
Abstract
The strategic engineering of an Ohmic junction at the Bi/Bi2O3 (BBO) interface is demonstrated to synergistically enhance photocatalytic CO2-to-methanol conversion through precisely modulated charge behavior and interfacial energy alignment. This metallic Bi-semiconductor Bi2O3 Ohmic junction with local surface plasmon resonance effect induces a robust built-in electric field that promotes the unidirectional electron transfer from Bi2O3 to Bi while suppressing charge recombination. Theoretical calculations and experimental evidence reveal that the interfacial Bi sites within the Ohmic junction predominantly facilitate CO2 adsorption and activation to form *COOH, whereas ensuing protonation steps are favored on metallic Bi sites on BBO Ohmic junction. Furthermore, the Ohmic junction enhances interfacial electron density and strengthens orbital hybridization between Bi 6p and O 2p orbitals, thereby reducing the activation energy of the rate-limiting *CO2 → *COOH step by 0.6 eV, enabling a CH3OH production rate of 610 μmol g-1 under light irradiation. The work deciphers the dual role of Ohmic junctions in simultaneously resolving bulk charge transport limitations and tailoring surface catalytic landscapes, establishing a universal paradigm for metal-semiconductor heterojunction photocatalyst design.
Similar content being viewed by others


Introduction
The photocatalytic reduction of CO2 into solar fuels like methanol represents a promising route to tackle both energy scarcity and carbon neutrality goals1,2,3,4,5. Nevertheless, the practical implementation of this photocatalytic reaction faces three fundamental challenges: rapid recombination of photogenerated charge carriers, high bond dissociation energy (≈750 kJ mol−1) for cleaving stable C=O bonds, and dependency on sacrificial agents for sustaining catalytic activity. Substantial research attention has focused on engineering the bulk phase configuration of photocatalysts, as this structural parameter fundamentally governs charge carrier separation dynamics, including constructing ultrathin architectures, introducing heteroatom dopants, and building heterojunctions6,7. Among these, Ohmic junctions formed, characterized by minimal contact resistance at material interfaces, offer a distinct advantage by enabling unimpeded charge injection while preserving catalytic sites8,9,10. However, achieving precise atomic-level modulation of Ohmic interfaces to concurrently enhance bulk charge migration and surface reaction kinetics remains a challenging.
Recent advances in Ohmic junction engineering, including metal-semiconductor systems (e.g., Bi/BiOCl, where the metallic Bi Nanoparticles formed a low-resistance contact with the n-type BiOCl photocatalyst to accelerate electron extraction)11,12 and semiconductor heterointerfaces (e.g., Mo2TiC2 MXene/ZnCdS ohmic junction)13, have demonstrated enhanced charge separation and catalytic activity in photocatalytic fields. To further improve the performance of Ohmic junction photocatalysts, leveraging the localized surface plasmon resonance (LSPR) effect presents a compelling approach. The LSPR effect confers unique advantages in photocatalytic CO2 reduction through multiple synergistic pathways: enhanced broadband light-harvesting capabilities, efficient hot-carrier manipulation, intensified electromagnetic near-fields, and thermally aided catalytic processes14,15. For example, Zhang et al. reported a plasmonic Fe-nanonecklace-hydrogenated titanium hybrid for advancing solar light-driven NH3 synthesis via a local heating effect-assisted photothermal pathway, circumventing the equilibrium limit of thermocatalysis16. He et al. synthesized anisotropic Ti3C2Tx MXene with strong LSPR features, which showed enhanced plasmonic charge carrier-induced photocatalytic water splitting activity17. These developments highlight the great potential of integrating LSPR effects into highly efficient Ohmic junction photocatalysts for CO2 photoconversion.
Herein, we demonstrate an atomic-level engineering strategy to construct Bi/Bi2O3 (BBO) Ohmic junctions with LSPR effect for high-efficiency CO2-to-methanol conversion. By in-situ growing metallic Bi nanodomains on nanosheets, we constructed an interfacial structure with aligned Fermi levels, thereby facilitating barrier-free electron migration from Bi2O3 to Bi. Combining X-ray absorption spectroscopy with density functional theory (DFT) simulations, we elucidate that the Ohmic contact facilitates electron accumulation on Bi and the polarized Biδ+ sites stabilize *CO2- intermediates via strong 6p-2p orbital hybridization (−ICOHP = 1.85 eV). This interaction reduces the energy barrier for *CO2 protonation by 0.6 eV and stabilizes *CHO intermediates through orbital hybridization. This synergistic configuration achieves a methanol production rate of 610 μmol g−1, surpassing most reported photocatalytic systems. Our work establishes a paradigm for designing Ohmic interfaces that unify ultrafast charge transport and targeted surface catalysis, overcoming longstanding efficiency bottlenecks in artificial photosynthesis.
Results
CO2 adsorption and activation mechanism over Bi/Bi2O3 Ohmic junction
Figure 1a systematically elucidated the interfacial electronic engineering and catalytic mechanisms of Bi/Bi2O3 (BBO) Ohmic junction for enhanced CO2 reduction. The BBO Ohmic junction revealed higher photocatalytic CO2 reduction activity to CH3OH than that of Bi or Bi2O3, which was ascribed to the interfacial interaction inducing fast separation of photogenerated carriers and the strong adsorption to *CO for its protonation reaction produces. To further understand the mechanism of CO2 activation and conversion on BBO, we analyzed detailed molecular orbital interactions. The results of the partial density of states (PDOS) indicated that during CO2 adsorption and activation on Bi2O3, hybridization occurred between the Bi 6px orbital and the C 2pz orbital (Fig. 1b and Supplementary Figs. 1–3). This leaded to partial electron transfer from the Bi 6px orbital to the 2π anti-bonding orbital of CO2, weakening the CO2 molecule and causing its linear structure to bend. This structural change drove CO2 adsorption and activation on the Bi2O3 surface. In the case of CO2 adsorption and activation on metal Bi of BBO (Fig. 1c, and Supplementary Figs. 4 and 5), the electronic states of Bi’s 6px, 6py, and 6pz orbitals near the Fermi level nearly vanished. Meanwhile, tightly overlapping and continuous hybrid peaks emerged in the electronic states of C 2px, 2py, and 2pz orbitals of CO2. This suggested significant electron transfer from 6px, 6py, and 6pz orbitals of Bi to the 2π anti-bonding orbital of CO2, facilitating CO2 adsorption and activation on metal Bi of BBO. The transferred electrons filled C 2px, 2py, and 2pz orbitals of CO2, resulting in degeneracy among these filled orbitals. This orbital degeneracy manifested as numerous continuous hybrid peaks in the corresponding PDOS. The projected crystal orbital Hamiltonian population (COHP) was also analyzed to understand the mechanism of *CO protonation. The rise of the antibonding state contributes to the bond strength, which can be seen from the computed integrated COHP (ICOHP) as shown in Fig. 1d, a more negtive ICOHP was observed for BBO Ohmic junction (−1.85 eV) than Bi2O3 (0.82 eV), indicating a much stronger bond coupling between BBO Ohmic junction and *CO, favoring the further protonation reaction.

a Reaction mechanisms of CO2 reduction on Bi, Bi2O3, and BBO Ohmic junction. b COHP for the Bi 6p-O 2p of absorbed *CO on the surface of Bi2O3, and BBO Ohmic junction. PDOS and IPDOS related to Bi 6p-O 2p interactions before (up) and after (down) CO2 adsorption on c Bi2O3 and d BBO Ohmic junction. e Differential Bader charge of BBO Ohmic junction. (f) Theoretical work function of the surface of Bi (012) and Bi2O3 (100). g The electron transfer mechanism of BBO Ohmic junction, thereby enabling the generation of CH3OH. Source data for Fig. 1b–d, f are provided as a Source Data file.
To clarify the interface charge transfer of BBO Ohmic junction, density functional theory calculation (DFT) calculations with Bader charge were calculated. Supplementary Fig. 6 exhibited the optimized crystal structure of Bi2O3 and BBO Ohmic junction. Figure 1e depicted the charge-density difference plots of BBO Ohmic junction, whereas the yellow and cyan areas represented electron density accumulation and deplete, respectively. Theoretical calculations indicated that the coupling of Bi and Bi2O3 promoted charge enrichment on Bi2O3 (Δq = 1.557 e), which suggested that Bi atoms can adjust the surrounding chemical microenvironment of Bi2O3, thus affecting the adsorption of intermediate species. To verity the heterojunction type in theory, the theoretical work functions of Bi and Bi2O3 were calculated to be 4.78 and 5.15 eV, respectively (Fig. 1f). The much smaller work function of Bi promoted the charge transfer from Bi to Bi2O3 until their consistent Fermi levels after the coupling of Bi and Bi2O3, and at the same time, inducing the generation of a strong built-in electric field and band bending (Fig. 1g). This phenomenon can accelerate the transfer and separation of photogenerated carriers for CO2 photoreduction to CH3OH (Supplementary Fig. 7).
Structural characterizations
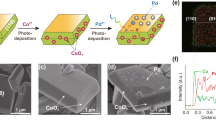
Figure 2a presents the synthetic procedures of BBO Ohmic junction via a hydrothermal and wet-chemical cascade strategy. The Bi19Br3S27 nanowires have been firstly synthesized by hydrothermal method, then followed by a NaBH4 reduction method to inducing the dissolution and crystallization of Bi species, and finally the BBO nanosheets were obtained. The X-ray diffraction (XRD) patterns of the samples were shown in Supplementary Fig. 8. The nanowire morphology of Bi19Br3S27 samples were shown in Fig. 2b and Supplementary Fig. 9. To validate the structure evolution of Bi19Br3S27 nanowires to form BBO nanosheets, the different reduction time for Bi19Br3S27 was conducted at the same conditions. Figure 2c–e and Supplementary Figs. 10–12 record the morphologies of BBO-2, BBO-4, and BBO-6. With the increasing of reduction time, the BBO samples exhibited a gradual increased nanosheet structure (Supplementary Figs. 13–15). Moreover, the progressive rise in Bi content of these samples indicated the existence of a rich bismuth environment (Supplementary Tables S2–4). The SEM, TEM and AFM tests of Bi2O3 were conducted. The scanning electron microscope (SEM) and transmission electron microscopy (TEM) images indicate that the Bi2O3 sample was composed of irregularly nanosheet (Supplementary Figs. S16a, b). The lattice spacings of 0.274 nm can be assigned to the (400) planes of Bi2O3 (Supplementary Fig. S16c). The AFM images and the corresponding height profile showed that the thickness of its nanosheet was ~5.06 nm (Supplementary Figs. S16d, e). These data provide a solid foundation for the subsequent comparison with BBO-4 composite materials. The excellent lattice matching and well-defined BBO-4 Ohmic junction were verified by high resolution TEM (HRTEM; Fig. 2f, g), demonstrating the success in fabricating the Ohmic junction. The high-quality coherent heterointerface with a 0.328 nm lattice fringe spacing belonging to the (012) plane of Bi and a 0.274-nm lattice fringe spacing attributing to the (400) plane of Bi2O3. Furthermore, the element composition of BBO-4 Ohmic junction was detected by energy dispersive spectroscopy (EDS) element mapping (Fig. 2h–j). Moreover, the Bi content was gradually increased as time extended, and the XRD patterns of BBO samples can be indexed to hexagonal Bi (JCPDS: 05-0519) and Bi2O3 (JCPDS: 22-0515) (Fig. 2k, and Supplementary Fig. 17) and no other phase structures were detected (Supplementary Fig. 18)18,19. To precisely resolve the phase composition, Rietveld refinement of the XRD patterns (Supplementary Fig. 19) was performed to determine the phase composition of the BBO catalysts. The refinement results confirm the exact phase composition. The Bi/Bi2O3 mass ratios for BBO-2, BBO-4 and BBO-6 were 53.65/46.35, 91.75/8.25, and 97.74 wt%/2.26 wt%, respectively. The Bi content increased with increasing reduction time. The above results showed there was on other phase structures. The Raman peaks of metallic Bi at 71 and 98 cm−1 corresponded to the asymmetric coupling vibration and symmetric vibration of Bi atoms within the metal (Supplementary Fig. 20). The Raman peaks of Bi2O3 at 326 and 465 cm−1 were attributed to the stretching of Bi–O bonds and the symmetric stretching of Bi–O–Bi in Bi–O octahedral units within Bi2O3. In the Raman spectrum of BBO-4, characteristic peaks of both metallic Bi and Bi2O3 were observed. The above results confirmed the chemical reduction can achieve the structure reconstruction of Bi19Br3S27 for BBO nanosheet formation.

a Schematic illustration of the synthesis of BBO. TEM image of b BBS, c BBO-2, d BBO-4, and e BBO-6. f High-solution TEM image, g lattice fringes, and h–j EDS mapping images of BBO-4. k XRD patterns of Bi, Bi2O3, BBO-2, BBO-4, BBO-6. Normalized XANES spectra at the l Bi-L3 edge and m corresponding Fourier transform R space fitting results. Source data for Fig. 2k–m are provided as a Source Data file.
As shown in Fig. 2l, the absorption edge of BBO-4 in the X-ray absorption fine structure (XAFS) Bi L3-edge was between the Bi foil and Bi2O3, meaning that the valence state was Biδ+ (0 < δ < 3), further confirming the existence of metal Bi. In addition, the absorption edge of Bi in the XAFS Bi L3-edge was between the Bi foil and Bi2O3, showing the occurrence of surface oxidation, which was accordance with the X-ray photoelectron spectroscopy (XPS) results. To obtain the quantitative structural parameters around Bi, Fourier transform of the least-squares curve fitting was performed on the EXAFS data of Bi, Bi2O3, and BBO-4 (Fig. 2m). BBO-4 exhibited a typical Bi/Bi2O3 structure. The presence of both Bi–O and Bi–O–Bi coordination shells confirmed the existence of Bi2O3 oxide, while the Bi-Bi metallic coordination indicated the presence of metallic Bi. In contrast, the Bi2O3 sample showed only a single Bi–O coordination shell, demonstrating its phase-pure Bi2O3 composition. The Bi metal sample, however, revealed both Bi-Bi and Bi-O bonds, suggesting the existence of surface oxidation. To investigate the electronic states and charge transfer of BBO-4, XPS analysis was employed. For Bi2O3, the XPS diffraction peaks at 159.00 eV (Bi 4f7/2) and 164.32 eV (Bi 4f5/2) were characteristic of Bi3+ species (Supplementary Fig. 21a)20,21. The characteristic Bi 4f peaks of Bi metals could be fitted into two pairs of doublets associated with Bi0 and Bi3+. The XPS signal of Bi oxidation states in Bi metals was derived from surface oxidation of the catalyst under exposure to air. For BBO-4, the Bi 4f signals deconvoluted into four peaks: two at 156.68 and 161.98 eV (assigned to Bi0 in metallic Bi) and two at 158.89 and 164.21 eV (attributed to Bi3+ in Bi2O3)22,23,24. Comparative analysis of binding energies across Bi and Bi2O3, BBO-4 revealed a 0.11 eV positive shift in the Bi0 binding energy for the heterojunction relative to pristine metallic Bi, indicating decreased electron density around Bi0. Conversely, the Bi3+ binding energy in BBO showed an 0.11 eV downshift compared to Bi2O3, signifying increased electron density around Bi3+. These XPS results collectively suggested electron transfer from metallic Bi to Bi2O3 within the heterojunction, consistent with the electron flow direction in Ohmic junction25,26,27. The O 1s XPS spectrum of BBO-4 was the same as that of pristine Bi2O3, with peaks at 529.52 and 530.85 eV, which were attributed to the chemical bonding of Bi–O and adsorbed oxygen (H–O), respectively (Supplementary Fig. 21b). The results were in accordance with the above work function analysis.
Local surface plasmon resonance effect and photocatalytic CO2 reduction over BBO
Prior to evaluating photocatalytic performance, we first investigated the material’s light utilization capability for photocatalytic applications. The UV-vis-NIR DRS (Fig. 3a) revealed that BBO-4 demonstrated significantly enhanced broad-spectrum light absorption (280–2100 nm) compared to its constituent materials (Bi and Bi2O3). This improved LSPR performance in the BBO Ohmic junction can be attributed to its unique nanosheet architecture. The abundant interfaces between metallic Bi and Bi2O3, along with their interfacial interactions, collectively modulated the LSPR intensity. Finite-difference time-domain simulations further corroborated the presence of a strong LSPR effect in BBO (Fig. 3b), demonstrating enhanced local electric field strength surpassing that of individual components28,29. Notably, the accumulation of LSPR-excited hot electrons at the interface spontaneously elevated the BBO surface temperature to 117.3 °C without external heating (Fig. 3b, and Supplementary Fig. 22). This LSPR-induced phenomenon simultaneously enhanced photon utilization efficiency and enabled localized photothermal energy conversion, thereby synergistically promoting photocatalytic activity. The corresponding bandgaps Eg of Bi2O3 was determined to be 2.51 eV, according to Tauc plots: (ahν)1/2 = A(hν- Eg) (Supplementary Fig. 23). Mott–Schottky analysis indicated flat potentials of −0.20 V (Bi) and −0.57 V (Bi2O3) versus NHE (Fig. 3c). Given that semiconductor conduction band potentials typically located at more negative values than their flat potentials, the BBO band alignment satisfied the thermodynamic requirements for CO2 photoreduction to CH3OH.

a UV-vis-NIR DRS spectra and b spatial distribution of electric fields enhanced by LSPR effect under the excitation wavelength of 800 nm of Bi, Bi2O3, and BBO, and surface temperature of BBO-4 under light irradiation with the increasing time c Mott–Schottky plots of Bi and Bi2O3. d Photocatalytic CO2 activities of the as-prepared samples. e Photocatalytic performance of BBO-4 under different conditions. f Cycling experiments of BBO-4 for photocatalytic CO2 reduction. The error bars represent the standard deviation derived from three repeated experiments. g Mass spectra for CO2 reduction of BBO-4 using 13CO2 as the reacting gas. h Photocatalytic CO2 reduction performances of BBO-4 compared with reported representative photocatalysts (the detailed reaction conditions were listed in Supplementary Table S5). Source data for a, c, d–h are provided as a Source Data file.
The photocatalytic CO2 reduction performances of synthesized catalysts were evaluated under full spectrum (Fig. 3d, and Supplementary Fig. 24). The primary products of Bi, Bi2O3, and Bi2S3 resulting from photocatalytic CO2 reduction were mainly CO. The precursor Bi19Br3S27 nanowires revealed the generation of CO, CH4, and CH3OH. The series of BBO derived from Bi19Br3S27 nanowires displayed enhanced photocatalytic activity for CH3OH generation. The optimal BBO-4 samples reached the highest value of 610 µmol g−1, substantially surpassing the rates observed with unmodified Bi, Bi2O3, and precursor Bi19Br3S27 nanowires. The liquild products were further detected by nuclear magnetic spectrum (NMR). As shown in Supplementary Fig. 25, the proton spectrum exhibited a methanol peak at δ 3.28 ppm, and a water peak at δ 4.7 ppm. Based on the (NMR) analysis of the liquid products from photocatalytic CO2 reduction, it is confirmed that the liquid product generated by the BBO-4 heterojunction photocatalyst was CH3OH. No impurity peaks or other liquid products were detected.
In contrast, no reduction products were observed under dark conditions, in the absence of a catalyst or Ar atmosphere (Fig. 3e). The above results of controlled experiments demonstrated the generation of products as a result of reduced CO2 in the photocatalytic system. N2 adsorption/desorption isotherms and the pore-size distributions of Bi, Bi2O3, and BBO-4 were characterized (Supplementary Fig. 26), suggesting surface area was not the influence factor on photocatalytic activity. As depicted in Fig. 3f, the yields of products were basically no significant change after four cycles, and its structure had no obvious agglomeration and transform (Supplementary Fig. 27), demonstrating the excellent stability of BBO-4. Furthermore, precise detection of 13CH4 (m/z = 17), 13CO peak (m/z = 29) and 13CH3OH (m/z = 33) utilizing 13C as the only carbon source, provided further evidence that CH3OH, CH4 and CO were derived from CO2 photoreduction (Supplementary Fig. 28, and Fig. 3g). To demonstrate the superior activity of BBO-4, we conducted comparative analyzes with over 20 different catalytic systems (Fig. 3h). The results conclusively showed that BBO-4 exhibited favorable photocatalytic performance among the reported photocatalyst.
Identification of CO2 photoreduction intermediates
To investigate the mechanistic pathway of photocatalytic CO2 reduction, we conducted in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) to monitor the dynamic evolution of adsorbed intermediates on pristine Bi, Bi2O3, and BBO-4 catalysts under light irradiation (Fig. 4a–f, and Supplementary Figs. 29–31). The characteristic peak at 1546 cm−1 (Fig. 4a) corresponds to combined bending and stretching vibrations of the *COOH intermediate on metallic Bi30,31,32, a feature consistently observed in both Bi2O3 and BBO-4 systems under illumination (Fig. 4b, c). Notably, comparative analysis revealed that BBO-4 exhibited weaker b-CO3− signal intensity but enhanced m-CO32− formation compared to pristine Bi and Bi2O3. This distinct behavior suggests that m-CO32− species generated through single oxygen-site adsorption at Bi centers serve as key intermediates in the CO2 reduction pathway on BBO-431,33,34. The diagnostic *CO intermediate (2076 cm−1) was detected across all three catalysts, with its presence indicating a critical branching point for subsequent hydrocarbon formation, particularly CH4 and CH3OH35,36,37. Temporal evolution studies (Fig. 4d–f) demonstrated progressive intensification of *COOH signals over 5–60 min, consistent with continuous CO2 activation and hydrogenation processes. Crucially, BBO-4 exhibited significantly stronger *CO adsorption compared to its constituent materials (Bi and Bi2O3), as evidenced by enhanced peak intensity. This pronounced adsorption capacity favored further hydrogenation steps over CO desorption, thereby directing the reaction pathway toward CH3OH products. Based on comprehensive analysis of these in situ DRIFTS results, we proposed a stepwise reaction mechanism for photocatalytic CO2 reduction on BBO-4 (Fig. 4g). The process initiates with CO2 chemisorption at Bi–O sites, followed by sequential protonation steps generating *COOH intermediates. Subsequent cleavage of C–O bonds yields adsorbed *CO species, which underwent progressive hydrogenation through surface-bound reactive hydrogen species to form CH4 and CH3OH.

Investigation of CH3OH generation during photocatalytic CO2 reduction. In situ ATR-FTIR experiments for (a, d) Bi, (b, e) Bi2O3, and (c, f) BBO-4 over 1000–2200 cm−1, 1500–1600 cm−1, and 2000–2200 cm-1 under full spectrum light illumination. g Proposed molecular mechanisms of CO2 adsorption and CO2-to-CH3OH photosynthesis on the surface of BBO-4 nanosheets. Source data for Fig. 4a–f are provided as a Source Data file.
Carrier dynamics
Through in situ Kelvin probe force microscopy (KPFM) coupled with controlled light illumination, we directly visualized the spatial-temporal dynamics of photoinduced charge carriers in BBO-4. Under dark conditions, surface potential line profiles revealed distinct differences among the materials: Bi2O3 and BBO-4 exhibited surface potentials of 0.50, 0.97 mV, respectively (Fig. 5a, b). Light irradiation induced dramatic surface potential reduction in BBO-4 (ΔV = 0.62 mV, Fig. 5c), demonstrating efficient photoinduced electron migration to the material surface under built-in electric field guidance. Notably, BBO-4 demonstrated the much more pronounced surface potential heterogeneity, suggesting enhanced charge redistribution capabilities. Thickness-normalized analysis (Fig. 5c, and Supplementary Fig. 14) quantified this effect, showing normalized potential gradients of 0.21 and 0.13 mV nm−1 for BBO-4 with or without light irradiation, respectively. However, the normalized potential difference of BBO-4 decreased to 0.08 mV nm−1 (Supplementary Fig. 32), indicating that the electric field can be optimized by appropriate Bi content1. Ohmic junction generated favorable localized electric fields that facilitate electron accumulation. This light-responsive behavior confirmed that BBO-4 Ohmic junction enhanced charge separation through two synergistic mechanisms: (1) creation of strong interfacial electric fields and (2) directional charge transport mediation, which were also supported by the photocurrent and electrical impedance spectra (Supplementary Fig. 33).

KPFM images and surface potential analysis of a Bi2O3 in the dark and b, c BBO-4 in the dark and under light irradiation, respectively (ΔA stands for differential absorbance). TA spectra and corresponding 3D contour plots of d, e Bi2O3 and g, h BBO-4. Normalized transient absorption kinetics for f Bi2O3 and i BBO-4. Source data for Fig. 5a–i are provided as a Source Data file.
To elucidate the interfacial charge transfer mechanisms in Ohmic junction systems, we conducted comparative femtosecond transient absorption (fs-TA) spectroscopy studies on Bi2O3 and BBO-4 heterostructures. Bi2O3 (Fig. 5d, e) and BBO-4 (Fig. 5g, h) demonstrated extended photoinduced absorption spanning 450–570 nm. The absorption intensity of BBO-4 was stronger than that of Bi2O3, indicating enhanced light-matter interactions in BBO-4 Ohmic junction system through interfacial electronic state hybridization. Figure 5f–i reveals the fs-TA kinetic profiles at 550 nm of Bi2O3 and BBO-4, and their signal can be well-fitted with triexponential results. The fs-TA spectra of Bi2O3 and BBO-4 were fitted by a tri-exponential function and the fitting parameters are τ1 = 0.191, τ2 = 6.53, τ3 = 1490 ps for Bi2O3, and τ1 = 0.366, τ2 = 44.1, τ3 = 994 ps for BBO-4. Photogenerated electron relaxation in the CB of Bi2O3 and BBO-4 occurred through three pathways: exciton-mediated states (τ1), defect trap states (τ2), and recombination of e- and h+(τ3). Notably, the lifetime components of BBO-4 (τ1 and τ2) were much longer than that of Bi2O3 (τ1 = 2.95 ps), which was attributed to the ineffective utilization of photoelectrons within a single photocatalyst, hindering efficient photocatalytic activity38,39. The shorter lifetime of τ3 (994 ps) for BBO-4 compared with Bi2O3 (τ3 = 1490 ps) was favorable to the interfacial charge transfer of BBO-4 Ohmic junction (Supplementary Fig. 34).
Reaction mechanism of CO2 photoreduction
The interfacial engineering strategy significantly enhances gas-solid interactions in the BBO-4 Ohmic junction, as evidenced by complementary gas adsorption analyzes. CO2 adsorption isotherms (Fig. 6a) demonstrated the adsorption capacities of Bi, Bi2O3 and BBO-4 were 0.3402, 0.4826, and 0.5632 cm3 g−1. This indicated that BBO-4 may provide more active sites for adsorbing and converting CO2 molecules during the photocatalytic CO2 reduction process, thus facilitating the occurrence of the reaction. The enhanced CO2 adsorption effect favored the adsorption and activation of CO2. The CO2 and CO TPD profiles (Fig. 6b, c) revealed intensified desorption signals spanning 100–450 °C, which was indicative of optimized physisorption/chemisorption site distribution. Moreover, the hydrophilicity of Bi2O3 and BBO-4 was also observed by contact angle test. The introduction of Bi reduced hydrophilicity of BBO-4, which was beneficial to inhibit the competing reaction of H2 generation (Fig. 6d, and Supplementary Figs. 35 and 36). The interfacial engineering of BBO-4 revealed improved CO2 activation capabilities through synergistic electronic modulation, as evidenced by comprehensive adsorption analyzes (Fig. 6e). Comparative structural studies revealed that CO2 chemisorption induced characteristic molecular distortion across all models: Bi2O3, BBO-4 surface, and BBO-4 interface. BBO-4 interface exhibited larger C=O bond elongation and smaller angular deformation compared with the surface of Bi2O3 and BBO-4, confirming effective CO2 polarization on BBO-4 Ohmic junction. The Bader charge analysis displayed that 0.381 e of BBO-4 interface transferred to CO2 during CO2 adsorption, which were much more than those of Bi2O3 (0.042 e) and BBO-4 surface (0.224 e). The CO2 adsorption energy of BBO-4 interface (−0.52 eV) was larger than that of Bi2O3 (−0.47 eV) and BBO-4 surface (−0.49 eV). These results confirmed that the interface effect was more conducive to CO2 activation in CO2 photoreduction process.

a CO2 adsorption isotherm curves, b CO2-TPD, c CO-TPD signals, and d contact angle measurements of Bi2O3 and BBO-4. e Calculated models, charge difference and adsorption energy of CO2 for Bi, Bi2O3, BBO (metallic Bi site), and BBO with interfacial site. Free energy diagram for CO2 photoreduction to CH3OH over the surface of f Bi, g Bi2O3 and h BBO with metallic Bi sites, and i the interfacial sites of BBO. Source data for a–c, f–i are provided as a Source Data file.
Figure 6f–i, and Supplementary Figs. 37–40 display the free energy profiles and the corresponding intermediate configurations. For the models of Bi, Bi2O3, BBO with metallic Bi site, and BBO with interfacial site, from a thermodynamic standpoint, COOH* formation represented a rate-limiting step in the CO2 reduction process with free-energy barriers of 2.07, 3.45, 1.76, and 1.51 eV, respectively. It is obvious that the Ohmic interface reduced the free-energy barrier of rate-determining step by 1.94 eV. The formation of Ohmic junction promoted charge redistribution at the interface, inducing the generation of built-in electric field, further accelerating the adsorption and activation of CO2. The *CO → CO (g) and *CO → *CHO were critical step which determine the selectivity of CO2 reaction. Due to the considerably lower free energy of *CO(g) compared to *CHO on both Bi and the metallic Bi sites in BBO, the formation of CO became the thermodynamically favored and primary product. For Bi2O3 and interface BBO, the *CHO formation energy was much lower than that of CO (g), suggesting the formation of *CHO was easier than that of CO (g). Moreover, the fabrication of the Ohmic junction interface greatly reduced the energy barrier of the rate-determining step. Experimentally and theoretically, BBO exhibited a better photocatalytic CO2 reduction performance into CO or CH3OH than Bi2O3.
Discussion
In summary, this work successfully constructed a BBO Ohmic junction photocatalyst via a sequential approach involving hydrothermal synthesis and in situ chemical reduction. The heterostructure exhibits enhanced broad-spectrum solar utilization, CO2 adsorption capacity, and photocatalytic performance for methanol synthesis. Mechanistic investigations indicate that interfacial Bi sites within the Ohmic junction play a dominant role in promoting CO2 adsorption and activation, while subsequent protonation reactions preferentially occurred on metallic Bi domains. The optimized catalyst achieved a remarkable methanol yield of 610 μmol g−1 under simulated solar irradiation, surpassing the performances of representative semiconductor photocatalysts and retaining stable activity across four consecutive reaction cycles. Theoretical calculations demonstrate that the BBO interface significantly lowers the energy barrier for *CO intermediate conversion, especially facilitating the pivotal *CO → *CHO transition during methanol formation. The enhancement in photocatalytic performance is attributed to the synergy between optimized charge transport mediated by the Ohmic junction and LSPR effects. This research not only presents a versatile strategy for designing high-performance heterojunction photocatalysts but also underscores the promise of plasmon-enhanced bismuth-based materials in advancing sustainable solar fuel generation via CO2 valorization.
Methods
Materials
Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O, 99%), sodium hydroxide (NaOH, ≥96%), sodium borohydride (NaBH4, ≥98%), sodium carbonate (Na2CO3, 99.95–100.05%), nitric acid (HNO3, 65%), bismuth(Iii) Bromide (BiBr3, 99.99%), Thiocarbamide (CH4N2S, 99%), Ethylenediaminetetraacetic acid (EDTA, 99.5%), ethanol (CH3CH2OH, 99.7%) were all purchased from Beijing InnoChem Science & Technology Co., Ltd. All chemical reagents were used without further purification, and ultrapure water was used throughout the experiment.
Preparation of Bi2S3
In a typical synthetic procedure, First, 0.1455 g of Bi(NO3)3·5H2O was dissolved in 20 mL of ethylene glycol under vigorous stirring for 15 min. Subsequently, 0.114 g of CH4N2S was added to the solution and stirred vigorously for another 15 min. The mixture was transferred to a 50 mL Teflon-lined stainless-steel autoclave, sealed, and heated in an oven at 180 °C for 24 h. After natural cooling to ambient temperature, the resulting precipitate was washed separately with deionized water and ethanol, then dried at 70 °C for 8 h to obtain Bi2S3.
Preparation of Bi nanosheets
Firstly, 2.4254 g of Bi(NO3)3·5H2O was weighed and dissolved in 50 mL of deionized water under vigorous stirring for 15 min. Subsequently, 4 g of NaOH was added to the solution, and vigorous stirring was continued for another 15 min. The mixture was transferred to a 50 mL Teflon-lined stainless-steel autoclave, sealed, and heated in an oven at 120 °C for 10 h. After natural cooling to ambient temperature, the resulting precipitate was washed separately with deionized water and ethanol, then dried at 60 °C for 8 h. Next, 0.2 g of the dried sample was dissolved in a 50 mL mixed solution of deionized water and ethanol (volume ratio of ethanol to deionized water = 1:1) under vigorous stirring for 30 min. Then, 2 g of NaBH4 was added to the mixture, and stirring was continued vigorously for an additional 2 h. The final precipitate was washed separately with deionized water and ethanol, followed by drying at 60 °C for 6 h to obtain metallic Bi.
Preparation of Bi2O3 nanosheets
Firstly, 5.21 g of Bi(NO3)3·5H2O was dissolved in 27 mL of 1 M nitric acid to form solution A. Separately, 6.78 g of Na2CO3 was dissolved in 107 mL of deionized water to prepare solution B. Solution B was then added dropwise into vigorously stirred solution A, followed by continuous vigorous stirring for 15 min. The mixture was allowed to stand at 60 °C for 12 h. The resulting precipitate was washed with deionized water and ethanol, respectively, and dried at 60 °C for 6 h. The dried sample was calcined at 350 °C with a heating rate of 10 °C min−1 for 30 min to obtain Bi2O3.
Preparation of Bi19Br3S27 nanorods
2.2436 g of BiBr3, 0.3502 g of CH4N2S (thiourea), and 0.2 g of EDTA were weighed and dissolved together in 60 mL of ethanol under vigorous stirring for 1 h. The mixture was transferred to a 100 mL Teflon-lined stainless-steel autoclave, sealed, and heated in an oven at 180 °C for 72 h. After natural cooling to ambient temperature, the resulting precipitate was washed separately with deionized water and ethanol, then dried at 80 °C for 4 h to obtain Bi19Br3S27.
Preparation of Bi/Bi2O3
Bi/Bi2O3 (BBO) was synthesized via an in-situ reduction method. Specifically, 0.2 g of Bi19Br3S27 was dissolved in 50 mL of a mixed solution of deionized water and anhydrous ethanol (volume ratio of ethanol to deionized water = 1:1) under vigorous stirring for 30 min. Subsequently, 2 g of NaBH4 was dissolved in 10 mL of deionized water, and the NaBH4 solution was slowly added dropwise into the Bi19Br3S27 suspension while maintaining vigorous stirring. After complete addition, stirring was continued for 2, 4, or 6 h. The resulting precipitate was washed five times with deionized water and dried at 60 °C for 6 h to obtain Bi/Bi2O3 (BBO-2, BBO-4, and BBO-6).
Photocatalytic CO2 reduction activity measurement
In a typical reaction, 20 mg of the catalyst was dispersed in a centrifuge tube containing 2 mL of deionized water and sonicated for 5 min. The dispersed catalyst suspension was then drop-cast onto a quartz plate using a pipette, and the quartz plate was dried in a vacuum oven at 60 °C until completely dry. The catalyst-loaded quartz plate was placed on a quartz column inside a photoreactor (400 mL), followed by the addition of 5 mL of deionized water and sealing of the reactor. High-purity CO2 (99.999%) was used as the carbon source. A multi-channel atmosphere controller evacuated and purged the sealed photoreactor with high-purity CO2 10 times to ensure a CO2-saturated environment. The sealed photoreactor was then irradiated under a 300 W xenon lamp for 4 h. Gas samples (1 mL) were collected hourly using a microsyringe and analyzed by gas chromatography (GC) equipped with thermal conductivity (TCD) and flame ionization (FID) detectors. The CH3OH product quantitative analysis was employed on an on-line gas chromatograph (GC-Agilent Technologies 8860, USA) equipped with thermal conductivity detector (TCD) and a flame ionized detector (FID). Furthermore, FID detector was connected with a HP-PLOT Q capillary column (Agilent, length 30 m, ID 0.53 mm, film 40 μm) for separation of C1-C6 paraffin and olefin hydrocarbons, alcohols and oxygenated compounds. The quantification of the products was based on the external standard and the use of a calibration curve.
Electrochemical and photoelectrochemical measurements
Transient photocurrent response, electrochemical impedance spectroscopy, and Mott–Schottky measurements were performed using a CHI760E electrochemical workstation (CHI, China) in a three-electrode quartz cell. A 300 W xenon lamp (MC-PF300C, Beijing MerryChange Technology Co., Ltd, China) served as the light source. The three-electrode system consisted of an Ag/AgCl reference electrode, a Pt wire counter electrode, and a working electrode made from the photocatalyst-coated fluorine-doped tin oxide (FTO, 1.5 × 2.2 cm). The electrolyte was 0.5 M Na2SO4 aqueous solution. The working electrode was fabricated as follows: 10 mg of the as-prepared photocatalyst was dispersed in 1 mL of ethanol, followed by the addition of 40 μL of Nafion solution. The mixture was ultrasonicated for 10 min. Then, 400 μL of the resultant suspension was drop-cast onto the FTO substrate and dried under near-infrared (NIR) irradiation.
Computational method
The first-principle calculations were carried out within DFT, using the Vienna Ab Initio Simulation Package code40. The interaction between atomic cores and valence electrons was described by the frozen-core projector augmented-wave method41, while the electron-electron exchange and correlation interactions were treated with the generalized gradient approximation in the form of the Perdew–Burke–Ernzerhof functional42. A kinetic energy cutoff of 500 eV was adopted to ensure full convergence of the calculations. Supplementary Data 1–4 for details on the models of Bi, Bi2O3, and Bi/Bi2O3 heterojunction with different sites have been built based on a collection of characterization results. Owing to the large cell of Bi (012)/Bi2O3(100) heterostructure, the Brillouin zone was sampled with the k-grid of 2 × 2 × 1. Structural relaxation was performed using the conjugate gradient (CG) method, by simultaneously minimizing the total energy and interatomic forces. The convergence threshold for the total energy was set to 10−5 eV. To properly describe van der Waals interactions, the dispersion-corrected DFT-D3 scheme was employed. In addition, a vacuum layer of 1.5 nm was introduced along the c-axis to avoid spurious interactions43. Gibbs free energies for each gaseous and adsorbed species were calculated at 298.15 K, according to the expression:
where EDFT is the electronic energy calculated for specified geometrical structures, EZPE the zero-point energy, S is the entropy, \(\hslash\) is the Planck constant, \(v\) is the computed vibrational frequencies, Θi is the characteristic temperature of vibration, k is the Boltzmann constant, and R is the molar gas constant. For adsorbates, all 3N degrees of freedom were treated as frustrated harmonic vibrations.
For each step, the ΔG is the difference of product or intermediate and reactant.
ΔG[CH3OH*] = G[CH3OH *] + G[H2O (g)]−(G[*] + G[CO2] + 6*G[H++e−])
where * is the substrate, U is the applied overpotential and e is elementary charge. In this study, U = 0 V versus reversible hydrogen electrode.
Data availability
All study data are included in the article and Supplementary Information. Source data are provided with this paper.
References
Yu, Y. T., Zhu, Z. J., Chen, F., Ma, T. Y. & Huang, H. W. Triggering asymmetric layer displacement polarization and redox dual-sites activation by inside-out anion substitution for efficient CO2 photoreduction. Adv. Mater. 36, 2413835 (2024).
Hou, W. D. et al. Radiation-synthesis of covalent bonding heterojunctions for selective solar-driven CO2 reduction. Mater. Today 84, 1–9 (2025).
Zhang, H. G. et al. Photocatalytic asymmetric C-C coupling for CO2 reduction on dynamically reconstructed Ruδ+-O/Ru0-O sites. Nat. Commun 16, 534 (2025).
Xu, Y. et al. Constructing asymmetric dual active sites through symbiotic effect for achieving efficient and selective photoreduction of CO2 to C2H4. Energy Environ. Sci. 17, 5060–5069 (2024).
Wu, Y. et al. Selective CO2-to-C2H4 photoconversion enabled by oxygen-mediated triatomic sites in partially oxidized bimetallic sulfide. Angew. Chem. Int. Ed. 62, e202301075 (2023).
Xu, Y. et al. Co single atom modulating the secondary coordination environment of Bi sites for boosting the adsorptive and catalytic capacity during CO2 photoreduction. Appl. Catal. B Environ. 340, 123230 (2024).
Wang, Y. et al. Lattice mismatch at the heterojunction of perovskite solar cells. Angew. Chem. Int. Ed. 63, 202405878 (2024).
Yang, Y. F. et al. Hierarchical Ohmic contact interface engineering for efficient hydrazine-assisted hydrogen evolution reaction. Adv. Mater. 36, 2307979 (2024).
Liu, L. Z. et al. Tunable interfacial charge transfer in a 2D-2D composite for efficient visible-light-driven CO2 conversion. Adv. Mater. 35, 2300643 (2023).
Wang, X. H. et al. “O-S” charge transfer mechanism guiding design of a ZnIn2S4/SnSe2/In2Se3 heterostructure photocatalyst for efficient hydrogen production. ACS Catal. 13, 1020–1032 (2023).
Liu, G. P. et al. Construction of Bi nanoparticles loaded BiOCl nanosheets Ohmic junction for photocatalytic CO2 reduction. Acta Phys. Chim. Sin. 40, 2306041 (2024).
Dai, W. L. et al. Constructing robust Bi active sites in situ on α‑Bi2O3 for Efficient and selective photoreduction of CO2 to CH4 via directional transfer of electrons. ACS Catal. 13, 2513–2522 (2023).
Fan, Y., Hao, X., Yi, N. & Jin, Z. Strong electronic coupling of Mo2TiC2 MXene/ZnCdS ohmic junction for boosting photocatalytic hydrogen evolution. Appl. Catal. B Environ. Energy 357, 124313 (2024).
Ren, L. T. et al. Cascaded *CO−*COH intermediates on a nonmetallic plasmonic photocatalyst for CO2-to-C2H6 with 90.6 % selectivity. Angew. Chem. Int. Ed. 63, e202404660 (2024).
Lu, C. H. et al. Constructing surface plasmon resonance on Bi2WO6 to boost high-selective CO2 reduction for methane. ACS Nano 15, 3529–3539 (2021).
Mao, C. L. et al. Beyond the thermal equilibrium limit of ammonia synthesis with dual temperature zone catalyst powered by solar light. Chem 5, 2702–2717 (2019).
Wu, Z. Y. et al. Anisotropic plasmon resonance in Ti3C2Tx MXene enables site-selective plasmonic catalysis. ACS Nano 19, 1832–1844 (2025).
Lv, L. et al. Coordinating the edge defects of bismuth with sulfur for enhanced CO2 electroreduction to formate. Angew. Chem. Int. Ed. 62, e202303117 (2023).
Liu, M. W. et al. Anisotropic dual S-scheme heterojunctions mimic natural photosynthetic system for boosting photoelectric response. Angew. Chem. Int. Ed. 63, e202407481 (2024).
Luo, L. et al. Selective photoelectrocatalytic glycerol oxidation to dihydroxyacetone via enhanced middle hydroxyl adsorption over a Bi2O3-incorporated catalyst. J. Am. Chem. Soc. 144, 7720–7730 (2022).
Feng, X. Z. et al. Bi2O3/BiO2 nanoheterojunction for highly efficient electrocatalytic CO2 reduction to formate. Nano Lett. 22, 1656–1664 (2022).
Jia, G. R. et al. Size Effects of highly dispersed bismuth nanoparticles on electrocatalytic reduction of carbon dioxide to formic acid. J. Am. Chem. Soc. 145, 14133–14142 (2023).
Gu, M. L. et al. Bismuth nanoparticles and oxygen vacancies synergistically attired Zn2SnO4 with optimized visible-light-active performance. Nano Energy 80, 105415 (2021).
Yu, L. H. et al. Twisted BiOCl Moiré superlattices for photocatalytic chloride reforming of methane. J. Am. Chem. Soc. 146, 32816–32825 (2024).
Huang, B. J. et al. Chemically bonded BiVO4/Bi19Cl3S27 heterojunction with fast hole extraction dynamics for continuous CO2 photoreduction. Adv. Powder Mater. 3, 100140 (2024).
Niu, R. R. et al. Black phosphorus/Bi19Br3S27 van der Waals heterojunctions ensure the supply of activated hydrogen for effective CO2 photoreduction. Appl. Catal. B Environ. 317, 121727 (2022).
Xu, F. Y. et al. Prolonging charge carrier lifetime via intraband defect levels in S-scheme heterojunctions for artificial photosynthesis. Angew. Chem. Int. Ed. 64, e202414672 (2025).
Wan, X. Y. et al. A nonmetallic plasmonic catalyst for photothermal CO2 flow conversion with high activity, selectivity and durability. Nat. Commun. 15, 1273 (2024).
Li, J. et al. Selective CO2 photoreduction into CH4 triggered by the synergy between oxygen vacancy and Ru substitution under near-infrared light irradiation. Adv. Sci. 11, 2405668 (2024).
Li, W. L. et al. Nitrogen-bridged S-N-Cu sites for CO2 photoreduction to ethanol with 99.5% selectivity in pure water. Angew. Chem. Int. Ed. 64, e202423859 (2025).
Tian, J. Q. et al. Enabling interfacial lattice matching by selective epitaxial growth of CuS crystals on Bi2WO6 nanosheets for efficient CO2 photoreduction into solar fuels. Angew. Chem. Int. Ed. 64, e202418496 (2025).
Huang, F. X., Wang, F., Liu, Y. & Guo, L. J. Cu-ZnS modulated multi-carbon coupling enables high selectivity photoreduction CO2 to CH3CH2COOH. Adv. Mater. 37, 2416708 (2025).
Zhang, J. J. et al. Strongly coupled interface in electrostatic self-assembly covalent triazine framework/Bi19S27Br3 for high-efficiency CO2 photoreduction. ACS Nano 19, 2759–2768 (2025).
Chen, Z. H. et al. Precision molecular engineering of carbon nitride for efficient and selective photoreduction of CO2 to C2H6 in pure water. Adv. Funct. Mater. 35, 2423213 (2025).
Li, X. D. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Jiang, J. W. et al. Understanding dual-vacancy heterojunction for boosting photocatalytic CO2 reduction with highly selective conversion to CH4. Appl. Catal. B Environ. 316, 121679 (2022).
Zhang, W. Y., Deng, C. Y., Wang, W., Sheng, H. & Zhao, J. C. Achieving almost 100% selectivity in photocatalytic CO2 reduction to methane via in-situ atmosphere regulation strategy. Adv. Mater. 36, 2405825 (2024).
Hu, P. Y., Zhang, J. J., Liang, G. J., Yu, J. G. & Xu, F. Y. Nonmetal plasmon-induced carrier backflow and prolonged lifetime for CO2 photoreduction. ACS Catal. 14, 15025–15035 (2024).
Deng, X. Y. et al. Ultrafast electron transfer at the In2O3/Nb2O5 S-scheme interface for CO2 photoreduction. Nat. Commun. 15, 4807 (2024).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558 (1993).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. DFT-D3-A dispersion correction for density functionals, Hartee-Fock and semi-empirical quantum chemical methods DFT-D3. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC) (No. 22308336) and the Joint Fund of Science and Technology R&D Plan of Henan Province (No. 242301420029).
Author information
Authors and Affiliations
Contributions
W.G. and Y.Z. contributed equally to this work. W.G., Y.Z., and J.T. performed most of the experiments and wrote the first version of the paper; Z.L. and B.L. helped to revise the paper; J.L. constructed and planned the whole project and revised the paper. All the authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ruonan Wang, Yong Xu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, W., Zhang, Y., Tian, J. et al. Atomic-scale interface engineering in Bi/Bi2O3 heterojunctions for selective CO2 photoreduction to methanol. Nat Commun 17, 983 (2026). https://doi.org/10.1038/s41467-025-67718-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67718-0
