicarbonitrile (Px-VPN), 2,3,5,6-tetrakis(3,6-di-tert-butylcarbazol-9-yl)-1,4-dicyanobenzene (4CzTPN-Bu), and 2-(2,6-diisopropylphenyl)-5,8-bis(9,9-dimethylacridin-10(9H)-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (DN)), the EML materials showed varying emissions. By selectively charging the array pixels, we successfully managed to regulate two-dimensional planar information (“P”, “E”, “R”, “S”, and “L”) and emission color (ranging from blue to red) (Fig. 4h and Supplementary Video 8). We believe this type of array device may expand the potential applications of organic PersL materials in multi-dimensional information storage technologies.
Discussion
In summary, we have proposed a host energy-level engineering strategy, demonstrating it to be an effective and versatile approach for modulating organic PersL. This strategy, centered on modifying the host materials within a host-guest molecular system, permits precise control over trap depths while maintaining the emission wavelength. A combination of TL measurements, ESR spectra, and DFT calculations reveals that upon UV light irradiation, an energy barrier attributed to the energy difference between the host and guest radical anions is created. The meta-stable species, which gain or lose electrons, need thermal stimulation or photo-stimulation to return to their original states, thus generating trap-induced PersL. In this sense, the luminescence dynamics of organic PersL is intricately linked to the trap depth and the working temperature. According to the host energy-level engineering, we have achieved a high-performance deep-blue emitter CPND@DPEPO, characterized by a deep trap of ~0.72 eV. This materials exhibits ultralong PersL emission lasting over 27 h and energy storage for 14 days, surpassing most previously reported organic long-lived luminescent materials.
Based on the superior electron storage capability of host-guest composites, we have developed a programmable information storage array device fabricated via vacuum deposition or spin-coating processing. This storage device is extendable to a variety of EML materials, provided they contain traps of a certain depth. By employing various host-guest composites as the EML materials, we have showcased storage devices with multicolor emission spanning from blue to red light. The proposed host energy-level engineering strategy has the potential to enhance our understanding of the mechanisms underlying trap depth modulation and luminescence characteristics. Furthermore, organic PersL materials, due to their controllable emission wavelengths, potential biocompatibility, and excellent processability, also have broad application prospects in fields of biological imaging and night indication. As shown in Supplementary Fig. 42, we utilized the high sensitivity of green emission to the human eye and the hour-level PersL from the DT@DPEPO to demonstrate its application in night indication signs. The 12-h duration meets the basic application duration requirement. However, their stability against moisture and oxygen remains limited, posing considerable challenges for applications in biological systems and complex environments. The introduction of host energy-level engineering lays a foundation for the development of organic PersL materials and is expected to facilitate cross-disciplinary breakthroughs and applications.
Methods
Synthesis of guest and host molecules
The guest molecules CPND and tCPND were synthesized via a palladium-catalyzed Suzuki cross-coupling reaction, with subsequent structural verification conducted through nuclear magnetic resonance (NMR) spectroscopy (Advance III 500 MHz NMR, Bruker), high-resolution mass spectrometry (FTICR MS, Bruker), and elemental composition analysis (Vario EL III, Elementar). The synthetic procedure outlined in Supplementary Note I was systematically validated using these complementary analytical techniques to ensure compound purity and structural integrity. All the host materials (TMPyPB, T2T, PPF, TSPO1 and DPEPO) were purchased from Xi’an Polymer Light Technology Corp and used without purification.
2,7-bis(4-(9H-carbazol-9-yl)phenyl)−1,8-naphthyridine (CPND)
Unless otherwise noted, all reagents used in the experiments were purchased from commercial sources without further purification. 2,7-dichloro-1,8-naphthyridine (ND) (99.52 mg, 0.5 mmol), 9-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-9H-carbazole (461.59 mg, 1.25 mmol), sodium carbonate (138.20 mg, 1.0 mmol), toluene, anhydrous ethanol and distilled water (50 mL, 2:2:1, v/v/v) were added and stirred for 1 h under a nitrogen atmosphere. After that, tetrakis(triphenylphosphine)palladium(0) (115.56 mg, 0.1 mmol) was quickly added and the mixture was heated to 110 °C under a nitrogen atmosphere for 24 h. After cooling to room temperature, the mixture was extracted with CH2Cl2, and the organic layer was washed with sodium chloride water and then dried over anhydrous Na2SO4 and concentrated. The crude product was purified by column chromatography on silica gel (normal hexane/dichloromethane = 2:3, v/v) to give CPND as a white solid (yield = 58%). This compound was further purified by temperature-gradient sublimation under vacuum.
9-(4-bromophenyl)-3,6-di-tert-butyl-9H-carbazole (tCP-1)
A mixture of 3,6-di-tert-butyl-9H-carbazole (2.79 g, 10.0 mmol), sodium tert-butoxide (1.92 g, 20.0 mmol) in 1,4-dioxane (50 mL) were stirred for 30 min at room temperature under a nitrogen atmosphere. After, copper (I) iodide (190.5 mg, 1.0 mmol), 1,10-phenanthroline (114.2 mg, 1.0 mmol), and 4-bromoiodobenzene (3.39 g, 12.0 mmol) were added and the mixture was heated to 100 °C under a nitrogen atmosphere for 36 h. After cooling to room temperature, 100 ml water was slowly added to the solution. The mixture was filtered, and the residue was washed with water and dried with anhydrous sodium sulfate. Then the crude product was purified by column chromatography on silica gel (petroleum ether/dichloromethane = 5:1, v/v) to give tCP-1 as a white solid (yield = 69%).
3,6-di-tert-butyl-9-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-9H-carbazole (tCP-2)
tCP-1 (434.4 mg, 1.0 mmol), bis(pinacolato)diboron (634.9 mg, 2.5 mmol), [1,1’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (Pd(dppf)Cl2, 73.2 mg, 0.1 mmol), and potassium acetate (KOAc, 294.4 mg, 3.0 mmol) were added to a dry three-neck flask under a nitrogen atmosphere. 1,4-dioxane (20 mL) was added into the mixture. Then the solution was stirred and heated to reflux at 100 °C for 12 h. After cooling to room temperature, 100 ml water was slowly added to the solution. The mixture was filtered, and the residue was washed with water and dried with anhydrous sodium sulfate. Then the crude product was purified by column chromatography on silica gel (normal hexane/ethyl acetate = 20:1, v/v) to give tCP-2 as a white solid (yield = 47%).
2,7-bis(4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)-1,8-naphthyridine (tCPND)
The synthesis method is similar to CPND. ND (99.52 mg, 0.5 mmol), 3,6-di-tert-butyl-9-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-9H-carbazole (601.86 mg, 1.25 mmol), sodium carbonate (138.20 mg, 1.0 mmol), toluene, anhydrous ethanol and distilled water (50 mL, 2:2:1, v/v/v) were added and stirred for 1 h under a nitrogen atmosphere. After that, tetrakis(triphenylphosphine)palladium(0) (115.56 mg, 0.1 mmol) was quickly added and the mixture was heated to 110 °C under a nitrogen atmosphere for 24 h. After cooling to room temperature, the mixture was extracted with CH2Cl2, and the organic layer was washed with sodium chloride water and then dried over anhydrous Na2SO4 and concentrated. The crude product was purified by column chromatography on silica gel (normal hexane/dichloromethane = 2:3, v/v) to give tCPND as a white solid (yield = 34%). This compound was further purified by temperature-gradient sublimation under vacuum.
Synthesis of CPND@DPEPO composites
A mixture of CPND and DPEPO was heated at 350 °C on a quartz substrate (2 × 2 × 0.1 cm³) within a controlled atmosphere glove box (O₂ and H₂O < 0.01 ppm). Following mechanical agitation, the homogenized mixture was rapidly quenched to ambient temperature. Since triplet excitons are easily quenched by moisture and oxygen, all samples must be encapsulated in a nitrogen environment using a UV-curable epoxy adhesive and a protective glass cover to isolate them from water and oxygen (Supplementary Fig. 43). The resulting samples were subsequently subjected to further tests under ambient atmospheric conditions at 18 °C. Identical fabrication protocols were applied to all host-guest composites to ensure procedural consistency.
Fabrication of devices by vacuum deposition process
ITO-coated glass substrates (Suzhou Fangsheng Optoelectronics) were cleaned with detergent, distilled water, acetone, isopropanol, dried in drying oven at 80 °C, and treated with plasma cleaner (Diener Femto) for 30 min before vacuuming thermal deposition. Thermal evaporation processes were initiated upon a base pressure of 6.6 × 10⁻⁴ Pa was achieved in the deposition chamber. Organic functional layers were sequentially deposited at controlled rates: 1–1.5 Å s−1 for host materials, 0.1 Å s−1 for Liq, and 3 Å s−1 for Al cathodes. Completed OLED architectures were used to fabricate electrically chargeable devices under inert atmosphere. The key parameters including current-voltage characteristics, luminescent intensity, and spectral radiant flux were systematically measured using a Keithley 2400 source meter and an absolute EQE measurement system (Hamamatsu C9920-12 equipped with Hamamatsu C10027-01 Photonic multichannel analyzer PMA-12). The preparation of the 4 × 4 array is consistent with the method of vacuum deposition mentioned above and the differences of them lie in the design of the ITO glass substrate and the shadow masks. The fabricated devices were capsulated into a compact sample holder (PR-SH-CSH1) to avoid the influence of oxygen and moisture. The array was driven by a DC power supply (ETM-605P) and each pixel could be independently manipulated with a multiplexing controller (PR-MPX-16C) for electrical information write-in. All the equipment was obtained from PURI Materials, Shenzhen.
Fabrication of devices by solution process
PEDOT:PSS solutions were filtered through poly(tetrafluoroethylene) syringe membranes (0.45 μm) and deposited via a spin-coating (4000 rpm, 30 s) method onto pre-cleaned ITO substrates, followed by thermal annealing at 140 °C for 15 min under ambient atmospheric conditions. A tCPND@DPEPO active layer (1–5 mg mL⁻¹ in DMF) was subsequently applied by spin-coating (3000 rpm, 60 s) onto the PEDOT:PSS layer and heated at 100 °C for 15 min. Finally, the substrates were transferred to an interconnected high-vacuum deposition system (base pressure 6.6 × 10−4 Pa) for thermal evaporation of DPEPO (40 nm) and Liq (1 nm)/ Al (120 nm) through shadow masks to complete the device.
Trap-induced Persl measurements
A THMS600E temperature stage (Linkam Scientific Instruments) was used to control the temperature of host-guest composites and the array devices. External excitation was implemented through UV irradiation (host-guest composite samples) or electric charging (devices). After ceasing the excitation source, a filter-attached PMT (R928P, Hamamatsu Photonics) and a luminance meter (LM-5, Evenfine) simultaneously monitored the PersL intensity. A multichannel spectrometer (QE-Pro, Ocean Optics) was utilized to acquire emission spectra. All instrumentation was operated through LabVIEW-controlled interfaces ensuring temporal synchronization and data acquisition precision.
Other photophysical properties measurements
Absorption spectra were acquired with a UV/visible/NIR spectrophotometer (UV−3600Plus, Shimadzu). Emission spectra were collected with fluorescence spectrometers (FLS980, Edinburgh or QE-Pro, Ocean Optics). ESR analysis was conducted on the X-band EPR spectrometer (EMXplus-9.5/12, Bruker). Photographs and videos of the samples and devices were taken with a digital camera (α7SIII, SONY).
DFT calculations
DFT calculations were executed with Gaussian 09. Geometries of all molecular systems were optimized using the hybrid B3LYP functional and def2-SVP basis set without symmetry constraints. The program optimizes the molecular geometry by iteratively adjusting atomic coordinates to locate the local minimum energy configuration at the ground state (S0). Subsequently, the FMOs based on the S0 geometry were depicted for the closed-shell and open-shell systems. HOMO/LUMO energies and orbital isosurfaces were visualized using Gauss View 6.0.
Data availability
Source data are provided with this paper. The source data that support the findings of this study have been deposited in the figshare repository (https://doi.org/10.6084/m9.figshare.30508355). Any requests on data and experiments can be also available from the corresponding authors. Source data are provided with this paper.
References
Liu, Y., Li, C., Ren, Z., Yan, S. & Bryce, M. R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 3, 18020 (2018).
Cai, X. & Su, S. Marching toward highly efficient, pure-blue, and stable thermally activated delayed fluorescent organic light-emitting diodes. Adv. Funct. Mater. 28, 1802558 (2018).
Wang, X. et al. Organic phosphors with bright triplet excitons for efficient X-ray-excited luminescence. Nat. Photonics 15, 187–192 (2021).
Abdi-Jalebi, M. et al. Maximizing and stabilizing luminescence from halide perovskites with potassium passivation. Nature 555, 497–501 (2018).
Wang, Z. et al. Color-tunable polymeric long-persistent luminescence based on polyphosphazenes. Adv. Mater. 32, 1907355 (2020).
Nie, F. & Yan, D. Bio-sourced flexible supramolecular glasses for dynamic and full-color phosphorescence. Nat. Commun. 15, 9491 (2024).
Chen, T., Ma, Y. & Yan, D. Single-component 0D metal-organic halides with color-variable long-afterglow toward multi-level information security and white-light LED. Adv. Funct. Mater. 33, 2214962 (2023).
Xing, C., Qi, Z., Ma, Y., Yan, D. & Fang, W. Dynamic ultralong phosphorescence and optical waveguiding switches in silver-organic complex via reversible single-crystal-to-single-crystal conversion. Angew. Chem.-Int. Edit. 64, e202502782 (2025).
Gu, F. et al. Visualization of photocuring and 4D printing with real-time phosphorescence. Nat. Commun. 16, 4173 (2025).
Wang, T. et al. Thermochromic aggregation-induced dual phosphorescence via temperature-dependent sp3-linked donor-acceptor electronic coupling. Nat. Commun. 12, 1364 (2021).
Tan, S., Jinnai, K., Kabe, R. & Adachi, C. Long-persistent luminescence from an exciplex-based organic light-emitting diode. Adv. Mater. 33, 2008844 (2021).
Dai, X. et al. Solution-processed, high-performance light-emitting diodes based on quantum dots. Nature 515, 96–99 (2014).
Yang, Z. et al. Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 46, 915–1016 (2017).
Jablonski, A. Efficiency of anti-Stokes fluorescence in dyes. Nature 131, 839–840 (1933).
Lower, S. K. & El-Sayed, M. A. The triplet state and molecular electronic processes in organic molecules. Chem. Rev. 66, 199–241 (1966).
Ju, H. et al. Polymerization-induced crystallization of dopant molecules: An efficient strategy for room-temperature phosphorescence of hydrogels. J. Am. Chem. Soc. 145, 3763–3773 (2023).
Ma, X., Wang, J. & Tian, H. Assembling-induced emission: an efficient approach for amorphous metal-free organic emitting materials with room-temperature phosphorescence. Acc. Chem. Res. 52, 738–748 (2019).
Wang, J. et al. A facile strategy for realizing room temperature phosphorescence and single molecule white light emission. Nat. Commun. 9, 2963 (2018).
Buck, J. T. et al. Spin-allowed transitions control the formation of triplet excited states in orthogonal donor-acceptor dyads. Chem 5, 138–155 (2019).
Parker C. A. and Hatchard C. G. Delayed fluorescence from solutions of anthracene and phenanthrene. Proc. R. Soc. Lond. A 269, 574–584 (1962).
Blasse, G. & McMillin, D. R. On the luminescence of bis (triphenylphosphine) phenanthroline copper (I). Chem. Phys. Lett. 70, 1–3 (1980).
Berberan-Santos, M. N. & Garcia, J. M. M. Unusually strong delayed fluorescence of C70. J. Am. Chem. Soc. 118, 9391–9394 (1996).
Zhao, W., He, Z. & Tang, B. Z. Room-temperature phosphorescence from organic aggregates. Nat. Rev. Mater. 5, 869–885 (2020).
An, Z. et al. Stabilizing triplet excited states for ultralong organic phosphorescence. Nat. Mater. 14, 685–690 (2015).
Bryden, M. A. & Zysman-Colman, E. Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 50, 7587–7680 (2021).
Ni, F., Huang, Y., Qiu, L. & Yang, C. Synthetic progress of organic thermally activated delayed fluorescence emitters via C-H activation and functionalization. Chem. Soc. Rev. 53, 5904–5955 (2024).
Liu, W. et al. High-efficiency stretchable light-emitting polymers from thermally activated delayed fluorescence. Nat. Mater. 22, 737–745 (2023).
Huang, T. et al. Enhancing the efficiency and stability of blue thermally activated delayed fluorescence emitters by perdeuteration. Nat. Photon. 18, 516–523 (2024).
Uoyama, H., Goushi, K., Shizu, K., Nomura, H. & Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012).
Xu, J. & Tanabe, S. Persistent luminescence instead of phosphorescence: History, mechanism, and perspective. J. Lumines. 205, 581–620 (2019).
Kabe, R. & Adachi, C. Organic long persistent luminescence. Nature 550, 384–387 (2017).
Li, W. et al. Organic long-persistent luminescence from a thermally activated delayed fluorescence compound. Adv. Mater. 32, 2003911 (2020).
Hamill, W. H. Debye-edwards electron recombination kinetics. J. Chem. Phys. 71, 140–142 (1979).
Ou, X. et al. High-resolution X-ray luminescence extension imaging. Nature 590, 410–415 (2021).
Chen, C. et al. Carbazole isomers induce ultralong organic phosphorescence. Nat. Mater. 20, 175–180 (2021).
Wang, G. et al. Dual-mechanism design strategy for high-efficiency and long-lived organic afterglow materials. J. Am. Chem. Soc. 146, 24871–24883 (2024).
Lin, C. et al. Charge trapping for controllable persistent luminescence in organics. Nat. Photonics 18, 350–356 (2024).
Zhuang, Y. et al. Trap depth engineering of SrSi2O2N2:Ln2+,Ln3+ (Ln2+ = Yb, Eu; Ln3+ = Dy, Ho, Er) persistent luminescence materials for information storage applications. ACS Appl. Mater. Interfaces 10, 1854–1864 (2018).
Liu, P., Zhang, Y., Li, B., Han, L. & Xu, Y. Trap depth engineering in MgGa2O4: Bi3+ for muticolor dynamic anti-counterfeiting, encryption and optical temperature sensing applications. Chem. Eng. J. 437, 135389 (2022).
Wang, L. et al. Engineering trap distribution to achieve multicolor persistent and photostimulated luminescence from ultraviolet to near-infrared II. Matter 6, 4261–4273 (2023).
Shizu, K. et al. Strategy for designing electron donors for thermally activated delayed fluorescence emitters. J. Phys. Chem. C 119, 1291–1297 (2015).
Cui, L.-S. et al. Fast spin-flip enables efficient and stable organic electroluminescence from charge-transfer states. Nat. Photonics 14, 636–642 (2020).
Liu, Y. et al. Space-confined donor-acceptor strategy enables fast spin-flip of multiple resonance emitters for suppressing efficiency roll-off. Angew. Chem.-Int. Edit. 61, e202210210 (2022).
Jin, J. et al. Synergetic modulation of steric hindrance and excited state for anti-quenching and fast spin-flip multi-resonance thermally activated delayed fluorophore. Angew. Chem.-Int. Edit. 63, e202401120 (2024).
Gao, R., Kodaimati, M. S. & Yan, D. Recent advances in persistent luminescence based on molecular hybrid materials. Chem. Soc. Rev. 50, 5564–5589 (2021).
Chen, T. & Yan, D. Long-persistent luminescence: The role of charge trap. Sci. Bull. 69, 1806–1808 (2024).
Bos, A. Thermoluminescence as a research tool to investigate luminescence mechanisms. Materials 10, 1357 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 52172156, 12474412), the Natural Science Foundation of Fujian Province (No. 2023J06005), the Natural Science Foundation of Guangdong Province (No. 2024A1515011197), and the Fundamental Research Funds for the Central Universities (No. 20720240057).
Author information
Authors and Affiliations
Contributions
C.Z., C.L., U.J., Y.Z., and R.-J.X. conceived the experiments. C.Z., C.L., R.Y., Y.W., Z.W., Y.L., W.D., and Y.Z. were primarily responsible for the experiments. C.Z. and C.L. contributed to DFT calculations. C.Z., C.L., J.U., Y.Z., and R.-J.X. prepared the paper. All authors contributed to the data analyses.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mei Pan and Dongpeng Yan for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhan, C., Lin, C., Yang, R. et al. Achieving trap-depth-tunable organic persistent luminescence through host energy-level engineering. Nat Commun 17, 824 (2026). https://doi.org/10.1038/s41467-025-67529-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67529-3
