
Abstract
The electrochemical co-reduction of CO2 and nitrate provides a sustainable route for urea synthesis via C–N coupling, yet kinetic limitations and poor intermediate interactions hinder urea yields. Here, we engineer a nano-confined CuRu bimetallic catalyst within mesoporous carbon hollow spheres (MCHS) to overcome these barriers. By spatially confining reactants and intermediates, the catalyst achieves a urea yield of 12.51 g h–1 gcat–1 at 250 mA cm–2, with 125-hour stability. In situ spectroscopy and computational analyses reveal that nano-confinement switches the C–N coupling pathway from the thermodynamically favored *COOH–*NH2 to kinetically driven *OCO–*NO intermediates, bypassing energy barriers. Precise pore-size engineering (4–11 nm) demonstrates that optimal confinement simultaneously enhances reactant transport and intermediate retention, boosting selectivity. This work establishes nano-confinement as a versatile approach for controlling multi-step electrocatalytic processes, enabling sustainable chemical synthesis.
Similar content being viewed by others

Introduction
The synthesis of organic compounds (urea, amines, and amino acids) through C‒N bond formation using CO2 and nitrogen sources (N2, NO, NO3–, NO2–) as the primary reactants has garnered significant attention1,2,3. The co-utilization of anthropogenic nitrogen pollutants and greenhouse gases to produce economically valuable products aligns with both environmental protection and carbon neutrality goals4,5. The electrochemical synthesis of urea from nitrate wastewater and CO2 under renewable electricity-driven conditions offers a green alternative to the conventional two-step Bosch–Meiser process (19.7 GJ/metric tonne urea), achieving 32% lower energy consumption (13.3 GJ/metric tonne urea) through single-step C‒N coupling while integrating greenhouse gas utilization and reduced pollutant emissions6,7.
The co-reduction of CO2 and NO3– for urea synthesis, involving a 16-electron transfer mechanism and a triphasic reaction system of gaseous and liquid reactants on solid electrode, is constrained by kinetic limitations and multi-phase interaction challenges, significantly limiting conversion efficiency and urea production yield. Designing multimetallic catalysts with distinct catalytic sites have been proposed as effective strategies for C–N coupling8,9,10,11,12,13,14,15. Yet the urea yield remains challenging to improve (typically below 2.4 g h–1 gcat–1 in H-type cells)16,17,18,19, primarily due to sluggish C–N coupling kinetics caused by limited CO2 adsorption capacity of metal catalysts20, inadequate local reactant concentrations (CO2/NO3–) under high current densities from poor mass transfer21,22,23, and disordered adsorption/conversion processes that hinder critical intermediate interactions along the urea formation pathway20. Enhancing the adsorption capacity of CO2/NO3–, accelerating their rapid conversion, and stabilizing critical intermediates constitute a direct strategy to boost catalytic efficiency and improve urea production yields.
Nano-confinement architecture design is a promising strategy to spatially confine reaction pathways within defined domains, which may thereby stabilize reactive intermediates and regulate active component dispersion24,25,26,27,28. Engineering spatial configurations to construct nano-confinement provides a pathway to overcome kinetic limitations of metal catalysts during reactant conversion. Previous studies have shown that structure-induced confinement effects enhance electrochemical CO2 reduction (CO2RR) through limiting the spatial mobility of electrocatalysts, stabilizing key intermediates, and regulating mass transfer between electrode and electrolyte29,30. To date, the nano-confinement effect on precisely controlling C‒N coupling pathways and urea yield remains unexplored. It is largely unknown how does nano-confinement affect the key C‒N coupling pathways and intermediate species, kinetics, and reactant/intermediate distribution in the microenvironment near catalysts. Moreover, tuning the dimension of nano-confinement structure may have impact on the adsorption-conversion thresholds during C‒N coupling, which is also needed to be systematically studied.
Carbon nanoreactors enable precise pore size regulation across broad nanoscale dimensions (2-50 nm), serving as stable conductive matrices while providing ideal template for nano-confined catalyst synthesis31. We prepared mesoporous carbon hollow spheres (MCHS) and solid carbon spheres (CS) as catalyst supports to investigate the effect of nano-confinement on C‒N coupling, using Cu and Ru as bimetallic catalytic sites (Fig. 1a). Benefited from CuRu synergy and nano-confinement effects, CuRu/MCHS achieved a urea yield rate of 12.51 g h–1 gcat–1 at a current density of 250 mA cm–2, along with long-term stability over 125 hours, demonstrating competitive performance compared with previous reports. In situ characterizations and theoretical calculations indicated that nano-confinement effect altered the key C‒N coupling routes from *NH2‒*COOH on CuRu/CS to *NO‒*OCO on CuRu/MCHS. A precise engineering of the pore sizes of CuRu/MCHS (4 nm, 7 nm, and 11 nm) revealed that the pore dimension governs urea selectivity and yield rate by modulating local concentrations and diffusion kinetics of reactants and intermediates.

a Schematic illustrating the design principles of confined and non-confined systems in urea electrosynthesis. b Illustration for the synthesis process and microstructure of MCHS, CS, CuRu/CS and CuRu/MCHS. c SEM image, d TEM, e HAADF-STEM and EDS elemental mapping profile of CuRu/CS. f SEM image, g HAADF-STEM image and h EDS elemental of a single CuRu/MCHS. i TEM images and the particle sizes distribution in a single sphere and j, k the TEM images of carbon shell of CuRu/MCHS.
Results and Discussion
Preparation of nano-confined bimetallic catalysts
The nano-confined MCHS support fabrication involved in situ synthesizing templates in a one-pot and surfactant-free process (Fig. 1b). Cu and Ru were further encapsulated within MCHS and CS to obtain CuRu/MCHS and CuRu/CS. The molar ratios of Cu:Ru in the as-synthesized products determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) were comparable with those of Cu:Ru precursors (24:1) (Table S1). Scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-angle annular dark-field scanning TEM (HAADF-STEM) and EDS mapping analyses of CuRu/CS showed a solid and pore-free structure (Figs. 1c–1e and Fig. S1). CuRu nanoparticles (average diameters of 9.16 ± 3.69 nm) were loaded in an amorphous form on the surface of the carbon sphere, with Cu uniformly dispersed on the carbon support and Ru distributed more loosely over the Cu-rich regions (Fig. S2). In contrast, the pore structure of MCHS shell walls was preserved after Cu and Ru encapsulation (Fig. 1f and Fig. S3). The Barrett-Joyner-Halenda (BJH) model indicated an average pore diameter of 7.55 nm. The Brunauer-Emmett-Teller (BET) surface area of CuRu/MCHS (721.7 m2 g–1) was lower than that of MCHS (1495.46 m2 g–1), attributing to the formation of metal clusters inside the pores and on the shell walls (Table S2). A single CuRu/MCHS sphere was 214.0 ± 4.0 nm in diameter and 52.0 ± 1.9 nm in shell thickness, with highly ordered radial channels (Fig. 1g and Table S3). The EDS mapping and line-scan indicated the high dispersion of Cu and Ru on MCHS (Fig. 1h), forming CuRu nanoclusters (average diameters of 2.08 ± 0.61 nm) dispersed throughout the channels and cavities (Figs. 1i-1k and Fig. S4). The lattice stripes with spacings of Cu(111) (0.208 nm), Ru(100) (0.233 nm) and Ru(002) (0.224 nm) crystal surfaces were identified in aberration-corrected HAADF-STEM images of a single nanocluster (Fig. S5). No characteristic lattice spacings of the CuRu alloy were observed32,33, indicating that Cu and Ru coexist primarily as adjacent or interfaced monometallic domains.
Raman analysis showed that the ratio of D and G band intensity (ID/IG) of CuRu/CS (0.97) was higher than that of CS (0.91), attributing to the increased defect density caused by the metal particles attached to the surface (Fig. 2a). The ID/IG value of CuRu/MCHS was 0.91, nearly identical to that of MCHS (0.92), suggesting that the introduction of CuRu nanoclusters on MCHS had little impact on its carbon defects34. Powder X-ray diffraction (XRD) patterns displayed the diffraction patterns of metallic Cu (111) (200), and (220) planes as well as Ru (100) (002) (101) and (110) planes (Fig. S6a). In the X-ray photoelectron spectroscopy (XPS) spectra, the Ru 3d signals were only observed in CuRu/CS (Figs. S6b and S6c). The peak intensities of Cu 2p and Ru 3p in CuRu/MCHS were significantly lower than those in CuRu/CS, further confirming that the metals were effectively encapsulated within the MCHS rather than on the surface (Figs. 2b and 2c). For both CuRu/MCHS and CuRu/CS, characteristic peaks of Cu0/1+, Ru0, and Ruδ+ were identified35. By quantifying the content of Cu0 and Cu1+ in the Cu LMM spectra (Fig. S6d), the high proportion of Cu0 was found in CuRu/MCHS (75.1%) and CuRu/CS (80.3%).

a Raman spectra. ID/IG represents the ratio of the D band to G band intensity. b Cu 2p XPS spectrum. c Ru 3p XPS spectrum. Normalized d Cu K-edge and e Ru K-edge XANES spectra. The k3-weighted Fourier-transform f Cu K-edge and g Ru K-edge EXAFS spectra, and without phase-corrected. Wavelet transforms of the k3-weighted Cu K-edge EXAFS signals for h CuRu/MCHS and i CuRu/CS and of the Ru K-edge of j CuRu/MCHS and k CuRu/CS.
In X-ray absorption near-edge structure (XANES), the Cu K-edge spectra showed that the pre-edge absorption energy of CuRu/MCHS and CuRu/CS were located between those of Cu foil and Cu2O references, implying the coexistence of metallic Cu (Cu0) and Cu (I) oxide (Cu+) phases (Fig. 2d). The minor Cu+ component likely originated from superficial oxidation during post-synthetic air exposure of the catalysts36. The Ru K-edge spectra of CuRu/MCHS and CuRu/CS were situated between Ru foil (0) and RuO2 (+4) (Fig. 2e). The k3-weighted Fourier transform Extended X-ray absorption fine structure (FT-EXAFS) oscillations (Fig. 2f and 2g) and the wavelet transforms (WT) (Fig. 2h–2k) further confirmed the coordination structure for Cu and Ru. Based on the fitting results (Fig. S7, Table S4 and S5), the coordination numbers (CN) of Cu‒Cu (2.6 Å, CN = 4.8) and Ru‒Ru (2.7 Å, CN = 1.6) bonds in CuRu/MCHS were lower than those in CuRu/CS (Cu‒Cu: 2.6 Å, CN = 8.0; Ru‒Ru: 2.7 Å, CN = 2.9). The lower coordination number in CuRu/MCHS reflected the ability of MCHS to restrict metal aggregation and growth, resulting in the presence of smaller nanoclusters, which was consistent with HAADF-STEM results. In combination with the AC HAADF-STEM images, these results strongly indicate that a Cu–Ru alloyed structure was not formed, or, if present, its proportion is negligible37,38. Cu and Ru are normally hard to form alloy due to distinct crystal structures, with Cu adopting a face-centered cubic (fcc) structure and Ru adopting a hexagonal close-packed (hcp) structure39,40.
Urea electrosynthesis under nano-confinement
We first investigated the urea electrosynthesis performance in 0.1 M KNO3 with gaseous CO2 in an H-cell. Electrochemical impedance spectra (EIS) and cyclic voltammetry (CV) curves showed that compared to CuRu/CS, CuRu/MCHS had lower charge transfer resistance (Rct), faster ion diffusion, higher double-layer capacitance (Cdl) (775 μF cm–2 vs. 491 μF cm–2) and electrochemical active surface area (ECSA) (36.9 cm2 vs. 23.4 cm2) (Fig. S8). As determined by linear sweep voltammetry (LSV) curves, the CuRu/MCHS exhibited the CO2/NO3– co-reduction performance with the largest current density of 40 mA cm–2 at –1.3 V (vs. RHE), surpassing CuRu/CS (22 mA cm–2) (Figs. S9a and S9b). Despite a small absolute offset after iR correction, the relative catalyst activity order was preserved, confirming that our comparative conclusions were robust to ohmic drop correction (Figs. S9c and S9d). Notably, with the introduction of CO2, the significant reduction in cathodic current density was attributed to the effective suppression of nitrate reduction (NO3RR) to other products and hydrogen evolution (HER), which promoted C–N coupling.
The introduction of nano-confinement structure significantly enhanced urea yield across a range of applied potentials between –0.7 and –1.1 V (vs. RHE). Further confirmation of urea synthesis was provided by 14N NMR and 15N isotope NMR (Fig. S10). The CuRu/MCHS catalysts exhibited a urea yield rate of 3.61 ± 0.22 g h–1 gcat–1 at –1.1 V (vs. RHE), approximately 2.5 times higher than that of CuRu/CS (1.43 ± 0.13 g h–1 gcat–1) (Fig. 3a). Urea quantification was cross-validated using both the enzymatic urease and high-performance liquid chromatography (HPLC) methods (Fig. S11)41, with the yields showing strong consistency and a marginal difference. This agreement confirms the accuracy and reliability of the measured urea yields. The optimal urea faradaic efficiency (FE) of CuRu/MCHS was 16.5 ± 1.2% at –0.7 V (vs. RHE), and 13.3 ± 1.3% at –1.1 V (vs. RHE) accompanied by a high NO3– conversion rate reaching 35.9% after only 30 minutes of electrochemical reduction, corresponding to a reaction rate of k = 0.0148 min–1 and 150 g-N h–1 gcat–1 (Figs. S12 and S13). In contrast, the NO3– conversion rate for CuRu/CS decreased to 23.4% with a lower reaction kinetics (k = 0.0089 min–1 and 98 g-N h–1 gcat–1). The urea FE also decreased to 7.2 ± 1.3% at –1.1 V (vs. RHE) and 7.6 ± 2.2% at –0.7 V (vs. RHE), attributing to the tendency toward side product formation. A higher working current density was observed from CuRu/MCHS (37 mA cm–2) than CuRu/CS (21 mA cm–2) at –1.1 V (vs. RHE) (Fig. S14).

a Urea yield rate of CuRu/MCHS and CuRu/CS at different potentials. b Potential-dependent FEs of primary products for CuRu/MCHS assessed in 0.1 M KNO3 + CO2. The proportion of Cu to Ru used in preparing CuRu/MCHS and CuRu/CS was fixed at a molar ratio of 24:1 (Cu24Ru1/MCHS). c Comparison of the performance for urea electrosynthesis by Cu/MCHS, Ru/MCHS, MCHS, Cu24Ru2/MCHS, Cu24Ru1/MCHS, Cu24Ru0.5/MCHS, Cu16Ru1/MCHS and Cu8Ru1/MCHS as electrocatalyst at the potentials of –0.7, –0.8, –0.9, –1.0 and –1.1 V (vs. RHE). d Stability testing of CuRu/MCHS in KNO3 + CO2 with –0.7 V (vs. RHE) (inset: Cycling stability performance of CuRu/MCHS in urea electrosynthesis at –1.1 V (vs. RHE)). e The photo and schematic diagram of the flow cell installation (inset: SEM image of the cross-sectional view of the cathode and the CuRu/MCHS catalyst layer). f Urea yield rate of CuRu/MCHS in the flow cell at the potential range from –0.7 V (vs. RHE) to –1.1 V (vs. RHE) (left axis) and the performance evaluated by chronopotentiometry (CP) testing form 50 mA cm–2 to 250 mA cm–2 (right axis). g FE of urea performance of CuRu/MCHS at various potential in flow cell. The tests in (a–d) were performed in an H-cell. The data in (a, b, f and g) are presented as mean values ± s.d. (n ≥ 3).
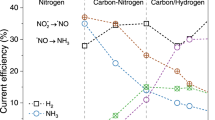
Detailed analysis of the primary products (urea, NH4+, NO2–, H2, CH4, N2, NH2OH, and HCOOH) revealed that CuRu/CS exhibited a diversified product distribution (Figs. S15 and S16). In contrast, CuRu/MCHS significantly suppressed the product of NO2– and promoted its further hydrogenation, leading to an effective decrease in the FE of NO2– (from 22.5 ± 2.2% to 10.5 ± 0.7%) at –1.1 V (vs. RHE) (Fig. 3b). Moreover, the formation tendency of the byproduct HCOOH was markedly suppressed, as evidenced by a decrease in FE from 12.5 ± 3.0% to 0.7 ± 0.1% at –1.1 V (vs. RHE). Within the tested potential range, CuRu/MCHS produced only trace amounts of N2, NH2OH, and HCOOH, with FEs all below 1%. Further evaluation of the performance of Cu/MCHS, Ru/MCHS, and CuRu/MCHS with varying molar ratios between Cu and Ru indicated that Ru/MCHS showed a strong tendency for H2 production, while Cu/MCHS favoured NO2– (26.9 ± 1.1%) and HCOOH (10.4 ± 2.2%) generation (Fig. S17). We have systematically evaluated the urea synthesis performance of CuRu/MCHS catalysts with varying Ru loadings (Fig. 3c). The urea production rate exhibited a distinct volcano-type dependence on Ru content, with the Cu:Ru = 24:1 ratio demonstrating the highest yield. This catalyst also achieved the highest turnover frequency (TOF) among all compositions (Table S6), confirming its intrinsic activity and optimal atomic utilization efficiency. These findings indicate that an optimal Ru content is critical for promoting the deep reduction of NO2– intermediates while appropriately regulating the *H supply—directing it toward C–N coupling rather than hydrogen evolution, thereby maximizing urea formation.
The current density of CuRu/MCHS in 20-cycle experiments under –1.1 V (vs. RHE) stayed constant in the range of 33–37 mA cm–2 (Fig. 3d inset). During long-term durability test over 125 hours, minor current density fluctuations (<0.01 A cm–2) were observed, demonstrating stable maintenance of catalytic activity (Fig. 3d). Additionally, the low amounts of Cu2+ and Ru4+ detected in the post-reaction electrolyte confirmed the robustness of Cu and Ru nanoclusters on MCHS (Table S7). The electrolyte pH exhibited only a moderate increase from 4.1 to 4.9 for CuRu/MCHS and to 4.7 for CuRu/CS after electrolysis. This limited change indicates that the substantial H+ consumption was effectively mitigated by the buffering capacity of the CO2-saturated solution (Fig. S18). The chemical valence states of Cu and Ru remained largely unchanged after the reaction (Fig. S19). The CuRu nanoclusters maintained a uniform dispersion with an average diameter of 2.39 ± 1.21 nm and showed no obvious aggregation. Moreover, the hollow cavity structure and mesoporous channels were well preserved (Fig. S20).
A gas diffusion electrode (GDE) flow cell was further applied to overcome the mass transport limitations in the H-cell (Fig. 3e). After loading onto the gas diffusion membrane, the CuRu/MCHS powders remained uniformly dispersed and maintained the three-dimensional spherical structure, facilitating the contact and diffusion of CO2/NO3– within the nanocatalyst (Fig. 3e inset). The operating current density in the flow cell was significantly increased compared to H-cell (Fig. S21a). Under constant voltage (CV) or constant current (CC) operation modes, the urea yield rate increased with higher potentials or current densities (Fig. 3f, Figs. S21 and S21b). Meanwhile, benefiting from the enhanced three-phase interfacial mass transfer effect, the flow cell increased the urea FE to 19.4 ± 4.5% at –1.0 V (vs. RHE) (Fig. 3g). Notably, at an industrial-level current density of 250 mA cm–2, CuRu/MCHS achieved a competitive urea yield rate of 12.51 g h–1 gcat–1. This value demonstrates competitive performance compared to previously reported bimetallic or carbon-based catalysts under similar conditions (Fig. S22a and Table S8)11,19. In addition, the urea partial current density (jUrea), a key indicator of actual production efficiency42, reached a maximum of 22.1 mA cm–2 at –1.0 V (vs. RHE), and maintained 16.07 mA cm–2 even at a total current density of 250 mA cm–2. These values demonstrate competitive performance, with most previous studies typically reporting values below 1–5 mA cm–2 (Fig. S22b). Note that our FEs were achieved under relatively higher current density conditions and are fully comparable to the performance of other state-of-the-art catalysts under similar high-current conditions12,43,44,45. The simultaneous realization of a competitive yield rate and high jUrea at 250 mA cm–2 validates the effective capability of our nano-confinement strategy in overcoming the fundamental challenge of mass transport limitations, thereby maximizing urea production under industrially relevant conditions.
Role of nano-confinement in intermediate switching
Attenuated total reflectance-surface-enhanced IR absorption spectroscopy (ATR-SEIRAS) measurements were conducted to determine intermediates. Compared to sole CO2RR or NO3RR (Fig. 4a), co-reduction of CO2 and NO3– on CuRu/MCHS showed new peaks at 1425 cm–1 and 1477 cm–1, which were identified for the C–N bond and N–C–N bond of urea (Fig. 4b)46. The characteristic peaks at 1890 cm–1 and 1408 cm–1 were assigned to *NO18,47 and *OCO48,49,50,51 species (Table S9), demonstrating their important correlation with urea generation. With increasing potentials from −0.9 V to −1.3 V (vs. RHE), the attenuated *OCO signal was attributed to its rapid consumption, concomitant with continuous accumulation of C–N and N–C–N bonds, consistent with electrocatalytic performance. Notably, characteristic vibrational peaks for *CO (2050 cm–1)16 in CO2RR and *NH2 (3345 and 1177 cm–1)52,53 in NO3RR were markedly attenuated during CO2 and NO3– co-electroreduction, though a weak but observable *NH2 signal was still detected on CuRu/MCHS (Fig. 4b). This suggests that the pathways of CO2 → *COOH → *CO was suppressed during C–N coupling, as further confirmed by the absence of a *COOH signal on CuRu/MCHS (Fig. 4c). The limited hydrogenation of CO2 to *COOH on CuRu/MCHS also correlates with the suppressed formation of HCOOH. In contrast, the CuRu/CS catalyst exhibited distinct signals of *COOH (1395 cm−1)54 and *NH2 (3345 and 1170 cm−1) intermediates (Fig. 4d). As the potential increased from −0.1 V to −1.5 V (vs. RHE), *NH2 species were gradually consumed while *COOH intermediates initially formed and progressively accumulated. This was in accordance with the appearance of C–N and N–C–N signals, yet with much lower intensities than CuRu/MCHS (Fig. 4e). The absence of *NO and *OCO signals on CuRu/CS indicated their rapid hydrogenation into *NH2 and *COOH, hindering the effective retention of pre-hydrogenation intermediates. Conversely, in CuRu/MCHS, the *NO species generated during NO3– reduction were preserved, enabling earlier coupling with *OCO. Note that the *NH2 generated were rapidly protonated to NH4+, which limited the steady-state surface concentration of *NH2 and thus its spectral intensity, despite the significant NH4+ product accumulation observed over the full electrolysis period.

a ATR-SEIRAS measurements on CuRu/MCHS under various potentials in electrolytes of 0.1 M KHCO3 + CO2 and 0.1 M KNO3 + Ar. ATR- SEIRAS spectra of CuRu/MCHS recorded from the open-circuit state to –1.7 V (vs. RHE) in a 0.1 M KNO3 electrolyte with CO2 bubbling between b 1000–4000 cm–1 and c 1350–1500 cm–1. The same spectra of CuRu/CS in the range of d 1000–4000 cm–1 and e 1350–1500 cm–1. f CO2-TPD spectra of CuRu/MCHS and CuRu/CS. g Reaction diagrams evaluated by periodic DFT of urea production on CuRu surface and corresponding atomic configurations. The gray, blue, red, white, pink and turquoise balls represent C, N, O, H, Cu and Ru atoms, respectively. h Illustration of urea synthesis process on CuRu/MCHS and CuRu/CS.
To further solidify the findings of adsorbed forms of CO2 and NO (*OCO and *NO) as key intermediates on CuRu/MCHS, CO2-temperature-programmed desorption (CO2-TPD) and utilization of gaseous NO as the nitrogen source for C–N coupling were respectively conducted. CO2-TPD showed CO2 adsorption at 429.2 °C with a large peak area on CuRu/MCHS, indicating its strong CO2 adsorption capacity (Fig. 4f)55. In contrast, the CO2 adsorption peak of CuRu/CS appeared at 482.6 °C, with a significantly lower intensity. These suggested that the nano-confinement structure enhanced CO2 chemical adsorption, providing more accessible CO2 adsorption states for C–N coupling. Additional NO-CO2 coupling tests were conducted using 0.1 M KHCO3 as the electrolyte, achieving urea yield of 2.69 g h–1 gcat–1 at –1.1 V (vs. RHE) (Fig. S23), suggesting the capability of CuRu/MCHS to utilize NO as the initial reactant for C–N coupling.
To reveal the mechanistic differences between the nano-confined and the non-confined system, density functional theory (DFT) calculations were performed (Fig. 4g). Based on the characterizations from XPS and EXAFS analysis, a structural model with a carbon layer as the support and Ru clusters loaded on Cu clusters was constructed to simulate the CuRu structures in the CuRu/MCHS and CuRu/CS systems. Adsorption energy analyses revealed that CO2 exhibited stronger bonding affinity with Cu (–1.58 eV) than Ru (–1.40 eV), whereas NO3– preferentially interacted with Ru (–1.65 eV) than Cu (–1.12 eV) (Fig. S24a). In the *OCONO path, the coupling between *OCO and *NO to *OCONO (i.e., the first C–N coupling step) required to overcome an energy barrier of 0.79 eV. In contrast, the *COOHNH2 path exhibited a smaller energy barrier of 0.64 eV during the first C–N coupling step (*COOH + *NH2 → *COOHNH2), yet with a much longer reaction path from *OCO and *NO (Table S10 and Table S11). The subsequent proton-coupled electron transfer (PCET) process was mainly limited by the hydrogenation of *CO2. After C–N bond formation, the energy barrier for the initial *CO2 hydrogenation was reduced from 0.67 eV (*OCO + *NH2 → *COOH + *NH2) to 0.33 eV (*OCONH2 → *COOHNH2), suggesting that urea was facilely produced after the first C–N coupling.
By comparing the reaction pathways on the pure Cu cluster model, the critical role of Ru in reducing the hydrogenation energy barriers during the PCET process was verified (Table S12). The incorporation of Ru clusters further facilitated the formation of *OCONHO and *OCONH intermediates following the initial C–N bond formation (Fig. S24b-d). On the pure Cu surface, the energy barrier for the *COOHNH2 → *CONH2 step was 0.50 eV, substantially lower than that on the CuRu surface (1.16 eV). However, the energy barrier for the first carbon-side hydrogenation step (*OCONH2 → *COOHNH2) increased to 0.61 eV on Cu (Fig. S25). In addition, a lower energy barrier was observed for CO2 reduction to *COOH on the pure Cu surface (–0.27 eV vs. 0.03 eV on CuRu), rationalizing the preferential formation of HCOOH on Cu/MCHS. These results indicate that the presence of Ru modulates the CO2 hydrogenation behavior on Cu, steering the pathway toward urea production instead of HCOOH.
ATR-SEIRAS and DFT collectively elucidated that with the nano-confined structure, CuRu/MCHS enabled earlier C–N coupling through the *OCONO pathway (Fig. 4h), eliminating four N-hydrogenation and one C-hydrogenation steps versus the *COOHNH2 pathway and suppressing byproduct formation (Fig. S26)56,57,58,59. This stems from the high kinetic driving force offered by the nano-confinement effect to break through the initial thermodynamic energy barrier of *OCO–NO coupling. Whereas in the non-confined structure of CuRu/CS, the energy barrier of *OCONO pathway forced it to go through the *COOHNH2 pathway, which underwent independent and long hydrogenation reduction reactions of CO2 and NO3–, thus leading to lower urea yield rates and selectivity.
Nano-confinement size effect on C–N coupling
To elucidate that whether different nano-confinement dimensions would lead to a similar promoting effect on C–N coupling, MCHS with other two different pore diameters (4.18 nm and 11.12 nm, labelled as CuRu/MCHS-4 and CuRu/MCHS-11) were synthesized in addition to the previously mentioned CuRu/MCHS (7.55 nm, labelled as CuRu/MCHS-7), by regulating the hydrolysis and nucleation processes of silica templates during fabrication (Fig. 5a and 5b). The variations in specific surface areas and pore volumes in three catalysts were attributed to differences in cavity and wall thickness (Table S2). The small differences in the average particle sizes of CuRu nanoclusters in CuRu/MCHS-4 (1.90 ± 0.48 nm), CuRu/MCHS-7 (2.08 ± 0.61 nm), and CuRu/MCHS-11 (2.56 ± 0.77 nm) were positively correlated with their pore size differences, which was attributed to the influence of the nano-confined space on metal nucleation (Fig. 5c and S27)60. EDS mapping (Fig. 5e and 5f), XPS (Fig. S28), and ICP-OES analyses (Table S1) verified that the CuRu nanoclusters on different supports were highly dispersed with consistent loadings and valence states, minimizing the impact of metal composition differences. The electrochemical parameters of Rct and ion diffusion ability were generally close among three catalysts (Fig. S29). Among three nano-confinement scales, CuRu/MCHS-7 exhibited the highest urea FEs and yield rates compared to CuRu/MCHS-4 and CuRu/MCHS-11 within the potential range of –0.7 V to –1.1 V (vs. RHE) (Figs. 5g and S30). CuRu/MCHS-7 also displayed higher jUrea and N-selectivity (7.50 mA cm–2 and 19.0%) than CuRu/MCHS-4 (3.32 mA cm–2 and 9.2%) and CuRu/MCHS-11 (4.83 mA cm–2 and 9.5%) at –1.1 V (vs. RHE) (Fig. 5h). Notably, NO3– conversion rates after 30 minutes of electrolysis at –1.1 V (vs. RHE) increased with pore size (26.9%, 35.9%, and 38.6% for 4, 7, and 11 nm, respectively), indicating that urea production was not solely governed by nitrate conversion (Fig. S31). In contrast, the larger-pore CuRu/MCHS-11 favored the conversion of NO3– to NO2–, with FE(NO2–) ranging from 23.5% to 13.2% between –0.7 and –1.1 V (vs. RHE), compared to 16.5%–10.6% for CuRu/MCHS-7 under the same conditions (Fig. 5i). This suggests that the enhanced NO3– conversion in CuRu/MCHS-11 was not effectively directed toward urea formation, which can be attributed to the diminished nanoconfinement effect impairing the retention of key intermediates and thus the efficiency of C–N coupling.

a Schematic illustration of CuRu/MCHS with different nano-confinement scales. b N2 adsorption–desorption isotherms of MCHS-4, MCHS-7, MCHS-11, CuRu/MCHS-4, CuRu/MCHS-7 and CuRu/MCHS-11 (inset: BJH pore size distribution profiles). TEM images of c CuRu/MCHS-4 and d CuRu/MCHS-11. HAADF-STEM images, and corresponding EDS elemental mapping profile of e CuRu/MCHS-4, f CuRu/MCHS-11. g Urea yield rate and h the comparison of various properties of three catalysts with different nano-confinement scales. The N-selectivity represents the molar proportion of urea to all N-containing products (urea, NO2– and NH4+). The C-selectivity represents the molar proportion of urea to all C-containing products (urea and CH4). HCOOH, N2, and NH2OH were detected in trace amounts or almost undetectable. i The comparison of FEs for urea, H2, NO2–, NH4+ and CH4 at various potentials with CuRu/MCHS-4, CuRu/MCHS-7 and CuRu/MCHS-11. The data in g and i are presented as mean values ± s.d. (n ≥ 3) and without iR correction.
Product distributions under varying potentials showed distinct hydrogenation selectivity among three pore sizes, suggesting the important role of *H in C–N coupling. Within the potential range of –0.7 V to –1.1 V (vs. RHE), the intermediate pore architecture of CuRu/MCHS-7 exhibited the highest urea FEs. The CuRu/MCHS-11 promoted HER to form H2, exhibited higher H2 FEs (14.4% ~ 43.3%) than CuRu/MCHS-4 (6.2% ~ 18.6%) and CuRu/MCHS-7 (11.7% ~ 32.4%). On the other hand, the NH4+ FEs (54.1% ~ 59.7%) of CuRu/MCHS-4 was higher than CuRu/MCHS-7 (36.4% ~ 42.5%) and CuRu/MCHS-11 (32.9% ~ 40.8%), suggesting the restricted smaller pores appeared to hinder C–N formation via favoring NO3– hydrogenation pathways to form NH4+.
To confirm the role of *H in urea synthesis, the catholyte was replaced by D2O61. The urea yield of CuRu/MCHS-7 decreased by 72% from 3.74 ± 0.25 to 1.00 ± 0.19 g h–1 gcat–1 at –1.1 V (vs. RHE) (Fig. 6a and Fig. S32), yielding a kinetic isotope effect (KIE, defined as the ratio of urea formation rates in H2O vs D2O) value of 3.6. This significant KIE ( > 1) confirms that proton-transfer events are involved in the rate-determining step of the C‒N coupling mechanism62. Combined with our DFT calculations, we propose that while the initial OCO‒NO coupling presents a substantial energy barrier, the subsequent hydrogenation steps along the *OCONO pathway involve sequential *H incorporation through PCET processes (Fig. 6b)63,64. The observed KIE likely arises from one or more of these PCET steps, where *H acts as a crucial hydrogen source for stabilizing key intermediates such as *OCONO and facilitating its conversion to urea. This interpretation aligns with the role of *H as an essential participant in the hydrogenation of N‒O and C‒O moieties during the later stages of the C‒N coupling pathway, rather than being limited solely to the initial Volmer step. To compare the production of *H from varied nano-confined structures, electron paramagnetic resonance (EPR) was performed using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the radical trapping reagent (Fig. 6c). Under identical pH conditions, CO2 introduction enhanced *H concentration in the reaction system. This may result from suppressed *H consumption by NO3RR in the presence of CO2 (Fig. S33). Pore size variations significantly modulated *H intensity, with CuRu/MCHS-7 exhibiting the strongest signal, followed by CuRu/MCHS-11 and CuRu/MCHS-4, aligning with the urea yield rates. Therefore, pore-size-dependent nano-confinement modulates *H generation and consumption dynamics, ultimately governing urea selectivity and hydrogenation product distributions.

a Urea yield rate in 0.1 M KNO3 with catholyte media of H2O and D2O, respectively. The data are presented as mean values ± s.d. (n ≥ 3). b A schematic diagram of proton sources and destinations in urea electrosynthesis with KNO3 and CO2 is presented, where pore size determines urea selectivity by modulating the binding pathways of *H. c, EPR spectra of three catalysts with different nano-confinement scales using DMPO as the radical trapping reagent in 0.1 M KNO3 under CO2, and of the CuRu/MCHS-7 catalyst in 0.1 M KNO3 under Ar. d Computed concentration and distribution of species: CO2, NO3–, *H and urea concentration. Three-dimensional MCHS with precisely tuned pore diameters (4, 7, and 11 nm) and a 2 nm catalytic layer were constructed to match with the structural features of CuRu/MCHS catalysts (Figs. S34, S35 and Table S13). The diffusion coefficients of solution species are provided in the Fig. S36 and Table S14. The computational model was fully immersed in the initial reactant environment (CO2/NO3–), maintaining consistent local concentrations across simulations. Colour scale, in mol L–1. e The production of *H, *NO, H2 and urea as a function of pore size. f Calculated CInterior/CExterior of CO2, NO3–, *H, *NO and urea in pore channel of the three catalysts. g Schematic diagram of the diffusion mechanism of C–N coupling on CuRu/MCHS catalyst and diffusion variations across three pore sizes.
To investigate how pore dimensions regulate the spatial distribution of reactants and critical intermediates during C–N coupling, we performed finite element simulation (FEM). The simulations visualized CO2 and NO3– diffusion through pore channels into the interior of the cavity, where surface catalytic reactions formed *NO and urea, achieving dynamic accumulation-depletion equilibrium. Concurrently, the intermediates and products (*H, *NO, H2, and urea) generated in the cavity diffused back into the electrolyte through the pore channels (Fig. 6d). The concentration of H2 and urea showed a positive correlation with the concentration of *H and *NO, highlighting the constructive role of nano-confinement catalysis of reaction intermediates (Fig. 6e). Concentration comparisons were conducted between the pore exterior and cavity interior (Fig. 6f). Enhanced CO2 and NO3– concentrations at the catalyst-electrolyte interface were observed with decreasing pore sizes, demonstrating improved enrichment effects near catalytic sites from more restrictive nano-confinement (i.e., 4 nm scale) (Fig. S37). However, the CO2/NO3– flux from the shell exterior into the cavity decreased with pore size reduction, indicating that the smaller apertures of 4 nm imposed diffusion limitations on reactant influx. Conversely, larger pore dimensions of 11 nm significantly enhanced the outward diffusion of intermediates and products (i.e., urea) from the cavity to the electrolyte, as evidenced by the CI/CE ratio of urea declined to1.34 at 11 nm versus 1.62 at 7 nm and 1.89 at 4 nm. The production rates of intermediates and products were slower at the smaller pore size of 4 nm due to excessive nano-confinement limiting CO2/NO3– transportation into the cavity, thus resulting in the direct reduction of NO3– to NH4+ within the pores of CuRu/MCHS-4. On the other hand, the cavity failed to retain the generated *H and *NO intermediates when the particle size increased to 11 nm, which consequently led to an increased observation of H2 of CuRu/MCHS-11.
Overall, the 7 nm pore architecture represents a kinetic optimum, arising from a synergistic balance among three pivotal factors (Fig. 6g). First, it established an optimal balance between mass transport and confinement. The 7 nm pores provided an effective compromise between efficient CO2/NO3– influx and effective retention of key intermediates (*H and *NO), avoiding the diffusion limitations observed in the 4 nm pores and the rapid intermediate leakage that occurred in the 11 nm pores. Second, it enabled highly efficient utilization of intermediates. The moderate confinement within the 7 nm cavities maintained a high local concentration of reactive species, maximized the generation and utilization of *H species (as confirmed by EPR results), and spatially confined them to preferentially participate in the hydrogenation steps of the *OCONO pathway, rather than being consumed by H2 evolution or excessive NH4+ formation. Third, it realized a synergistic kinetic effect of confinement. The 7 nm structure provided a kinetic optimum by harmonizing reactant supply, intermediate stabilization, and product diffusion, thereby delivering the highest urea selectivity and production rate. Collectively, this multi-scale regulation enhances the local availability of reactive species, lowers the kinetic barriers for C‒N coupling, and steers the reaction pathway toward urea formation, demonstrating that precise pore-size engineering provides a powerful lever for optimizing complex electrocatalytic processes.
In summary, we have demonstrated that a nano-confinement architecture effectively overcame kinetic and diffusion limitations in the co-reduction of CO2 and NO3–, achieving a 2.5-fold enhancement in urea yield compared to non-confined systems. This work establishes a nano-confinement strategy for the rational design of C‒N coupling electrocatalysts. First, the synergy between CuRu bimetallic nanoclusters and the nano-confined environment enhances the adsorption of reactants and intermediates, enabling high urea production rates at industrial-scale current densities. Second, the confined pore channels and cavities facilitate the overcoming of C–N coupling energy barriers, shifting the key bonding step from the conventional COOH–NH2 pathway to the earlier and kinetically favored OCO‒NO intermediate coupling. Third, precise engineering of pore sizes optimizes the diffusion and distribution of reactants and intermediates, balancing reaction kinetics and significantly improving urea productivity and selectivity. Further efforts should prioritize mitigating side reactions and enhancing urea recovery. To enhance FE, strategies including the precise modulation of the active-site microenvironment to suppress hydrogen evolution and regulate intermediate adsorption, optimization of the metal active centers with tailored electronic structures, more advanced reactor designs that enhance local reactant concentrations, could be built upon our nano-confinement platform to substantially boost selectivity65,66.
The findings of this study not only established a strategy for urea electrosynthesis through nano-confinement-engineered bimetallic catalysts but also delivered a universal design blueprint to address kinetic limitations in C–N coupling. This strategy would provide a versatile platform for the synthesis of other high-value C–N coupling products, including amines and amino acids, which are of critical importance in agriculture, pharmaceuticals, and biomaterials. Tailored nano-confinement dimensions and engineered metal active sites stabilize reactive intermediates and control the local reaction microenvironment. The proposed catalytic design paradigm offers a promising approach to overcoming the intrinsic kinetic limitations associated with multi-step C–N bond formation and the complex triple-phase interfacial transport, which remain longstanding challenges in electrosynthetic processes.
Methods
Materials
All reagents were commercially available and used without further purification. Tetrapropyl orthosilicate (TPOS, C12H28O4Si, 97%), tetraethyl orthosilicate (TEOS, C8H20O4Si, 97%), resorcinol (C6H6O2, analytical grade), xanthydrol (97%), ammonium iron (Ⅲ) sulfate dodecahydrate aqueous and urease were obtained from Macklin. Formaldehyde aqueous solution (37 wt%), NH3·H2O (25 wt%), sodium acetate, glacial acetic acid, NaOH, HCl, methanol, and ethanol were purchased from Sinopharm Chemical Reagent. 1,10-phenanthroline (99%) was obtained from Energy Chemical. Ruthenium (Ⅲ) trichloride (Ru content 45–55%), copper nitrate hydrate (99.99%), urea-15N2(99 atom%), and potassium nitrate-15N (99 atom%) were obtained from Aladdin. Dimethyl sulfoxide-d6 (D, 99.9%) was purchased from Cambridge Isotope Laboratories, Inc. Deuterium oxide (D2O, 99.9 atom% D) was obtained from J&K Scientific.
Synthesis of mesoporous carbon hollow spheres (MCHS) and carbon spheres (CS)
MCHS was prepared using a one-pot synthesis strategy and partially optimized67. Specifically, ethanol, ultrapure water, and NH3·H2O were mixed in a 7:1:1 ratio (total volume of 83 mL), and stirred for 15 minutes. Then, 3.46 mL of tetrapropyl orthosilicate (TPOS) was added, and the mixture was stirred for an additional 15 minutes. Afterward, 0.4 g of resorcinol and 0.56 mL of formaldehyde (37 wt%) were introduced. The mixture was stirred for 24 hours, and the resulting precipitate was separated by centrifugation and dried overnight in a vacuum oven. The obtained powder was then calcined at 700 °C for 5 hours under a nitrogen atmosphere, followed by etching with a 20 wt% NaOH solution to remove the silica. The preparation method of CS was consistent with MCHS-7, but omitted the etching step. Other mesoporous hollow carbon spheres with different pore sizes (MCHS-4 and MCHS-11) were prepared by modifying either the molar ratio of TPOS/TEOS (with the total silicon content fixed at 12 mmol) or the volume ratio of EtOH/H2O, while all other experimental conditions remained constant. To synthesize MCHS-11, the ethanol-to-water volume ratio was adjusted to 7:2 (originally 7:1). For MCHS-4, the molar ratio of TPOS/TEOS was precisely controlled at 0.25 under fixed total silicon content (12 mmol).
Synthesis of CuRu/MCHS and CuRu/CS
The CuRu bimetallic loading on MCHS and CS was performed via a simple impregnation method. Specifically, 50 mg of MCHS-7 and Cu(NO3)2 were thoroughly mixed and dispersed in anhydrous ethanol. Subsequently, RuCl3 solution (4 mg Ru mL–1) was added dropwise to the suspension, followed by stirring overnight. After evaporating the anhydrous ethanol, the composite was thermally treated at 500 °C for 2 h under a 5% H2 atmosphere. The amounts of Cu and Ru added to prepare CuRu/MCHS and CuRu/CS were determined based on a molar ratio of 24:1. For other catalysts, the preparation was adjusted according to the desired molar ratio of Cu to Ru.
Catalyst characterization
SEM images were obtained with Gemini 560. Transmission electron micrography (TEM) and high-resolution TEM (HR-TEM) images were taken from JEM-F200 electronic microscopy (JEOL). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) images were taken from JEM-ARM300F2 (JEOL). X-ray diffraction (XRD) patterns were recorded on Rigaku SmartLab SE diffractometer (Cu Kα). XPS measurements were performed on Thermo Scientific K-Alpha spectrometer. Raman spectra were obtained on HORIBA Scientific LabRAM HR Evolution Raman spectrometer equipped with a 532 nm excitation laser. Inductively coupled plasma-optical emission spectrometer (ICP-OES) test were performed on an Agilent 5110 (OES). Brunauer-Emmett-Teller (BET) using a Quantachrome EVO and the pore size distributions were measured on the N2 desorption isotherm using cylindrical pore model (BJH method). Nuclear magnetic resonance (NMR) spectroscopy data were collected in Agilent DD2-600 with a liquid helium ultralow-temperature detection probe. The high performance liquid chromatography-fluorescence detection (HPLC-FLD) spectroscopy data were collected using a Waters Acquity Arc HPLC system with Inertsil ODS-4 column (4.6 mm *250 mm, 5 μm). The corresponding mobile phase, column temperature, and detected wavelengths under fluorescence mode were ammonium acetate aqueous solution (0.02 M)–acetonitrile (25:75), 35 °C, with excitation at 213 nm and emission at 308 nm. The ion chromatography (IC) data were collected in Dionex Aquion (Thermofisher Scientific). X-ray absorption spectroscopy (XAFS) data of Ru K-edge were collected at the BL14W1 station in Shanghai Synchrotron Radiation Facility (SSRF). XAFS spectroscopy of Cu K-edge were carried out using the RapidXAFS 2 M (Anhui Absorption Spectroscopy Analysis Instrument Co., Ltd.) by transmission mode at 20 kV and 20 mA, and the Si (553) spherically bent crystal analyzer with a radius of curvature of 500 mm was used for Cu. Normalization, transformation from energy into k-space, subtraction of a spline background, and shell fits were performed with Athena and Artemis. Calculation details can be found in Supplementary Note 1.
Electrochemical measurements
All the data was collected by using CHI660E as the electrochemical workstation. 2 mg catalyst were ultrasonically dispersed in a mixed solution (280 μL of ethanol and 20 μL Nafion solution). The dispersed catalyst ink was dropped onto 1 cm2 of hydrophobic carbon paper. A Platinum tablet (1.5*1.5 cm2) and an Ag/AgCl electrode were used as counter and reference electrodes, respectively. The reaction was operated in a 25 °C water bath. All electrolytes were prepared using ultrapure water and freshly made prior to use. The electrocatalytic activity measurements were performed in a sealed two-compartment H-cell separated by a proton-conductive membrane (Nafion 117). The membrane was pretreated by boiling in ultrapure water for 1 h, then 5% H2O2 at 80 °C for 1 h, 0.5 M H2SO4 at 80 °C for 2 h, and finally boiling in water for 6 h. The introduction of an additional carbon source (0.1 M KHCO3 + 0.1 M KNO3) decreased both the urea yield and FE compared to 0.1 M KNO3 alone (Fig. S38). Therefore, considering factors such as pH, the stoichiometric ratio of carbon to nitrogen atoms, and avoid introducing non-target reaction pathways, 0.1 M KNO3 was selected as the electrolyte for the co-reduction of CO2 and NO3–. The 0.1 M KNO3 was applied to electrocatalysis of sole NO3– reduction. The 0.1 M KHCO3 was applied to electrocatalysis of sole CO2 reduction. The electrolysis cell was purged with Ar or CO2 for 30 min before testing, and the gas was continuously supplied during the experiment with a flow rate of 25 mL min–1. The flow rates of the electrolyte solution were all 60 mL min–1 in flow cell. The pH was 7.6 ± 0.1 in 0.1 M KNO3 electrolyte, and 4.1 ± 0.1 in carbon dioxide saturated 0.1 M KNO3 solution. All potentials measured were calibrated to vs. RHE (reversible hydrogen electrode) using the following formula.
where \({E}_{{{{\rm{Ag}}}}/{{{\rm{AgCl}}}}}^{0}\) is the equilibrium potential of Ag/AgCl electrode. The working electrode was activated and stabilized by cyclic voltammetry prior to testing with a 20-turn sweep under a potential window of 0 to –2 V (vs. Ag/AgCl). Linear sweep voltammetry (LSV) and electrochemical impedance spectra (EIS) were conducted in H-cell. The electrolyte was bubbled with Ar or CO2 for 30 min before the test. LSV curves were performed to choose the appropriate potential range for the catalysts. The sweeping range was from 0 to –2.0 V (vs. Ag/AgCl) at a scan rate of 5 mV s–1 in electrolyte with CO2/Ar flowing. The EIS were recorded over a frequency range of 1 M Hz to 0.1 Hz at open-circuit voltage with 5 mV. The data were corrected with iR compensation, where i is the current and R is the uncompensated ohmic resistance of the electrolyte determined by high frequency resistance in EIS. The electrochemical active surface area (ECSA) was estimated by measuring the capacitive current associated with double-layer charging from the scan-rate dependence of cyclic voltammograms (CVs). The potential window of CVs was in a non-Faradic potential range under different scan rates with 20, 30, 40, 50, 60 and 70 mV s–1. All current densities were normalized by geometric area, and the geometric area of all tested working electrodes was 1×1 cm2. The linear slope was equivalent to twice of the double-layer capacitance Cdl, and the electrochemical active surface area (ECSA) of catalyst was calculated by the following equation and normalized to geometric area: ECSA = Rf×S = (Cdl/Cs)×S. S stands for the real surface area of the smooth metal electrode, which was generally equal to the geometric area of carbon paper electrode (in this work, S = 1.0 cm2). The roughness factor Rf was estimated from the ratio of double-layer capacitance Cdl. The catalyst loading was maintained at 0.02 mg cm–2. Cs was the specific capacitance under identical electrolyte conditions, 21 μF cm–2 was used in this work68.
Product detection and quantification
A gas sampling bag was employed to collect the emitted gases throughout the reaction process. The quantitative analysis of N2, H2, CO, and CH4 was conducted by gas chromatography (GC) with a thermal conductivity detector (TCD) and flame ionization detectors (FID). Urea concentration was determined via urease decomposition method. The final solution was reacted for 40 min at 37 °C in a shaker. Urea was decomposed by urease into CO2 and two NH4+ molecules. High performance liquid chromatography with fluorescence detection (HPLC-FLD) was then used for secondary verification of the urea yield. After the reaction, Ar was continuously purged through the electrolyte for 30 min to remove the dissolved CO2. In a 40 °C derivatization bath, 0.5 mL of urea standard solution in 0.1 M KNO3 or the electrolyte were sequentially mixed with 0.4 mL of methanol solution, 50 μL of xanthydrol solution, and 50 μL of 1% hydrochloric acid solution to undergo a derivatization reaction. The produced NH4+ was spectrophotometrically quantified by the indophenol blue method, with absorbance measured by an ultraviolet-visible spectroscopy (UV-Vis). The coloring agents were as follows: (A) 0.15 M phenol and 0.47 mM potassium nitroprusside, (B) 0.13 M NaOH and 0.002 M NaClO solution. Nitrite ions were spectrophotometrically quantified using the Griess reagent. N2H4 was quantified using the Watt and Chrisp method: 5.99 g of para-(dimethylamino) benzaldehyde was dissolved in a mixture of 300 mL ethanol and 30 mL concentrated HCl to prepare the color reagent. Subsequently, 1 mL of the N2H4 color reagent was added to 2 mL of the electrolyte solution, and the mixture was allowed to stand for 1 h before absorbance measurement at 450 nm using a UV-Vis spectrophotometer. The quantification of NH2OH was based in its reduction of Fe3+ to Fe2+ which then forms an orange complex with 1,10-phenanthroline. 1.2 mL of a 1 M aqueous acetate buffer (0.048 M sodium acetate with 0.952 M glacial acetic acid), 60 μL of 15 mM ammonium iron (Ⅲ) sulfate dodecahydrate aqueous solution and 0.6 mL of 5 mM aqueous 1,10-phenanthroline solution were added into 1.2 mL of the electrolysis solution. The mixture was allowed to stand for 50 min before absorbance measurement at 510.5 nm using a UV-Vis. HCOOH was quantitatively analyzed by ion chromatography (IC).
15NO3 – isotope labelling experiments
1H NMR analysis (600 MHz) was performed to detect 14N-urea and 15N-urea after electrolysis at −1.1 V (vs. RHE) for 0.5 hours. A 900 μL aliquot of the diluted electrolyte or standard solution was mixed with 100 μL of DMSO-d6 (99.9 atom% D). For NMR measurements, the mixture was transferred into a 5 mm NMR tube and sealed.
FE and urea yield calculation
The FE of liquid products was calculated by using the following formula:
where \({C}_{{{{\rm{Product}}}}}\) is the measured concentration of the corresponding product (μg mL–1), \(V\) is the total volume of electrolyte (mL), \(F\) is the Faraday constant (96,485 C mol–1), \(Q\) is the total charge passed through the working electrode (C), n is the number of electrons for generation of one corresponding product, \(M\) is the relative molecular mass of products.
The FE of gas products was calculated by using the following formula:
where \(P\) is the pressure (1.013\(\times\)105 Pa), \({V}_{{{{\rm{Total}}}}}\) is the total volume of gas collected (m3), \(R\) is the ideal gad constant (8.314 m3 Pa mol–1 K–1), \(T\) is the reaction temperature (298 K). The yield rate for urea synthesis could be calculated as following formula:
where \({C}_{{{{\rm{Urea}}}}}\) is the concentration of generated urea, \(V\) is the volume of the electrolyte (L), \(t\) is the reduction time (h) and \({m}_{{{{\rm{cat}}}}.}\) is the catalyst loading mass. The partial current density of corresponding product (j) refers to the average current density for corresponding production during the whole electrochemical process, which can reveal the reaction kinetics. The j of corresponding product was calculated by using the following formula:
where \(t\) is the electrolysis time and \(G\) is the geometric area of the working electrode.
Electron paramagnetic resonance (EPR) measurement
EPR spectrum was conducted on a Bruker A300 spectrometer. Active hydrogen produced during the electrolysis process was trapped using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) in a 100 mL electrolyte solution containing 20 mM DMPO.
CO2 temperature programmed desorption (TPD) measurement
A 50 mg catalyst sample was loaded into a quartz tube and pretreated under a helium atmosphere by heating from room temperature to 200 °C at a ramp rate of 10 °C min–1, followed by calcination for 1 h. The sample was then cooled to 50 °C, exposed to CO2 for 30 min, and purged with helium for 30 min to remove weakly adsorbed species. After the TCD signal stabilized for 1 h, the sample was heated to 700 °C at a rate of 10 °C min–1 for desorption, with the desorbed species monitored using TCD.
ATR-SEIRAS measurement
Attenuated Total Reflectance-Surface-Enhanced IR Absorption Spectroscopy (ATR-SEIRAS) was performed using a Nicolet iS50 FTIR spectrometer equipped with an MCT detector and a reflectance unit for the electrochemical cell at an incidence angle of 60°. The homemade in situ electrochemical cell consisted of two chambers with three electrodes; the anode and cathode chambers were each filled with 10 mL of electrolyte. A 1 cm2 Pt sheet and Ag/AgCl were used as the counter electrode and reference electrode, respectively. For the working electrode, 30 μL of catalyst ink (5 mg mL–1) was deposited onto the Si crystal and air-dried before testing. The detector was cooled with liquid nitrogen for at least 30 min prior to testing to stabilize the signal. A background infrared spectrum was then recorded at open-circuit potential (OCP). All tests were conducted via chronoamperometry, and spectral data were collected after 1 min of reaction. All spectra were processed as absorbance units defined by with a spectral resolution of 4 cm–1. All spectra were given by absorbance –log (R/R0) with a spectral resolution of 4 cm–1 for each curve.
DFT modeling approach
DFT calculations were performed using the Vienna Ab-Initio Simulation Package (VASP) 5.4.4 code incorporating the projector augmented wave (PAW) scheme. The projector augmented wave method pseudopotentials with the Perdew–Burke–Ernzerhof generalized gradient approximation (GGA-PBE) functional, which were provided in the VASP database, were used to describe the electron–ion interactions. The plane-wave cutoff energy was set to be 400 eV. For structure optimizations, the total energy convergence was set to be smaller than 1 × 10–5 eV and the force convergence was set to be lower than 0.03 eV Å–1 (0.05 eV Å–1 for transition states calculations using CI-NEB method) on the atoms. The DFT-D3 method of Grimme was used to include the van der Waals corrections. It should be noted that DFT cannot effectively simulate catalytic activity involving the 7 nm nanoconfinement effect of the CuRu/MCHS catalyst. To better consider the potential role of the MCHS support structure, we included the carbon support in the computational model. The observed differences in the valence states and structural configurations of CuRu metals between CuRu/MCHS and CuRu/CS are minimal and are predominantly attributable to variations in Cu coordination numbers arising from disparities in nanocluster dimensions. It is scientifically untenable to ascribe the entirety of catalytic activity to such minor discrepancies in metal coordination. To systematically elucidate the reaction pathways of CuRu bimetallic systems in urea electrosynthesis and the structural influence of nano-confined carbon supports on reaction dynamics, a simplified averaged model was adopted for analysis (Supplementary Data 1).
The computational hydrogen electrode (CHE) model was adopted to calculate the Gibbs free energy change (∆G) for each elementary step as follows:
where ΔE represents the electronic energy contribution directly derived from DFT calculations. ΔEZPE and TΔS denote the contributions of zero–point energy and entropy (at 298.15 K), respectively. The vibrational frequency for intermediates can be computed accordingly (Tables S10 and S12).
FEM methods
The finite element method (FEM) simulation was performed by utilizing COMSOL multiphysics 6.2 software with secondary current distribution and transport of diluted species (tds) physics modules. We use the Nernst-Planck equations and the Fick’s Rule to solve the mass transport of solution species including the effects of diffusion, packing, migration as well as reaction, which incorporate a mean-field continuum description of steric effects. The electrode surface reaction follows the Tafel equation. The simulation parameters remained consistent across all three models, with only the spatial geometric area varied to isolate its influence. The simulations incorporated CO2 and NO3– co-diffusing to the catalyst surface under electric field, while H2O electrolysis supplied *H for the synthesis process. The diffusion coefficient (\(D\), m2 s–1) of urea was analyzed from the slope of mean square displacement (MSD) curves by Einstein relationship. Molecular dynamics simulations are performed with urea parameterized using the Generalized AMBER Force Field (GAFF) and water molecules modeled with the opc3 explicit solvent, conducted at 298.15 K. For more detailed information about the calculations, refer to Supplementary Note 2.
Data availability
All data supporting the results and discussions of this study are available in this Article and its Supplementary Information. Source data are provided with this paper.
References
Liu, C., Gao, Y. & Zhang, B. Organonitrogen electrosynthesis from CO2 and nitrogenous sources in water. Nat. Synth. 3, 794–796 (2024).
Song, X. et al. One-step formation of urea from carbon dioxide and nitrogen using water microdroplets. J. Am. Chem. Soc. 145, 25910–25916 (2023).
Zhu, X., Zhou, X., Jing, Y. & Li, Y. Electrochemical synthesis of urea on MBenes. Nat. Commun. 12, 4080 (2021).
Liu, J. et al. Electrocatalytic upgrading of nitrogenous wastes into value-added chemicals: A review. Mater. Today 73, 208–259 (2024).
Li, H. et al. Ligand engineering towards electrocatalytic urea synthesis on a molecular catalyst. Nat. Commun. 15, 8858 (2024).
Muhyuddin, M. et al. Electrochemical urea production using carbon dioxide and nitrate: state of the art and perspectives. Energy Environ. Sci. 17, 3739–3752 (2024).
Luo, Y. et al. Selective electrochemical synthesis of urea from nitrate and CO2 via relay catalysis on hybrid catalysts. Nat. Catal. 6, 939–948 (2023).
Song, X. et al. Urea Synthesis via Coelectrolysis of CO2 and Nitrate over Heterostructured Cu-Bi Catalysts. J. Am. Chem. Soc. 146, 25813–25823 (2024).
Zhao, C. et al. Tailoring activation intermediates of CO2 initiates C−N coupling for highly selective urea electrosynthesis. J. Am. Chem. Soc. 147, 8871–8880 (2025).
Hu, Q. et al. Pulsed co-electrolysis of carbon dioxide and nitrate for sustainable urea synthesis. Nat. Sustain. 7, 442–451 (2024).
Xu, M. et al. Kinetically matched C–N coupling toward efficient urea electrosynthesis enabled on copper single-atom alloy. Nat. Commun. 14, 6994 (2023).
Zhang, X. et al. Identifying and tailoring C–N coupling site for efficient urea synthesis over diatomic Fe–Ni catalyst. Nat. Commun. 13, 5337 (2022).
Wang, F. et al. Selective urea electrosynthesis from nitrate and CO2 on isolated copper alloyed ruthenium. ACS Energy Lett 9, 4624–4632 (2024).
Du, W. et al. Synergistic Cu single atoms and MoS2-edges for tandem electrocatalytic reduction of NO3− and CO2 to urea. Adv. Energy Mater. 14, 2401765 (2024).
Chen, K. et al. Urea electrosynthesis from nitrate and CO2 on diatomic alloys. Adv. Mater. 36, 2402160 (2024).
Meng, N. et al. Oxide-derived core-shell Cu@Zn nanowires for urea electrosynthesis from carbon dioxide and nitrate in water. ACS Nano 16, 9095–9104 (2022).
Xiang, J., Qiang, C., Shang, S., Guo, Y. & Chu, K. Relay catalysis of isolated rhodium-alloyed copper boosts urea electrosynthesis from nitrate and CO2. ACS Nano 18, 29856–29863 (2024).
Wei, X. et al. Oxygen vacancy-mediated selective C−N coupling toward electrocatalytic urea synthesis. J. Am. Chem. Soc. 144, 11530–11535 (2022).
Zhao, Y. et al. Efficient urea electrosynthesis from carbon dioxide and nitrate via alternating Cu–W bimetallic C–N coupling sites. Nat. Commun. 14, 4491 (2023).
Wei, X. et al. Lattice oxygen-driven Co-adsorption of carbon dioxide and nitrate on copper: A pathway to efficient urea electrosynthesis. J. Am. Chem. Soc. 147, 6049–6057 (2025).
Yang, P.-P. et al. Protecting copper oxidation state via intermediate confinement for selective CO2 electroreduction to C2+ fuels. J. Am. Chem. Soc. 142, 6400–6408 (2020).
Li, Q. et al. Ultralow coordination copper sites compartmentalized within ordered pores for highly efficient electrosynthesis of n-propanol from CO2. J. Am. Chem. Soc. 147, 6688–6697 (2025).
Liu, X., Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S.-Z. Mechanism of C−N bonds formation in electrocatalytic urea production revealed by ab initio molecular dynamics simulation. Nat. Commun. 13, 5471 (2022).
Grommet, A. B., Feller, M. & Klajn, R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 15, 256–271 (2020).
Gu, J. et al. Synergizing metal-support interactions and spatial confinement boosts dynamics of atomic nickel for hydrogenations. Nat. Nanotechnol. 16, 1141–1149 (2021).
Dong, B. et al. In situ quantitative single-molecule study of dynamic catalytic processes in nanoconfinement. Nat. Catal. 1, 135–140 (2018).
Kang, Y. et al. Unveiling the spatially confined oxidation processes in reactive electrochemical membranes. Nat. Commun. 14, 6590 (2023).
Shen, Z. et al. Self-enhanced localized alkalinity at the encapsulated Cu catalyst for superb electrocatalytic nitrate/nitrite reduction to NH3 in neutral electrolyte. Sci. Adv. 10, 9325 (2024).
Tang, Y.-F. et al. Strong effect-correlated electrochemical CO2 reduction. Chem. Soc. Rev. 53, 9344–9377 (2024).
Lu, Y. et al. Confinement catalysis of reaction intermediates in Ag@Cu2O cascade nanoreactors toward boosted electrochemical C–C coupling. ACS Catal 14, 14744–14753 (2024).
Tian, H., Liang, J. & Liu, J. Nanoengineering carbon spheres as nanoreactors for sustainable energy applications. Adv. Mater. 31, 1903886 (2019).
Liu, Y. et al. Boosting the Volmer step by synergistic coupling of dilute CuRu nanoalloy with Cu/Ru dual single atoms for efficient and CO-tolerant alkaline hydrogen oxidation. Chinese J. Catal. 72, 266–276 (2025).
Chen, S. et al. Engineering Cu/Ru heterointerface-shelled nanocavities by the kirkendall effect for highly efficient nitrate electroreduction to ammonia. J. Am. Chem. Soc. 147, 36494–36507 (2025).
Kalbac, M. & Kavan, L. The influence of doping on the Raman intensity of the D band in single walled carbon nanotubes. Carbon 48, 832–838 (2010).
Zhang, J. J., Lou, Y. Y., Wu, Z., Huang, X. J. & Sun, S. G. Spatially separated Cu/Ru on ordered mesoporous carbon for superior ammonia electrosynthesis from nitrate over a wide potential window. J. Am. Chem. Soc. 146, 24966–24977 (2024).
Wang, X. et al. Embedding oxophilic rare-earth single atom in platinum nanoclusters for efficient hydrogen electro-oxidation. Nat. Commun. 14, 3767 (2023).
Pan, D. et al. Rapid synthesis of ruthenium–copper nanocomposites as high-performance bifunctional electrocatalysts for electrochemical water splitting. Small 20, 2404729 (2024).
Chen, F.-Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Zuo, Y. et al. Ru–Cu Nanoheterostructures for Efficient Hydrogen Evolution Reaction in Alkaline Water Electrolyzers. J. Am. Chem. Soc. 145, 21419–21431 (2023).
Huang, B. et al. Solid-solution alloying of immiscible Ru and Cu with enhanced CO oxidation activity. J. Am. Chem. Soc. 139, 4643–4646 (2017).
Huang, Y. et al. Unveiling the quantification minefield in electrocatalytic urea synthesis. Chem. Eng. J. 453, 139836 (2023).
Dutta, N. et al. A guideline to determine faradaic efficiency in electrochemical CO2 reduction. ACS Energy Lett 9, 323–328 (2024).
Meng, N. et al. Oxide-Derived Core–Shell Cu@Zn Nanowires for Urea Electrosynthesis from Carbon Dioxide and Nitrate in Water. ACS Nano 16, 9095–9104 (2022).
Liu, Y. et al. C-Bound or O-Bound Surface: Which one boosts electrocatalytic urea synthesis?. Angew. Chem. Int. Ed. 62, e202300387 (2023).
Zhai, S. et al. Brass phase determining selectivity in urea electrosynthesis from CO2 and nitrate. ACS Catal 15, 3276–3283 (2025).
Li, Y. et al. Sequential co-reduction of nitrate and carbon dioxide enables selective urea electrosynthesis. Nat. Commun. 15, 176 (2024).
Chen, K. et al. Atomically dispersed W1–O3 bonded on Pd metallene for cascade NO electroreduction to NH3. ACS Catal 13, 9550–9557 (2023).
Zhu, S. et al. Direct observation on reaction intermediates and the role of bicarbonate anions in CO2 electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 139, 15664–15667 (2017).
Liu, X. et al. High C-selectivity for urea synthesis through O-philic adsorption to form *OCO intermediate on Ti-MOF based electrocatalysts. Adv. Funct. Mater. 34, 2400892 (2024).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal 7, 606–612 (2017).
Zhu, S. et al. CO2 electrochemical reduction as probed through infrared spectroscopy. ACS Energy Lett 4, 682–689 (2019).
Cheng, M. et al. Rectifying heterointerface facilitated C−N coupling dynamics enables efficient urea electrosynthesis under ultralow potentials. Angew. Chem. Int. Ed. 64, e202413534 (2025).
Gao, Y. et al. Tandem catalysts enabling efficient C−N coupling toward the electrosynthesis of urea. Angew. Chem. Int. Ed. 63, e202402215 (2024).
Huang, D.-S. et al. Electrosynthesis of urea by using Fe2O3 nanoparticles encapsulated in a conductive metal–organic framework. Nat. Synth. 3, 1404–1413 (2024).
Bi, J. et al. High-rate CO2 electrolysis to formic acid over a wide potential window: An electrocatalyst comprised of indium nanoparticles on chitosan-derived graphene. Angew. Chem. Int. Ed. 62, e202307612 (2023).
Chen, X. et al. Amorphous bismuth-tin oxide nanosheets with optimized C−N coupling for efficient urea synthesis. J. Am. Chem. Soc. 146, 13527–13535 (2024).
Lv, C. et al. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat. Sustain. 4, 868–876 (2021).
Dai, Z. et al. Surface engineering on bulk Cu2O for efficient electrosynthesis of urea. Nat. Commun. 16, 3271 (2025).
Roy, P., Pramanik, A. & Sarkar, P. Dual-silicon-doped graphitic carbon nitride sheet: An efficient metal-free electrocatalyst for urea synthesis. J. Phys. Chem. Lett. 12, 10837–10844 (2021).
Wu, Y. et al. Composite mesostructures by nano-confinement. Nat. Mater. 3, 816–822 (2004).
Zhang, J. et al. Isotopic labelling of water reveals the hydrogen transfer route in electrochemical CO2 reduction. Nat. Chem. 17, 334–343 (2025).
Liu, Y. & McCrory, C. C. L. Modulating the mechanism of electrocatalytic CO2 reduction by cobalt phthalocyanine through polymer coordination and encapsulation. Nat. Commun. 10, 1683 (2019).
Feng, J. et al. Modulating adsorbed hydrogen drives electrochemical CO2-to-C2 products. Nat. Commun. 14, 4615 (2023).
Zhao, Z. et al. Dynamical barrier and isotope effects in the simplest substitution reaction via Walden inversion mechanism. Nat. Commun. 8, 14506 (2017).
Mukherjee, J. et al. Understanding the site-selective electrocatalytic Co-reduction mechanism for green urea synthesis using copper phthalocyanine nanotubes. Adv. Funct. Mater. 32, 2200882 (2022).
Lai, W. et al. Design strategies for markedly enhancing energy efficiency in the electrocatalytic CO2 reduction reaction. Energy Environ. Sci. 15, 3603–3629 (2022).
Du, J. et al. Synergistic effect of intercalation and EDLC electrosorption of 2D/3D interconnected architectures to boost capacitive deionization for water desalination via MoSe2/mesoporous carbon hollow spheres. Water Res 235, 119831 (2023).
Yang, F. et al. Bismuthene for highly efficient carbon dioxide electroreduction reaction. Nat. Commun. 11, 1088 (2020).
Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundation of China (No. 52341001, 52400098, 52370120), National Key R&D Program of China (2025YFE0207100), Natural Science Foundation of Zhejiang Province (No. LZ25B070003), and the State Key Laboratory of Water Pollution Control and Green Resource Recycling Open Research Fund (PCRRF25040).
Author information
Authors and Affiliations
Contributions
X.Wu. supervised the project. X.Wu. and J.Du. conceived and designed the experiments. J.Du. conducted the material synthesis and characterizations, the electrochemical experiments, the FEM calculations, and analysed the data. Y.Wu. performed the DFT calculation. X.Wu. and J.Du. wrote the manuscript. D.Xu helped with the discussion of the FEM calculation. M.Liu., H.Liang, Z.Wu., and G.Lu. provided helpful suggestions and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The author declares no competing interests.
Peer review
Peer review information
Nature Communications thanks Ke Chu, Pranab Sarkar, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Du, J., Wu, Y., Fang, S. et al. Nano-confinement engineering boosts C–N coupling for urea electrosynthesis. Nat Commun 17, 1008 (2026). https://doi.org/10.1038/s41467-025-67741-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67741-1
