
Abstract
Protein Language Models (PLMs) trained on large databases of protein sequences have proven effective in modeling protein biology across a wide range of applications. However, while PLMs excel at capturing individual protein properties, they face challenges in natively representing protein-protein interactions (PPIs), which are crucial to understanding cellular processes and disease mechanisms. Here, we introduce MINT, a PLM specifically designed to model sets of interacting proteins in a contextual and scalable manner. Using unsupervised training on a large curated PPI dataset derived from the STRING database, MINT outperforms existing PLMs in diverse tasks relating to protein-protein interactions, including binding affinity prediction and estimation of mutational effects. Beyond these core capabilities, it excels at modeling interactions in complex protein assemblies and surpasses specialized models in antibody-antigen modeling and T cell receptor-epitope binding prediction. MINT’s predictions of mutational impacts on oncogenic PPIs align with experimental studies, and it provides reliable estimates for the potential for cross-neutralization of antibodies against SARS-CoV-2 variants of concern. These findings position MINT as a powerful tool for elucidating complex protein interactions, with significant implications for biomedical research and therapeutic discovery.
Similar content being viewed by others


Introduction
The success of large language models in natural language processing—where complex semantic and syntactic relationships are learned from sequences of words—has inspired their application to protein sequences. By treating amino acid sequences as “sentences,” protein language models (PLMs) can implicitly learn structural and functional patterns, enabling predictions of protein folding, mutational effects, and antibody optimization without explicit structural labels1,2,3,4,5,6,7,8,9. However, within cellular environments, proteins rarely act in isolation; instead, they form extensive interaction networks essential for processes such as signal transduction, metabolic pathways, and cellular structural stability. Hence, a comprehensive understanding of protein biology requires moving beyond isolated protein sequences to consider the complex interactions between multiple proteins. Despite this, even PLMs that predict high-resolution structures have thus far been limited to learning patterns from single protein sequences8,10.
Since almost every PLM is trained using a self-supervised objective on single chains, models often struggle to effectively capture protein-protein interactions (PPIs). Previous approaches that have used PLMs to predict PPIs11,12,13,14,15,16 utilized representations generated from each protein sequence without incorporating the contextual information from the interacting partners. This independence causes critical interaction-specific features to be overlooked, as the representations of each protein sequence are generated in isolation. Furthermore, this approach becomes increasingly impractical for PPIs that involve complex multi-sequence (2+) interactions, such as those seen in antibody-antigen or TCR-epitope-MHC complexes17,18. While concatenating input sequences has been proposed as a workaround, it risks degrading embedding quality by treating all sequences as a single unified entity, potentially masking distinct sequence-specific features19. To address these challenges, we propose Multimeric INteraction Transformer (MINT), an extension of the single-sequence paradigm that allows PLMs to learn distributions of sets of interacting protein sequences. We hypothesize that this approach will enable PLMs to produce context-aware representations that more accurately capture the nuances of PPIs.
MINT contains two new conceptual advances that enable it to model PPIs effectively. The first modification is of the popular Masked Language Modeling (MLM) objective, so that our model can learn from the interacting protein chains present in STRING-DB, a comprehensive and high-confidence database filtered to contain information on 96 million experimental and predicted PPIs20. By training a PLM on this vast corpus of interactions, we aim to uncover deeper representations of not only individual proteins but also how sets of proteins function together in concert. To the best of our knowledge, no existing PLM has been trained extensively on STRING-DB using a self-supervised pre-training objective. Our second innovation is the adaptation of the model architecture and the training of the popular PLM ESM-210 to handle multiple inputs of protein sequences at once. We present a strategy that allows us to fine-tune ESM-2 on STRING-DB by adding a cross-attention module that explicitly extracts inter-sequence information21.
We comprehensively benchmarked MINT against widely used PLMs across multiple PPI prediction tasks. MINT consistently outperformed baseline models in binary interaction classification, binding affinity prediction, and mutational impact assessment, achieving a new state-of-the-art AUPRC of 0.69 on the gold-standard dataset constructed by Bernett et al.22, and delivering a 30% improvement in predicting binding affinity changes upon mutation on the SKEMPI dataset23. Furthermore, MINT demonstrated superior performance in antibody modeling, exceeding antibody-specific baselines on property prediction tasks from the Fitness Landscapes for Antibodies (FLAB) benchmark24 by over 10%. It also outperformed AbMap19 in predicting binding affinity changes in SARS-CoV-2 antibody mutants, achieving a 14% performance gain in the setting where only 0.5% of the data was available for training. In TCR-epitope modeling, MINT surpassed state-of-the-art models like PISTE17 and AVIB-TCR25 with minimal fine-tuning. We show how MINT can model the important task of predicting variant effects in PPIs. In oncogenic PPI (oncoPPI) analysis, MINT effectively distinguished between pathogenic and non-pathogenic interactions, with its predictions matching 23 of 24 previously experimentally validated mutational effects. Similarly, in the prediction of antibody cross-neutralization against SARS-CoV-2 variants, MINT achieved high precision-recall performance, capturing shifts in neutralization profiles across Omicron sub-variants and demonstrating an 80% accuracy in identifying antibodies with consistent neutralization capabilities. By treating multimeric interactions as interdependent sets of sequences rather than isolated groups, MINT provides a unified approach to computational PPI modeling, offering a powerful framework for studying disease mechanisms, guiding therapeutic design, and advancing immunological research.
Results
Overview of the MINT model and training
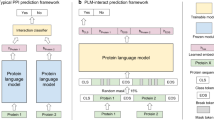
PLMs have been successfully applied to predict the structural, functional, and evolutionary attributes of proteins1,3,4,5,6,7. However, encoding PPIs poses unique challenges, as PLMs are traditionally trained to model single protein sequences independently. To address these challenges, prior approaches have involved passing each sequence through the PLM separately and then concatenating the embeddings, or alternatively, embedding interacting proteins as a single continuous sequence (Fig. 1a)16. However, these methods introduce limitations, such as the loss of inter-residue context and an oversimplified treatment of multi-sequence interactions.

a Existing PLMs either process multiple interacting proteins either by concatenating the output embeddings (left) or concatenating the input tokens (right). The former involves making multiple passes through the PLM to generate embeddings for each sequence independently and then concatenating them. The latter treats interacting sequences as a single sequence and generates the embeddings for the concatenated sequence. b MINT treats multiple interacting sequences as separate entities and generates embeddings in a contextual manner that conserves cross-sequence relationships and maintains scalability. This enables it to learn from the vast number of physical PPIs from STRING-DB20 using a modified version of the masked language modeling (MLM) loss. c The workflow and architecture of MINT. Each protein sequence is tokenized using the ESM-2 tokenizer10, and special tokens are added for the start and end of the sequence. Note here that we add these special tokens for each interacting sequence, maintaining sequence identity. Our architecture involves adding cross-attention blocks to the base ESM-2 model. This results in the output representations of each token being affected by the tokens in the same sequence and those in the interacting sequences. Each block is repeated L times, where L is 33 for MINT. d A non-exhaustive list of protein types, PPI properties, and research questions that can be evaluated using MINT. We benchmark it on general protein complexes, antibodies, and TCR-Epitope-MHC interactions against other PLMs. We then provide examples of the types of analysis that can be done using MINT by predicting oncogenic PPIs and SARS-CoV-2 antibody cross-neutralization using experimentally labeled data. Created in BioRender. Ullanat, V. (2025) https://BioRender.com/d80o431.
To address these limitations, we developed MINT, a PLM specifically designed to model PPIs by enabling the simultaneous input of multiple interacting protein sequences (Fig. 1b). Building on the 650-million-parameter ESM-2 architecture, MINT introduces a cross-chain attention mechanism that preserves inter-sequence relationships and scales effectively to interactions with more than two chains. Whereas the self-attention mechanism in ESM-2 utilizes rotary positional encoding to capture intra-sequence positional relationships, MINT applies self-attention solely within chains and incorporates additional attention blocks for cross-chain interactions without rotary encoding10. This modification ensures that each token representation in our model captures contextual information across all input sequences (Fig. 1c). Further architectural details and pseudocode are provided in the “Methods” Section “MINT architecture”.
To train MINT, we curated a dataset from the STRING database, which includes 2.4 billion physical PPIs and 59.3 million unique protein sequences. By applying clustering and diversity measures, we refined this dataset to 96 million high-quality PPIs involving 16.4 million unique sequences (“Methods” Section “STRING dataset construction”). This dataset serves as the foundation for training MINT using a MLM objective augmented with cross-attention26. Unlike traditional MLM, where token prediction is conditioned solely on intra-sequence context, MINT leverages cross-chain representations to capture co-evolutionary constraints imposed by interacting residues (Fig. 1c, “Methods” Section “MINT architecture”). Model training followed a multiphase approach, beginning with the initialization of attention and embedding weights from ESM-2. This warm-start approach allowed us to preserve foundational sequence-level knowledge while optimizing cross-chain interactions. The model was trained with a masking scheme and hyperparameters closely aligned with ESM-2, with the objective function incorporating both sequence-specific and interaction-specific signals (“Methods” Section “MINT training”). Performance during training was assessed using the perplexity metric (“Methods” Section “MINT training”) on a validation set obtained from a random split of the curated dataset, and compared against the base ESM-2 models (Supplementary Note 2). MINT exhibited lower perplexity than the sequence-concatenated ESM-2 baseline, indicating improved modeling of PPIs. To further examine MINT’s generalization capacity, we performed supervised PPI prediction on a held-out STRING validation set that was excluded from pretraining, achieving superior performance over the ESM-2-650M baseline (Supplementary Note 3).
Together, these architectural and training innovations enable MINT to overcome the inherent limitations of existing PLMs in learning PPI rules. We demonstrate versatility in handling diverse protein sequence inputs, including general complexes, antibody heavy and light chains, T-Cell Receptor (TCR) regions, and peptides, without restrictions on the number of sequences processed concurrently (Fig. 1d). This capability allows MINT to capture intricate features relevant to disease-specific interactions and mechanisms, such as mutational impacts in cancer and the cross-neutralization potential of SARS-CoV-2 antibodies.
Comprehensive benchmarking of MINT on PPI prediction
PPI prediction is fundamental to understanding cellular processes, offering insights into disease mechanisms and therapeutic targets. Although structure-based models such as AlphaFold7,27 provide detailed predictions, they face challenges with scalability and accuracy for non-interacting pairs15. Sequence-based methods, in contrast, offer efficiency and flexibility but fall short in explicitly encoding PPIs. To address these gaps, we benchmarked MINT against widely used PLMs on supervised tasks including binary interaction classification, binding affinity prediction, and mutational impact assessment. We use standard dataset splits and provide detailed descriptions of dataset construction, splitting protocols, and measures to mitigate potential information leakage in “Methods” Section “Benchmarking tasks”.
We initially assessed the performance of MINT in predicting the presence or absence of interactions between protein chains, as well as in binary interaction prediction. This evaluation was conducted using three distinct datasets, including a comprehensive dataset curated by Bernett et al.22 that employs rigorous sequence similarity thresholds to mitigate data leakage, alongside two additional datasets derived from the PEER benchmark, which specifically focus on binary predictions of PPIs present in human and yeast systems12. Next, we evaluated how well MINT can predict absolute binding affinity by leveraging binding data for protein-protein complexes from PDB-Bind, where each complex can have a variable number of protein chains28. Both the binary interaction prediction and binding affinity prediction tasks involve a single interacting group of protein sequences, and an overview of the prediction process is illustrated in Fig. 2a.

a Overview of the model framework for downstream tasks involving the prediction of properties for a single interacting group of sequences. This analysis involves the gold-standard dataset (Gold-standard PPI)22, the human (HumanPPI) and yeast (YeastPPI) datasets from the PEER benchmark12,67, and the PDB-Bind affinity (PDB-Bind) dataset28. We generate embeddings for the baseline PLMs and MINT and use a Multi-Layer Perceptron (MLP) to predict either the binary interaction or the binding affinity. b Overview of the model framework for downstream tasks involving the prediction of properties for mutation effect analysis, involving wild-type and mutated sequence groups. This analysis involves the datasets from SKEMPI23 and the binary prediction of PPI binding after mutation (MutationalPPI)29. We generate embeddings for the baseline PLMs and MINT for both wild-type and mutant sequence groups separately, and then aggregate them to obtain a final embedding. Then, we use an MLP to predict either the binary interaction or the change in binding affinity. c–h Results for all benchmarking tasks against baseline models with three experimental repeats each; data are presented as mean values ± s.d. c Human PPI, d Yeast PPI, e Gold-standard PPI, f Mutational PPI, g SKEMPI, h PDB-Bind. AUPRC area under the precision-recall curve, PCC Pearson correlation coefficient. Source data are provided as a Source Data file. Created in BioRender. Ullanat, V. (2025) https://BioRender.com/2knnohq.
We also evaluated the ability of MINT to resolve mutational effects on binding on two tasks. The first contained data from SKEMPI—a database documenting changes in binding affinity due to mutations in one of the interacting proteins—for which we evaluated the models with previously established cross-validation splits by protein complex23. The second task in this context involved predicting whether two human proteins remain bound after a mutation occurs in one of them, thereby testing the model’s sensitivity to sequence alterations that impact binding outcomes29. These tasks involve encoding the wild-type protein pairs and the mutant protein pairs, as described in Fig. 2a.
In each task, MINT’s embeddings were utilized to train lightweight predictor models, facilitating equitable comparisons with baseline pre-trained language models (PLMs) such as ESM-1b30, ESM-2 (comprising both 650M and 3B parameters)10, ProGen (3B parameters)31, and ProtT5 (3B parameters, specifically trained on the UniRef and BFD databases)32 (“Methods” Section “Benchmarking tasks”). We compared two baseline embedding strategies: (1) concatenating independently computed embeddings from the PLM and (2) concatenating interacting sequences as a single input to the PLM (Fig. 1). For all tasks except PDB-Bind, the first strategy of concatenating embeddings worked better for all baseline models. For the PDB-Bind task, we evaluated only the second method of concatenation since each input can contain a variable number of interacting sequences, making it impractical to perform a variable number of PLM calls. We show the results for the best performing embedding strategy in Fig. 2c. Results for both embedding strategies are available in Supplementary Table 1.
MINT consistently outperformed all baseline PLMs across all tasks (Fig. 2c). On the gold-standard dataset constructed by Bernett et al., MINT achieved a new state-of-the-art performance with an area under the precision-recall curve (AUPRC) of 0.69, demonstrating its ability to excel under challenging conditions and to capture non-trivial interaction signals missed by traditional PLMs. In the other binary PPI prediction tasks, MINT achieved an improvement of 11% over the second-best baseline in yeast interactions and 3% over the average baseline for human interactions. On the SKEMPI dataset for predicting binding affinity changes upon mutation, our model achieved a 32% improvement, representing the highest performance for any model using only sequence information. Notably, MINT consistently outperformed the base ESM-2 model, indicating that the architectural enhancements and fine-tuning on STRING-DB enable the learning of essential PPI signatures absent in ESM-2. Furthermore, its performance advantages over larger general-purpose PLMs like ProGen and ProtT5 underscore the importance of task-specific adaptations over mere increases in model size. Finally, inference time benchmarking (Supplementary Note 4) showed that MINT also achieves competitive runtime efficiency compared to the baselines while outperforming them in predictive accuracy.
Performance on antibody modeling tasks
Building on its superior performance in general PPI tasks, we extended MINT to domain-specific challenges in antibody and immune modeling to test its ability to generalize beyond traditional protein interaction datasets. Antibodies are essential for the adaptive immune system’s ability to recognize and neutralize pathogens. They are symmetrical, Y-shaped molecules composed of two heavy chains and two light chains33. Antibody-antigen interactions are also uniquely difficult to model because CDR regions are highly variable and are not as constrained by evolution34. Here, we show how MINT can be used to predict the binding characteristics of antibodies to antigens by leveraging its ability to jointly model the heavy and light chains, thereby enabling richer representations suitable for various downstream tasks such as binary interaction and binding affinity prediction. We compare MINT with three deep-learning-based approaches specific to antibody modeling, evaluating our model on the same datasets and employing the same evaluation strategies used in each approach.
We compared MINT against IgBert and IgT5—two models fine-tuned on extensive datasets comprising two billion unpaired and two million paired sequences of antibody light and heavy chains18. These models explicitly “learn the language” of antibodies, encoding heavy and light chains either jointly as pairs or separately in an unpaired manner. We assessed their performance on four supervised property prediction tasks using the FLAB dataset, which includes experimentally measured binding energy data from Shanehsazzadeh et al.35, Warszawski et al.36, Koenig et al.37, and expression data from the latter study. To ensure a faithful comparison, we followed the tenfold cross-validation procedure outlined by Kenlay et al.18, fitting a ridge regression model on embeddings from MINT (Fig. 3a, “Methods” Section “Benchmarking tasks”). Performance was evaluated by measuring the coefficient of determination (R2) between the predicted and actual values. MINT achieves a performance boost over the antibody-specific baselines on all four datasets, with an increase of over 10% on three of them (Fig. 3b). Moreover, it is able to perform equally well in predicting antibody binding affinity and expression tasks. This performance gain demonstrates that pretraining on a diverse corpus of PPI data enhances generalizability to antibody-specific tasks.

a Overview of the model framework for downstream tasks from the FLAB benchmark for evaluating model performance on antibody sequences24. MINT treats the heavy and light chains as separate sequences, enabling better representations. We follow previous work in embedding the antibody sequences and training a Linear Regression (LR) model under a tenfold cross-validation setting to predict either the expression levels or the binding affinity values18. b Results for the antibody-specific PLMs and MINT across the four datasets in the FLAB benchmark. Bars show mean R2 values (coefficient of determination), with error bars denoting ± s.d. across ten outer folds of cross-validation. Models were trained using linear least squares with L2 regularization, with the regularization parameter chosen by nested fivefold inner cross-validation. Sample sizes (n) are the number of independent antibody-antigen pairs: Binding (n = 422, n = 2048, n = 4275) and Expression (n = 4275). Baseline models include AbLang, AntiBERTy, IgBert, IgBert-unpaired, IgT5, and IgT5-unpaired. c Overview of the model framework for the prediction of binding affinity changes for m396 antibody mutants against the SARS-CoV-2 virus38. We follow previous work (AbMap) in embedding the antibody sequences and training a Linear Regression (LR) model using different fractions of the dataset, testing on the rest19. d Results for different configurations of the AbMap model and MINT across dataset splits of 20, 5, and 0.5% training data. Bars show the Spearman rank correlation in the test set. AbMap-E uses ESM-1b embeddings, and AbMap-P uses ProtBert embeddings, each evaluated with MLP and ridge regression predictors19. Source data are provided as a Source Data file. Created in BioRender. Ullanat, V. (2025) https://BioRender.com/y5e0lbu.
We further evaluated the performance of MINT in comparison with AbMap, a transfer learning framework fine-tuned specifically for antibodies by focusing on their hypervariable regions and trained on antibody-specific structural and binding specificity datasets19. Both models were assessed on a mutational variation prediction task involving the prediction of changes in binding affinity (ddG) for m396 antibody mutants against the SARS-CoV-2 virus, using experimental data from Desautels et al.38 (Fig. 3c). The m396 antibody originally targets the receptor-binding domain (RBD). Of the SARS-CoV-1 spike protein39; mutants were generated to evaluate potential binding to the SARS-CoV-2 RBD. Approximately 90,000 mutants were generated in silico, with binding efficacy estimated through ddG scores computed using five energy functions: FoldX Whole, FoldX Interface Only40, Statium41, Rosetta Flex, and Rosetta Total Energy42. Adhering to the dataset splitting strategy employed by the AbMap method, we trained on designated subsets of the data and evaluated performance on the remaining portions using the Spearman rank correlation (“Methods” Section “Benchmarking tasks”). Again, MINTachieves comparable or better performance compared to AbMap across all data-splitting instances (Fig. 3d). Crucially, our model achieves a 14% increase in performance when trained on just 0.5% of the samples, suggesting that it can be used for in-silico antibody design even with a small number of training data.
Learning the language of TCR-Epitope-MHC interactions with minimal finetuning
The interactions between T cell receptors (TCRs), epitopes, and major histocompatibility complex (MHC) molecules are central to the human immune response, allowing the immune system to identify and respond to pathogens, cancer cells, and other threats. TCRs on the surface of T cells recognize specific antigenic peptides (epitopes) that are presented by MHC molecules on antigen-presenting cells. This recognition is the first step in activating T cells, which is critical for orchestrating an immune response43. Several deep learning-based approaches have been developed to model the interactions between TCR, epitopes, and MHC molecules, primarily focusing on binary predictions of TCR-epitope or MHC-epitope binding (first-order interactions)44,45. More recently, methods have been designed to consider all three entities simultaneously, enabling the prediction of TCR-epitope-MHC binding (second-order interactions)17. Additionally, some techniques aim to enhance specificity by predicting the interface residues between interacting TCR-epitope pairs46. MINT offers a flexible framework to model these tasks by accommodating the various input sequences. However, the short length of epitopes and the common practice of considering only the Complementarity-determining Region 3 (CDR3) sequences of MHC and TCRs in most datasets present challenges for its direct application, since MINT is not trained on peptides. To address these limitations, we minimally fine-tuned the final layer of MINT to learn TCR-epitope-MHC interactions across the three task types mentioned above (Supplementary Note 5).
For the first-order tasks, we use the TCR-epitope binding prediction benchmark from Therapeutics Data Commons (TDC), which focuses on predicting whether a TCR binds to an epitope44. The interaction data is originally sourced from the IEDB47, VDJdb48, and McPAS-TCR49 databases. We compare popular TCR-Epitope prediction models, namely AVIB-TCR25, MIX-TPI50, Net-TCR251, PanPep52, TEINet53, and TITAN54. We only use TCR CDR3-beta and epitope sequences as input to MINT to maintain a fair comparison with all baseline models (Fig. 4a). MINT obtains an average AUROC score of 0.581, surpassing the best baseline, which has an AUROC of 0.576 (Fig. 4b). These results demonstrate that minimal fine-tuning of MINT can effectively model TCR-epitope interactions, even for protein types not encountered during its pre-training.

a Overview of the first-order interaction prediction task involving TCR-CDR3 and epitope sequences as input44. Since MINT has not been trained on short sequences, we finetune its last layer (red block in the architecture) to capture the language of TCR-epitope interactions. We then apply an MLP model to predict whether binding occurs or does not occur. b Performance of MINT for predicting binary binding compared to published TCR-epitope specific models. Bars represent mean AUROC values, with error bars indicating ± s.d. across five independent training runs with different random seeds. Baseline results for TITAN, TENet, PanPep, NET-TCR2, MIX-TPI, and AVIB-TCR are taken directly from ref. 44 and represent reported performance on the same dataset under identical evaluation. AUROC area under the receiver operating characteristic curve. c Overview of the second-order interaction prediction task involving TCR-CDR3, HLA-CDR3, and epitope sequences as input. Again, we finetune the last layer of MINT (red block in the architecture) to capture the language of TCR-epitope-HLA interactions. We then apply an MLP model to predict whether binding occurs or does not occur. d Results on the second-order binary binding predictions across different dataset splits on the cancer dataset used for evaluation. Bars show AUROC values for each dataset split. Baseline models (ImRex, TEIM, pMTNet, PISTE) are single reported results from ref. 17 (n = 1; shown as single dots, no error bars). MINT results are averaged over multiple independent runs (n = 3 for random split, n = 5 for the Unified peptide and Reference TCR split); individual runs are shown as dots; error bars indicate mean ± s.d. e Visualization of the TCR-epitope interface prediction task using MINT. We input the TCR-CDR3 and epitope sequences, embed them using MINT, and train a downstream Convolutional Neural Network (CNN) model to predict the interface using a contact map. f Results for the TCR-epitope interface prediction task. Shown are distributions of AUPRC values between predicted and true flattened contact maps for MINT and TEIM46. Each dot represents a unique TCR-epitope pair from the test set (n = 76). Boxplots display the median (center line), interquartile range (box), and range (whiskers). Results are reported for two evaluation conditions: test sets with unseen CDR3 sequences and test sets with unseen epitopes. AUPRC area under the precision-recall curve. Source data are provided as a Source Data file. Created in BioRender. Ullanat, V. (2025) Ullanat, V. (2025) https://BioRender.com/8mx00km.
For the second-order interaction prediction, we train our model on the dataset of human TCR, Epitope, and Human leukocyte antigen (HLA) interactions, where HLA is the human version of MHCs (Fig. 4c). This binding data was originally procured from the McPAS-TCR, VDJdb, and pMTnet55 databases and curated in the PISTE model work17. The evaluation set used consists of TCR-Epitope-HLA triplets obtained from studies of different cancer types. We used all three dataset splitting schemes as described in the same work and show the performance of the finetuned MINT with PISTE, pMTnet, and other first-order models. MINT achieves the highest performance across all dataset splits by leveraging its ability to process multiple protein chains (Fig. 4d). These results highlight MINT’s ability to process multiple protein chains and achieve state-of-the-art performance on complex multi-protein interaction tasks. The scalability of MINT to handle an increasing number of protein chains further emphasizes its utility for modeling intricate biological systems.
For the interface prediction task, we used the dataset constructed in the TEIM work46, with the TCR-Epitope structure data originally sourced from STCRDab56 (Fig. 4e). We restrict ourselves to the splitting strategies where the test set contains novel epitopes or novel TCRs that are not seen during training, and each split results in a total of 122 data points. MINT matches the performance of TEIM on the unseen TCR-CDR3 split and performs better on the unseen epitope split (Fig. 4f). These results underscore MINT’s capacity to infer structural relationships between TCRs and epitopes without extensive pretraining and its robust performance with small datasets, suggesting that its internal protein sequence representations are readily transferable to specialized tasks.
Leveraging MINT for predicting mutation-induced perturbations in oncogenic PPIs
After establishing the superior performance of MINT against popular PLMs on general PPI prediction tasks and against domain-specific models on antibody and TCR-Epitope-MHC tasks, we highlight the types of analyses that can be achieved with MINT. We first applied it to estimate the pathogenicity of missense mutations affecting PPIs in cancer, focusing on cancer-associated mutations that disrupt PPIs, which we refer to as oncoPPIs. Disease-associated germline and somatic mutations are enriched at PPI interfaces, indicating their potential role in disrupting PPI networks. Cheng et al.57 identified 470 potential oncoPPIs linked to patient survival and drug responses and experimentally validated the perturbation effects of 24 somatic missense mutations across 13 oncoPPIs using Yeast Two-Hybrid (Y2H) assays, hypothesizing their functional impact on cancer progression. Existing computational approaches distinguishing pathogenic from non-pathogenic interactions typically focus on single protein sequences, employing biophysical and evolutionary constraints or leveraging protein embeddings (e.g., EVE58, AlphaMissense59, ESM-1b3). However, in oncoPPIs or any other pathogenic PPIs, mutations rarely significantly affect protein structure but alter protein-partner binding60,61. Therefore, we took advantage of MINT’s ability to effectively model multiple interacting proteins and trained it on a dataset containing information on whether a mutation in a human protein sequence disrupts its binding to another human protein or not29. We then predicted the mutational effects on the experimentally validated oncoPPIs from Cheng et al., as outlined in Fig. 5a. Since the training dataset is quite small, we performed multiple repetitions of the training runs, or model ensembling, and computed a binding score that is representative of the proportion of times that the model predicts binding over all repetitions (“Methods” Section “Case studies”).

a Outline of the analysis to predict the experimentally validated mutational effects on binding in oncoPPIs. The oncoPPI discovery was conducted by Cheng et al.57 to identify 24 mutational effects in 13 different oncoPPIs through computational predictions and experimental Y2H assays. We used MINT embeddings and a trainable MLP on a dataset containing wild-type and mutant human protein pairs to predict whether the mutation results in a loss of binding or not, in a manner similar to Fig. 2b. We then used the trained model to predict on the oncoPPI dataset across the 24 mutational effects. Since the training dataset is small, we use an ensemble of 100 trained models and their consensus to compute the predicted binding score, which is the fraction of times a particular oncoPPI is assigned a value of 1 (binding conserved) or 0 (binding lost). b Predicted binding scores and true mutational effects from Y2H assays for all 24 mutations. Interacting protein names, missense mutation information (wild-type residue and position in green, mutant residue in red), and the predicted binding scores are shown. Homodimers are indicated with identical icons, whereas heterodimers are indicated with distinct icons. Green check marks and red crosses denote correct and incorrect predictions, respectively. Using a threshold of 0.68 (“Methods” Section “Case studies”), MINT correctly distinguished 23 of 24 mutations. Source data are provided as a Source Data file. Created in BioRender. Ullanat, V. (2025) https://BioRender.com/e80g506.
We show the real mutational effects and model predicted scores for all of the experimentally validated oncoPPIs from Cheng et al. in Fig. 5c. Using a threshold value computed in an unsupervised manner (“Methods” Section “Case studies”), MINT’s predictions match the mutational effects for 23 out of 24 PPIs. Our model is able to correctly assign the ALOX5-MAD1L1 interaction as being lost after the M146K mutation in ALOX5, and both of these proteins have been implicated in tumor progression in several types of cancer62. Interestingly, our analysis correctly predicts a destabilizing mutation for the HOMEZ-EBF1 interaction, and previous ZDOCK analysis has shown that the mutation occurs at the binding interface, potentially disrupting the salt bridge and hydrogen bond at that position13. Out of all 24 mutations, only one oncoPPI, the ARHGDIA-RHOA pair, was incorrectly assigned. Our results indicate that MINT has the ability to effectively model mutational effects in oncoPPIs and can be used to provide insights into how mutations disrupt PPIs in cancer.
MINT predicts antibody cross-neutralization against SARS-CoV-2 variants
Next, we consider the task of estimating the cross-neutralization of antibodies against different SARS-CoV-2 variants. SARS-CoV-2 undergoes a very high rate of mutation, leading to significant changes in transmissibility, immune evasion, and severity between strains. One highly mutated version of the virus is the Omicron variant, which was able to evade immunity from several neutralizing antibodies produced by natural infection and vaccination against previous variants63. Using experimental methods like deep mutational scans to predict immune escape for each newly emerging variant, such as Omicron, is time-consuming, expensive, and intractable. Computational methods such as EVEScape, which use pre-pandemic data, do not directly model antibody-spike protein interactions and thus fall short in predicting how existing antibody titers might target emerging or potential variants5,6. To address these limitations, we utilized MINT in conjunction with the CoV-AbDab database64 to encode antibodies—both naturally produced and vaccine-induced—and the SARS-CoV-2 spike proteins. The resulting embeddings were used to train an MLP model to predict whether an antibody-spike protein pair results in neutralization. Training was conducted using data from early pandemic variants (wild-type, Alpha, Beta, and others). For evaluation, we predicted the cross-neutralization capabilities of antibodies targeting Omicron sub-variants using a normalized score that reflects the likelihood of neutralization (Fig. 6a, “Methods” Section “Case studies”). This approach mirrors a realistic pandemic scenario in which existing antibody titers or designed antibodies need to be evaluated for their cross-neutralization capabilities against newly emerging variants. Figure 6b highlights the composition of the evaluation dataset, showing the distribution of data points for each Omicron sub-variant and their origins, categorized as vaccine-induced or infection-derived.

a Procedure for predicting antibody cross-neutralization ability against SARS-CoV-2 variants. First, we extract data from the CoV-AbDab database64 and filter entries to include antibodies produced in response to early SARS-CoV-2 variants (wild-type, Alpha, Beta, Gamma, etc.) and those that target their receptor-binding domain (RBD). For evaluation, we obtain the entries for these antibodies against different Omicron sub-variants (BA.1, BA.2, BA.4, BA.5). The inputs to MINT are the heavy and light chain sequences, along with the RBD sequence. We train an MLP on embeddings generated by MINT to predict the presence or absence of neutralization for each antibody-RBD pair. We then evaluate MINT's performance against the Omicron sub-variants to validate the neutralization ability. b The dataset composition for the constructed evaluation set across all four sub-variants of Omicron shows the proportion of entries across different antibody origins. c Distribution of the predicted normalized score for each sub-variant of Omicron across antibodies of all origin types. We group it by the actual neutralization profiles (neutralizing or non-neutralizing). We also show AUPRC values for each sub-variant. d Distribution of the normalized score for each sub-variant of Omicron across vaccine-induced antibodies only, grouped by actual neutralization values. e Normalized score values for 10 antibodies against different variants of Omicron, along with their experimentally-derived IC50 values from Liu et al.66. We divide the IC50 values into non-neutralizing (= 10000 ng/ml), weakly-neutralizing ( > = 1000 ng/ml and < 10000 ng/ml), and neutralizing categories ( < 1000 ng/ml). We do the same for the predicted normalized scores: non-neutralizing (negative scores), weakly-neutralizing (positive scores less than 0.10), and neutralizing (positive scores greater than 0.10). Source data are provided as a Source Data file. Created in BioRender. Ullanat, V. (2025) https://BioRender.com/uy52u6w.
MINT demonstrated robust predictive performance, achieving high area under the precision-recall curve (AUPRC) values for the first three Omicron sub-variants (Fig. 6c). Notably, predictions for vaccine-induced antibodies outperformed those for infection-derived antibodies, with higher AUPRC scores observed across all variants (Fig. 6d). This enhanced performance likely reflects the greater homogeneity of vaccine-induced antibody repertoires compared to those generated through natural infection65. To further validate the model, we compared MINT’s normalized scores against IC50 (Half maximal inhibitory concentration) binding affinity values reported in experimental studies. Using BBIBP-CorV vaccine-induced antibodies evaluated in Liu et al.66, we observed an 80% hit rate for correctly identifying antibodies with consistent neutralization across Omicron sub-variants (Fig. 6e). Importantly, the model captured transitions in neutralization capacity along the evolutionary trajectory of Omicron, from BA.1 to BA.5, for antibodies such as Liu 14-5G, Liu 16-1C, and Liu 14-1C. Hence, MINT is able to discern nuanced shifts in neutralization profiles associated with variant evolution.
Discussion
In this study, we introduced MINT, a protein language model designed to encode PPIs by simultaneously processing multiple interacting protein sequences. Unlike traditional PLMs that treat sequences independently, MINT incorporates cross-chain attention mechanisms to capture inter-sequence relationships. This architectural innovation preserves inter-residue context and overcomes limitations associated with previous PLMs that either concatenate embeddings or treat interacting proteins as a single continuous sequence. Our comprehensive benchmarking demonstrated that MINT significantly outperforms existing PLMs across a diverse set of supervised downstream tasks involving PPIs. In the context of antibody modeling, we showed that MINT’s training on a broad corpus of PPI data enabled it to generalize more effectively to antibody-related tasks. On TCR-epitope tasks, we highlighted MINT’s flexibility and its potential to be adapted to tasks involving protein types not present in its original training set.
We applied MINT to biologically relevant case studies to showcase its practical utility. MINT effectively modeled interacting protein sequences implicated in cancer and successfully differentiated between mutations that disrupt PPIs and those that do not. In the realm of infectious diseases, MINT accurately assessed the cross-neutralization potential of antibodies against emerging SARS-CoV-2 variants. The model’s predictions closely matched experimental data, capturing nuanced shifts in neutralization profiles associated with variant evolution and highlighting its potential in guiding vaccine design and therapeutic interventions.
Despite these advancements, there are limitations to our approach. Firstly, MINT is currently only trained on pairs of sequences from STRING using the modified MLM approach. However, our architecture can support pre-training or further finetuning on a dataset of complexes with a variable number of interacting sequences. Next, while MINT demonstrates strong overall performance across multiple PPI-related tasks, the degree of improvement over baseline PLMs varies. On datasets with high sequence redundancy or saturated classification tasks, such as HumanPPI, MINT achieves only modest performance gains—a trend observed with other recent PLMs, including those that incorporate explicit structural information67. These marginal improvements may reflect an overall saturation in standard benchmarks rather than a shortcoming unique to MINT, but they nonetheless limit the immediate impact on such tasks. MINT’s performance also is inherently linked to the quality and diversity of the training data. Without finetuning, biases or gaps in PPI datasets could affect their generalizability, especially for underrepresented protein classes like peptides or interactions involving non-model organisms. For such cases, we recommend parameter-efficient fine-tuning approaches16 that leverage domain-specific data while minimizing computational costs. These strategies can make MINT more accessible for resource-constrained research groups and enable broader impact across biomedical applications.
Finally, although MINT achieves strong performance on many PPI tasks, recent advances suggest that incorporating additional data modalities, particularly protein structure, could further expand its capabilities, as shown by SaProt67 and ESM-368. Structural modeling approaches such as AlphaFold3 have set a new benchmark for high-resolution complex prediction, yet they focus on generating 3D coordinates rather than providing direct functional outputs like PPI classification, affinity estimation, or mutational impact; moreover, their runtimes can be prohibitive for large-scale studies27. Future iterations of MINT could merge the scalability of sequence-based methods with the complementary structural insights from models like AlphaFold3, enabling better generalization across diverse PPI landscapes.
MINT represents an advancement in protein language modeling by effectively capturing complex inter-sequence dependencies crucial for accurate PPI prediction. Its versatility across multiple tasks, including antibody modeling and TCR-epitope-MHC interactions, underscores its potential as a powerful tool in biomedical research. By facilitating a deeper understanding of protein interactions and their implications in disease mechanisms, MINT holds promise for accelerating therapeutic discoveries and advancing personalized medicine.
Methods
Here we describe the following methodological details: (1) curation and processing of training datasets, (2) architectural design and implementation of MINT, (3) datasets and workflows for downstream benchmarking tasks, and (4) datasets and workflows used in case studies.
STRING dataset construction
We start with 2.4 billion PPIs comprising 59.3 million unique protein sequences classified as physical links from the STRING database. We use mmseqs69 to cluster the protein sequences at a 50% sequence similarity threshold, resulting in 15.6 million unique clusters. We then apply a filtering scheme that keeps only one PPI between any two clusters. This is done to ensure that PPIs between proteins belonging to the same two clusters are not repeated, so that our model can learn interactions between diverse protein types. This results in 382 million PPIs comprising 29 million unique sequences. Next, we set aside 250,000 PPIs for validation. For the training set, we apply further filtering such that no cluster in the training set is seen in the validation set. This is done to limit data leakage between the training and validation sets. Finally, we end up with 95.8 million training PPIs comprising 16.4 million unique protein sequences. We provide an illustration of the dataset construction in Supplementary Fig. S1 along with alternative splitting strategies we considered in Supplementary Note 1.
MINT architecture
We use ESM-2 as our backbone PLM10. This is due to its strong empirical performance, architectural flexibility, and broad adoption within the protein modeling community70. Its modular architecture enables straightforward modifications, which we leverage to integrate cross-chain attention mechanisms for modeling multi-chain proteins in MINT.
Each layer of MINT receives as input an embedding of the sequences \({{\bf{x}}}\in {{\mathbb{R}}}^{L\times F}\), a self-attention mask \({{{\bf{m}}}}^{att}\in {{\mathbb{R}}}^{L\times L}\), and a padding mask \({{{\bf{m}}}}^{pad}\in {{\mathbb{R}}}^{L\times L}\), where L is the total length of the input comprising multiple sequences, and F is the feature dimension (F = 1280 in MINT). The main attention mechanism in MINT is the MultiHeadAttention module, as used in ESM-210. We adapt this module to output both the pre-softmax attention matrix and the value tensor, since our architecture requires combining attention weights from both intra-sequence (self-attention) and inter-sequence (cross-attention) computations prior to the softmax operation. A key design choice in our implementation is that both self-attention and cross-attention are realized via the same MultiHeadAttention module, but with different attention masks. Intra-sequence (self-attention) restricts attention to within each individual sequence, and inter-sequence (cross-attention) allows attention across different sequences (chains). Although both steps use the same MultiHeadAttention module, the different masking patterns effectively control whether attention is intra- or inter-sequence. This approach allows us to avoid introducing a separate MultiHeadCrossAttention module, while still enabling cross-chain interactions when required. We present further details for the transformer block as pseudocode in Algorithm 1.
Algorithm 1
Single functional transformer block of MINT
1: function TransformerLayer(x, mpad, matt)
2: x ← LayerNorm(x)
3: \({{\bf{s}}},{{{\bf{v}}}}^{self}\leftarrow {{\rm{MultiHeadAttention}}}({{\bf{x}}},{{{\bf{m}}}}^{pad},{{\rm{before}}}\_{{\rm{softmax}}}={{\rm{True}}})\)
4: \({{\bf{c}}},{{{\bf{v}}}}^{cross}\leftarrow {{\rm{MultiHeadAttention}}}({{\bf{x}}},{{{\bf{m}}}}^{pad},{{\rm{before}}}\_{{\rm{softmax}}}=True)\)
5: \({{{\bf{A}}}}_{ij}^{h}\leftarrow \left\{\begin{array}{l}{{{\bf{s}}}}_{ij}^{h},if\,{{{\bf{m}}}}_{ij}^{att}=1\\ {{{\bf{c}}}}_{ij}^{h},{{\rm{otherwise}}}\end{array}\right.\)
6: \({{{\bf{A}}}}_{ij}^{h}={{\rm{Softmax}}}_{j}({{{\bf{A}}}}_{ij}^{h})\)
7: d = Dropout(A)
8: \({{{\bf{p}}}}_{ij}^{self}\leftarrow \left\{\begin{array}{l}{{{\bf{d}}}}_{ij},if\,{{{\bf{m}}}}_{ij}^{att}=1\\ 0,{{\rm{otherwise}}}\end{array}\right.\)
9: \({{{\bf{p}}}}_{ij}^{cross}\leftarrow \left\{\begin{array}{l}{{{\bf{d}}}}_{ij},if\,{{{\bf{m}}}}_{ij}^{att}=0\\ 0,{{\rm{otherwise}}}\end{array}\right.\)
10: z = pselfvself + pcrossvcross
11: \({{{\bf{x}}}}^{out}={{\rm{Mean}}}_{h}({{\rm{Linear}}}({{{\bf{z}}}}_{ij}^{h}))\)
12: xout = x + xout
13: return xout
14: end function
MINT training
We train MINT using the MLM objective as defined in the main text. Formally, for a given set of input amino acids in a sequence tokenized to be x = x1x2…xn, where \(x\in {{\mathbb{R}}}^{L\times F}\), the tokens that are masked \(M\in {{\mathbb{R}}}^{L}\), and the output vector \({{\bf{h}}}\in {{\mathbb{R}}}^{L\times F}\) (corresponding to xout from the last layer of the model), the MLM loss is described as:
During model training, we consider 15% of tokens randomly sampled from the sequence, similar to ESM-210. For those 15% of tokens, we change the input token to a special masking token with 80% probability, a randomly chosen alternate amino acid token with 10% probability, and the original input token (i.e., no change) with 10% probability. We take the loss to be the whole batch average cross-entropy loss between the model’s predictions and the true token for these 15% of amino acid tokens.
We set the Adam optimizer with parameters β1 = 0.9, β2 = 0.98, and ϵ = 10−8, along with an L2 weight decay of 0.01. The learning rate was increased over the first 2000 steps to a peak of 4e − 4, then gradually reduced to one-tenth of the peak value over 90% of the training. To process large proteins efficiently, we cropped long sequences to random 512-token lengths and used special “BOS” and “EOS” tokens to mark the beginning and end of proteins. We use a batch size of 2, and we accumulate gradients every 32 batches, resulting in an effective batch size of 64. The models were trained on NVIDIA A100 80GB and NVIDIA RTX A6000 GPUs in a distributed data parallel manner. We use Pytorch Lightning (https://lightning.ai/docs/pytorch/stable/) to handle the complete training and validation loops. We trained MINT for 4 million iteration steps.
We use the perplexity metric (\({e}^{{L}_{MLM}}\)) on the validation set to measure how well the model is learning across the training run (Supplementary Fig. S3). We compare this with the perplexity of ESM-2 models as a baseline on the validation set in two settings: the first where the inputs are treated as separate protein sequences and the second where the two protein sequences are concatenated. ESM-2 with single protein chains as input achieves a perplexity of 5.41 on the validation set, while concatenating achieves a perplexity of 5.16. Our MINT model achieves a validation perplexity of 4.84 at the end of its training run. We also provide perplexity values across training runs for alternative configurations of MINT initialization and training in Supplementary Note 2 and Supplementary Fig. S2.
Benchmarking tasks
We compile a list of all datasets used across all tasks (general PPI, antibody, and TCR-epitope), along with information about task type, evaluation metric used, and references in Table 1. For the antibody and TCR-epitope tasks, we use the evaluation metric used by baseline methods for consistency. For all the tasks, we use sequence-level embeddings from MINT (or a baseline PLM) to train a downstream model (MLP, ridge regression, or CNN). Specifically, we extract the residue-level embedding h from the final layer for each group of interacting sequences, \({{\bf{h}}}\in {{\mathbb{R}}}^{L\times F}\), where L is the total length of the input comprising multiple sequences and F is the feature dimension. We average over the sequence length L to get a sequence-level embedding of shape \({{\mathbb{R}}}^{F}\) for each sequence. For tasks involving mutational effect prediction, we extract the sequence-level embedding for both the wild-type and mutant sequence, and use the difference between these embeddings as the input to the downstream model. The subsections describe task-specific methodologies.
General PPI tasks
We briefly discuss the construction of training-test splits in each benchmarking dataset. Unless specified, we use the final train-test splits as provided by the authors of each work.
-
1.
Gold-Standard PPI: This dataset was designed explicitly to prevent models from leveraging sequence similarity as a shortcut for PPI prediction. Using SIMAP2 bitscores, the human proteome was first partitioned into training, validation, and test sets to minimize pairwise sequence similarity across splits. CD-HIT was then applied with a 40% sequence identity threshold both within and across splits to ensure non-redundancy. As a result, no similar protein sequences exist in both training and test data, making this benchmark particularly stringent22.
-
2.
Yeast-PPI: The dataset was initially constructed by Guo et al.71 using a protocol where negative PPI pairs are constructed using proteins from different subcellular locations. Protein redundancy was then removed in two stages: first, a 90% sequence identity threshold was used to filter similar sequences across the entire dataset; then, a 40% identity threshold was applied across train/validation/test splits to prevent redundancy between them12.
-
3.
Human-PPI: We use Pan et al.’s72 dataset with positive interactions from HPRD and negative examples derived from different subcellular compartments. The data splitting mirrors the yeast PPI protocol, with initial redundancy removal at 90% identity and a further 40% cutoff applied between the resulting train/validation/test splits (using an 8:1:1 ratio). This setup supports evaluation of generalization to dissimilar protein sequences12.
-
4.
SKEMPI: To mitigate the risk of information leakage from structurally similar complexes, we split the dataset by protein complex into three mutually exclusive folds. Each fold contains unique complexes, ensuring that mutations from the same protein complex do not appear in both training and test sets. We follow Luo et al.73 and report results via threefold cross-validation so that each data point is tested exactly once.
-
5.
PDB-Bind and MutationalPPI: For these tasks, we apply tenfold cross-validation, with folds split by protein complex rather than by individual sequences. This strategy ensures that all chains involved in a complex are assigned to the same fold, preventing any component of a test complex from appearing in training data.
We evaluate two embedding strategies for the baseline PLMs. The first strategy involves making two calls to the PLM, generating the sequence-level embeddings separately, and concatenating these embeddings for all input sequences. The other involves concatenating the input sequences and generating a sequence-level embedding from the PLM. For all experiments except SKEMPI, we perform 3 repetitions of the training and evaluation and report the mean and standard deviation values. For SKEMPI, we calculate the mean and standard deviation for each fold and aggregate according to the following heuristic:
Where ni, μi, and σi are the number of entries, the mean, and the standard deviation for each fold i.
The objective function used for training the regression tasks is the Mean Squared Error loss, and for the binary classification tasks, we use the Binary Cross-Entropy (BCE) loss. Training was conducted using a 2-layer MLP with a hidden size of 640 for all experiments. We chose this number because it is half of the smallest embedding dimension across all models. We train all models for 100 epochs and choose the epoch with the best validation metric for test set evaluation. For datasets with validation splits, we use fivefold inner cross-validation to choose the best model.
Antibody tasks
FLAB
This dataset includes the binding energy datasets from Shanehsazzadeh et al. (2023)35, Warszawski et al. (2019)36, and from Koenig et al. (2017)37, as well as the corresponding expression data from this most recent study. We use the sequence-level embedding over both antibody chains from MINT. We then follow18 to perform the training experiments. This involves running a linear least squares fit with L2 regularization on the mean embedding representation features, evaluated using tenfold cross-validation. To choose the regularization hyperparameter λ from the set 1, 10−1, …, 10−6, 0, we perform an additional fivefold inner cross-validation. We use the best-performing model across each fold on the held-out testing fold and average the computed R2 across all folds.
SARS-CoV2 binding
We use sequences from ref. 38 and split the data according to ref. 19. We use the wild-type m396 sequence from PDB (PDB ID: 2G75) and feed that into MINT separately from the mutant sequences. We extract the sequence-level embedding for both wild-type and mutant sequences and take the difference between the two as the final embedding. We then train a ridge regression model using an α value of 0.01 on these embeddings.
TCR-Epitope-MHC tasks
We train the last layer of MINT and the downstream projector module (MLP or CNN) for the TCR-Epitope-MHC tasks.
TDC-Tchard
We use the “TDC.tchard” dataset from TDC-244, which contains 5 different folds of train-test splits. We train the model using a batch size of 48 and an initial learning rate of 1e−4 for 15 epochs. We choose the best epoch as the one that has the highest validation AUROC value and report those metrics. Then, we take the weighted average of the AUROC across all splits (the product of the AUROC and the test set size) to report as the final AUROC value, as suggested by TDC-2 benchmarking.
TCR-Epitope-HLA
For this task, we use the evaluation strategy as described in ref. 17. Specifically, we generate embeddings from MINT for the TCR-CDR3, HLA-CDR3, and epitope sequences and train an MLP model for binary interaction prediction using the BCE loss. We perform three repetitions and report the mean and standard deviation values in our results.
TCR-epitope interface prediction
We procure and use the dataset splits as constructed in ref. 46. For the sake of simplicity, we skip the pre-training task of TCR-epitope interaction prediction employed in the baseline model, TEIM. We also do not use the epitope autoencoder features as in TEIM. We do, however, use the same training setup after generating the embeddings for the TCR-CDR3 and epitope sequences using MINT. This entails training a three-layer CNN model to predict the contact map. We then use a BCE loss over the predicted and true contact maps to train the model.
Case studies
oncoPPI prediction
We used the dataset from ref. 29 that contains labels indicating whether two human proteins continue to bind after a missense mutation as our main training set. For the evaluation of mutations on oncoPPIs, we extracted the experimentally validated mutational effect data from Cheng et al.57 and retrieved the sequence data from UniProt. Following Cheng et al.57, we converted the Y2H score (range of 0–4) to a 0 (non-binding) or 1 (binding) based on a cutoff of 2. For example, a Y2H score of 4 (yeast colony grows) for a mutated oncoPPI would map to a 1 (binding conserved).
The MLP is trained on the difference between wild-type and mutated PPI embeddings from MINT (in a manner similar to 2b). Since the training dataset is very small, we performed 100 repetitions of a training and evaluation run. We then calculated a binding score Sbinding that is defined as:
where yi is the output of the model for entry i in the evaluation set, and N = 100 repetitions.
To determine an optimal threshold for classification using only the predicted binding score, we fitted the data using a Gaussian Mixture Model. This method assumes that the scores belong to two distinct groups (binding vs non-binding) and models them as a mixture of two Gaussian distributions. The threshold for classification was identified as the point where the two Gaussian distributions overlap most significantly. In our case, where the two distributions have similar variances, the threshold is approximated as the midpoint between their means, resulting in a value of 0.68. Hence, binding scores below 0.68 would be classified as non-binding, while those above would be classified as binding. We provide the AUROC curves for this analysis in Supplementary Fig. S4.
SARS-CoV-2 Cross-neutralization prediction
We first download the entire CoV-AbDab database64. We then filter out non-antibody entries and entries that do not have a sequence associated with them. We then keep antibodies that fall into one of these origins: “B-cells SARS-CoV2 WT”, “Convalescent Patient (Unvaccinated)”, “B-cells; SARS-CoV1 Human Patient; SARS-CoV2 Vaccinee”, “B-cells; SARS-CoV2 WT Convalescent Patients”, “B-cells; SARS-CoV2 WT Vaccinee (BBIBP-CoV)”, “B-cells; SARS-CoV2 WT Vaccinee”, “B-cells; SARS-CoV2 WT Human Patient”, “B-cells; Unvaccinated SARS-CoV2 WT Human Patient”, “B-cells; SARS-CoV2 Gamma Human Patient”, “B-cells; SARS-CoV1 Human Patient”, “B-cells (SARS-CoV2 Beta Human Patient)”. Next, we only keep antibodies that bind to the RBD of the SARS-CoV-2 spike protein. Finally, we split the data to ensure that only neutralization values against early variants (Wild-type, Alpha, Beta, Delta, Epsilon, Gamma, Eta, Iota, Lambda, and Kappa) are included in the training set. The evaluation set contains only Omicron sub-variants, namely BA.1, BA.2, BA.4, and BA.5. This results in 3365 data points in the training set and 1052 entries for evaluation. We downloaded the spike protein sequences for all included variants and sub-variants from Expasy (https://viralzone.expasy.org/9556).
During training, we generate sequence-level embeddings from MINT for the antibody heavy-chain, light-chain, and the RBD domain of the spike protein. We train an MLP on these embeddings to predict whether the antibody is able to neutralize that spike protein using the BCE loss. In the training set, antibodies may be repeated since they have neutralization data against multiple variants. After training, we test the model on the evaluation set and record the raw outputs. Since the raw outputs across the different sub-variants may have different scales, we use quantile normalization to normalize the scores across the sub-variants to calculate the “Normalized score”. We retrieve the IC50 values from ref. 66.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We retrieved physical PPI training data for MINT from STRING-DB20. We obtained the gold-standard PPI dataset from https://figshare.com/articles/dataset/PPI_prediction_from_sequence_gold_standard_dataset/21591618/322, the HumanPPI dataset from https://github.com/westlake-repl/SaProt67, and the YeastPPI dataset from PEER (https://miladeepgraphlearningproteindata.s3.us-east-2.amazonaws.com/ppidata/yeast_ppi.zip)12. The SKEMPI entries were downloaded from https://life.bsc.es/pid/skempi223 and the PDB-Bind dataset from https://www.pdbbind-plus.org.cn/28. The Mutational PPI data were obtained from https://github.com/jishnu-lab/SWING/tree/main/Data/MutInt_Model29. The FLAB antibody datasets are available at https://github.com/Graylab/FLAb/tree/main/data24, and the SARS-CoV-2 binding datasets at this link: https://www.biorxiv.org/content/10.1101/2020.04.03.024885v1.supplementary-material38. The TCR-epitope task from TDC-2 was downloaded from (https://tdcommons.ai/)44. The TCR-epitope-HLA data were retrieved from https://github.com/Armilius/PISTE/tree/main/data17, and the TCR-epitope interface prediction data were obtained from https://github.com/pengxingang/TEIM46. We obtained experimentally validated oncoPPI data from https://github.com/ChengF-Lab/oncoPPIs57. Finally, we obtained SARS-CoV-2 neutralization data from https://opig.stats.ox.ac.uk/webapps/covabdab/64. Source data for all figures is provided with this paper. Source data are provided with this paper.
Code availability
The code used to develop MINT, perform the analyzes, and generate results in this study is publicly available and has been deposited at https://github.com/VarunUllanat/mint under the MIT License. The publication release is deposited on Zenodo at https://doi.org/10.5281/zenodo.1717487574.
References
Bepler, T. & Berger, B. Learning protein sequence embeddings using information from structure. Int. Conf. Learn. Represent. (2019).
Bepler, T. & Berger, B. Learning the protein language: evolution, structure, and function. Cell Syst. 12, 654–669 (2021).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 118, e2016239118 (2021).
Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. Adv. Neural Inf. Process. Syst. 34, 29287–29303 (2021).
Thadani, N. N. et al. Learning from prepandemic data to forecast viral escape. Nature 622, 818–825 (2023).
Hie, B., Zhong, E. D., Berger, B. & Bryson, B. Learning the language of viral evolution and escape. Science 371, 284–288 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. nature 596, 583–589 (2021).
Wu, R. et al. High-resolution de novo structure prediction from primary sequence. Preprint at https://www.biorxiv.org/content/10.1101/2022.07.21.500999v1 (2022).
Singh, R., Sledzieski, S., Bryson, B., Cowen, L. & Berger, B. Contrastive learning in protein language space predicts interactions between drugs and protein targets. Proc. Natl. Acad. Sci. USA 120, e2220778120 (2023).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Sledzieski, S., Singh, R., Cowen, L. & Berger, B. D-SCRIPT translates genome to phenome with sequence-based, structure-aware, genome-scale predictions of protein-protein interactions. Cell Syst. 12, 969–982 (2021).
Xu, M. et al. Peer: a comprehensive and multi-task benchmark for protein sequence understanding. Adv. Neural Inf. Process. Syst. 35, 35156–35173 (2022).
Charih, F., Biggar, K. K. & Green, J. R. Assessing sequence-based protein–protein interaction predictors for use in therapeutic peptide engineering. Sci. Rep. 12, 9610 (2022).
Sledzieski, S., Devkota, K., Singh, R., Cowen, L. & Berger, B. TT3D: leveraging precomputed protein 3d sequence models to predict protein–protein interactions. Bioinformatics 39, btad663 (2023).
Singh, R., Devkota, K., Sledzieski, S., Berger, B. & Cowen, L. Topsy-turvy: integrating a global view into sequence-based PPI prediction. Bioinformatics 38, i264–i272 (2022).
Sledzieski, S. et al. Democratizing protein language models with parameter-efficient fine-tuning. Proc. Natl. Acad. Sci. USA 121, e2405840121 (2024).
Feng, Z. et al. Sliding-attention transformer neural architecture for predicting T cell receptor–antigen–human leucocyte antigen binding. Nat. Mach. Intell. 6, 1216–1230 (2024).
Kenlay, H. et al. Large scale paired antibody language models. Preprint at https://arxiv.org/abs/2403.17889 (2024).
Singh, R. et al. Learning the language of antibody hypervariability. Proc. Natl. Acad. Sci. USA 122, e2418918121 (2025).
Szklarczyk, D. et al. The string database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic acids Res. 51, D638–D646 (2023).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 30 (2017).
Bernett, J., Blumenthal, D. B. & List, M. Cracking the black box of deep sequence-based protein–protein interaction prediction. Brief. Bioinforma. 25, bbae076 (2024).
Jankauskaitė, J., Jiménez-García, B., Dapkūnas, J., Fernández-Recio, J. & Moal, I. H. Skempi 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 35, 462–469 (2019).
Chungyoun, M., Ruffolo, J. A. & Gray, J. J. Flab: benchmarking deep learning methods for antibody fitness prediction. Preprint at https://www.biorxiv.org/content/10.1101/2024.01.13.575504v1 (2024).
Grazioli, F. et al. Attentive variational information bottleneck for TCR–peptide interaction prediction. Bioinformatics 39, btac820 (2023).
Devlin, J. et al. BERT: Pre-training of deep bidirectional transformers for language understanding. Proc. NAACL-HLT. 1, 4171–4186 (2019).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold3. Nature 630, 493–500 (2024).
Liu, Z. et al. Forging the basis for developing protein–ligand interaction scoring functions. Acc. Chem. Res. 50, 302–309 (2017).
Siwek, J. C. et al. Sliding Window Interaction Grammar (SWING): a generalized interaction language model for peptide and protein interactions. Nat. Methods 22, 1707–1719 (2025).
Rao, R. M. et al. MSA Transformer. In Proceedings of the 38th International Conference on Machine Learning. (eds Meila, M. & Zhang, T.) 8844–8856 (PMLR, 2021).
Madani, A. et al. Progen: language modeling for protein generation. Preprint at https://arxiv.org/abs/2004.03497 (2020).
Elnaggar, A. et al. Prottrans: towards cracking the language of life’s code through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2021).
Greenfield, E. A. Antibodies: A Laboratory Manual, Second Edition (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar], 2014).
Gabrielli, E. et al. Antibody complementarity-determining regions (cdrs): a bridge between adaptive and innate immunity. PLoS ONE 4, e8187 (2009).
Shanehsazzadeh, A. et al. Unlocking de novo antibody design with generative artificial intelligence. Preprint at https://www.biorxiv.org/content/10.1101/2023.01.08.523187v4 (2023).
Warszawski, S. et al. Optimizing antibody affinity and stability by the automated design of the variable light-heavy chain interfaces. PLoS Comput. Biol. 15, e1007207 (2019).
Koenig, P. et al. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc. Natl. Acad. Sci. USA 114, E486–E495 (2017).
Desautels, T., Zemla, A., Lau, E., Franco, M. & Faissol, D. Rapid in silico design of antibodies targeting SARS-CoV-2 using machine learning and supercomputing. Preprint at https://www.biorxiv.org/content/10.1101/2020.04.03.024885v1 (2020).
Zhu, Z. et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 104, 12123–12128 (2007).
Delgado, J., Radusky, L. G., Cianferoni, D. & Serrano, L. Foldx 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics 35, 4168–4169 (2019).
Leaver-Fay, A. et al. Scientific benchmarks for guiding macromolecular energy function improvement. In Methods in Enzymology, Vol. 523, 109–143 (Elsevier, 2013).
Barlow, K. A. et al. Flex ddg: Rosetta ensemble-based estimation of changes in protein–protein binding affinity upon mutation. J. Phys. Chem. B 122, 5389–5399 (2018).
Peters, B., Nielsen, M. & Sette, A. T cell epitope predictions. Annu. Rev. Immunol. 38, 123–145 (2020).
Velez-Arce, A. et al. Signals in the cells: multimodal and contextualized machine learning foundations for therapeutics. NeurIPS Workshop on AI for New Drug Modalities (2024).
Yoo, S., Jeong, M., Seomun, S., Kim, K. & Han, Y. Interpretable prediction of SARS-CoV-2 epitope-specific TCR recognition using a pre-trained protein language model. IEEE/ACM Trans. Comput. Biol. Bioinforma. 21, 428–438 (2024).
Peng, X. et al. Characterizing the interaction conformation between T-cell receptors and epitopes with deep learning. Nat. Mach. Intell. 5, 395–407 (2023).
Vita, R. et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Shugay, M. et al. Vdjdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427 (2018).
Tickotsky, N., Sagiv, T., Prilusky, J., Shifrut, E. & Friedman, N. Mcpas-tcr: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929 (2017).
Yang, M. et al. Mix-tpi: a flexible prediction framework for TCR–PMHC interactions based on multimodal representations. Bioinformatics 39, btad475 (2023).
Montemurro, A. et al. Nettcr-2.0 enables accurate prediction of TCR-peptide binding by using paired tcrα and β sequence data. Commun. Biol. 4, 1060 (2021).
Gao, Y. et al. Pan-peptide meta learning for T-cell receptor–antigen binding recognition. Nat. Mach. Intell. 5, 236–249 (2023).
Jiang, Y., Huo, M. & Cheng Li, S. Teinet: a deep learning framework for prediction of TCR–epitope binding specificity. Brief. Bioinforma. 24, bbad086 (2023).
Weber, A., Born, J. & Rodriguez Martínez, M. Titan: T-cell receptor specificity prediction with bimodal attention networks. Bioinformatics 37, i237–i244 (2021).
Lu, T. et al. Deep learning-based prediction of the T cell receptor–antigen binding specificity. Nat. Mach. Intell. 3, 864–875 (2021).
Leem, J., de Oliveira, S. H. P., Krawczyk, K. & Deane, C. M. Stcrdab: the structural T-cell receptor database. Nucleic Acids Res. 46, D406–D412 (2018).
Cheng, F. et al. Comprehensive characterization of protein–protein interactions perturbed by disease mutations. Nat. Genet. 53, 342–353 (2021).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95 (2021).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Sahni, N. et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660 (2015).
Fragoza, R. et al. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 10, 4141 (2019).
Wang, Y. et al. Alox5 exhibits anti-tumor and drug-sensitizing effects in MLL-rearranged leukemia. Sci. Rep. 7, 1853 (2017).
Fan, Y. et al. SARS-CoV-2 omicron variant: recent progress and future perspectives. Signal Transduct. Target. Ther. 7, 1–11 (2022).
Raybould, M. I., Kovaltsuk, A., Marks, C. & Deane, C. M. Cov-abdab: the coronavirus antibody database. Bioinformatics 37, 734–735 (2021).
Cho, A. et al. Anti-SARS-CoV-2 receptor-binding domain antibody evolution after mRNA vaccination. Nature 600, 517–522 (2021).
Liu, Y. et al. Inactivated vaccine-elicited potent antibodies can broadly neutralize SARS-CoV-2 circulating variants. Nat. Commun. 14, 2179 (2023).
Su, J. et al. Saprot: protein language modeling with structure-aware vocabulary. Preprint at https://www.biorxiv.org/content/10.1101/2023.10.01.560349v1 (2023).
Hayes, T. et al. Simulating 500 million years of evolution with a language model. Science eads0018 (2025).
Steinegger, M. & Söding, J. MMSEqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Chen, J.-Y. et al. Evaluating the advancements in protein language models for encoding strategies in protein function prediction: a comprehensive review. Front. Bioeng. Biotechnol. 13, 1506508 (2025).
Guo, Y., Yu, L., Wen, Z. & Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic acids Res. 36, 3025–3030 (2008).
Pan, X.-Y., Zhang, Y.-N. & Shen, H.-B. Large-scale prediction of human protein-protein interactions from amino acid sequence based on latent topic features. J. Proteome Res. 9, 4992–5001 (2010).
Luo, S. et al. Rotamer Density Estimator is an unsupervised learner of the effect of mutations on protein–protein interaction. Proc. ICLR (2023).
Ullanat, V., Jing, B., Sledzieski, S. & Berger, B. Learning the language of protein-protein interactions. varunullanat/mint: Publication release https://doi.org/10.5281/zenodo.17174876 (2025).
Acknowledgements
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 1R35GM141861 and by a research gift from Quanta Computer. B.J. was partially supported by the Department of Energy Computational Science Graduate Fellowship under Award Number DESC0022158. S.S. was partially supported by the NSF Graduate Research Fellowship under Grant No. 2141064. We would also like to acknowledge Aditya Parekh and Anish Mudide for their helpful discussions and comments.
Author information
Authors and Affiliations
Contributions
B.J., S.S., and B.B. conceptualized the project. V.U. and B.J. constructed the training pipeline for MINT. V.U. and B.J. ran the training. V.U. performed downstream computational analysis, including model benchmarking and case studies. B.B. designed and led the study. All authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nimisha Ghosh, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ullanat, V., Jing, B., Sledzieski, S. et al. Learning the language of protein-protein interactions. Nat Commun 17, 1199 (2026). https://doi.org/10.1038/s41467-025-67971-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67971-3
