
Abstract
The development of efficient acidic CO2 reduction reaction (CO2RR) to high value-added chemical HCOOH at high current density (>1 A cm-2) is crucial. However, addressing high potential, low Faradaic efficiency (FE), and poor stability simultaneously under acidic conditions remains challenging. Here, we construct small PdCx interstitial compounds and engineer the interstitial atoms to modulate the catalyst’s soft acid strength, thereby overcoming the triple challenge of “low overpotential-high current density-high stability” in acidic CO₂RR to HCOOH. Density functional theory (DFT) calculations and experimental characterization reveal that interstitial carbon infusion modulates Pd’s soft acid strength and weakens the Pd-O bond energy to form and desorb HCOOH, circumventing CO poisoning of the catalyst. Meanwhile, interstitial carbon infusion optimizes electronic structures, enhancing OCHO intermediate coverage as well as the effective retention and utilization of *H. This effect suppresses hydrogen evolution reaction (HER) while enhancing HCOOH selectivity. The optimized PdC0.13/CNT achieves >95% FEHCOOH and demonstrates stability in a proton exchange membrane (PEM) electrolyzer, maintaining 1000 mA cm-2 operation for 500 hours at 1.8 V cell voltage.
Similar content being viewed by others
Introduction
Carbon dioxide (CO2), a major greenhouse gas, intensifies global warming and climate change1,2,3,4,5. The conversion of CO2 into high-value products like formic acid (HCOOH) through electrocatalytic reduction (CO2RR), powered by surplus renewable energy, presents a promising sustainable strategy6,7,8,9. While recent advances focus on optimizing CO2RR for HCOOH production10,11,12,13,14, alkaline electrolytes suffer from carbonate formation due to CO2 reaction with OH-, leading to low CO2 utilisation15,16,17,18 and electrolyte contamination via HCOO- and HCO3- anion crossover19,20. Acidic electrolytes effectively suppress carbonate formation21,22,23 while utilizing H+ as the proton source, eliminating OH- generation and creating an industrially favorable environment for high-current-density HCOOH production24,25. Current research enhances acidic CO2RR performance through local microenvironment modulation, tensile strain engineering, elemental doping, and mass transfer optimization26,27,28,29, yet challenges persist in achieving low operating potentials, >95% HCOOH Faradaic efficiency (FE), and >500-hour stability at >1 A cm-2 in the electrolyzer. Meanwhile, the key mechanisms of modulation at the molecular level remain unclear30,31,32.
Developing high-performance acidic CO2RR electrocatalysts demands innovative design strategies. Pd-based catalysts are promising due to their ability to form *OCHO intermediates at low potentials33,34,35. However, we observe that the Pd (111) surface readily forms *COOH, a key intermediate in the CO₂ → CO reaction pathway (Fig. 1a)36,37. Even though the formation energy of the *OCHO (0.55 eV) is similar to *COOH (0.48 eV) (Supplementary Fig. 1), for Pd-*OCHO, the Pd-O bond (381 kJ mol-1) and C-O bond (380 kJ mol-1) have comparable bond energies, hindering subsequent desorption of *HCOOH38. It has been reported that softening acidic metal sites weakens metal-oxygen interactions, thereby promoting product desorption and enhancing selectivity39. On the other hand, altering the electronic structure can modify the formation energy of key intermediates and regulate their coverage40. Simultaneously, high intermediate coverage enhances *H utilization efficiency and suppresses the hydrogen evolution reaction (HER)41. These insights guide the design of Pd-based catalysts that synergistically regulate soft acid strength and elevate *OCHO coverage to achieve CO-resistant, HER-suppressed, and selective HCOOH production.

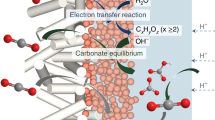
a On a typical Pd surface, the C-O bond is easier to cleave, thus producing CO. b On a carbon infusion Pd surface, the Pd-O bond is weakened, thus producing HCOOH. (Green: Pd, gray: C, red: O and white: H, same as below.).
In this study, we engineered interstitial atoms to modulate the soft acid strength of the catalyst, thereby enabling stable regulation of the Pd-O bond strength. This work demonstrates that a dual mechanism simultaneously suppressing H₂ and CO formation is crucial for achieving high performance in acidic CO2RR to HCOOH using Pd-based electrocatalysts. The interstitial carbon infusion strategy induces electron delocalization from Pd to carbon atoms while increasing the softness of Pd catalytic sites, weakening the Pd-O bond energy to favor the HCOOH formation pathway (Fig. 1b). The weakened Pd-O bond effectively promotes HCOOH desorption. Concurrently, interstitial carbon infusion optimizes electronic structure, lowering *OCHO formation energy and enhancing *OCHO coverage, thereby preventing catalyst poisoning and instability. The higher HER energy barrier enhances the retention and utilization efficiency of *H, with retained *H serving as a “proton reservoir” for CO2 hydrogenation, ensuring high selectivity and efficient HCOOH synthesis. The optimized PdC0.13/CNT achieves FEHCOOH of 95.1 ± 1.3% at 1000 mA cm⁻² and demonstrates stable 500-hour operation in a proton exchange membrane (PEM) electrolyzer at this current density (cell voltage: 1.8 V).
Results and Discussion
In this study, surfactant-free PdCx/CNT catalysts were synthesized via a solvent-free microwave method (Supplementary Fig. 2). Scanning electron microscopy (SEM) image reveals smooth carboxylated CNT surfaces (Supplementary Fig. 3). X-ray diffraction (XRD) analysis (Fig. 2a) confirmed Pd/CNT peaks matching the standard Pd reference (PDF #46-1043), with characteristic (111), (200), (220), and (311) planes at 40.1°, 46.7°, 68.1°, and 82.1°, respectively. The peak of the interstitial PdC0.13/CNT is shifted to a lower angle, indicating lattice expansion from carbon infusion. SEM and transmission electron microscopy (TEM) images (Fig. 2b, c) confirmed small (~5 nm) PdC0.13 nanoparticles uniformly dispersed on CNTs. High-resolution transmission electron microscopy (HRTEM) reveals a lattice spacing of 0.24 nm corresponding to Pd (111) planes (Fig. 2d), while the energy dispersive X-ray spectroscopy (EDS) elemental mapping verifies homogeneous Pd and C distribution (Fig. 2e). In order to visually demonstrate the infusion of C atoms in PdC0.13 as deduced from the XRD analysis, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) measurements were performed. Line-scanning of the selected region in Fig. 2f further show some isolated C atoms in the Pd lattice interstitials with low imaging contrast, which is characteristic of light elements with low atomic numbers. The lattice spacing expands to 0.24 nm due to the infusion of C (Fig. 2g). The fast Fourier transform (FFT) image shows that the PdC0.13 sample has (111) crystalline facet, which is consistent with previous characterization (inset of Fig. 2f). In conclusion, the interstitial compound PdC0.13/CNT was successfully synthesized. In addition, PdCx/CNT with varying interstitial carbon content was synthesized by modulating cellulose mass (Supplementary Fig. S4, 5). The lattice parameters and compositions at different mass of reactants are given according to formula and Supplementary Table 142. Increasing carbon content induces progressive negative XRD peak shifts (Fig. 2h), consistent with lattice expansion. In addition, the state change of Pd in interstitial PdCx/CNT with different carbon contents was further investigated by X-ray photoelectron spectroscopy (XPS). The XPS spectra of interstitial PdC0.13 nanoparticles present two main peaks at 336.47 eV and 341.76 eV, corresponding to the Pd 3d5/2 orbitals and Pd 3d3/2 orbitals of the zero-valence state, respectively, and the peak positions are obviously shifted to higher binding energies (Fig. 3a and Supplementary Fig. 6)43,44. This is due to the fact that the electronegativity of Pd (2.20) is smaller compared to C (2.55), and electron transfer occurs between the two elements. As the carbon content increases, the peaks move more towards the high binding energy. In addition, the energy band structure of interstitial PdC0.13/CNT was characterised by valence band spectroscopy (Supplementary Fig. 7). As previously reported, the valence band of interstitial PdC0.13/CNT becomes relatively narrower. This is due to the increase of Pd-Pd distance and the change of electronic structure45. The successful infusion of carbon can modulate the Pd acid site to a softer state, thereby modulating subsequent product formation39.

a The XRD pattern of Pd/CNT and PdC0.13/CNT. b SEM image, c TEM image and d HRTEM image of PdC0.13/CNT. (insert: lattice spacing). e Corresponding elemental mapping image of Pd0.13/CNT. f AC HAADF-STEM image and corresponding FFT pattern of PdC0.13/CNT. g Line-scanning intensity profile obtained from the area highlighted with cyan lines in (f). h The XRD pattern of PdCx/CNT.

a The XPS spectra of PdCx/CNT. b Pd K-edge XANES and (c) EXAFS spectra of PdC0.13. The wavelet transform of the EXAFS spectra of (d) PdC0.13 (e) PdO and (f) Pd-foil. The intensity color scale depicts the magnitude of the wavelet transform (a.u.) ΔR denotes the phase shift.
We further employed X-ray absorption spectroscopy (XAS) to investigate the electronic structures and local coordination environments of Pd sites in these synthesized catalysts. In order to precisely characterize the electronic structure of PdC0.13, Fig. 3b shows that the k-edge X-ray absorption near-edge structure (XANES) spectrum of PdC0.13 exhibits a valence state that is tightly bound to the Pd foil, which confirms that the major valence state of Pd in PdC0.13 is zero valence state46. This conclusion is consistent with the XPS results. Subsequent extended X-ray absorption fine structure (EXAFS) analysis identifies a distinct Pd-C scattering peak at 1.5 Å and a characteristic peak at 2.82 Å attributable to lattice relaxation of Pd-Pd bonds following carbon intercalation (Fig. 3c). This is the same conclusion as the successful infusion of interstitial carbon in Fig. 2f, g. Detailed quantitative parameters are provided in Supplementary Table 2. The EXAFS fitting data unambiguously confirm carbon atoms predominantly reside in subsurface interstitial sites rather than on the surface, evidenced by: (1) a well-defined Pd-C coordination shell (R = 2.09 ± 0.05 Å characteristic of interstitial carbon, distinct from surface-adsorbed species with R > 2.5 Å); (2) a Pd-C coordination number (CN) of 0.93 ± 0.2 confirming bulk lattice incorporation (surface confinement would yield near-zero CN); and (3) carbon-induced lattice expansion elongating Pd-Pd bonds from 2.73 Å (pure Pd) to 3.01 Å while reducing CN from 12 to 8.03. Wavelet transform (WT) analysis further validates these findings, showing a pronounced Pd-C intensity maximum at 1.5 Å. The Pd-Pd WT peak shifts from 2.7 Å (Pd-foil) to 3.01 Å (PdC0.13) due to lattice expansion (Fig. 3d–f)47. This confirms stronger long-range Pd-Pd interactions and more dispersed electron clouds, leading to a more delocalized electronic structure of Pd (i.e., softer metal sites). Complete EXAFS fitting parameters and k-space data are provided in Supplementary Fig. 8-9. The Pd-foil reference (R = 2.73 Å, CN = 12, R-factor = 0.002) in Supplementary Table 2 serves as the essential benchmark for quantifying distortions. Therefore, it can be concluded that the carbon infused into the lattice exists in the subsurface interstitials rather than at the surface, and that the infusion of interstitial carbon modulates the electronic structure and softness of Pd.
The CO2RR performance of PdCx/CNT was evaluated in a two-compartment H-cell with a standard three-electrode system (Supplementary Fig. 10a). After optimization of carbon content, electrolyte pH, and K+ concentration, it is determined that PdC0.13/CNT exhibits the competitive performance in 0.5 M K2SO4 (pH = 3 ± 0.03 adjusted by H2SO4, CK+ = 1.0 M) electrolyte (Supplementary Fig. 10b–d). As shown in Fig. 4a, the current density of PdC0.13/CNT is significantly reduced in the Ar-saturated electrolyte, indicating that interstitial carbon infusion effectively suppresses the HER side reaction. On the contrary, under CO2-saturated condition, PdC0.13/CNT shows a substantial increase in current density compared to Pd/CNT at all applied potentials, indicating enhanced CO2RR activity. Next, detailed rotating disk electrode (RDE) experiments were performed in the same acidic electrolyte (pH = 3 ± 0.03) to elucidate the transport behavior of CO2 and H+ on the surface of the PdC0.13/CNT catalyst. The diffusion-limiting mechanism of CO2 reduction is evaluated by measuring the LSV profiles of PdC0.13/CNT and Pd/CNT at different rotational speeds (0-2400 rpm) under CO2 atmosphere. At 0 rpm, the CO2RR onset potential of PdC0.13/CNT is advanced by 50 mV compared with that of Pd/CNT, and the CO2RR onset potential of PdC0.13/CNT is consistently advanced by 100 mV for all other rotational speeds (Fig. 4b-c). Moreover, PdC0.13/CNT exhibits higher current density relative to Pd/CNT at all applied potentials. These are attributed to the enhanced CO2RR capacity, so the rate of CO2 addition to the catalyst surface is accelerated, which is the described enhanced mass transfer of CO2. Similarly, we performed a complete RDE test under Ar atmosphere. By measuring the LSV curves of PdC0.13/CNT and Pd/CNT at different speeds (0-2400 rpm), we evaluate the diffusion limiting mechanism of H+ reduction (Supplementary Fig. 11). At 0 rpm, the HER onset potential of PdC0.13/CNT is delayed by 75 mV compared with that of Pd/CNT and is consistently delayed by 50 mV for PdC0.13/CNT at all other speeds. In addition, a current density plateau corresponding to diffusion-limited hydronium ion reduction is found in all polarization curves. Notably, PdC0.13/CNT exhibits a smaller plateau current density than Pd/CNT, possibly due to the weakening of H+ reduction at the cathode surface, resulting in a weakening of H+ mass transfer. In addition, for Pd/CNT, an increase in secondary current density is observed at ~-1.13 V vs. RHE, consistent with the onset of water reduction. In contrast, PdC0.13/CNT initiates water reduction at a more negative potential (-1.2 V vs. RHE). The above confirms that PdC0.13/CNT improves CO2RR selectivity and activity by inhibiting competitive HER. Subsequently, the liquid and gaseous products after one-hour electrolysis at different voltages were analyzed. CO and H2 were determined by gas chromatography (GC), and the associated gas calibration curves as well as quantitative data are shown in Supplementary Fig. 12 and Supplementary Table 3-4. HCOOH liquid product were detected using 1H NMR analysis and the NMR results are shown in Supplementary Fig. 13. The final quantification of the FE distribution of the products of the catalysts with and without interstitial carbon infusion show that PdC0.13/CNT is dominated by HCOOH, whereas the gaseous products CO and H2 are the main components in Pd/CNT (Supplementary Fig. 14). This is attributed to the inhibition effect of PdC0.13/CNT on HER and the advantage of preferential formation of HCOOH. For Pd/CNT without interstitial carbon (Supplementary Fig. 15), FEHCOOH increased from 12.0 ± 1.4% at -0.7 V vs. RHE to a maximum of 16.1 ± 2.0% at -0.9 V vs. RHE and then decreases due to HER enhancement. At -0.9 V vs. RHE, the optimum current density of PdC0.13/CNT catalyst for CO2RR reaches about -89.8 mA cm-2. PdC0.13/CNT maintained FEHCOOH > 85% across all applied potentials, peaking at 95.3 ± 1.3% at -0.9 V vs. RHE (Fig. 4d and Supplementary Table 5), demonstrating competitive performance compared to previously reported catalysts. The 5-fold increase in FEHCOOH for PdC0.13/CNT compared to Pd/CNT (95.3 ± 1.3% vs. 16.1 ± 2.0%) suggests that the Pd-O bond weakens to form HCOOH. Compared to Pd/CNT, PdC0.13/CNT achieves higher partial current densities for HCOOH at all potentials. Additionally, PdC0.13/CNT has the highest current density (JHCOOH) in the HCOOH part at -0.9 V vs. RHE, reaching -85.9 mA cm-2 (Supplementary Fig. S16). Furthermore, PdC0.13/CNT achieves the highest HCOOH cathode energy efficiency (CEE) compared to Pd/CNT. The maximum CEE reaches 64.0%, which is more than five times that of Pd/CNT (Supplementary Fig. S17). These results confirm that the infusion of interstitial carbon enhances the ability of PdC0.13/CNT to electrolyze CO2 into HCOOH.

a LSV curves with iR-corrected (95%), where R was measured to be 1.1 ± 0.03 Ω. Under varied rotating speed, LSV curves of b PdC0.13/CNT and c Pd/CNT in CO2-saturated electrolyte. R was measured to be 2.7 ± 0.2 Ω (iR-corrected (95%)). d FEHCOOH of different catalysts at various potentials. e Tafel slope. f CO2-TPD spectra of PdC0.13/CNT and Pd/CNT. The error bars represent the standard deviation for at least three independent measurements.
Tafel slope and electrochemical impedance spectroscopy (EIS) measurements were conducted to further investigate the role of PdC0.13/CNT in CO2RR under acidic conditions. As shown in Fig. 4e, the Tafel slope of PdC0.13/CNT (535.3 mV dec-1) is lower than that of Pd/CNT (757.9 mV dec-1). This indicates that the CO2RR kinetics is accelerated after the infusion of interstitial carbon. EIS Nyquist plots (Supplementary Fig. S18) reveal that PdC0.13/CNT exhibits lower charge transfer resistance compared to Pd/CNT, suggesting enhanced electron transfer efficiency at the electrode-electrolyte interface during CO2RR. The electrochemical double layer capacitance (Cdl) is calculated based on cyclic voltammetry (CV) at different scan rates (Supplementary Fig. S19-20). The Cdl value of 13.9 mF cm-2 for PdC0.13/CNT is higher than that of Pd/CNT, which suggests that the former can expose more active sites. The above electrochemical data demonstrate the intrinsic catalytic activity and fast interfacial reaction kinetics of PdC0.13/CNT. The strength of CO2 binding to the electrocatalyst is subsequently determined by corroborating the intrinsic properties of the catalyst through the temperature-programmed desorption (TPD) technique. As shown in Fig. 4f, PdC0.13/CNT is required to reach 630 °C for desorption of CO2, higher than that of Pd/CNT. The desorption temperature is positively correlated with the binding strength of the catalyst, which is more favorable for the subsequent reaction process48.
The long-term electrolytic stability of PdC0.13/CNT is a key indicator for evaluating the catalytic performance and practical applicability of CO2RR. Five-cycle tests demonstrated high stability in acidic medium at -0.9 V vs. RHE, with negligible fluctuations in FEHCOOH (average 94.9%) and current density (Supplementary Fig. S21). The relevant FE of gas products are listed in Supplementary Table 4-5. In order to verify that the catalyst has the potential for long-term operation, its stability over 24 h was initially explored. In the stability test, PdC0.13/CNT maintains a stable current density of -88.9 mA cm-2 (Supplementary Fig. S22a) and there is almost no decay of FEHCOOH after the reaction (maintained at 94.9%, Supplementary Fig. S22b). Therefore, the catalyst has the potential for longer reaction times, which will be followed by long stability tests. The pH of the electrolyte remained constant after the reaction, ensuring a stable electrocatalytic environment (Supplementary Fig. S23). The CO stripping test is an effective means of verifying the catalyst’s tolerance to CO deactivation. The CO oxidation onset potential of PdC0.13/CNT is negatively shifted by 96 mV (0.493 V) compared with that of Pd/CNT (0.589 V) (Supplementary Fig. S24), suggesting resistance to CO poisoning. The electronic modulation induced by the infusion of carbon weakens the intrinsic CO binding strength, which is consistent with the lower d-band center of PdC0.13/CNT (Supplementary Fig. S25)49,50. Subsequently, we complement the CO reduction experiments: a chronoamperometry method (-0.9 V vs. RHE) is performed in a CO-saturated acidic electrolyte (0.5 M K₂SO₄, pH = 3 ± 0.03) to monitor the current decay kinetics. Pd/CNT shows a rapid decay of the current (due to CO poisoning of the active site). PdC0.13/CNT, on the other hand, is able to maintain a stable current for several hours, directly verifying the tolerance of working in CO environment (Supplementary Fig. S26). Post-test physical characterization shows that PdC0.13/CNT retained its original structure, with particle size (~5 nm) and Pd (111) lattice spacing (0.24 nm) unchanged (Supplementary Fig. S27). After the reaction, the positions of the XRD diffraction peaks of PdC0.13/CNT coincide exactly with those before the reaction, and the peak intensities do not change significantly. In addition, the half-height widths of the diffraction peaks of the samples before and after the reaction also remained basically the same (Supplementary Fig. 28a). XPS analysis confirms stable composition and valence states of PdC0.13/CNT, with negligible Pd dissolution detected in solution (Supplementary Fig. 28b and Supplementary Table 6). Together, the above characterization results demonstrate that H in solution is not infused into the lattice. To further confirm the absence of H in the lattice after the reaction, H2-TPD can verify the ability of H to enter the lattice51. H can enter the lattice of Pd/CNT, and the decomposition of β-PdHx occurs at ~60 °C, confirming the presence of lattice H (Supplementary Fig. 29). However, this peak disappears in the patterns for PdC0.13/CNT containing carbon interstitial atoms, indicating that the subsurface layer is occupied by carbon interstitial atoms. These results collectively confirm the structural and electrochemical stability of PdC0.13/CNT under operating conditions.
Further in situ Fourier transform infrared spectroscopy (FTIR) experiments were carried out to elucidate the reaction pathways and the role of PdC0.13/CNT in the enhancement of CO2RR performance in acidic media. Intermediates are analyzed in order to relate the concentration of adsorbed substances to the reaction kinetics. As shown in Fig. 5a, b, a prominent *OCHO intermediate peak is observed near 1380 cm-1 - a key characteristic peak for the efficient production of HCOOH compared to *COOH. At the same time, almost no signal of *CO was detected, suggesting that PdC0.13/CNT favors Pd-O bond weakening to form HCOOH. In addition, the *COOH signal at the corresponding position of Pd/CNT is related to the linear adsorption stretching mode of *COL at ~2031 cm-1. Pd/CNT preferentially undergoes C-O bond breakage, and thus detects a more obvious *CO19,52,53. The *OCHO signal intensity increases steadily with applied potential from -0.7 to -0.9 V vs. RHE, which is attributed to the enhanced intermediate coverage facilitated by the abundant active sites of PdC0.13/CNT. At potentials above -0.9 V vs. RHE, there is a slight decrease in *OCHO intensity along with a slight loss of FE, which is attributed to competition with HER. These findings confirm that PdC0.13/CNT inhibits HER and CO toxicity by stabilizing the formation and coverage of crucial intermediates.

In situ FTIR spectra of a PdC0.13/CNT and b Pd/CNT at different potentials. Free energy profile of CO2 electroreduction to c CO and d HCOOH for the Pd (111) and PdC0.13 (111) at −0.9 V vs. RHE. e Dissociation energy of C-O and Pd-O bond on PdC0.13 (111). f The free energy profiles of Heyrovsky step in HER on Pd and PdC0.13.
Density Functional Theory (DFT) calculations were performed to further elucidate the mechanistic role in the electrocatalytic formation of HCOOH from CO2RR by PdC0.13/CNT. Notably, if CO2 adsorbs on the surface of Pd (111), it tends to form *COOH and thus CO poisoning the catalyst. *OCHO readily forms on the PdC0.13 (111) surface, serving as a key intermediate for subsequent HCOOH formation while minimizing CO poisoning of the catalyst (Supplementary Fig. 1). The applied potential for the Gibbs free energy calculations was set based on -0.9 V vs. RHE for DFT calculations using the computational hydrogen electrode method54. Free energy diagrams of CO2RR steps for these catalysts are shown in Fig. 5c-d. Compared to the PdC0.13, the Pd shows further reduced free energy (-0.30 eV) to form the critical *COOH, which undergoes an exothermic reaction from CO2 (Fig. 5c). In contrast, PdC0.13 exhibits a thermodynamically non-spontaneous energy barrier (0.05 eV) during the *COOH formation step. Regarding *CO generation, both catalysts produce *CO via an exothermic reaction pathway. The desorption process on PdC0.13 shows a more negative free energy, making the third step (*CO desorption) more readily achievable. This aligns with prior characterization indicating that CO exhibits weak binding energy on the PdC0.13 surface, thereby avoiding adverse effects. As shown in Fig. 5d, both catalysts spontaneously form OCHO and HCOOH intermediates at -0.9 V vs. RHE. The infusion of carbon favors the *OCHO step, significantly promoting subsequent *HCOOH desorption. Although *OCHO can also form on the Pd surface, its desorption after forming *HCOOH exhibits a markedly increased energy barrier (0.12 eV). The results indicate that the PdC0.13 catalyst exhibits stronger *OCHO adsorption and more readily dissociated *HCOOH, making the CO₂RR process more spontaneous and maximizing HCOOH selectivity. The dissociation energy results also align with the reaction pathway free energy. Although *OCHO may form on the Pd surface, the comparable bond energies of Pd-O (381 kJ mol-1) and C-O (380 kJ mol-1) bonds hinder efficient desorption of HCOOH, making CO formation the primary pathway. After carbon infusion, the Pd-O bond dissociation energy in *OCHO (2.36 eV) is significantly lower than the C-O bond energy (3.19 eV) (Fig. 5e). Protonation of *OCHO further weakens Pd-O bond interactions (2.28 eV), greatly facilitating HCOOH formation and desorption while enhancing product selectivity (Supplementary Fig. 31). This aligns with DFT calculations and characterization results: carbon infusion renders Pd sites in a softer state, thereby weakening oxygen interactions and ultimately improving product desorption capacity and FE. It is worth mentioning that by calculating the Gibbs free energy of *H adsorption on PdC0.13 and Pd, it is found that the infusion of interstitial carbon enhances the adsorption of *H (-0.58 eV), which is higher than that of pure Pd (-0.45 eV). This suggests that HER is effectively suppressed, which is conducive to improving the selectivity of the product HCOOH. Notably, the Volmer step on the surface of PdC0.13 is exothermic and has the lowest energy barrier (-0.58 eV), which favors the generation of *H (Supplementary Fig. 32). Subsequently, the Heyrovsky step energy barriers for both catalysts were calculated. As shown in Fig. 5f, the energy barrier after carbon introduction is 0.35 eV, lower than that of the Pd site without interstitial carbon (0.49 eV), indicating that the PdC0.13 catalyst exhibits an advantage in suppressing the HER. Based on these results, it can be concluded that PdC0.13 not only enhances the retention of *H, but also effectively inhibits the competitive HER. Additionally, we further investigated the influence of interstitial carbon on H* regulation in the CO₂RR to HCOOH at practical operating potentials via Bode plots. Supplementary Fig. 33 displays the Bode phase plots of PdC0.13/CNT and Pd/CNT in 0.5 M K2SO4 (pH = 3 ± 0.03) solution under applied potentials of -0.7 V to -1.0 V vs. RHE. When interstitial carbon is infused, the peak intensity is significantly lower than that of Pd/CNT. As the negative voltage increases, its peak frequency decreases markedly. This result further confirms that interstitial carbon slows down the Heyrovsky step and suppresses H₂ generation, thereby enabling the intermediate to readily capture H* and form HCOOH. Based on the above results and discussion, it can be concluded that the infusion of interstitial carbon confers significant advantages for charge transfer to produce HCOOH during the CO₂RR process while effectively preventing the HER, thereby ensuring high FE for HCOOH production. Combined with the high coverage of *OCHO in the in situ FTIR spectra, it can be seen that the retained *H can act as a “proton reservoir” for the hydrogenation of CO2. The easily weakened Pd-O bond, stable and enhanced *OCHO coverage, HER inhibition and inherent CO tolerance collectively improve the activity, selectivity, and stability of acidic CO2-to-HCOOH conversion. These theoretical insights align with experimental observations, providing atomic-scale rationale for the catalytic performance trends in CO2RR. The relevant Pd and PdC0.13 adsorption configurations are shown in Supplementary Figs. 34–36. Although our DFT calculations provide valuable insights, they have limitations in fully capturing the real catalytic environment, such as the effects of solvents and surface dynamics, which may influence reaction pathways and energetics.
The reaction system is scaled up using a PEM electrolyser with a gas diffusion electrode (GDE) to assess its industrial applicability (Fig. 6a and Supplementary Fig. 37a). Operation in an acidic electrolyte (0.5 M K2SO4, pH = 3 ± 0.03 adjust by H2SO4) is challenging due to the corrosive cathodic environment and increased HER competition. It is worth mentioning that the anodic oxygen evolution reaction is replaced by a Pt-Ru black-catalyzed hydrogen oxidation reaction (HOR)2. This not only reduces the total voltage, but also results in higher current density at lower overpotential. More importantly, the generation of harmful hydrogen peroxide, which may damage or even destroy the PEM membrane, is avoided. As a result, the membrane of the PEM reactor is more durable and have a longer service life. Clearly, PdC0.13/CNT is capable of acidic CO2 electrolysis at 600–1200 mA cm-2 (Fig. 6b), which corresponds to a cell voltage range of 1.28–2.06 V. Similarly, the FE of the product is quantified after 1 h of electrolysis. FEHCOOH greater than 75% are achieved in this range, peaking at 95.1 ± 1.3% at 1000 mA cm-2 (Fig. 6c). Notably, PdC0.13/CNT can stably operate for 500 h at 1.8 V (1000 mA cm-2) (Fig. 6d), demonstrating competitive performance in the acidic CO₂RR to HCOOH with high FE and long-term stability (Supplementary Table 3). During the long-term stability tests, the electrolyte is changed every 24 h to avoid liquid and gas buildup at the cathode. Even after 500 hours, the PdC0.13/CNT electrocatalyst maintains nearly constant chemical stability (FE decay of ~6.8%) (Fig. 6e). Despite the highly proton-conductive environment, the PEM electrolyzer exhibits sustained high FEHCOOH and reduced H₂ yield. This is precisely due to *H retention and utilization (*OCHO coverage enhancement) as well as weakened HER kinetics. The GC data, molar amounts and production rates of H2 and CO are shown in Supplementary Table 7–8. After 24 h of electrolysis, pH measurement confirms that there are minimal changes in the anode and cathode regions (Supplementary Fig. 37b), thus maintaining a stable catalytic environment. A techno-economic analysis was conducted using results from a two-compartment electrolyser to estimate a plant-gate levelized cost for HCOOH production using the PdC0.13/CNT electrocatalysts, evaluating the commercial potential. As shown in Supplementary Fig. 38a, the plant-gate levelized cost is significantly lower than the lowest market price in the paired CO2RR-HOR system, demonstrating a considerable advantage over the conventional CO2RR-OER system. The CO2RR-HOR system exhibits significantly higher energy efficiency (EE) compared to the CO2RR-OER system. Under conditions of current density varying from 600 to 1000 mA cm-2, the EEs of the CO2RR-HOR system range from 64.3% to 54.5%, indicating that most of the electrical energy in the system was utilized to generate HCOOH (Supplementary Fig. 38b). The CO2RR-HOR system can effectively decrease the required voltage and energy consumption (EC) for HCOOH production, with an average EC reduction of 37.6% compared to the CO2RR-OER system (Supplementary Fig. 38c). As shown in Supplementary Fig. 39, the CO2RR-HOR system demonstrates competitive profitability compared to traditional systems at different current densities, enabling it to better buffer against fluctuations in electricity costs and market prices. These results demonstrate the industrial feasibility of interstitial carbon-infused PdC0.13/CNT in acidic CO2 electroreduction.

a Schematic illustration of the PEM reactor. b Current dependent cell voltage of PdC0.13/CNT in PEM electrolyzer without iR-correction. c FE of various products at different current density. d Long-term stability testing and FEHCOOH at 24 h interval. e FE of the three products after 500 h. The error bars represent the standard deviation for at least three independent measurements.
In conclusion, we developed an electrocatalyst design for acidic CO2 electroreduction to HCOOH, achieving enhanced activity, selectivity, and stability through elevate intermediate coverage and softened acidic strength of Pd sites. By optimizing the interstitial carbon ratio, surfactant-free PdC0.13/CNT demonstrated competitive catalytic performance, delivering a FEHCOOH of 95.1 ± 1.3% at 1000 mA cm−2 in a PEM electrolyzer. Remarkably, PdC0.13/CNT exhibits high stability, enabling stable operation for 500 hours at 1000 mA cm−2 (cell voltage: 1.8 V). Combined experimental and theoretical analyses revealed that interstitial carbon incorporation weakens Pd-O interactions and softens the acidic strength of Pd sites, promoting *HCOOH desorption to favor HCOOH production. Although PEM electrolyzer favors proton conduction, optimizing hydrogen utilization efficiency as a proton reservoir enhances *OCHO coverage and suppresses the HER, enabling efficient and stable HCOOH production. The discovery of a dual mechanism simultaneously suppressing H₂ and CO formation provides novel guiding principles for achieving high performance using Pd-based electrocatalysts in acidic CO₂RR to HCOOH. By engineering interstitial atoms to modulate the catalyst’s soft acid strength, it pioneers a new research direction enabling stable regulation of chemical bond strengths between the catalyst and intermediates under high current densities.
Methods
Materials
Palladium (II) acetylacetonate (Pd, 97%, Macklin), potassium sulphate (K2SO4, AR ≥ 99%, Aladdin), cellulose (25 μm, Macklin), concentrated sulphuric acid (98%, Sinopharm Chemical Reagent Co. Ltd.), carboxylated multi-walled carbon nanotubes (>95%, Aladdin). Platinum ruthenium black (Pt 50%, Ru 50%) was purchased from Aladdin. Nafion solution (5%) was purchase from Sigma-Aldrich. High pure H2 (99.999%) and CO2 (99.999%) come from Qingdao Deyi Gas Company. The deionized water in the experiment is always ultrapure water (18.2 MΩ·cm).
Synthesis of PdC0.13/CNT
All catalysts were synthesized via a solvent-free microwave-assisted method. Specifically, Pd(acac)₂ (10 mg), cellulose powder (10 mg), and carboxylated carbon nanotubes (10 mg) were combined in a mortar and thoroughly ground to form a homogeneous mixture. This mixture was then transferred into a 10 mL quartz vial and irradiated in a household microwave oven (Midea, Model PM2001) at a power of 1 kW for 30 s. The reaction was initiated under ambient temperature and atmospheric pressure. After irradiation, the product was washed with ethanol and isolated by centrifugation. Finally, the obtained solid was dried overnight in an oven at 60 °C, yielding a black powder.
Synthesis of other catalysts
Pd/CNT, PdC0.07/CNT, PdC0.20/CNT and PdC0.26/CNT were synthesised as described above. The difference is that the mass of cellulose was replaced with 0 mg, 5 mg, 15 mg and 20 mg, respectively.
Calculation of the lattice hydrogen atoms
Where λ is the X-ray wavelength (0.154 nm), θ is the angle in the XRD, (hkl) is the face plane of Pd, \(\alpha {Pd}\), \(\alpha {{Pd}}_{C}\) is the lattice constant, x is the number of hydrogen atoms.
Characterization
SEM images were obtained by Hitachi, S-8200. TEM and HRTEM of the catalyst were tested using FEI Tecnai-G2 F30 at an accelerating voltage of 80 KV. To confirm the atomic dispersion of metal atoms in the samples, AC-HAADF-STEM (FEI Themis Z and Titan Cubed Themis G2 60-300) were used. Powder XRD spectra were recorded on an X’Pert-Pro MPD diffractometer with Cu Kα radiation at 40 KV and 40 mA. XPS analysis was performed with an Axis Supra spectrometer using a monochromatic Al Kɑ source at 15 mA and 14 kV. The TPD experiments were conducted with a Micromeritics Auto Chem II 2920 instrument. The composition of as-prepared samples was collected by the inductively coupled plasma-atomic emission spectroscopy (ICP-AES, Agilent 8800). The catalysts after the durability test were sonicated in ethanol and then collected for the next step of characterization.
X-ray absorption spectra were carried out at 9-BM beamline of Advanced Photon Source (Argonne in America), The X-ray beam size at horizontal 1 mm*1 mm vertical. The resolution of beamline ΔE/E = 1*10-4, and the photon fux (photons/sec) was 1*1011@15 keV per second. The spectra were processed and analyzed by the software codes Athena. The normalized, energy-calibrated Pd K-edge XANES spectra were obtained using standard data reduction techniques with ATHENA and ARTEMIS software. The EXAFS oscillations χ(k) as a function of photoelectron wave number k was extracted by following standard procedures. For the samples, a k-range of 3-10.0 Å-1 was used and curve fittings were carried out in R-space within an R range of 1.0-4.0 Å for k2-weighted χ(k) functions. The obtained EXAFS data were treated using the ATHENA module of the IFEFFIT software packages, and the parameter fitting was performed in the ARTEMIS55 module by using following equation:
in which Nj refers to the number of neighbors in the jth atomic shell, S02 refers to the amplitude reduction factor, Fj(k) refers to the effective curved-wave backscattering amplitude, Rj refers to the back scatterer factor, λ refers to the mean free path, ϕj(k) refers to the phase shift, σj refers to the Debye-Waller parameter of the jth atomic shell.
Electrochemical measurements
All electrochemical tests were performed on a CHI660E electrochemical workstation (Chenhua, Shanghai) and Gamry Reference 3000. The catalyst ink for working electrode was prepared by dispersing 2 mg of catalyst in a mixed solution of 30 μL Nafion (0.5 wt%), 500 μL ethanol and 470 μL water followed by sonication for 30 minutes. The homogeneous ink was carefully dropped onto the carbon paper (1 cm2) to obtain the working electrode with a desirable loading of 0.2 mg cm-2 (0.19 mgPd cm-2). All experiments were carried out at room temperature (25 oC). The electrochemical measurements of CO2RR were conducted via a three-electrode setup with pre-reduction catalyst, Ag/AgCl electrode and platinum sheet electrode as working electrode, reference electrode and counter electrode, respectively. The thermodynamic potential corresponding to Ag/AgCl was determined by the average potential where the current approaches zero. The electrochemical measurement was conducted in a typical H-type cell with the proton exchange membrane (Nafion 117, thickness:183 µm) and each chamber contained 40 mL 0.5 M K2SO4 solution (pH adjusted to 3 ± 0.03 by H2SO4). The electrolyte is prepared as needed and stored at room temperature away from light. Before electrochemical measurement, the electrolyte in the cathodic compartment was purged with CO2 for 30 min. All potentials were reported with respect to the RHE by the following equation:
In 0.5 M K2SO4 solution (pH adjusted to 3 ± 0.03 by H2SO4) saturated with CO2, LSV was used to test and evaluate the CO2RR performance of the catalyst at a sweep rate of 10 mV s-1. All polarization curves were corrected for 95% iR, where R was measured to be 1.1 ± 0.03 Ω. The durability test was performed in 0.5 M K2SO4 solution (pH adjusted to 3 ± 0.03 by H2SO4) using chronoamperometry. EIS measurements were carried out in a 0.5 M K2SO4 solution (pH adjusted to 3 ± 0.03 by H2SO4) pumped into pure CO2 gas at -0.9 V vs. RHE with a frequency range from 100 kHz to 0.1 Hz and an amplitude of 5 mV.
Preparation of PEM electrolyzer
The electrolyzer (with an electrode exposure area of 2 × 2 cm²) was a two-electrode system with the reference electrode removed, where one gas diffusion layer (YLS-30T, GDL) with cathodic catalyst and the other with Pt-Ru black were used as cathode and anode, respectively. First, Nafion 117 membranes were treated with hydrogen peroxide and 0.5 M sulfuric acid at 80 °C for 1 h, respectively. Next, a homogeneous catalyst ink was prepared by mixing 20 mg of catalyst, 20 µl of 1% by weight PTFE emulsion, 2 ml of ethanol, and 100 μl of 5% ethanol solution of Nafion, followed by ultrasonication for 0.5 hours. The ink was then sprayed onto the GDL as a cathode electrode via an air gun with a mass loading of 0.5 mg cm-². During the cathodic reaction, the rear of the gas diffusion electrode was permeated with high-purity carbon dioxide at a flow rate of 20.0 sccm, which was controlled by a gas flow meter used in the electrolysis process. The cathodic electrolyte flowed over the catalyst surface. The pH of the cathodic electrolyte was adjusted by sulfuric acid and potassium sulfate to adjust 3 ± 0.03. In the anodic reaction (HOR), 20 mg of Pt-Ru black catalyst, 20 µl of 1% wt. PTFE emulsion, 2 ml of ethanol, and 100 μl of Nafion in 5% ethanol solution were mixed to prepare a homogeneous catalyst ink, which was subsequently ultrasonicated for 0.5 hours. The ink was then sprayed onto the GDL as an anode electrode via a gas gun with a mass loading of 0.5 mg cm-². The rear of the gas diffusion electrode was permeated with high purity wet hydrogen at a flow rate of 20.0 sccm, which was also controlled by a gas flow meter used in the electrolysis process. All electrochemical measurements were carried out on a Gamry reference 3000 electrochemical workstation. All electrochemical data presented in this work were not corrected by iR compensation in the two-electrode system.
CO stripping curves were performed in 0.5 M K2SO4 (pH adjusted to 3 ± 0.03 by H2SO4) solution. Before the test, the solution was passed to saturation under N2 atmosphere. Then, CO was bubbled into the cell for 15 min while the potential of the working electrode was held at a constant potential of 0.1 V vs. RHE. Subsequently, it was transferred to N2-saturated 0.5 M K2SO4 (pH adjusted to 3 ± 0.03 by H2SO4), and CO stripping was recorded by CV curves at a scan rate of 50 mV s-1 at potentials ranging from 0-1.05 V vs. RHE.
Calculation of the FE of liquid product HCOOH: The products were characterized by ¹H NMR spectroscopy (AVAN CE NEO, 400 MHz). A 500 µL sample from the H-type cell was directly diluted with 100 µL of a DMSO-d6 and D₂O mixture (1:1000, v/v) for analysis. The concentration of the reduced product HCOOH is calculated as follows.
NMR sample contains:
All spectra were acquired under consistent parameters (e.g., scan number) and the resultant ¹H-NMR signals were integrated and normalized relative to DMSO. The concentration of the reduced product HCOOH is calculated as follows.
Where \({C}_{x}\) is the concentration of HCOOH; \({{I}}_{x}\) and \({I}_{{std}}\) are the NMR signal integrals of HCOOH and the internal standard (DMSO), respectively; \({N}_{{std}}\) and \({N}_{x}\) represent the numbers of protons in DMSO and HCOOH; and \({C}_{{std}}\) is the concentration of DMSO. Subsequently, the FE for HCOOH (the exclusive liquid product) was determined according to the equation below:
Where “\({V}_{{NMR\; tube}}\)” is 600 µL (500 µL NMR sample + 100 µL DMSO-d6 and D₂O mixture), “\({V}_{{electrolyte}}\)” is 40 mL, “\({V}_{{NMR\; sample}}\)” is 500 µL, “\(Z\)” is the electron transfer number for the product formation, and “\(F\)” is Faraday constant (96485.3 C mol-1). Furthermore, I indicates the net circuit current (A), and t is the total time in seconds.
Calculation of the FE, concentration and productivity of H2 and CO: Gaseous products from the cathode compartment were subjected to periodic headspace sampling and subsequent gas chromatographic (GC) analysis. The product gas stream from the H-cell was directed to the GC, where samples from a saturated pre-chamber were introduced into the injection port. A GC7900 chromatograph, featuring dual detection with a thermal conductivity detector (TCD) and a flame ionization detector (FID), was employed to determine the yields of H₂ and CO across the range of applied potentials.
The concentration of gaseous species (i) was quantified based on the detected signals, using the formula:
Where F is the Faraday constant; Z refers to the electron transfer number per product; V, v, and t are the gas volume, flow rate, and reaction time, respectively; p, T, and R denote atmospheric pressure, temperature, and the ideal gas constant; and Q stands for the total hourly charge.
Calculation of the half-cell energy conversion efficiency: The half-cell energy conversion efficiency (also called cathodic energy efficiency, CEE) is calculated using the equation:
where “\({E}_{{HCOOH}}\)” is the standard potential of the HCOOH formation (-0.199 VRHE). “\({{FE}}_{{HCOOH}}\)” is the Faradaic efficiency of HCOOH. “\({E}_{{cathode}}\)” is the applied potential vs. RHE.
In situ FTIR measurement
The intermediate products during the CO2RR process were detected by in-situ FTIR through Thermo iS50 FT-IR with a liquid-nitrogen-cooled MCT-A detector. The in-situ FTIR curves were collected by the method of internal reflection. The pre-reduction catalyst was used as working electrode, Ag/AgCl electrode and platinum sheet electrode were worked as reference electrode and counter electrode respectively. All the tests were conducted in CO2 saturated 0.5 M K2SO4 solution (pH adjusted to 3 ± 0.03 by H2SO4). The applied potential was stepped positively from -0.7 V to -1.1 V (vs. RHE) with an interval of 100 mV. The upward bands represent the formation of products, the downward bands represent the consumption of reactants.
Calculation Setup
We carried out all the DFT calculations in the Vienna ab initio simulation (VASP 6.3.0) code. The exchange-correlation is simulated with PBE functional and the ion-electron interactions were described by the PAW method. The vdWs interaction was included by using empirical DFT-D3 method. The 4×4 supercell of four layered Pd (111) (a = b = 11.005 Å, c = 23 Å; α = β = 90°, γ = 120°) and the C doped Pd (PdC0.13 (111)) were used to investigate the CO2RR and HER. The atoms in the upper two layers of these surfaces are allowed to move freely while the bottom two layers are fixed to simulate the surface of structure. The Monkhorst-Pack-grid-mesh-based Brillouin zone k-points are set as 3×3×1 for all surface with the cutoff energy of 450 eV. Taking the PdC0.13 (*OCHO and *COOH) system as an example to test the energies calculated at different K points, it can be seen that the energy of the system almost reaches equilibrium at 3×3×1. Moreover, the adsorption energy difference (<2 meV/atom) of different intermediates at this point satisfies the established selection criteria. Considering the computational resource usage and computational accuracy, the 3×3×1 chosen in this work is reasonable (Supplementary Fig. 40). The convergence criteria are set as 0.02 eV A-1 and 10-5 eV in force and energy, respectively.
The free energy calculation of species adsorption (∆G) is based on following model.
Herein ΔE, ΔEZPE, and ΔS respectively represent the changes of electronic energy, zero-point energy, and entropy of the intermediate. The ΔH0→T refers to the change in enthalpy when heating from 0 K to T K. The entropy of H+ + e- pair is approximately regarded as half of H2 entropy in standard condition. The binding energy was calculated as Ebinding = Esubstrate + Eadsorbate − Etotal, where the Esubstrate, Eadsorbate and Etotal were the DFT energy of the catalyst substrate, the adsorbate and the total system.
The band center can be determined using the following formula:
The given equation represents the calculation formula for the band center. In this equation, ε represents the energy level, and norbitals (ε) corresponds to the density of states (DOS) of the orbitals (such as s, p, d and f) in the material. The optimized configuration of Pd and PdC0.13 are shown in Supplementary Data 1, 2.
Data availability
The data generated in this study are provided in the Supplementary Information and Source Data file. Source data are provided with this paper.
References
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Woldu, A. R. et al. Insights into electrochemical CO2 reduction on SnS2: main product switch from hydrogen to formate by pulsed potential electrolysis. Angew. Chem. Int. Ed. 62, e202301621 (2023).
Kang, X. et al. Electro-reduction of carbon dioxide at low over-potential at a metal–organic framework decorated cathode. Nat. Commun. 11, 5464 (2020).
Zhang, H. et al. Efficient capture and separation of CO2-boosted carbon neutralization enabled by tailorable metal-organic frameworks: a review. EcoEnergy 1, 217–247 (2023).
Yang, S. et al. Halide-guided active site exposure in bismuth electrocatalysts for selective CO2 conversion into formic acid. Nat. Catal. 6, 796–806 (2023).
Wang, H. et al. Exfoliated 2D layered and nonlayered metal phosphorous trichalcogenides nanosheets as promising electrocatalysts for CO2 reduction. Angew. Chem. Int. Ed. 62, e202217253 (2023).
Huang, L. et al. Pressure dependence in aqueous-based electrochemical CO2 reduction. Nat. Commun. 14, 2958 (2023).
Rabiee, H., Yan, P., Wang, H., Zhu, Z. & Ge, L. Electrochemical CO2 reduction integrated with membrane/adsorption-based CO2 capture in gas-diffusion electrodes and electrolytes. EcoEnergy 2, 3–21 (2024).
Hu, L. et al. A scalable membrane electrode assembly architecture for efficient electrochemical conversion of CO2 to formic acid. Nat. Commun. 14, 7605 (2023).
Cobb, S. J., Dharani, A. M., Oliveira, A. R., Pereira, I. A. C. & Reisner, E. Carboxysome-inspired electrocatalysis using enzymes for the reduction of CO2 at low concentrations. Angew. Chem. Int. Ed. 62, e202218782 (2023).
Cao, X. et al. Strong p-d orbital hybridization on bismuth nanosheets for high performing CO2 electroreduction. Adv. Mater. 36, 2309648 (2024).
Shi, Y. et al. Unveiling hydrocerussite as an electrochemically stable active phase for efficient carbon dioxide electroreduction to formate. Nat. Commun. 11, 3415 (2020).
Deng, Y. et al. Operando spectroscopic analysis of axial oxygen-coordinated single-Sn-atom sites for electrochemical CO2 reduction. J. Am. Chem. Soc. 145, 7242–7251 (2023).
Fan, J. et al. Immobilized tetraalkylammonium cations enable metal-free CO2 electroreduction in acid and pure water. Angew. Chem. Int. Ed. 63, e202317828 (2024).
Zi, X. et al. Breaking K+ concentration limit on Cu nanoneedles for acidic electrocatalytic CO2 reduction to multi-carbon products. Angew. Chem. Int. Ed. 62, e202309351 (2023).
Li, H. et al. Tailoring acidic microenvironments for carbon-efficient CO2 electrolysis over Ni-N-C catalyst in a membrane electrode assembly electrolyzer. Energy Environ. Sci. 16, 1502–1510 (2023).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Shen, H. et al. Acidic CO2-to-HCOOH electrolysis with industrial-level current on phase engineered tin sulfide. Nat. Commun. 14, 2843 (2023).
Qin, H.-G. et al. Surface-immobilized cross-linked cationic polyelectrolyte enables CO2 reduction with metal cation-free acidic electrolyte. Nat. Commun. 14, 5640 (2023).
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Sheng, X., Ge, W., Jiang, H. & Li, C. Engineering Ni-N-C catalyst microenvironment enabling CO2 electroreduction with nearly 100% CO selectivity in acid. Adv. Mater. 34, 2201295 (2022).
Zhu, C. et al. Selective CO2 electroreduction to multicarbon products exceeding 2 A cm-2 in strong acid via a hollow-fiber Cu penetration electrode. Energy Environ. Sci. 17, 510–517 (2024).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Zhang, L. et al. Atomically dispersed Ni-Cu catalysts for pH-universal CO2 electroreduction. Adv. Mater. 35, 2209590 (2023).
Xu, K. et al. Favoring CO intermediate stabilization and protonation by crown ether for CO2 electromethanation in acidic media. Angew. Chem. Int. Ed. 62, e202311968 (2023).
Zhong, Y. et al. An artificial electrode/electrolyte interface for CO2 electroreduction by cation surfactants self-assembly. Angew. Chem. Int. Ed. 59, 19095–19101 (2020).
Chen, X. et al. Activating inert non-defect sites in Bi catalysts using tensile strain engineering for highly active CO2 electroreduction. Nat. Commun. 16, 1927 (2025).
Cheng, D. et al. Structure sensitivity and catalyst restructuring for CO2 electro-reduction on copper. Nat. Commun. 16, 4064 (2025).
Zhang, Z.-M. et al. Probing electrolyte effects on cation-enhanced CO2 reduction on copper in acidic media. Nat. Catal. 7, 807–817 (2024).
Cheng, Y. et al. Microenvironment tailoring for electrocatalytic CO2 reduction: Effects of interfacial structure on controlling activity and selectivity. J. Am. Chem. Soc. 147, 12438–12448 (2025).
Min, X. & Kanan, M. W. Pd-catalyzed electrohydrogenation of carbon dioxide to formate: High mass activity at low overpotential and identification of the deactivation pathway. J. Am. Chem. Soc. 137, 4701–4708 (2015).
Abdinejad, M. et al. Insertion of MXene-based materials into Cu-Pd 3D aerogels for electroreduction of CO2 to formate. Adv. Energy Mater. 13, 2300402 (2023).
Zhou, Y. et al. Mesoporous PdAg nanospheres for stable electrochemical CO2 reduction to formate. Adv. Mater. 32, 2000992 (2020).
Vasileff, A. et al. Selectivity control for electrochemical CO2 reduction by charge redistribution on the surface of copper alloys. ACS Catal 9, 9411–9417 (2019).
Yoo, J. S., Christensen, R., Vegge, T., Nørskov, J. K. & Studt, F. Theoretical insight into the trends that guide the electrochemical reduction of carbon dioxide to formic acid. ChemSusChem 9, 358–363 (2015).
Kong, S. et al. Delocalization state-induced selective bond breaking for efficient methanol electrosynthesis from CO2. Nat. Catal. 6, 6–15 (2023).
Liu, Z. et al. Switching CO2 electroreduction toward ethanol by delocalization state-tuned bond cleavage. J. Am. Chem. Soc. 146, 14260–14266 (2024).
Chen, H., Zhang, B., Liang, X. & Zou, X. Light alloying element-regulated noble metal catalysts for energy-related applications. Chin. J. Catal. 43, 611–635 (2022).
Xue, H., Zhao, Z.-H., Liao, P.-Q. & Chen, X.-M. “Ship-in-a-bottle” integration of ditin(IV) sites into a metal-organic framework for boosting electroreduction of CO2 in acidic electrolyte. J. Am. Chem. Soc. 145, 16978–16982 (2023).
Guo, R. et al. PdCx nanocrystals with tunable compositions for alkyne semihydrogenation. J. Mater. Chem. A 7, 4714–4720 (2019).
Wang, Z. et al. The PdHx metallene with vacancies for synergistically enhancing electrocatalytic N2 fixation. Chem. Eng. J. 450, 137951 (2022).
Zhang, D. et al. Advanced ultrathin RuPdM (M=Ni, Co, Fe) nanosheets electrocatalyst boosts hydrogen evolution. ACS Cent. Sci. 5, 1991–1997 (2019).
Shi, Y. et al. Solution-phase synthesis of PdH0.706 nanocubes with enhanced stability and activity toward formic acid oxidation. J. Am. Chem. Soc. 144, 2556–2568 (2022).
Guilherme Buzanich, A. et al. BAMline-A real-life sample materials research beamline. J. Chem. Phys. 158, 244202 (2023).
Timoshenko, J. & Roldan Cuenya, B. In Situ/operando electrocatalyst characterization by X-ray absorption spectroscopy. Chem. Rev. 121, 882–961 (2021).
Li, Z. et al. Electron-rich Bi nanosheets promote CO2⋅- formation for high-performance and pH-universal electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. 62, e202217569 (2023).
Chen, S. et al. Engineering support and distribution of palladium and tin on MXene with modulation of the d-band center for CO-resilient methanol oxidation. Angew. Chem. Int. Ed. 61, e202209693 (2022).
Xia, Z. et al. Cavities-induced compressive strain in unique nanotubes boosts the C1 pathway of ethanol oxidation electrocatalysis. ACS Nano 19, 7379–7390 (2025).
Yang, Y. et al. Breaking scaling relationships in alkynol semi-hydrogenation by manipulating interstitial atoms in Pd with d-electron gain. Nat. Commun. 13, 2754 (2022).
Zheng, M. et al. Electrocatalytic CO2-to-C2+ with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936–14944 (2022).
Wang, P. et al. Boosting electrocatalytic CO2-to-ethanol production via asymmetric C-C coupling. Nat. Commun. 13, 3754 (2022).
Hansen, H. A., Varley, J. B., Peterson, A. A. & Norskov, J. K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 4, 388–392 (2013).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52272222 (L.W.)), Taishan Scholar Young Talent Program (tsqn201909114 (L.W.), tsqn201909123 (J.L.)), University Youth Innovation Team of Shandong Province (202201010318 (J.L.)). The author would also like to thank Shiyanjia Lab (www.shiyanjia.com) for the support of DFT calculations.
Author information
Authors and Affiliations
Contributions
Y.Y., Z.W., and W.W. contributed equally to this work. L.W. and J.L. supervised the research. J.L. conceived the research. Y.Y. and Z.W. designed the experiments. Y.Y. and W.W. performed most of the experiments and data analysis. J.H., T.D., and H.L. prepared the electrodes and helped with electrochemical measurements. G.J., Q.L., and A.K. helped analyze physical characterization data. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Mohammadreza Karamad, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Y., Wang, Z., Wang, W. et al. Small PdCx interstitial compound for efficient acidic CO2 electroreduction to formic acid. Nat Commun 17, 1181 (2026). https://doi.org/10.1038/s41467-025-67949-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67949-1
