
Abstract
The reversible interconversion between spiropyran and merocyanine constitutes one of the most intensively investigated photochromic systems, enabling diverse applications in stimuli-responsive materials and optical imaging. Here, we report pyridine-substituted merocyanines (PMCs) as a class of photochromic molecules. Upon visible light irradiation, these compounds undergo an uncommon photoinduced C-N bond formation to yield spiroindolizine (SIZ) structures, accompanied by the color fading. The SIZs can thermally revert to the colored PMC form. This photochromism is marked by rapid response, high photo-switching ratio, robust water compatibility, and fatigue resistance. Furthermore, we have synthesized quinoline-substituted merocyanines and found that they can be bidirectionally switched by 660 nm and 500 nm light to form SIZ and PMC structures. Finally, we showed the photochromism of these molecules in solid matrices and demonstrated a light-controlled sequential switching function, highlighting their potential for advanced photonic applications.
Similar content being viewed by others
Introduction
Photochromic molecules represent a versatile class of smart materials capable of reversibly altering color and physicochemical properties upon light exposure. Owing to their distinctive photo-responsive characteristics, they have been applied across various fields of chemical and biological research, including photo-patterning, activity modulation and fluorescence switching1,2,3,4,5,6. To date, significant progress in photochromic molecules research has led to the development of diverse organic molecular photoswitches, such as azobenzenes7,8, spiropyrans9,10, diarylethenes11, indigoids12, hydrazones13,14 and Stenhouse compounds15,16,17. They can be classified into two main categories according to their structural changes: Z-E isomerization (e.g., azobenzene, indigoid) and covalent bond formation/cleavage (e.g., spiropyran, diarylethene, Stenhouse compound). These systems exhibit distinct advantages and have been applied in various fields. For instance, azobenzenes possess simple and diverse chemical structures, making them widely used in supramolecular chemistry and light-controlled biological regulation7,8. Spiropyrans undergo more pronounced structural changes upon light stimulation, thereby leading to higher contrast on spectral and physicochemical properties10. In addition, the isomerization of spiropyrans can be regulated by pH and metal ions, enabling their use in the design of complicated molecular logic gates9. Diarylethenes, on the other hand, exhibit excellent fatigue resistance and thermal bistability, which makes them highly suitable for multiple switching cycles and particularly the applications in molecular electronics and photochromic devices11. However, these molecules still exhibit certain limitations, such as relying predominantly on ultraviolet (UV) light for photoisomerization, insufficient water solubility, or ineffective switching and poor fatigue resistance in aqueous media. Thus, the development of additional photochromic molecules continues to be of substantial scientific and practical significance. Ideally, the desirable photochromic molecules should feature facile synthesis, high switching ratio, responsiveness to visible light, robust fatigue resistance and broad-spectrum matrix (aqueous and solid) compatibility.
From the perspective of chemical transformations, spiropyrans undergo reversible C-O bond cleavage to form merocyanine whereas diarylethenes rely on reversible C-C bond transition in the photocyclization processes (Fig. 1A). Both systems have been extensively explored as prototypical molecular photoswitches. In contrast, photoinduced reversible C-N bond transformations remain exceedingly rare (Fig. 1B), and most of these examples have only demonstrated photochromic behavior in organic solutions18,19,20,21,22,23,24,25. The discovery of additional photoinduced C-N bond transformations can expand the structural diversity of photoswitches.

A Photochromic reaction of spiropyran and diarylethene. B Reported examples of photochromism involving reversible C-N bond. C Structures of PMCs and their photoisomerization mechanism. D Absorption spectra of PMC1-5 in water before and after irradiation with white light-emitting diode (LED) for 10 s.
In this work, we report a distinct class of photochromic molecules featuring reversible C-N bond formation (Fig. 1C). These compounds adopt a pyridyl-substituted cyanine (PMC) scaffold, in which light irradiation induces the pyridyl nitrogen to attack the C2 atom of the indolium moiety, leading to a cyclization reaction and formation of a spiroindolizine (SIZ) structure, accompanied by a drastic spectral variation. This transformation exhibits fast kinetics, high switching ratio, excellent fatigue resistance and water compatibility. Furthermore, we also synthesized quinoline-substituted derivatives and characterized their photochromic behavior under two wavelengths. Finally, we demonstrated the photo-responsiveness of PMCs in various solid-state matrices and constructed a light-controlled sequential switch. This study expands the scope of molecular photoswitches and provides a molecular platform for diverse applications.
Results
Pyridine-substituted merocyanines
PMC1-5 were synthesized by coupling 2-formylpyridine analogues to an indolium moiety (Figure S2). Density functional theory (DFT) calculation showed that the amino-substitution significantly decreased the energy gap between HOMO and LUMO of PMC1-3 ( ~ 4.5–4.8 eV, Figure S3) compared to that of PMC4 and PMC5 ( ~ 5.5 eV). Time dependent DFT revealed distinct intramolecular charge transfer (ICT) features during excitation process. For PMC1-3, charge density difference (CDD) on the amino-substituted pyridine is around 43%, implying a significant electron depletion during excitation (S0 → S1). In contrast, CDDs on the pyridine moieties of PMC4 and PMC5 are only −5.9% and 1.4%, respectively (Figure S4). Therefore, the pyridine moieties can not act as the electronic donor in PMC4 and PMC5. Experimentally, PMC1-3 exhibited absorption peaks at 462-530 nm (Fig. 1D) and fluorescence at 566-607 nm; whereas both PMC4 and PMC5 having relatively weak charge transfer effects displayed shorter absorption and fluorescence wavelengths (Table 1 and Figures S5–S9).
Unlike imidazole-biradicals and heterocycle-substituted diarylethenes which only showed the photochromic property in organic solvent, PMCs displayed photochromic behaviors in water and rapidly faded upon irradiation with white LED (Fig. 1D), generating blue-shifted absorption peaks at 280-419 nm. This spectroscopic change was consistent with our envision that PMCs may undergo cyclization to form spiroindolizine structures under light irradiation. The photostationary state distributions (PSD) of the SIZ products were all above 95%, indicating almost completed transitions (Table 1). Photoisomerization quantum yields of PMCs were between 5.4% and 12% (Table 1), which is comparable to those of spiropyrans and other photochromic systems (Table S1).
In the absence of light stimulation, the spectra of SIZs gradually returned to those of PMCs in a temperature-dependent manner, from which we calculated the activation energy between 15–21 kcal/mol (Table 1). Spectral tests in organic solvents also supported PMCs as T-type (thermally reversible type) photochromism (Figures S5–S9). Notably, PMC1-3 exhibited almost complete thermal recovery in all solvents (Figures S5–S7). In contrast, PMC4 showed a weak absorption peak at 372 nm and a stronger peak at 280 nm, suggesting that it can spontaneously convert to SIZ4 in solutions (71% SIZ4 in CH3OH/H2O by HPLC; 47% SIZ4 in CD3Cl by 1H NMR assay, Figure S10). It was noted that PMC4 and PMC5 can only retrieve half of their initial absorbance in water in the first recovery process (Figures S8F and S9F), suggesting their relatively low stability in water (Figure S11). We attributed this to the side reaction of PMC4 and PMC5 without the electron-donating amino groups, because the lack of electron-donating groups (e.g., amino) usually leads to instability26,27.
As mentioned above, the simple molecular structure of PMCs endows their photochromism with good water compatibility, which may be advantageous for various applications in aqueous environments. Then, taking PMC2 as an example, we studied its photochromic property in water. As shown in Fig. 2, under the irritation of 500 nm LED, PMC2 converts to SIZ2 in less than 10 s with PSD of 99.4% and the photoisomerization quantum yield is 8.4%. Then, SIZ2 fully returns to PMC2 (98.9%) at room temperature after about 500 min (Fig. 2A–C). Temperature increase can accelerate this transformation from half-life of 250 min at 25 °C to 12.4 min at 50 °C, with the corresponding rate constants of 0.0076 min−1 and 0.056 min−1 (Fig. 2C). Although irradiation with short wavelengths (310 nm, 365 nm or 405 nm) can accelerate the SIZ-to-PMC conversion, the resulting photostationary state remains far from complete recovery (< 10%) of PMC2 (Figure S12), which may be attributed to the simultaneous absorption of PMC and SIZ at the stimulating wavelength. Considering the structure differences between PMC and SIZ, we measured their partitions in octanol-water system. It was found that 98% of PMC2 existed in water phase (oil-water partition coefficient, logP ~ −1.75), further verifying the good water solubility of PMC2; after irritation, only 36% of SIZ2 existed in water phase (logP ~ 0.25), suggesting a typical photo-induced phase-transfer behavior (Fig. 2E).

A Absorption spectra of PMC2 before (black) and after (red) 500 nm LED irradiation for 10 s, and then thermally recovered (room temperature without LED) for 10 h (blue). B Photo-response kinetic curve of PMC2. Arrow indicates the operation of turning on LED lamp. C Thermal recovery kinetic curves of PMC2 at 25 °C (black) and 50 °C (red). D Cyclic stability of PMC2 in water under repeated light stimulations (500 nm LED for 10 s) and thermal recovery (room temperature for 10 h). E Photographs illustrating the photochromism and concomitant phase transfer of PMC2 upon 500 nm LED irradiation. F Photochromism of PMC2 to SIZ2 in pH 1-12 buffer solution.
Then, we compared the photochromic properties of PMC2 and the most intensively studied spiropyran 1,3,3-trimethylindoline-6-nitrobenzopyran (6-nitro-BIPS) in water. Fatigue resistance is a crucial property for photochromic molecules. We tested the fatigue resistance of 6-nitro-BIPS in water by repeated irradiations at 365 nm and 500 nm. It was found that 6-nitro-BIPS degraded greatly after several cycles (Figure S13), which may be ascribed to the involvement of the triplet state under the UV 365 nm excitation28,29. In contrast, PMC2 exhibited excellent reversibility and fatigue resistance (the absorbance at 517 nm retains above 95% after 10 cycles) in water under repeated irradiation and thermal recovery (Fig. 2D). The possible explanation may be that the photoisomerization of PMC2 is driven by 500 nm light, which possesses lower photon energy and therefore reduces the likelihood of photochemical degradation. Whether this photoreaction involves a triplet-state pathway remains to be clarified. Additionally, previous reports have shown that the ring-opening/closing switching of spiropyrans is strongly influenced by pH and the presence of metal ions30,31,32. However, the photoisomerization of our PMC2 molecule could proceed efficiently within the pH range of 4–10 (Fig. 2F, Figure S14) and was not affected by common metal ions (Figure S15), demonstrating its good resistance to environmental interferences. Even in more acidic media (pH < 4), PMC2 with a slightly blue-shifted absorption peak still exhibited efficient photoisomerization, though strongly basic media (pH > 10) largely decreased the photoisomerization. The above results indicated that PMC represents a robust photochromic system with various advantages over traditional spiropyran systems.
Photochromic mechanism
The structures of pyridine-substituted merocyanines (PMC1, PMC2, PMC3 and PMC5) were unambiguously determined to be a planar molecule through single-crystal X-ray diffraction (Figure S16). We attempted to prepare single crystals of the corresponding SIZs but were unsuccessful. On the other hand, we used DFT calculations to determine the geometries of the SIZ isomers (Figure S3). The optimized structures of SIZs reveal a C-N bond length of ~1.53 Å, which is slightly longer than a typical C-N bond20,23,24.
Further, we employed NMR spectroscopy to investigate the structural information on the PMC2-SIZ2 transformation (Figures S17 and S18). As shown in Fig. 3A, the proton signals at 8.28 ppm (H4, doublet, J = 15.3 Hz) and 7.72 ppm (H5, doublet, J = 15.3 Hz) confirm that the predominant species is the trans-alkenyl merocyanine. These protons shift to 7.35 ppm (H4, d, J = 6.5 Hz) and 6.85 ppm (H5, d, J = 6.4 Hz) upon irradiation, and are assigned as cis-alkenyl hydrogens, indicating the photoinduced trans-cis isomerization of the ethylene bond. Meanwhile, the chemical shift of the C2 (i.e., Cp in Figures S17 and S18) in the indole moiety shifts from 180.7 ppm to 111.4 ppm (Fig. 3B), indicating a transformation from an imine carbon to a saturated spiro-carbon (Cspiro). Notably, in SIZ2, the resonance peaks of indole 1-methylene protons (H10) broaden into very wide peaks (Fig. 3A), possibly suggesting the formation of a chiral center at the spiro carbon33. More definitive evidence is provided by the heteronuclear multiple bond correlation (HMBC) spectra, which typically detects 2J or 3J correlation. In 1H-13C HMBC spectra of SIZ (Fig. 3B), Cspiro displays a correlation with the H1 proton in pyridine. In 1H-15N HMBC spectra (Fig. 3C), a strong coupling appears between the Npyridine and H5 in SIZ2. The emergence of these two correlations strongly supports the formation of the Cspiro-Npyridine bond, enabling 3JHC and 3JHN correlations in SIZ.

A 1H NMR spectral evolution of PMC2 in CD2Cl2 before (up) and after (down) irradiation. B 1H-13C HMBC spectral evolution of PMC2 in CD2Cl2 before (left) and after (right) irradiation. C 1H-15N HMBC spectral evolution of PMC2 in CD2Cl2 before (up) and after (down) irradiation. Irradiation was conducted with white LED for 120 s. The 3JHC and 3JHN correlations are indicated by red bonds and blue ovals for clarity. More NMR spectra can be seen in Figures S17 and S18.
Quinoline-substituted merocyanines
Encouraged by the above findings, we anticipated that quinoline-substituted merocyanine may undergo photochromic reaction under longer wavelength irradiation due to extended conjugation. Hence, we studied the photochromic behavior of two quinoline-substituted merocyanines (PMC6 and PMC7) theoretically and experimentally. DFT calculations reveal that PMC6 and PMC7 possess relatively low HOMO-LUMO gap (~4.1 eV) and exhibit pronounced excited-state charge transfer characteristics (CDD > 48%, Figure S19). Under 660 nm LED irradiation, the absorption peaks of the PMCs around 610 nm fade rapidly and weak peaks around 436 nm emerge, implying the generation of SIZ6 and SIZ7 (Fig. 4A, B). The PSD of SIZ6 and SIZ7 is 90.1% and 95.8% (Table 1), respectively. Unlike SIZ1-5 that can thermally return to PMCs, SIZ6 and SIZ7 show thermostability. No significant absorption peak of PMCs was observed even when SIZ6 and SIZ7 were kept in dark for 72 h (Fig. 4C, D). However, exposure to 365 nm, 500 nm and white LED light could retrieve PMC6 and PMC7 rapidly, indicating that quinoline-substituted merocyanines can function as a P-type photochromic system (Fig. 4C, D and Figures S20–23). Thus, here we can manipulate the molecular transition between PMC and SIZ forms using 500 nm and 660 nm light. Moreover, PMC6 and PMC7 exhibited fast response, > 90% switching ratio and impressive fatigue resistance (Fig. 4E, F).

A, B Photo-switching transition of PMC6 and PMC7 under 660 nm and then 500 nm. Photographs show the color change during the PMC-to-SIZ transition. C, D The photo-induced and thermal-induced recovery kinetic curves of PMC6 (C) and PMC7 (D). E, F Cyclic stability of PMC6 (E) and PMC7 (F) in CH2Cl2 under alternative irradiation (500 nm for 30 s; 660 nm for 30 s).
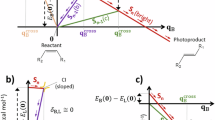
To elucidate the distinct photochromic behaviors of PMC2 and PMC6 (T-type and P-type, respectively), the reaction energy profiles associated with the PMC-SIZ isomerization were investigated using DFT calculations. The Gibbs free energies of PMCs are lower than those of their corresponding SIZs (Fig. 5 and Figures S24-S28), which is consistent with their negative photochromic character. The only exception is PMC4, which exhibits a higher Gibbs free energy than SIZ4, explaining its spontaneous conversion to SIZ4 as observed in chromatographic and NMR analyses (Figure S10). The photoisomerization process from PMC to SIZ can be simplified into two steps: the C = C bond trans-cis isomerization and the subsequent C-N bond formation. The former step involves a higher energy barrier and proceeds through an excited state to a cis-configured intermediate, whereas the latter has a relatively low barrier and can occur on the ground state (Fig. 5). In the reverse process, the isomerization begins with the cleavage of the C-N bond to yield a cis-configured intermediate, followed by cis-trans isomerization of the C = C bond to form the PMC structure. As illustrated in the reverse process in Fig. 5, the cis-trans isomerization step for SIZ2 proceeds with a relatively low energy barrier (~ 24.2 kcal/mol), allowing for efficient thermal recovery to PMC2 (Fig. 5A). In contrast, SIZ6 encounters a significantly higher energy barrier (~ 32.1 kcal/mol) during the cis-trans isomerization (Fig. 5B), which drastically slows the thermal relaxation rate, calculated to be ~105-fold slower than that of SIZ2. Consequently, the SIZ6-to-PMC6 transformation predominantly requires photochemical activation rather than occurs spontaneously via thermal recovery. Similarly, the energy barrier calculations also provided a good explanation for the isomerization mechanisms of PMC1, PMC3, and PMC7 (Figures S24, S25 and S28). However, for PMC4 and PMC5, the transition states of trans-cis isomerization were difficult to converge, suggesting that these compounds may follow an alternative reaction pathway, probably due to the absence of ICT effects in PMC4 and PMC5 (Figures S26 and S27).

Energies are referenced to the PMC isomer (set as 0.0 kcal mol-1). A Energy profile for the PMC2-SIZ2. B Energy profile for the PMC6-SIZ6. All values represent unscaled zero-point corrected relative Gibbs free energies at 298.15 K. TS = transition state.
Photochromism in solid matrices
The applicability of molecular photoswitches critically depends on their ability to undergo photochromic transitions in the solid matrices1,34,35. Photo-responsive materials are particularly important for applications in optoelectronic devices, data storage, and smart coatings. Accordingly, we investigated the photochromic behavior of PMC2 embedded in polymethyl methacrylate (PMMA) as well as absorbed in silica gel, and filter paper (Fig. 6A and Figure S29). Upon irradiation with 500 nm light, the red color of PMC2 faded, and the complete recovery of the red color was observed at room temperature within 2 h; the recovery process with an apparent activation energy of 13.2 kcal/mol (Figure S30) can further be accelerated by heating. This reversible color change could be repeated over many cycles without any noticeable degradation (Figure S31). Furthermore, PMC7 embedded in PMMA exhibited P-type photochromism modulated by 500 and 660 nm light, consistent with its behavior in solution (Fig. 6B and Figures S32 and S33).

A Photochromic and thermal recovery behavior of PMC2-PMMA film. B Photochromic behavior of PMC7-PMMA film. C Light-controlled sequential switch in PMC2-PMC7-PMMA film (PMC2: 0.5‰, PMC7: 1‰; w/w). The corresponding absorption spectra of the films are shown alongside.
Finally, we demonstrated the application of PMCs by constructing a light-controlled sequential switch. As shown in Fig. 6C, a PMMA sheet co-loaded with PMC2 and PMC7 initially appears brown (PMC2-PMC7). Sequential irradiation first with 500 nm light and then with 660 nm light resulted in an orange color (SIZ2-SIZ7), which is attributed to successive conversion of PMC2 to SIZ2 and PMC7 to SIZ7. In contrast, reversing the order of irradiation (first with 660 nm followed by 500 nm) produced a dark purple color (SIZ2-PMC7) via an orange transition (PMC2-SIZ7), as 500 nm light not only converted PMC2 to SIZ2 but also reverted SIZ7 back to PMC7 (Fig. 6C). The same switching sequence was observed in solution (Figure S34), corroborating the photochromic pathway. This behavior constitutes a sequential switch, in which the sequence of two input signals dictates the system’s output36. Such a strategy could be extended to the development of more sophisticated photo-controlled materials.
Discussion
The discovery of additional photochromic molecules is essential for advancing the design of intelligent molecular systems and functional materials37. Over the past decades, numerous photochromic molecules have been developed and widely applied in various fields such as stimulating-response material and photo-modulating biological systems38. Most of the known systems rely on well-established mechanisms such as Z-E isomerization or the reversible cleavage/formation of C-O or C-C bonds (Table S1). In contrast, photochromism based on reversible C-N bond formation remains exceedingly rare, and the previous examples have been largely limited to organic solvents18,19,20,21,22,23,24,25. In this context, PMCs are particularly noteworthy, as they undergo visible-light-induced C-N bond formation even in aqueous media. This behavior highlights both their distinctive mechanistic features and potential for applications in environments inaccessible to conventional photochromic systems.
Similar to classical spiropyran systems, substituent effects also play a crucial role39 in determining the photochromic properties of PMCs. The introduction of an electron-donating amino group enhances molecular stability and imparts the ICT feature to the molecule. This effect extends the absorption into the visible region, enabling efficient photoisomerization under mild visible-light irradiation (a significant advantage for photonic and biological applications). In contrast, electron-withdrawing substituent destabilizes the merocyanine framework and suppresses the ICT, leading to a pronounced hypochromic shift to the UV region. Compared with spiropyrans, the photoisomerization of PMCs is driven by visible light rather than UV irradiation, which, together with the electron-donating substituent effect, improves both stability and reversibility of PMCs. Moreover, by substituting the pyridine (T-type) with a quinoline moiety, the PMCs can be transformed into P-type photochromic molecules, in which the back reaction is triggered by another light rather than thermal relaxation. It might be envisioned that further tuning the thermal equilibrium position via structural substitution may obtain a positive photochromic system, which would broaden the application scope of these molecules.
Additionally, although our preliminary studies suggest that metal ions do not markedly alter the photochromic behavior of PMCs, the presence of the pyridyl coordination site may open the possibility of distinct metal-ligand interactions40. The substantial structural difference between PMC and SIZ could be harnessed to design light-responsive ion chelator/channels or even photoactivatable therapeutic agents. These opportunities underscore the broader implication of our findings, not only in materials chemistry but also in chemical biology.
In summary, we have developed a series of pyridine- and quinoline-substituted merocyanines that exhibit fast, reversible, and fatigue-resistant photochromic property in both solution and solid matrices, featuring a reversible photoinduced C-N bond formation/cleavage. By tailoring the substitution patterns, we achieved tunable absorption wavelength and different photochromic types (T-type and P-type), as elucidated through detailed spectroscopic analyses and DFT calculations. Notably, the combination of PMC2 and PMC7 facilitated the construction of a light-controlled sequential switch, wherein the order of dual-wavelength inputs determined distinct optical outputs. This study highlights the potential of PMCs as versatile molecular photoswitches for applications in optical information processing and smart materials. We anticipate that the C-N bond photochromic strategies and functional capabilities disclosed in this study will inspire further exploration of photo-responsive molecular systems.
Methods
Syntheses
For all syntheses, and characterization, please see Supplementary Information.
Preparation of single crystals
Single crystals for X-ray diffraction analysis were obtained by dissolving the solid in CH2Cl2, followed by dropwise addition of low-solubility solvents (ether, n-hexane, or ethyl acetate) to induce crystal formation via solvent diffusion. The mixture was left undisturbed at room temperature for 2-3 days to form single crystal.
Fluorescence quantum yield measurements
Fluorescence quantum yields were determined by the relative method41. PMC solutions were prepared at a series of concentrations (ensuring absorbance below 0.05). Both absorption and emission spectra were recorded in 1 cm quartz cuvettes. Parallelly, the corresponding reference fluorophores were measured: fluorescein (0.85 in 0.1 M NaOH) for PMC142, resorufin (0.74 in H2O) for PMC2 and PMC343, quinine sulfate (0.54 in 0.05 M H2SO4) for PMC4 and PMC544, and Cy5 (0.27 in pH7.4 phosphate buffer) for PMC6 and PMC745. The integrated fluorescence intensity was plotted against the absorbance (I vs. A) linearly, and the slope was compared with that of the reference fluorophore. The relative fluorescence quantum yields were calculated by:
where S is the slope of the I vs. A linear regression, Φ is the quantum yield, and n is the refractive index of the solvent. The subscripts s and r denote sample and reference respectively.
Photochromism measurements
Generally, 3 mL of PMC solution (10 μM) in a 1-cm quartz cell was irradiated with LED light for 10-30 s and the UV-vis absorption spectra were determined before and after irradiation.
Determination of photoisomerization quantum yield
Because of the wide distribution of the absorption spectra of PMC1-7, it is hard to use single photochromic standard to determine the relative quantum yield. Hence, the photoisomerization quantum yield was determined by combining kinetic analysis with photon flux calculation. A 3-mL of PMC solution was placed in 1×1 cm quartz cuvette and fully irradiated with an LED light and continuous stirring. The time-dependent absorption spectra were recorded and fitted with a single-exponential decay function to obtain the isomerization reaction rate constant kiso. The absorbed photon flux per unit area (Iabs) was calculated by differentiating the incident and transmitted photon fluxes determined using a power meter. The photoisomerization quantum yield (Φiso) was then calculated using the following equation:
where C0 is the initial concentration, V is the solution volume, NA is Avogadro’s constant, and A is the illuminated area.
Thermal recovery kinetics
The thermal recovery process was carried out by keeping the SIZ solution either under normal laboratory lighting or in a dark enclosure. During the study, no observable effect was detected for the normal laboratory lighting compared with dark condition. The absorbance evolution of the thermal recovery was analyzed using a first-order kinetic model:
where A₀ is the initial absorbance, k is the rate constant, and t is elapsed time.
Determination of apparent activation energy
The apparent activation energy was determined using the Arrhenius equation. Sample solutions were kept in dark at different temperatures (25, 37 and 50 °C), and the absorption spectra were recorded at defined time intervals. The time-dependent spectral changes were fitted to obtain the rate constant k at each temperature. A plot of lnk vs. 1/T was linearly fitted, from which the apparent activation energy (Ea) was derived by:
where A is a pre-exponential factor, R is the gas constant and T is the absolute temperature.
Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations
All calculations were carried out using Gaussian 16 (Rev. C.01) at the M06-2X/def2-TZVP level46,47. Solvent effect (CH₂Cl₂) was included using the solvent model based on density. In the vertical transition calculations, we calculated 10 excited states. During the excited state structure optimization, the lowest three excited states were computed. Frequency analyses confirmed that all optimized ground- and excited-state minima exhibited no imaginary frequencies. Transition states (TS1 and TS2) were optimized using opt(ts, calcfc, noeigentest) and verified by a single imaginary frequency together with intrinsic reaction coordinate calculations. Charge-density difference (CDD) analyses were performed with Multiwfn48,49.
Solid matrix preparation
Silica gel plates and filter papers were immersed in 100 μM PMC2 solution of acetonitrile for 10 min, and then dried to afford the functionalized materials.
PMMA (500 mg) was dissolved in CHCl3 (5 mL) under continuous stirring at 50 °C for 30 min. PMC2 stock solution was then added dropwise to the PMMA solution to achieve a PMC2:PMMA mass ratio of 1:1000 (w/w). The mixture was subsequently sonicated for 30 min to ensure homogeneous dispersion. Then the solution was cast into a glass beaker, and allowed to evaporate in oven at 50 °C for 48 h. Finally, the dried polymer film was further annealed at 40 °C under vacuum for 24 h to remove any residual solvents.
Preparation of the composite film: PMMA (500 mg) was dissolved in CHCl3 (5 mL) under continuous stirring at 50 °C for 30 min. Subsequently, PMC2 and PMC7 stock solutions were added dropwise into the PMMA solution to achieve a mass ratio of PMC2:PMC7:PMMA = 0.5:1:1000 (w/w). The resulting mixture was then sonicated for 30 min to ensure homogeneous dispersion. The solution was poured into a glass beaker and allowed to evaporate in an oven at 50 °C for 48 h. Finally, the dried polymer film was annealed at 40 °C under vacuum for 24 h to remove any residual solvents.
Data availability
The data generated in this study are provided in Source Data file. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre. Deposition numbers for compounds: PMC1 (2455320), PMC2 (2455316), PMC3 (2455317), PMC5 (2455318), PMC6 (2455319). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via (https://www.ccdc.cam.ac.uk/structures/). Coordinates for computational experiments are provided as Supplementary Data 1. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Mollick, S. & Tan, J. C. Organic solid-state photochromism using porous scaffolds. Nat. Rev. Mater. 10, 519–535 (2025).
Becker, J. et al. Optical control of TRPM8 channels with photoswitchable menthol. Angew. Chem. Int. Ed. 64, e202416549 (2025).
Eisenreich, F. et al. A photoswitchable catalyst system for remote-controlled (co)polymerization in situ. Nat. Catal. 1, 516–522 (2018).
Welleman, I. M., Hoorens, M. W., Feringa, B. L., Boersma, H. H. & Szymanski, W. Photoresponsive molecular tools for emerging applications of light in medicine. Chem. Sci. 11, 11672–11691 (2020).
Gao, M. et al. New molecular photoswitch based on the conformational transition of phenothiazine derivatives and corresponding triplet emission properties. J. Am. Chem. Soc. 147, 2653–2663 (2025).
Zhang, X. et al. Optical control of gene expression using a DNA G quadruplex targeting reversible photoswitch. Nat. Chem. 17, 875–882 (2025).
Rifaie-Graham, O. et al. Photoswitchable gating of non-equilibrium enzymatic feedback in chemically communicating polymersome nanoreactors. Nat. Chem. 15, 110–118 (2023).
Cardano, F., Marquez Garcia, R. & Szymanski, W. Manipulation of chemistry and biology with visible light using tetra-ortho-substituted azobenzenes and azonium ions. Angew. Chem. Int. Ed. 64, e202423506 (2025).
Kortekaas, L. & Browne, W. R. The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome. Chem. Soc. Rev. 48, 3406–3424 (2019).
Sassmannshausen, T. et al. Wavelength selective photocontrol of hybrid azobenzene-spiropyran photoswitches with overlapping chromophores. Angew. Chem. Int. Ed. 63, e202314112 (2024).
Ai, Q., Lan, K., Li, L., Liu, Z. & Hu, X. Beyond photochromism: alternative stimuli to trigger diarylethenes switching. Adv. Sci. 11, 2410524 (2024).
Petermayer, C. & Dube, H. Indigoid photoswitches: visible light responsive molecular tools. Acc. Chem. Res. 51, 1153–1163 (2018).
Shao, B., Qian, H., Li, Q. & Aprahamian, I. Structure property analysis of the solution and solid-state properties of bistable photochromic hydrazones. J. Am. Chem. Soc. 141, 8364–8371 (2019).
Shao, B., Fu, H. & Aprahamian, I. A molecular anion pump. Science 385, 544–549 (2024).
Helmy, S. et al. Photoswitching using visible light: a new class of organic photochromic molecules. J. Am. Chem. Soc. 136, 8169–8172 (2014).
Fang, L. et al. Ultrafast and reversible photoswitching in bulk polymers enabled by octupolar molecule design. Angew. Chem. Int. Ed. 63, e202402349 (2024).
Puthoff, D., Kuttiyil, H. & Peterson, J. A. Stenhouse salts: visible light photoswitches for protic environments. J. Am. Chem. Soc. 146, 34008–34013 (2024).
Peng, P., Strohecker, D. & Liao, Y. Negative photochromism of a TCF chromophore. Chem. Commun. 47, 8575–8577 (2011).
Shi, Z., Peng, P., Strohecker, D. & Liao, Y. Long-lived photoacid based upon a photochromic reaction. J. Am. Chem. Soc. 133, 14699–14703 (2011).
Hatano, S., Horino, T., Tokita, A., Oshima, T. & Abe, J. Unusual negative photochromism via a short-lived imidazolyl radical of 1,1’-binaphthyl-bridged imidazole dimer. J. Am. Chem. Soc. 135, 3164–3172 (2013).
Mutoh, K. & Abe, J. Fast photochromism of helicene-bridged imidazole dimers. Chem. Sci. 15, 13343–13350 (2024).
Ito, H., Mutoh, K. & Abe, J. Bridged-imidazole dimer exhibiting three-state negative photochromism with a single photochromic unit. J. Am. Chem. Soc. 145, 6498–6506 (2023).
Hamatani, S., Kitagawa, D. & Kobatake, S. Diarylethene photoswitches undergoing 6π azaelectrocyclic reaction: disrotatory thermal cycloreversion of the closed-ring isomer. J. Phys. Chem. Lett. 14, 8277–8280 (2023).
Sacherer, M. & Dube, H. Second generation zwitterionic aza-diarylethene: photoreversible C-N bond formation, three-state photoswitching, thermal energy release, and facile photoinitiation of polymerization. Angew. Chem. Int. Ed. 64, e202415961 (2025).
Wang, X. Y. et al. Light-triggered regionally controlled n-doping of organic semiconductors. Nature 642, 599–604 (2025).
Abeyrathna, N. & Liao, Y. Stability of merocyanine-type photoacids in aqueous solutions. J. Org. Chem. 30, e3664 (2017).
Dhahri, N., Taoufik, B. & Goumont, R. Kinetics of alkaline hydrolysis of p-substituted benzylidenemalononitriles in 50% aqueous acetonitrile: substituent effects and quantification of the electrophilic reactivity. J. Phys. Org. Chem. 27, 484–489 (2014).
Malkin, Y. N., Krasieva, T. B. & Kuzmin, V. A. Quantitative study of the photostability of spiropyrans. J. Photochem. Photobiol. A 49, 75–88 (1986).
Görner, H., Atabekyan, L. S. & Chibisov, A. K. Photoprocesses in spiropyran-derived merocyanines: singlet versus triplet pathway. Chem. Phys. Lett. 260, 59–64 (1996).
Kortekaas, L., Chen, J., Jacquemin, D. & Browne, W. R. Proton-stabilized photochemically reversible E/Z isomerization of spiropyrans. J. Phys. Chem. B 122, 6423–6430 (2018).
Tian, Z. et al. Spirooxazine to merooxazine interconversion in the presence and absence of zinc: approach to a bistable photochemical switch. J. Phys. Chem. A 114, 11900–11909 (2010).
Kubinyi, M. et al. Metal complexes of the merocyanine form of nitrobenzospyran: Structure, optical spectra, stability. J. Mol. Struct. 1000, 77–84 (2011).
Becker, E. D. et al. High Resolution NMR: Theory And Chemical Applications, Ch. 2 (Elsevier Ltd. Press 1999).
Han, Z., He, M., Wang, G., Lehn, J. M. & Li, Q. Visible-light-driven solid-state fluorescent photoswitches for high-level information encryption. Angew. Chem. Int. Ed. 63, e202416363 (2024).
Xie, Y. et al. Hydrogen bond-associated photofluorochromism for time-resolved information encryption and anti-counterfeiting. Angew. Chem. Int. Ed. 64, e202414846 (2025).
Andreasson, J. et al. All-photonic multifunctional molecular logic device. J. Am. Chem. Soc. 133, 11641–11648 (2011).
Zhang, J., Zou, Q. & Tian, H. Photochromic materials: more than meets the eye. Adv. Mater. 25, 378–399 (2013).
Kobauri, P., Dekker, F. J., Szymanski, W. & Feringa, B. L. Rational design in photopharmacology with molecular photoswitches. Angew. Chem. Int. Ed. 62, e202300681 (2023).
Balmond, E. I. et al. Comparative evaluation of substituent effect on the photochromic properties of spiropyrans and spirooxazines. J. Org. Chem. 81, 8744–8758 (2016).
Benchimol, E., Tessarolo, J. & Clever, G. H. Photoswitchable coordination cages. Nat. Chem. 16, 13–21 (2024).
Liu, D. K. et al. Near-infrared II cyanine fluorophores with large stokes shift engineered by regulating respective absorption and emission. Nat. Commun. 16, 4911 (2025).
Gong, Q. Y., Shi, W., Li, L. H. & Ma, H. M. Leucine aminopeptidase may contribute to the intrinsic resistance of cancer cells toward cisplatin as revealed by an ultrasensitive fluorescent probe. Chem. Sci. 7, 788–792 (2016).
Gao, X. H., Li, X. H., Li, L. H., Zhou, J. & Ma, H. M. A simple fluorescent off-on probe for the discrimination of cysteine from glutathione. Chem. Commun. 51, 9388–9390 (2015).
Zhou, J. et al. Detection of misdistribution of tyrosinase from melanosomes to lysosomes and its upregulation under psoralen/ultraviolet A with a melanosome-targeting tyrosinase fluorescent probe. Anal. Chem. 88, 4557–4564 (2016).
Resch-Genger, U., Grabolle, M., Cavaliere-Jaricot, S., Nitschke, R. & Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 5, 763–775 (2008).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists. Multiwfn. J. Chem. Phys. 161, 082503 (2024).
Acknowledgements
We thank the financial support from the NSF of China (22474147, 22174148, 22374153, 22474144, 22174147). We thank Dr. Qian Li for 2D NMR analysis. We also thank Dr. Tongling Liang and Shuang You for single-crystal structure analysis.
Author information
Authors and Affiliations
Contributions
W.S. and Y.L. conceived the project. H.L. designed and performed most experiments, analyzed the data, and wrote the original manuscript. F.S., Z.H., S.L. (Sijia Lu), and Q.T. carried out partial experiments. S.L. (Shiquan Lin) performed partial theoretical calculations. X.L. and H.M. contributed to manuscript revision. W.S. supervised the research and finalized the manuscript. All authors discussed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, H., Liu, Y., Sun, F. et al. Photochromism of pyridine-substituted merocyanine through reversible C-N bond formation. Nat Commun 17, 904 (2026). https://doi.org/10.1038/s41467-025-67626-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67626-3
