
Abstract
Detection of somatic mutations in cell-free DNA (cfDNA) is challenging due to low variant allele frequencies and extensive DNA degradation. Here we develop a benchmarking strategy using longitudinal patient-matched cfDNA samples from individuals with colorectal and breast cancer. Samples with high and ultra-low levels of tumor-derived DNA are combined into controlled dilution series that preserve the properties of authentic cell-free DNA, including each patient’s germline and blood-cell mutation backgrounds. Using deep whole-genome (150x) and exome (2,000x) sequencing, we define a reference set of ~37,000 single nucleotide variants and ~58,000 indels to benchmark nine somatic variant callers across varying ctDNA levels and sequencing depths. We also explore machine learning–based tuning of individual callers and identify features that improve accuracy in cfDNA. This benchmarking resource clarifies the detection limits of current approaches and provides practical guidance for selecting somatic variant calling methods in liquid biopsy applications.
Similar content being viewed by others


Introduction
Analysis of cell-free DNA (cfDNA) using high-throughput Next-Generation Sequencing (NGS) has emerged as a valuable approach in precision oncology1. cfDNA comprises DNA fragments that are released into the blood plasma through cell death or active secretion2,3. In a cancer patient, a fraction of cfDNA, known as circulating tumor DNA (ctDNA), originates from tumor cells4,5, offering a minimally-invasive approach to access actionable tumor information with a simple blood draw6. This strategy has demonstrated potential in monitoring treatment response7, detecting minimal residual disease8, molecularly stratifying patients for targeted treatments9, and identifying therapy-acquired resistance10. Moreover, recent methods have demonstrated how the cumulated signal from hundreds to thousands of somatic mutations identified with broad Whole Exome (WES) or Whole Genome (WGS) Sequencing can increase cancer detection sensitivity11,12,13,14,15. Key to such clinical applications is the ability to accurately identify cancer somatic mutations, specifically Single Nucleotide Variants (SNVs) and short Insertions/Deletions (INDELs), from blood plasma16,17. However, most existing methods for somatic variant calling have not been designed for or benchmarked on plasma samples, leaving method choice challenging for end users.
Several factors contribute to higher noise levels in plasma cfDNA as compared to genomic DNA (gDNA) from tissue biopsies. One key factor is the low fraction of ctDNA present in most plasma samples, resulting in cancer mutations being present at very low variant allele frequencies (VAFs). cfDNA also exhibit higher levels of degradation as well as non-uniform depth of coverage due to nucleosome wrapping18. Other biological factors further complicate mutation detection, such as Clonal Hematopoiesis of Indeterminate Potential (CHIP)19,20 and potential for subclonal variation in multi-focal disease. Variant callers designed for tumor tissue samples typically do not account for these factors. While recent methods have incorporated novel strategies tailored to cfDNA mutation calling21,22,23, the performance of these methods have not been independently tested and compared to existing variant callers designed for tumor tissue.
Previous studies24,25,26 have benchmarked variant callers designed for tumor tissue samples using variant reference datasets generated for synthetic27 and real28 tumors. However, such large-scale reference datasets are currently lacking for plasma samples. Unfortunately, existing strategies used in tumor-tissue benchmarks are not directly applicable in the setting of cfDNA. For example, tumor-tissue benchmarks may utilize dilution series generated from mixing of tumor DNA with matched-normal DNA samples24,25,26. This strategy cannot be adopted for cfDNA since these samples have different error and fragmentation characteristics as compared to their matched normal samples comprising gDNA from white-blood cells. Recently, the FDA-led Sequencing Quality Control Phase 2 (SEQC2) project conducted a benchmark of cfDNA assays and somatic variant calls from four commercial providers29,30,31. However, this benchmark study focused on narrow targeted sequencing panels and plasma-only mutation calling, using nuclease-degraded DNA from cell lines as a surrogate for cfDNA. Thus, there is a need for approaches to generate large-scale and genome-wide reference datasets for variant calling using bona-fide cfDNA samples.
In this study, we addressed these research gaps to develop a comprehensive benchmarking framework for comparison of 9 somatic mutation callers applied to cfDNA samples from cancer patients. We created a comprehensive reference dataset using longitudinal plasma samples from breast cancer (BRCA) and colorectal cancer (CRC) patients. For each patient, we identified timepoints with high and ultra-low ctDNA levels, establishing controlled dilution series preserving cfDNA fragmentation patterns as well as patient-specific germline and somatic haematopoiesis variant backgrounds. We then used deep WGS (150x) and ultra-deep WES (2,000x) to construct a reference set of ~37,000 Single Nucleotide Variants (SNVs) and ~58,000 Insertions/Deletions (indels). We evaluated and compared the performance of 9 somatic variant calling methods in plasma-vs-normal mode. We systematically evaluated the impact of ctDNA levels and sequencing depth on method accuracy, revealing guidelines for method choice depending on use case. Finally, we investigated the potential for fine-tuning individual methods using machine learning, revealing features that enable improved accuracy on cfDNA data.
Results
A large-scale cfDNA somatic mutation reference dataset
Benchmarking of variant callers in tumor tissue often make use of dilution series generated through in silico mixing of sequencing data from a tumor and a matched-normal DNA sample (e.g., buffy-coat)24,25,26. However, this strategy is ill-suited for plasma samples, as cfDNA (plasma) and gDNA (matched normal) have markedly different error and fragmentation characteristics. We reasoned that a cfDNA benchmark dataset instead could adopt in silico mixtures of patient-matched plasma samples, preserving cfDNA fragmentation patterns as well as patient-specific germline and somatic haematopoiesis variant backgrounds (Fig. 1a, b). With this approach, a sample with very high ctDNA levels could be used to establish ground-truth mutations with high confidence for each patient (Fig. 1c, d).

a Plasma samples with high (orange) and low (blue) ctDNA levels are identified from longitudinal samples obtained from an individual cancer patient. b Patient-specific benchmark dilutions series are created through in-silico mixing of low and high ctDNA level samples. Signal-to-noise ratios are step-wise lowered by decreasing signal (series A) or increasing noise (series B). c Nine somatic mutation callers, including seven tissue-based and two cfDNA-based methods, are applied for each benchmark dilution sample in tumor-normal mode. Performance and feature analysis are conducted to establish practical guidelines. d High-confidence cancer mutations (pseudo ground-truth) are established from a consensus over variant call methods applied to the high ctDNA level sample. ctDNA circulating tumor DNA. cfDNA cell-free DNA. WGS whole genome sequencing. WBCs white blood cells.
To create a comprehensive cfDNA benchmarking dataset based on such a strategy, we first performed deep targeted sequencing ( ~ 5000x) and low-pass WGS ( ~ 5x) of longitudinal plasma samples from a cohort of colorectal and breast cancer patients (Fig. 2a). In these samples, we estimated ctDNA/tumor fractions (TF) using a consensus based on copy number aberrations (ichorCNA, lpWGS) and VAFs (targeted sequencing). We identified 4 patients (1 BRCA, 3 CRC) with samples that exhibited very high ( ~ 40%) and low ctDNA levels ( ~ 1%) at different timepoints (Methods, Supplementary Table 1). We confirmed that ctDNA levels were minimal ( < 3%) in all low-TF samples by inspecting VAFs for mutations identified with high confidence in the matched high-TF sample (Fig. 2b).

a Sample selection: Timelines for the four candidate cancer patients (IDs = 986, 1014, 123, 412), with the timepoints for the selected high-TF sample (orange) and low-TF sample (blue) indicated. TF levels were profiled longitudinally using shallow WGS (ichorCNA) and targeted sequencing (VAFs of cancer mutations). b VAFs of cancer mutations in low and high-TF plasma samples, sequenced with both deep WGS and deep targeted sequencing. WGS: whole genome sequencing, ctDNA circulating tumor DNA, TF tumor fraction, VAF variant allele frequency.
To establish dilution series, the 4 sample pairs were profiled with 150x and 75x WGS in the low and high TF sample, respectively. The low-TF sample was profiled at higher depth in order to create dilution series with lower ctDNA levels. We profiled the matched normal sample (buffy coat) for each patient using deep WGS ( ~ 150x) to adhere to guidelines32 advising sequencing the plasma and matched normal samples at comparable depth to limit variant calling errors due to CHIP. We created two types of WGS dilution series (Fig. 1b, and Supplementary Table 2): Series A was constructed with decreasing TF (i.e., decreasing signal) at fixed 150x effective depth of coverage, varying TF from ~25% down to ~2%. Series B was created by increasing non-ctDNA reads from the low-TF sample (i.e., increasing noise) while keeping a fixed amount of reads (70x) from the high-TF sample, thereby increasing the effective coverage from 70x to 250x in this series. In summary, both series comprise samples of high-to-low signal-to-noise ratios. However, this is achieved by decreasing signal in series A and increasing noise in series B.
Generation of ground truths labels using consensus calling
We applied 9 SNV callers and 8 short INDEL callers to the protein-coding regions (exome) in plasma-normal mode (Fig. 1c, Table 1 and Methods). Seven of these are methods designed for tumor tissue (Freebayes33, Mutect234, Strelka235, VarDict36, Varscan237, SMuRF38, VarNet39) and two are methods designed for plasma cfDNA samples (ABEMUS21, SiNVICT23).
We used the high-TF sample from each patient to generate ground truth variant calls using a majority-voting scheme (Fig. 1d, Methods). We reasoned that a majority-voting approach would be an unbiased and conservative approach to define the ground truth sets under the expectation that the different methods often yield independent errors. We explored this assumption in tumor tissue mutation benchmark datasets from ICGC (Alioto et al. 2015). We conducted an evaluation of distinct ensemble voting schemes (‘k of N’) in this dataset and found that a majority-voting approach yielded the best balance of accuracy and sensitivity (Supplementary Fig. 1).
When a matched tumor tissue was not available, we hypothesized that the high-ctDNA (high-TF) plasma sample could serve as a proxy to derive high-confidence truth labels. Accordingly, for each patient we constructed ground-truth variant sets from the high-TF plasma by consensus across methods (Fig. 1d; Methods). We validated this assumption in the patient with matched tissue (patient 986): the high-TF plasma contained 206 SNVs, while deep WGS of the primary (T1) and metastatic (M1) tissues contained 95 and 120 SNVs, respectively. The overlaps with plasma were 47 (T1∩plasma) and 54 (M1∩plasma), both highly significant after Bonferroni correction (Fisher’s exact test p < 10⁻⁶⁸ and p < 10⁻⁷⁴; Supplementary Fig. 2a), supporting the reliability of the plasma-derived ground truth. Tissue-specific (T1- or M1-only) calls typically had low VAFs, consistent with subclonality, and were rarely detectable in plasma (Supplementary Fig. 2b, c).
In the benchmark dilution series, the VAFs of the ground-truth mutations expectedly decreased linearly as a function of dilution level (Supplementary Fig. 3). Expectedly, some ground-truth variants were not detected at high dilution levels due to random sampling in the Series A dilution dataset (Fig. 1b). During benchmarking, we only considered ground-truth variants supported by at least one variant read in a diluted sample, thereby omitting counting of non-sampled ground-truth variants as false-negatives.
Variant calling accuracy at 150x sequencing depth
We considered two use cases when comparing the mutation callers: (i) unbiased genome-wide discovery, and (ii) clinical targeted genotyping. In the discovery use case, we evaluated the extent that methods offer a good trade-off between precision and recall when applied to an entire genome or exome11,12,13,14,15. In the clinical genotyping use case, the key challenge is often the ability to detect if known actionable mutations are present in a given sample40,41. In this setting, we therefore evaluated the sensitivity of methods at a given fixed and relaxed precision level. In both use cases, we analysed the of impact of the two distinct dilution series with lower signal and higher noise in each of the four patients (Fig. 1b, and Supplementary Fig. 5).
In the discovery setting, all methods expectedly demonstrated lower accuracy when the signal-to-noise increased. Strelka2 and SMuRF exhibited the best performance for SNV calling across all ctDNA ranges, both when samples were diluted with lower signal and higher noise (Fig. 3a). The AUPRC of these two methods were about twice as high as the next-best methods across most dilution levels. The performance of individual methods were largely consistent across the four patients (Fig. 3c, and Supplementary Fig. 6a, b). For INDEL calling, VarScan and SMuRF consistently demonstrated highest accuracy, with Strelka2 performing well only in samples with highest signal-to-noise levels (Supplementary Fig. 5). We observed consistent results when performing analysis at the whole genome level, with high concordance of method rankings between exome and whole genome calling (Supplementary Fig. 8). However, the AUPRC values were almost 50% lower for all methods in the whole genome setting, highlighting the increased difficulty in calling mutations in many non-coding regions.

a, b Exome variant calling accuracy for each method, when applied to distinct dilution series: decreasing signal series (A), and increasing noise series (B). Methods were evaluated in two settings: a unbiased discovery analysis focusing on the AUPRC metric, and (b) clinical genotyping focusing on sensitivity at a fixed precision level. Vertical error bars show the standard error of the mean (s.e.m.) of the averaged metric curves, horizontal error bars show the s.e.m. of the ctDNA fraction estimates among patients (n = 4). SNV ground-truth counts (n) per patient are shown in the small box: CRC_986 n = 206, CRC_1014 n = 361, CRC_123 n = 453, BRA_412 n = 285 (pooled N = 1305). Here, n denotes the number of true somatic mutations in the benchmark for that patient. c Precision-Recall curves for Strelka2, the best performing method in the discovery setting, shown separately for each patient. The reference undiluted high ctDNA sample is also displayed as the first sample of the dilution series. Gray lines represents iso-F1 curves. SNV Single Nucleotide Variant. TF Tumor Fraction, ctDNA circulating tumor DNA. AUPRC Area Under Precision Recall Curve.
In the clinical genotyping setting, three methods (VarScan, VarDict and Mutect2) demonstrated highest sensitivity (Fig. 3b). VarScan generally showed highest accuracy, except when TF dropped below 5% in series A (Fig. 3b). Mutect2 and VarDict were able to outperform VarScan in that latter case. For INDELs, VarScan showed best performance of all callers across both dilution series (Supplementary Fig. 5).
Surprisingly, both cfDNA-specific callers (ABEMUS and SiNVICT) included in this study exhibited poor accuracy in these benchmarks (Fig. 3a, b). Overall, variant callers developed for tumor tissue variant discovery also showed highest accuracy for variant calling in plasma samples at intermediate sequencing depth (75–200x).
We hypothesize that the observed ranking could reflects three design choices: (i) evidence representation (local haplotype assembly vs. pile-up counting); (ii) background-error priors (use of matched normal, panel-of-normals, and sequence-context/read-position/orientation-bias models); and (iii) training/calibration, including whether scoring is rule-based or machine-learned and the domain on which it was trained. Methods that couple assembly with rich priors or learned scoring (e.g., Strelka2, Mutect2 with artifact filters, SMuRF) better preserve precision as ctDNA levels decline, yielding higher AUPRCs in the discovery analysis. In contrast, pile-up–centric callers (VarScan, VarDict) prioritize recall via simpler thresholds and statistical tests, which benefits the fixed-precision clinical genotyping readout but produces an earlier precision drop in discovery. cfDNA-specific tools tuned for UMI-enabled, ultra-deep targeted panels (ABEMUS, SiNVICT) may be disadvantaged on the intermediate-depth exome/genome data used here, consistent with calibration to a different library design. Indel accuracy tracks the availability of local realignment/assembly and realignment-aware features, potentially explaining VarScan and SMuRF’s stronger recall and Strelka2’s more conservative performance as signal decreases.
Variant calling accuracy at 2000x sequencing depth
To evaluate the impact of increased sequencing depth on variant calling accuracy, we re-sequenced the samples from one CRC patient using ultra-deep WES at 2000x sequencing depth (Methods, Supplementary Table 3). We generated consensus ground truth calls using the high-TF ultra-deep WES sample and obtained 739 SNVs and 1300 INDELs.
In the discovery analysis setting emphasizing overall variant calling accuracy, the ranking of callers remained similar between the 150x and 2000x sequencing. The main exception was ABEMUS whose performance was improved, and similar to Strelka2 and SMURF, in the ultra-deep sequencing setting (Fig. 4a, and Supplementary Fig. 9). In the clinical analysis setting, emphasizing variant calling sensitivity, we observed notable differences in method performance with ultra-deep sequencing. Here, VarScan and ABEMUS demonstrated highest sensitivity, maintaining recall above 50% across all ctDNA ranges and noise levels (Fig. 4b, and Supplementary Fig. 9). In contrast, VarDict and Mutect2 exhibited a drop in sensitivity at 2000x (20–40%) compared to 150x (40–75%, Fig. 4b, and Supplementary Fig. 9). Overall, our findings demonstrate that somatic mutation caller accuracy depends on factors such as use case and sequencing depth. Orthogonally, to test whether ABEMUS’s gains could reflect PoN usage rather than caller logic, we applied a unified, caller-agnostic Panel-of-Normals post-filter to the deep-WES dataset (Supplementary Fig. 10). PoN filtering improved VarScan (ΔAUPRC + 0.033; Δsensitivity at 3% precision +0.043) and VarDict (ΔAUPRC + 0.026; Δsensitivity at 3% precision +0.129). In contrast, it had only a marginal effect on FreeBayes (ΔAUPRC + 0.002) and produced no measurable changes for Mutect2, Strelka2, SMuRF, VarNet, or SiNVICT. While the relative performance of VarScan was notably improved at low ctDNA levels, the relative ranking of the remaining methods was largely unchanged.

Callers’ performance on Single Nucleotide Variants in exonic regions in dilution series (A) at two different fixed coverage levels: 150x (left, derived from Whole Genome Sequencing samples) and 2000x (right, derived from Whole Exome Sequencing samples). Callers are evaluated in two application settings: a discovery analysis with the Area Under Precision-Recall Curve metric with a zoomed version of the ctDNA fraction range [1-5%] and (b) clinical analysis with the sensitivity at a precision fixed to 3%. ctDNA: circulating tumor DNA.
Fine-tuning tumor tissue-designed callers for cfDNA
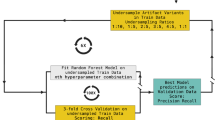
Our benchmark revealed that variant callers designed for tumor tissue analysis can outperform callers specifically designed for cfDNA. We therefore investigated to what extent these tissue-based variant callers could be further fine-tuned to improve performance on cfDNA. We leveraged the generated cfDNA variant calling benchmark dataset to train cfDNA-tuned versions of each variant caller. We trained Random Forest classifiers using each caller’s predicted variant score combined with other auxiliary features provided in the Variant Call Format (VCF) output. VarNet was excluded from this analysis due to its absence of companion features due to its image-based convolutional neural network approach. We trained models on the combined 150x benchmark dataset based on the three CRC patients using leave-one-subject-out cross-validation. Notably, this approach resulted in a marked improvement in the precision of Mutect2, positioning it as the top-performing method at 150x for AUPRC, surpassing both Strelka2 and SMuRF (Fig. 5a, b). The cfDNA-tuned FreeBayes also demonstrated improvements in both AUPRC and sensitivity but remained in the mid-tier of method rankings. Strikingly, fine-tuning on cfDNA data did only yield improvements in sensitivity for the remaining methods (Fig. 5c). This underscores the need for innovative approaches to account for cfDNA-specific sequencing properties in order to further enhance sensitivity.

a We fine-tuned and evaluated the performance of tissue-based variant calling methods using a Random Forest model and leave-one-patient-out cross-validation (150x sequencing depth). The averaged Precision-Recall curves display both the performance of the initial base callers designed for tumor tissue (dashed lines) and of the corresponding cfDNA-tuned methods (colored lines) on the unseen patient data. b Gain in AUPRC between the naive and the corresponding cfDNA-tuned caller. c Gain in sensitivity at 3% precision between the naive and the corresponding cfDNA-tuned caller. The error bands for (a–c) show the standard error of the mean (s.e.m.) of the three metrics values created by permutations of the three CRC patients. d Bar-plots display for each cfDNA-tuned caller the median features importance based on the Mean Decrease in Impurity to predict pseudo ground truth labels, points show values for cross-validation evaluation. The prediction-score feature of the naïve model is shown in bold; features are color-coded by category. Full feature names and definitions are provided in Supplementary Table 5. cfDNA: cell-free DNA, CRC: colorectal cancer, AUPRC: Area Under Precision-Recall Curve.
To further explore which companion features enabled performance gains on cfDNA, we ranked features according to their importance, determined as the mean of the impurity decrease within the Random Forest models fitted during cross-validation (Fig. 5d, Methods). This analysis revealed that, for all callers, except FreeBayes and SMuRF, the standard prediction score is the most important feature. However, the cfDNA-tuned version of Freebayes utilized many other features, including mapping quality, strand bias, VAF, read counts, and base quality. The cfDNA-tuned Mutect2, exhibiting significantly improved precision, incorporated multiple features derived from the matched normal sample. Notably, the NALOD feature, defined as the likelihood of an artifact to be present in buffy coat with same VAF as in plasma, may aid in filtering potential CHIP variants. The cfDNA-tuned version of Strelka2 incorporated the variant position in the read, quality scores, and mapping quality in both plasma and the matched normal. The cfDNA-tuned SMuRF relies heavily on its own standard prediction score, as well as those of Strelka2, Mutect2, and VarDict, with additional incorporation of features such as the quality score of Strelka2 and Mutect2’s mapping quality and matched normal genotype likelihood. In contrast, VarDict and VarScan did not rely on companion features in the ctDNA-tuned versions, suggesting minimal improvement based on the existing auxiliary features used by these methods.
Overall, this analysis highlights the fine-tuning potential of each method designed for tumor tissue for further improved performance for cfDNA variant calling. Inspection of feature importance yields further insights for future cfDNA-specific software development.
Guidelines for accurate whole genome somatic variant calling in cfDNA
Our benchmark dataset allowed us to conduct an exhaustive performance analysis of tumor-agnostic cfDNA variant calling approaches. Our findings led us to derive practical guidelines for users to optimize somatic mutation calling in cfDNA (Fig. 6). Notably, our experiments showed that for unbiased mutation discovery analysis ( ~ 100x coverage), Strelka2 and SMuRF provided the most accurate SNV calls, while VarScan yielded the most accurate INDELs calls. For clinical analysis, emphasizing sensitivity and traditionally conducted at high depth ( > 1000x), VarScan and ABEMUS achieved the highest sensitivity. Furthermore, our analysis highlighted Mutect2 and VarDict in the mid-range of caller rankings across all cfDNA experiments. Moreover, Mutect2 also demonstrated superior performance in whole genome analyses at low ctDNA concentrations (Supplementary Fig. 8), and showed potential for improved precision following cfDNA-specific fine-tuning (Fig. 5a-b).

Top-performing method depending on the analysis setting: (left) unbiased discovery analysis, (right) clinical analysis; (top) intermediate sequencing depth ( ~ 150x), (bottom) deep sequencing ( ~ 2000x).
Discussion
The emergence of cfDNA analysis using NGS has offered promising results for precision oncology, enabling non-invasive and repeatable profiling of tumor molecular information. Indeed, accurate tumor-agnostic detection of cancer somatic mutations from blood samples has important use cases such as early non-invasive genotyping of tumors, discovery and analysis of tumor evolution and treatment resistance, as well as early cancer detection. However, the absence of cfDNA benchmark datasets and unbiased evaluation of existing methods presents an unmet need to the field. Here, we tackle this challenge with three main contributions: the creation of a large-scale benchmark dataset using longitudinal patient-matched sample pairs; the evaluation and comparison of variant callers in both discovery and clinical analysis settings; and the evaluation of the fine-tuning potential of existing methods using a machine-learning approach.
First, we presented a large-scale benchmark dataset created from real CRC and BRCA plasma samples for somatic mutation calling. Our dataset encompasses two widespread cancer types and contains in total close to 100,000 high-quality SNVs and short INDELs. This dataset is complementary to the SEQC2 studies29,30. The SEQC2 studies focus on narrow targeted assays from molecular diagnostic companies, yielding in total only about 100 cancer mutations for benchmark studies. Moreover, the SEQC2 studies use mock cfDNA samples created from cell-lines31. To mimic cfDNA degraded fragments, the reference cell lines were mixed in vitro at known ratios and enzymatically sheared and size selected. While this approach gives the advantage of creating well-characterized and verified ground truths using orthogonal approaches, most of the ground truths are likely germline mutations rather than bona fide cancer somatic mutations. Indeed, 91% of verified mutations overlapped annotated germline mutations in dbSNP42. Our proposed dataset, therefore, provides a complementary large-scale dataset of bona fide cancer mutations derived from real cfDNA samples, particularly in genome/exome-wide sequencing settings with a matched normal sample. Nevertheless, our in‑silico high/low-TF dilution design also has key limitations. For example, the low-TF sample imposes a lower limit for the testable range of mutation VAFs. Additionally, our design does not allow for modeling of pre‑analytical factors (e.g., extraction or storage‑induced artefacts) that could influence cfDNA analyses.
Our analysis led us to derive practical guidelines for users to optimize their choice of somatic mutation calling for cfDNA. Importantly, the benchmark demonstrated that distinct methods were superior in the unbiased discovery and clinical setting emphasizing sensitivity. Moreover, method performance varied further when considering SNV and INDEL calling as well as depth of sequencing (150 vs 2,000x). Finally, using a machine learning approach, we evaluated the fine-tuning potential of each method for further improving performance for cfDNA variant calling. Inspection of feature importance yielded insights for future cfDNA-specific software development.
In addition to performance metrics, the practicality of implementation and hardware requirements are crucial considerations when deploying software applications. Therefore, we meticulously documented the design and implementation strengths and weaknesses of each method and accompanying software (Table 1). Methods designed for tumor tissue were in general easy to run on large-scale datasets. As for cfDNA-specific methods, ABEMUS requires a panel of at least ten normal buffy coat samples sequenced with the same platform and at the same depth in order to build the background error model. Such panels are rarely available in practice. SiNVICT does not output a probability score but several lists of calls with different filters applied, which may be inconvenient for users. We recommend that it is crucial for mutation callers to provide default parameters for users to be able to run on new samples and report a probability score for each mutation call, rather than just a binary label.
The constructed dataset and benchmark methodology present certain limitations and opportunities for further improvement. First, the ultra-low ctDNA sample used to dilute the high ctDNA sample was not entirely cancer-free cfDNA. However, it remained the best proxy for a patient-matched non-cancer cfDNA sample. Despite accounting for the presence of residual ctDNA when estimating the resulting TF, there might still be slight genetic differences between the high and ultra-low ctDNA samples due to treatment or tumor evolution during the time gap. Treatment-related mutations can only emerge when the ultra-low ctDNA sample follows the high ctDNA sample. This scenario is observed in two of the four studied patients (Patients 986 and 123) with time gaps of 1 year and 3 months, and 3 months, respectively. Although this may introduce some noise in the benchmark dataset, the impact is mitigated by the ultra-low levels of ctDNA.
The newly created dataset and benchmarking strategy introduced in this study may also benefit future research in evaluating callers of other molecular alterations, such as structural variants and copy number alterations. It could also be used as a training or validation set for development of novel mutation callers. Overall, our study provides a comprehensive framework to improve and standardize cancer mutation detection in blood, supporting the development of next-generation liquid biopsy assays and algorithms for precision oncology.
Methods
Patient sample preparation
Plasma and patient-matched buffy coat samples were isolated from whole blood within six hours from collection by centrifuge 300 × g 10 min at room temperature. Upper plasma layer was further centrifuged at 9720 × g for 10 min at 4 oC and subsequently stored the supernatant at −80 oC. Middle Buffy coat layer from first spin was removed and stored directly at −80 oC until further process. Tissue was cut into pieces immediately after collection from Operation Theater and snap freeze with Liquid nitrogen before storage at −80 oC.
DNA extraction
Different kits were used for DNA extraction: Plasma(cfDNA) used Qiagen QIAamp Circulating Nucleic Acid Kit; Buffy coat (gDNA) used Qiagen QIamp DNA Blood Midi and tissue used Qiagen AllPrep DNA/RNA Mini Kit. DNA was further quantified with Invitrogen Quant-iT™ PicoGreen ® dsDNA Reagent.
Library preparation and NGS sequencing
Up to 100 ng of tissue or Buffy coat gDNA and 4-50 ng of cfDNA were used for library prep. Tissue DNA & Buffy coat gDNA was sheared to 200 bp by Covaris LE220 Focused-ultrasonicator with the following condition: Target BP(Peak) 200, Duty Factor 30%, Peak Incident Power (W) 450, Cycles per Bust 200, Treatment Time 175 sec. Sheared DNA subsequently was cleaned-up with 1.4X Agencourt AMPure XP beads (Beckman Colter, A63881). Purified DNA and cfDNA (without shearing) were further processed with KAPA Hyper Prep Kit (Roche, KK8504) for libraries generation using library adapters with a random 8-mer proximal to the library index site (Oligos synthesized by IDT). Hybridization capture was done using an IDT xGen Exome (ver 1) Hyb Panel and reagents as per manufacturer’s instruction (xGen® Hybridization and Wash Kit, IDT 1080577). Libraries were quantified with KAPA Library Quantification Kits (KAPA, KK4854) and sent for 100x coverage for Whole Genome Sequencing and 1000x or 200x coverage for Whole Exome Sequencing. Sequencing was done Paired-end (2x151bp) on an illumina Novaseq 6000 S4.
Variant calling in cfDNA deep targeted sequencing (~5000x) samples
The deep targeted sequencing cfDNA samples were analysed using the bcbio-nextgen pipeline43, including read alignment with BWA mem, PCR duplicate marking with biobambam, as well as recalibration and realignment with GATK. Somatic variant calling was performed using MuTect and VarScan with default parameters, and all calls were annotated with Variant Effect Predictor. Variants were removed if they were outside coding regions. The inferred VAFs were either from one of the two callers if the variant was missed by one caller, or the mean if the variant was called by both callers. Variants from HLA-A, KMT2C and MUC17 were filtered because the majority of variants in these genes were also found by at least one caller at ≥0.005 VAF in buffy coat sequencing. The list of known cancer driver mutations of a given patient is composed of the union of mutations identified in at least one cfDNA sample.
ctDNA tumor burden estimation in shallow WGS (~5x) samples
We ran ichorCNA44 with the same parameters as indicated in the authors’ Wiki page, except for parameter chrs set to c(1:22) to analyse autosome only to reduce complexity and includeHOMD set to True to include HOMozygous Deletions as large bin size is used (1 Mb), consistent with parameter usage description in the documentation.
Selecting cfDNA samples of patients with an exceptional timeline
Building the benchmark cfDNA dataset required precisely identifying relevant patients to send to a costly deep WGS procedure and post-verification of those samples. To identify suitable patients, we performed shallow WGS ( ~ 5x cov) on multiple timepoints. This allowed us to estimate tumor fraction (TF) using the ichorCNA44 method, describe in the previous paragraph. Additionally, ultra-deep targeted sequencing ( ~ 5,000x cov) was conducted to track the VAFs of known CRC driver mutations, which were identified in at least one timepoint, as described above. To classify a sample as ‘high ctDNA’, both the ichorCNA44 TF estimate and the VAFs of known CRC mutations needed to exceed 20%, as illustrated in Fig. 2a. We compared the ichorCNA44 TF estimate with four purity estimation methods designed for tumor tissue samples, adapted for high TF values, to ensure the obtained estimates were consistent. For a sample to be categorized as ‘ultra-low ctDNA’, the ichorCNA44 TF estimate has to be 0%, indicating the TF is below 3% since ichorCNA44 detection limit is TF = 3%. Additionally, the VAFs of known mutations had to range from less than 1% to up to 3%, as depicted in Fig. 2a.
ctDNA tumor burden estimation in deep WGS (~100x) samples
We first ran on all initial deep WGS cfDNA samples the cfDNA-specific ichorCNA44 TF estimator. We also reported for each initial sample the median VAF of known cancer driver mutations (Supplementary Table 1). For the high ctDNA samples, we also ran the four tumor purity estimators designed for tissue samples: THetA245, TitanCNA46, AbsCN-seq47 and PurBayes48. As the amount of tumor-derived reads in those high ctDNA samples is comparable to what is observed in a tissue sample, they are expected to give values comparable to ichorCNA44 TF. Those tumor purity estimators were applied in tumor-normal mode on cfDNA and using somatic mutations and copy number alterations called by SMuRF38 and CNVkit49, respectively, via the bcbio-nextgen43 workflow. All high ctDNA samples included in the study had comparable tissue and plasma-based ctDNA fractions. The TF of the high ctDNA samples was set as the mean between ichorCNA44 TF and the median of the four purity estimates. For ultra-low TF samples, as we observed a discrepancy between the ichorCNA44 TF estimated on matched deep VS shallow WGS samples, we relied on the mutations’ VAF to estimate TF. The TF of the ultra-low ctDNA samples was set as TF = 2 x medianVAF. The intermediate and final values of TF estimates for each initial sample is summarized in Supplementary Table 1. The study thus includes three matched pairs of high ctDNA and ultra-low ctDNA samples with the following estimated TFs. For patient 986, TFhigh = 32.6% and TFlow = 0%. For patient 1014, TFhigh = 40.3% and TFlow = 2.56%. For patient 123, TFhigh = 62.1% and TFlow = 3.15%. For patient 412, TFhigh ∼40%, TFlow ∼0%.
Verifying deep WGS candidate cfDNA samples of patients
Once the candidate samples were sequenced with deep WGS, we verified our conditions on TF estimate and mutations’ VAFs were satisfied on the obtained sample and provided reliable TF estimates for each sample in Supplementary Table 1. To verify the condition on the TF estimate, we applied the TF estimation methods on the deep WGS samples. We obtained expected elevated values for high ctDNA samples (range TF = [23-65%]) and then set the TF estimate of high ctDNA samples as the mean between ichorCNA44 TF and the median of 4 DNA purity estimates. For ultra-low TF samples, we observed a discrepancy between the ichorCNA44 TF estimates obtained on the deep WGS (range TF = [3-8%]) compared to the matched shallow WGS (TF = 0%). Thus, we decided to rely on the mutations’ VAF to estimate TF in those ultra-low TF samples. To check the condition on mutations’ VAFs is satisfied, Fig. 2b reports the VAFs of mutations attributed to this patient obtained in the deep WGS samples. As desired, the VAFs are concentrated in the upper left corner of the scatter plot, indicating a pronounced prevalence in the high ctDNA sample and minimal incidence in the ultra-low ctDNA sample. To set a confident TF estimate for the ultra-low ctDNA samples, TF was set as 2 x medianVAF.
Benchmark dilution series creation
Samples were processed using the bcbio-nextgen43 pipeline. Sequencing reads were aligned to the GRCh37 reference human genome using BWA-MEM v0.7.17. Samtools v.1.9 was used to perform in silico mixture series from the high and ultra-low ctDNA samples with ‘samtools view -s’ and ‘samtools merge’ commands. The command ‘samtools depth -a’ was used to estimate average depth of coverage.
Germline mutation calling
Genome Analysis Toolkit (GATK)50 was run as part of the bcbio-nextgen42 pipeline to call germline mutations on the matched normal sample of each plasma sample. Those germline calls were systematically removed from analysis to focus on cancer somatic mutations.
Somatic mutation calling methods
Each caller was applied with default recommended parameters and following the authors’ guidelines unless otherwise specified. For the five callers designed for tumor tissue samples Freebayes v1.3.5, Mutect2 v2.2, Strelka2 v2.9.10, VarDict v1.8.2 and VarScan 2.4.4, the bcbio-nextgen43 framework v1.2.9 was used to generate somatic mutation calls. Calling was run in tumor-normal mode, without INDEL recalibration. Duplicates were marked using biobambam v2.0.87. We lowered the VAF threshold ‘min_allele_frequency’ (default 10%) to adapt it to the low VAF mutations harbored in cfDNA. Thus, the VAF cutoff was set to 1% for 150x series and 0.01% for 2,000x series in order to match the limit of detection of a single supporting variant. We further recovered in the post-processing of output VCF files the low VAF calls that were rejected just because of the low VAF. The four other callers, SMuRF, VarNet, ABEMUS and SiNVICT, were run independently. SMuRF v2.0.12 was applied using output of the 5 previous callers. VarNet v1.1.0 was run after marking and removing duplicates using Picard. ABEMUS required a panel of normal WBC samples sequenced with comparable depth of coverage and, more importantly, sequenced with the same sequencing assay and platform to build a background error model. Although authors recommended at least 10 samples, only 8 such samples from other CRC patients could be obtained from the studied cohort. For SiNVICT, we lowered the minimum required read depth parameter min-depth (default 100) for a given call to 5 in ~100x WGS and to 50 in ~1,000x WES. Mutation callers were run on High Performance Computing hardware provided by A*STAR Computational Resource Center on Red Hat Enterprise Linux 8.1 (Optpa).
Pseudo ground truth construction and threshold selection
The threshold was fixed a priori based on external calibration on ICGC tissue and used uniformly for evaluation. Six tissue-applicable callers were run on ICGC. For consensus k = 2..6, precision, recall, F1 and FDR were computed against ICGC ground truth. We selected the minimal k maximizing recall (elbow at 4/6). We mapped proportionally to the 8-caller cfDNA panel (5/8; 4/7 indels).
Influence analysis (k-of-N consensus)
We quantified the influence of each caller under a k-of-N consensus rule. A caller “supports” a variant if it reports that variant after applying the uniform filters described above. For a given dataset and threshold k:
For each variant, count the number of supporting callers.
Let D(k) be the number of variants supported by at least k callers (i.e., variants that would pass a k-of-N consensus).
For caller i, let Ni(k) be the number of variants supported by exactly k callers including caller i (i.e., variants for which i is pivotal: without i, support would drop below k).
Define the influence of caller i at threshold k as influencei(k) = Ni(k) / D(k). If D(k) = 0, influencei(k) is set to 0.
Influence was computed per patient and per variant class (SNVs and indels separately). When reporting pooled results, we averaged influences across patients, weighting by D(k) unless stated otherwise.
Somatic mutation caller evaluation metrics
For this binary classification problem (mutation or no mutation for each considered genomic locus), the following notations are used: True Positive (TP), True Negative (TN), False Positive (FP), False Negative (FN) and the following wide-spread metrics are considered:
• Precision = TP/(TP + FP)
• Recall (or Sensitivity) = TP/(TP + FN)
• F1−score = 2 Precision × Recall / Precision + Recall (harmonic mean of the Precision and Recall)
• Area Under the Precision Recall Curve (AUPRC) which is often use in rare-event binary classification problem. Its interpretation though is a bit harder than classical Area Under the Receiver Operating Characteristic (ROC) curve (AUC) as a random estimator AUC is 0.5 while a random estimator AUPRC depends on the proportion of positive events. For instance, if there are 1% true mutations in the dataset, the random AUPRC is 0.01.
The Precision Recall Curve (PR curve) helps visualizing the performance of the individual algorithms under different variant score thresholds (Freebayes log-odds score ODDS, MuTect2 tumor log-odds score TLOD, Strelka2 tumor phred score SomaticEVS, VarDict SSF score, VarScan SSC score, ABEMUS odd score afcase/(afcase+filter.pbemcoverage) or the SiNVICT caller however, the cut-off threshold is an input parameter. The method does not output any score but reports 6 lists of calls corresponding to the successive application of 6 different filters. Thus, we assigned a score for each call equal to the number of occurrences of this given call across all lists divided by 6. Therefore, a call present even after applying all filters will be considered very confident with probability score equals to 1, while a call only present in the first list, thus caught by the Poisson model, will be considered of small confidence with a probability score equals to ⅙=0.167.
The implementation of Precision, Recall, AUPRC, PR curve provided by the scikit- learn51 v.0.21.3 library was used in our analysis.
Benchmark analysis and visualization
Results analysis and visualization was performed in Python v3.7.10. For feature analysis using Random Forest, the scikit-learn51 v.0.21.3 library was used. The Venn diagrams were created using the nVennR package in R v4.0.4.
Panel-of-normals post-filtering
To compare callers fairly, we took the PoN logic from ABEMUS and turned it into a small, standalone post-filter that can be applied to any VCF (Mutect2, Strelka2, VarDict, VarScan, FreeBayes, etc). Using unmatched normal WGS BAMs processed like our cohort, we summarize each covered site across normals to learn:
-
a position- and allele-specific background error rate (how often spurious signal appears at that exact base), and
-
a coverage-aware allelic-fraction (AF) threshold (the percentile of AF seen in normals within depth bins, so low-depth sites have stricter cutoffs than high-depth ones).
How filtering works (post-hoc on VCFs):
-
1.
Basic quality: require minimum total depth and a minimum number of alternate reads.
-
2.
AF threshold: require AF ≥ the bin-specific cutoff learned from normals.
-
3.
Per-base error test: keep the variant only if its observed alternate reads are unlikely under the site’s background error (FDR-controlled).
If per-allele counts or PoN coverage are missing, we apply steps 1–2 and skip step 3. Multi-allelic sites are checked per allele.
As a result, we obtain an caller-agnostic PoN screen that removes recurrent artefacts and caller-specific biases, enabling a fair, apples-to-apples comparison.
Model training and evaluation
We evaluated several supervised classification algorithms using the scikit-learn v0.21.3 Python library. The models tested included decision trees (using Gini impurity), random forests (100 estimators), gradient boosting (100 estimators), logistic regression with L1 (Lasso), L2 (Ridge), and ElasticNet penalties (liblinear or saga solvers), and a multilayer perceptron (MLP) with one hidden layer of 100 neurons, ReLU activation, a learning rate of 0.001, batch size of 200, and a maximum of 100 iterations. Models were trained on the combined 150× WGS dataset from three colorectal cancer patients. We applied a leave-one-subject-out cross-validation strategy for the train/test split. We used stratified 10-fold cross-validation to preserve class ratios and report average precision averaged across folds. Performance results are presented in Supplementary Table 4.
Feature importance analysis
We used features other than standard read counts available in each variant caller’s VCF file to train the models. Feature counts per caller were: FreeBayes (37), Mutect2 (21), Strelka2 (19), VarDict (9), VarScan (5), and SMuRF (30). To interpret the random forest classifier, we computed feature importance using the Mean Decrease in Impurity (MDI). This metric quantifies the reduction in Gini impurity associated with each feature across all splits and trees in the ensemble. While informative, MDI is known to favor high-cardinality features and primarily reflects performance on the training set, rather than generalizability. Most important input features and their definitions are listed in Supplementary Table 5 and summarized in Fig. 5.
Ethics & inclusion statement
Participants were recruited at the National Cancer Center Singapore (NCCS) under SingHealth Centralized Institutional Review Board approvals 2018/2709, 2018/2795, 2018/3046, 2019/2401 and 2012/733/B, and at the National University Health System (NUHS). Written informed consent was obtained from all participants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated in this study are available from the European Genome-Phenome Archive (EGA; dataset ID EGAS50000001313, (https://ega-archive.org/studies/EGAS50000001313). The data are available under restricted access and can be released for a 2-year period subject to a data-transfer agreement, with an expected processing time of approximately 2 months. Source data are provided with this paper.
Code availability
All scripts for data generation and analysis, together with the configuration files required to run the variant-calling pipelines, are available at GitHub: https://github.com/skandlab/cfdna_snv_benchmark.
References
Schwarzenbach, H., Hoon, D. S. B. & Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437 (2011).
Stejskal, P. et al. Circulating tumor nucleic acids: biology, release mechanisms, and clinical relevance. Mol. Cancer 22, 15 (2023).
Hu, Z. et al. The main sources of circulating cell-free DNA: apoptosis, necrosis and active secretion. Crit. Rev. Oncol. Hematol. 157, 103166 (2021).
Thierry, A. R. et al. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 35, 347–376 (2016).
Bronkhorst, A. J. et al. Characterization of the cell-free DNA released by cultured cancer cells. Biochim Biophys. Acta 1863, 157–165 (2016).
Heitzer, E., Haque, I. S., Roberts, C. E. S. & Speicher, M. R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet 20, 71–88 (2019).
Salvianti, F. et al. Circulating tumour cells and cell-free DNA as a prognostic factor in metastatic colorectal cancer: the OMITERC prospective study. Br. J. Cancer 125, 94–100 (2021).
Abbosh, C. et al. Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA. Nature 616, 553–562 (2023).
Rolfo, C. et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): a statement paper from the IASLC. J. Thorac. Oncol. Publ. Int Assoc. Study Lung Cancer 13, 1248–1268 (2018).
McCoach, C. E. et al. Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non–small cell lung cancer. Clin. Cancer Res. 24, 2758–2770 (2018).
Zviran, A. et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 26, 1114–1124 (2020).
Widman, A. J. et al. Machine learning guided signal enrichment for ultrasensitive plasma tumor burden monitoring. 30, 1655–1666 (2022).
Wan, J. C. M. et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med. 12, eaaz8084 (2020).
Bae, M. et al. Integrative modeling of tumor genomes and epigenomes for enhanced cancer diagnosis by cell-free DNA. Nat. Commun. 14, 1–15 (2023).
Wan, J. C. M. et al. Genome-wide mutational signatures in low-coverage whole genome sequencing of cell-free DNA. Nat. Commun. 13, 4953 (2022).
Hodson, R. Precision oncology. Nature 585, S1–S1 (2020).
Kumar-Sinha, C. & Chinnaiyan, A. M. Precision oncology in the age of integrative genomics. Nat. Biotechnol. 36, 46–60 (2018).
Zhu, G. et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat. Commun. 12, 2229 (2021).
Hu, Y. et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin. Cancer Res. 24, 4437–4443 (2018).
Spoor, J. et al. Liquid biopsy in esophageal cancer: a case report of false-positive circulating tumor DNA detection due to clonal hematopoiesis. Ann. Transl. Med. 9, 1264 (2021).
Casiraghi, N. et al. ABEMUS: platform-specific and data-informed detection of somatic SNVs in cfDNA. Bioinforma. Oxf. Engl. 36, 2665–2674 (2020).
Li, S. et al. Sensitive detection of tumor mutations from blood and its application to immunotherapy prognosis. Nat. Commun. 12, 4172 (2021).
Kockan, C. et al. SiNVICT: ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics 33, 26–34 (2017).
Bohnert, R., Vivas, S. & Jansen, G. Comprehensive benchmarking of SNV callers for highly admixed tumor data. PloS One 12, e0186175 (2017).
Fang, L. T. et al. Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole-genome sequencing. Nat. Biotechnol. 39, 1151–1160 (2021).
Xiao, W. et al. Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat. Biotechnol. 39, 1141–1150 (2021).
Ewing, A. D. et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 12, 623–630 (2015).
Alioto, T. S. et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 6, 10001 (2015).
Deveson, I. W. et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat. Biotechnol. 39, 1115–1128 (2021).
Gong, B. et al. Ultra-deep sequencing data from a liquid biopsy proficiency study demonstrating analytic validity. Sci. Data 9, 170 (2022).
Jones, W. et al. A verified genomic reference sample for assessing performance of cancer panels detecting small variants of low allele frequency. Genome Biol. 22, 111 (2021).
Chan, H. T., Chin, Y. M., Nakamura, Y. & Low, S.-K. Clonal hematopoiesis in liquid biopsy: from biological noise to valuable clinical implications. Cancers 12, 2277 (2020).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907, https://doi.org/10.48550/arXiv.1207.3907 (2012).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Lai, Z. et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 44, e108 (2016).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012).
Huang, W. et al. SMuRF: portable and accurate ensemble prediction of somatic mutations. Bioinformatics 35, 3157–3159 (2019).
Krishnamachari, K. et al. Accurate somatic variant detection using weakly supervised deep learning. Nat. Commun. 13, 4248 (2022).
Tie, J. et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage ii colon cancer. N. Engl. J. Med. 386, 2261–2272 (2022).
Rothwell, D. G. et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat. Med. 25, 738–743 (2019).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311 (2001).
Guimera, R. V. bcbio-nextgen: automated, distributed next-gen sequencing pipeline. EMBnet. J. 17, 30 (2011).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8, 1324 (2017).
Oesper, L., Satas, G. & Raphael, B. J. Quantifying tumor heterogeneity in whole-genome and whole-exome sequencing data. Bioinforma. Oxf. Engl. 30, 3532–3540 (2014).
TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data - PubMed. https://pubmed.ncbi.nlm.nih.gov/25060187/ (2023).
Bao, L., Pu, M. & Messer, K. AbsCN-seq: a statistical method to estimate tumor purity, ploidy and absolute copy numbers from next-generation sequencing data. Bioinforma. Oxf. Engl. 30, 1056–1063 (2014).
Larson, N. B. & Fridley, B. L. PurBayes: estimating tumor cellularity and subclonality in next-generation sequencing data. Bioinforma. Oxf. Engl. 29, 1888–1889 (2013).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLOS Comput Biol 12, e1004873 (2016).
der O’Connor V. A Genomics in the Cloud [Book]. https://www.oreilly.com/library/view/genomics-in-the/9781491975183/ (2023).
Pedregosa, F. et al. Scikit-learn: machine learning in python. J. Mach. Learn Res. 12, 2825–2830 (2011).
Acknowledgements
This work was supported by the Singapore Ministry of Health’s National Medical Research Council under its OF-IRG program (OFIRG21nov-0083), with support by the A*STAR Computational Resource Center through the use of its high performance computing facilities.
Author information
Authors and Affiliations
Contributions
A.S., W.L.S., and H.C. designed the study. I.T. and Y.S.Y. provided samples and clinical information. P.M.W., Y.T.L., A.G., and P.P. performed the sequencing experiments. S.T. designed and ran the in-house VarDict-based mutation calling pipeline on the deep targeted cfDNA samples. H.C. created the in silico benchmark dilution series and built the pseudo ground truth labels. H.C. performed mutation calling on deep WES/WGS with assistance from N.L.S. and K.K. H.C. performed data analysis and prepared the figures. A.S. and H.C. wrote the manuscript, with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Carrié, H., Sim, N.L., Wong, P.M. et al. Comprehensive benchmarking of methods for mutation calling in circulating tumor DNA. Nat Commun 17, 1082 (2026). https://doi.org/10.1038/s41467-025-67842-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67842-x
