
Abstract
Li-ion batteries, widely used in electronic devices, electric vehicles, and aviation, demand high energy density, fast charging capabilities, and broad operating temperature ranges. Computations combined with experiments have gained increasing attention for electrolyte development. However, the inherent complexity of electrolytes poses a significant challenge. Classical molecular dynamics often fails due to inaccuracies in force field parameters, while ab initio calculations are limited by high computational costs. Recently, machine learning molecular dynamics has emerged as an efficient and accurate alternative. However, its application is hindered by limited transferability of machine learning potentials. In this work, we developed a universal machine learning potential for electrolytes using an iterative training approach on randomly composed datasets, enabling the accurate computation of key properties for a broad range of electrolytes via molecular dynamics. Furthermore, coordination dynamics analysis of Li ion, by quantifying the coordination lifetime, provides a direct, quantitative measure of solvation strength. The universal machine learning potential for electrolytes facilitates the prediction and optimization of electrolyte properties, offering a powerful tool for electrolyte design.
Similar content being viewed by others


Introduction
Given the increasing public concerns regarding energy and environmental issues, significant efforts have been dedicated to the development of safer Li-ion batteries (LIBs) with improved energy density (Fig. 1a)1,2,3,4,5,6,7. The performance of LIBs is limited by various electrolyte properties, including density, solvation structure8,9,10, viscosity11,12, ionic conductivity13, operating temperature window14, and electrochemical stability window3,15, etc. The solvation structure of electrolytes plays a critical role in determining their interfacial stability, ion diffusion properties, and fast-charging capabilities. For example, weakly solvating electrolytes, which feature reduced coordination between Li+ and solvent molecules, can enhance ionic conductivity16 and promote the formation of stable anion-derived solid-electrolyte-interphases on electrodes17. Understanding and optimizing solvation of Li+ is therefore key to designing high-performance electrolytes for advanced LIBs.

a Illustration of the applications and basic components of LIBs, along with a brief overview of the key properties of electrolytes. b The high-dimensional chemical space of electrolytes, illustrating the process of component optimization through property-based screening. c Illustration of the workflow of training a uMLP for electrolytes and accelerating electrolyte optimization through property calculation via MLMD simulations.
Therefore, significant research efforts have focused on electrolyte design, emphasizing the regulation of solvation structures through a synergistic approach that combines experimental and computational methods to enhance battery performance3,16,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32. For example, Yamada et al. applied classical molecular dynamics (CMD) to obtain the solvation structure of Li ion and carried out experiments to characterize the Li deposition potential32. Machine learning algorithms are utilized to uncover that the radial distribution functions of cations and anions are the determining factor for the Li deposition potential in different electrolytes.
However, computational chemistry methods still face many limitations, with the most notable limitations being that CMD is restricted by the accuracy of force field parameters. The accuracy of the solvation structures obtained from CMD is limited by the accuracy of force field parameters33,34. Note that variations in the force field parameters can result in different simulation results35. To accurately compute the solvation structure and electrochemical properties of electrolytes, ab initio molecular dynamics simulations (AIMD) have been employed to study the battery electrolytes36,37,38,39,40,41,42,43,44,45. For instance, Salanne et al. utilized AIMD simulations to investigate water-in-salt electrolytes and discovered the formation of polymer-like nanochains with short Li-Li distances, which has never been reported in CMD simulations42. However, the computational expense of AIMD restricts the system size to ∼1000 atoms and the time scale to less than 100 ps, significantly limiting its application in electrolyte design.
In order to address the issue of low computational efficiency in AIMD, machine learning molecular dynamics (MLMD) has been introduced to accelerate simulations of complex systems46,47,48,49,50,51,52,53,54,55. By constructing accurate mappings, that is, machine learning potential (MLP), from local atomic environments to potential energies and atomic forces, nanosecond-level molecular dynamics simulations of larger models can be achieved without sacrificing ab initio accuracy48,50. Using a concurrent learning workflow56,57, it becomes feasible to iteratively construct MLP training datasets, facilitating efficient construction of MLPs. This approach enables accurate simulations of complex systems such as solid-state electrolytes58,59, and liquid electrolytes41,60,61. However, the complexity of the MLP training process and the limited transferability to new systems hinder high-throughput calculations with MLMD.
Thus, it is highly desirable to train universal MLP (uMLP) models showing good transferability to different systems. To further push to the limit of covering all elements across the periodic table, foundation models, such as open large atomic model (OpenLAM)62, foundation model of atomistic materials chemistry (MACE)63, and general reactive machine learning potential64 and so on, have been proposed with the integration of multi-task training methods. The underlying idea is to reduce the size of datasets and training cost for a wide range of complex systems by exploiting the expressiveness of representation and transferability of the MLP models. However, these models may show reduced accuracy for specific applications and lower efficiency for MLMD simulations, as too many elements and a vast of chemical configurations need to be considered. As such, additional finetuning or distillation processes are often required for specific systems. Alternatively, it is possible to train uMLP models of certain types of systems for downstream applications. This is the focus of this work, and note that due to the diverse composition of electrolytes and complexity of configurational space (Fig. 1b), it is very challenging to construct adequate datasets to train uMLP for electrolytes. Although several MLPs, such as BAMBOO65, PhyNEO electrolyte66, and SevenNet-067, have been developed to simulate electrolytes with MLMD, their scope is limited to fewer than 20 types of solvents and salts, falling short of capturing the full complexity of the electrolyte chemical space.
In this study, as illustrated in Fig. 1c, a uMLP for electrolytes is developed using the Deep Potential (DeePMD)52 framework and the concurrent learning approach implemented in the ai2-kit68. To comprehensively cover the chemical space of electrolytes, an electrolyte generator is utilized with a database of over 2300 solvents and 20 salts, allowing for the creation of diverse systems with randomized compositions. MLMD simulations with the uMLP are then employed to systematically compute solvation structures and key properties of electrolytes, including density, viscosity, ionic conductivity, and operating temperature ranges. Furthermore, a approach is introduced to estimate ionic conductivity by quantifying the coordination lifetime (\(\bar{\tau }\)) of Li+ ions, which directly links shorter \(\bar{\tau }\)-"indicative of weak solvation-”to faster ion transport and higher ionic conductivity. The uMLP significantly accelerates the initial screening of electrolyte candidates, minimizing both time and computational resources, while also enabling a deeper understanding of ionic transport mechanisms. Overall, property calculations through MLMD the with uMLP for electrolytes provide a powerful tool for electrolyte design.
Results and discussion
Training uMLP for electrolytes
To construct a uMLP for electrolytes, it is essential to enhance the efficiency of sampling within the high-dimensional chemical space of electrolytes (Fig. 1b). Thus, concurrent learning scheme56,69, implemented in ai2-kit68, is applied. As shown in Fig. 1c, an electrolyte generator is applied to generate a series of electrolytes with random compositions. Thus, the solvent database includes solvents developed by Chen and Zhang et al.18, as well as commonly used solvent molecules, forming a comprehensive solvent molecular database containing over 2300 solvent molecules. This database covers a wide range of elemental types, including C, H, O, N, B, S, F, Cl, Br, I, P, and Si, and encompasses various solvent types such as carbonates, ethers, nitriles, heterocycles, sulfones, and more. Similarly, commonly used Li-salts (LiTFSI, LiFSI, LiBETI, LiFTFSI, LiOAc, CF3COOLi, LiPF6, LiClO4, LiBF4, Li2SO4, LiNO3, LiCl, LiBr, LiI, LiDFOB, LiSCN, LiSO4CH3, LiSO3CH3, LiDCA, etc.), are selected to construct a salt database. In addition, ionic liquids and salts with cations such as Na+, K+, Mg2+, Ca2+ and Zn2+ are also included to enhance the diversity and transferablity of the database. ∼1,000,000 electrolytes with random combinations and concentrations were constructed and pre-equilibrated using CMD, with their compositions created by randomly selecting solvent molecules and salt anions, varying the solvent-salt ratio (from 30:1 to 1:1), and incorporating one to six solvent or salt types. These systems were divided into multiple batches to serve as initial structures for subsequent exploration with MLMD and 2000 structures were randomly selected for DFT calculations to construct the initial datasets for training an ensemble of MLP. Then, an MD sampler is used to sample electrolytes with different compositions at various temperatures and pressures, with the MLP updated in each iteration. Then, based on the ensemble of MLPs, the model deviation57 of MD trajectories is calculated. Structures with high model deviation are selected as new structures and fed into the DFT calculator to update the training datasets. After several iterations of concurrent learning, an accurate uMLP for electrolytes can be constructed. Note that the universality of the uMLP for electrolytes is ensured by the randomly composed electrolytes mentioned above; moreover, by constructing datasets at different concentrations and employing an iterative training approach, one can effectively collect MLP training datasets41,60,70 that accurately covers the range from pure solvent to highly concentrated electrolyte. This aligns with the observations of Goodwin et al. that sampling high-entropy compositions is more effective than pure-system sampling and allows for a robust MLP71. Furthermore, not all electrolytes used for MLMD exploration are included in the datasets due to the transferability of local environment descriptors, which effectively reduces the size of the dataset.
Evaluation of accuracy of uMLP
With 91 iterations of concurrent learning, ∼160,000 configurations are collected to construct the uMLP for electrolytes using DeepPot-SE52. Although there are existing works training potentials based on hybrid functionals70,72, DFT calculations are performed with the Perdew-Burke-Ernzerhof (PBE) functional73 with Grimme D374(PBE-D3), which could ready provide accurate solvation structure70,75 and dynamics properties65,76 for electrolytes. Notably, the size of the dataset is similar to those reported for electrolyte-specific MLPs65,66, yet significantly smaller than those required for universal MLPs62,63,64. The predicted errors of potential energies, atomic forces, and virial tensors are shown in Fig. 2a. The root-mean-square-error (RMSE) of predicted potential energies is ∼0.005 eV/atom. Although the RMSE of predicted potential energies is slightly higher than previously reported MLP accuracy, the predicted atomic forces, with an RMSE of around 160 meV/Å, and virial tensors, with an RMSE of about 5.0l meV/atom, are accurate enough to reproduce MD trajectories with ab initio accuracy. Note that the cutoff radius (Rcut) for local environments is set to 6.0 Å, which shows similar accuracy compared to Rcut = 8.0 Å (Figure S1), as suggested in a previous publication77. Additionally, as shown in the insets of Fig. 2a, the proportion of higher errors is very small, primarily resulting from unphysical structures, such as configurations with atomic distances smaller than 1 Å. The large force values (± 40 eV/Å) primarily originate from the extreme conditions explicitly sampled in our simulations, including high temperatures (about 800 K), high pressure (100 bar), and configurations involving chemical bond breaking. Inclusion of these structures is essential for achieving robust performance in MLMD simulations. As shown in Figure S2a, due to the complexity of the electrolyte compositions, the training datasets encompass a diverse range of modeling systems with varying sizes, spanning from 400 to 4000 atoms. Systems containing more than 3000 atoms are a consequence of including large solvent molecules and the fraction of such dataset is small. Furthermore, the training datasets contain 19 frequently used elements in electrolytes (Figure S2b), ensuring coverage of a variety of different solvation environments. For further demonstration of the training data set’s diversity, the local environments of Li+ ions are described using the Smooth Overlap of Atomic Positions (SOAP)78 descriptor and then projected into a low-‘dimensional similarity map via Uniform Manifold Approximation and Projection (UMAP). In separate plots, we use color coding to indicate the number of O, N, and F atoms within 2.5 Å and the number of C, S, P, B, and Cl atoms within 3.5 Å of each Li+ ion. As shown in Figure S2c, the Li+ environments cluster into multiple distinct regions; the varied elemental distributions around Li+ highlight the substantial structural diversity and the complexity of our training data set. To further illustrate the accuracy of the MLP, potential energies, atomic forces, and virial tensors for previously reported glyme-LiTFSI datasets60 are computed using the uMLP and compared with corresponding DFT values. As shown in Figure S3, the predicted values exhibit similar accuracy to those in the training datasets. Notably, these datasets are not included in the training datasets, demonstrating the good transferability of the uMLP for electrolytes. To demonstrate the transferability and accuracy of the uMLP for electrolytes, over 13,000 five-ps MLMD trajectories (10,000 steps) of electrolytes with various compositions are generated using the uMLP. The final configurations from these trajectories are computed using DFT to construct the validation sets. It should be noted that the validation sets consist of a wide range of complex electrolytes, with compositions ranging from binary to nonary and concentrations spanning from dilute to highly concentrated and not included in the training datasets. As shown in Fig. 2b, the predicted potential energies, atomic forces, and virial tensors of the validation set exhibit similar accuracy to those of the training datasets, indicating that the training datasets are sufficiently representative of the chemical space of liquid electrolytes. Note that the ranges of atomic force and virial tensors of validation sets are narrower than those of training datasets, which is due to the mild simulation conditions (300K and 1 Bar).

a The predicted errors of potential energies, atomic forces and virial tensors on training datasets. The distribution of prediction errors is presented in the corresponding inset. b The predicted errors of potential energies, atomic forces and virial tensors on validation datasets. The distribution of prediction errors is presented in the corresponding inset. c, d Validation of uMLP transferability for an unseen co-solvent (TFAONP, c) and an unseen anion (DFOP−, d) showing that the errors in atomic forces and virial tensors of 100 configurations randomly selected from 1 ns MLMD simulations are comparable to those of the training datasets. The C, H, O, N, P, F and Li atoms are in gray, white, red, blue, tan, green and pink, respectively.
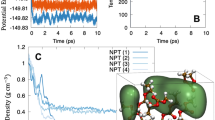
Although the uMLP for electrolytes demonstrates good accuracy in predicting potential energies, atomic forces, and virial tensors, we further validate the accuracy of MLMD trajectories by comparing them with results obtained from DFT and AIMD. Three electrolyte models with different sizes and compositions are constructed, and over 1 ns MD trajectories with the NpT ensemble are generated using the MLP (Figure S4). Configurations are selected every 10 ps along the 1 ns MLMD trajectories, and the corresponding potential energies, atomic forces, and virial tensors are computed using DFT. As shown in Figure S4, for all three systems, the predicted atomic forces and virial tensors show good agreement with the DFT values, and the corresponding RMSEs are smaller than those of the training datasets, indicating that the MLP can drive MD simulations with ab initio accuracy. However, the predicted potential energies for each system exhibit small systematic errors79,80,81. Overall, the predicted atomic forces and virial tensors are sufficiently accurate to generate MLMD trajectories with ab initio accuracy.
To demonstrate the accuracy of computed structural properties, MLMD trajectories of a ternary electrolyte composed of ethylene carbonate (EC), diethyl carbonate (DEC), and LiPF6 are generated using both AIMD and MLMD. As shown in Figure S5a, the simulation model contains 1520 atoms, making AIMD simulations computationally expensive and limiting the generated trajectories to less than 1 ps. The radial distribution functions (RDFs) are computed from the AIMD and MLMD trajectories and plotted for comparison The peaks and intensities of RDFs for Li+ − ODEC and Li+ − OEC computed from MLMD trajectory (Figure S5b and c) show good agreement with the AIMD results. However, the RDF of \({{{{\rm{Li}}}}}^{+}-{{{{\rm{F}}}}}_{{{{{\rm{PF}}}}}_{6}^{-}}\) from long time scale of MLMD simulation is very different from the AIMD results. For comparison, a 5 ps MLMD trajectories are generated with the same initial configurations as AIMD (Figure S5d). The RDF of \({{{{\rm{Li}}}}}^{+}-{{{{\rm{F}}}}}_{{{{{\rm{PF}}}}}_{6}^{-}}\) from AIMD and 5 ps MLMD are are nearly identical. That means the contribution of \({{{{\rm{PF}}}}}_{6}^{-}\) to the first coordination shell of Li+ is replaced by the solvent molecules (EC or DEC) as the MLMD simulation extends over a long time scale.
The transferability of uMLP is crucial to its predictive capability82. As shown in Fig. 2c, a 1 ns MLMD simulation was performed for electrolytes containing (4-nitrophenyl) 2,2,2-trifluoroacetate (TFAONP) as the co-solvent, which was not included in the training dataset. By conducting DFT validations on 100 randomly selected configurations from the MLMD trajectory, we found that the prediction errors of atomic forces and virial tensors are comparable to those in the training datasets. This result highlights the good transferability of the uMLP for electrolytes to new solvent molecules. Then, to validate the transferability of the uMLP for electrolytes to different salts, a 1 ns MLMD simulation was carried out on an electrolyte containing lithium difluorobis(oxalato)phosphate (LiDFOP) as the Li-salt, and 100 configurations were randomly selected for DFT calculations. It should be noted that DFOP− was not included in the training datasets, yet the predicted errors in atomic forces and virial tensors show good agreement with those of the training datasets (Fig. 2d). To further evaluate the transferability of the uMLP for electrolytes across different Li salts, we systematically analyzed its performance on diverse electrolyte systems (Figure S6), including datasets of electrolytes with 40 PC molecules and 10 LiFSI derivatives, where the F atoms on the FSI anion were substituted with different fluoroalkyl groups (Figures S6a-c), and a dataset representing a 0.1 M LiETFSI solution in EC/DEC (v/v = 1:1)83 (Figure S6d). The predicted potential energies, atomic forces, and virial tensors show similar accuracy to the training datasets and are in good agreement with DFT calculations, highlighting the uMLP’s ability to effectively handle variations in anionic chemistry.
Based on the above analysis, the uMLP for electrolytes not only ensures accuracy in reproducing potential energies, atomic forces, and virial tensors from DFT calculations but also maintains accuracy in computing structural properties of electrolytes. Furthermore, it extends the time scale of AIMD simulations without loss of accuracy. Therefore, the uMLP for electrolytes demonstrates good accuracy and transferability, enabling the computation of electrolyte properties using MLMD simulations.
Structure and properties
Computing electrochemical properties through MLMD is the initial step to evaluate the performance of electrolytes. The first requirement is to confirm whether accurate densities can be obtained from MLMD simulations. The performance of electrolytes is determined by solvation structures and some key properties such as diffusion coefficients, viscosity, and operating temperature ranges. Therefore, we first computed the densities and RDFs for some electrolytes. The solvation structures of Li+ significantly influence the desolvation processes of Li+ during charging and discharging processes. However, since solvation structures cannot be directly obtained from experiments, those derived from MLMD simulations using the uMLP are compared with results from small dataset-based MLMD and AIMD. Subsequently, diffusion coefficients and viscosity are obtained from long-time-scale MLMD simulations. Recently, Fan, Wang and co-workers developed an electrolyte with high performance across a wide operating temperature range16. As suggested, the operating temperature ranges of electrolytes can be determined by measuring the temperature intervals exhibiting high ionic conductivity. To demonstrate the ability of uMLP for electrolytes, the ionic conductivity of a variety of electrolytes at different concentrations and temperatures are computed with MLMD and compared with experimental measurements from the work of Fan, Wang and co-workers16.
Density
As shown in Figure S7a and b, the densities of electrolytes and some frequently used solvents, such as carbonates, esters, and nitriles, are averaged over 1 ns MLMD simulations with the NpT ensemble. The computed densities from MLMD simulations show good agreement with experimental values, with errors less than 5%. Note that 5% is already sufficiently accurate, and the residual errors primarily arise from the limitations of the PBE-D3 functional. Further improvements in functional accuracy could lead to even more precise calculations. Thus, the uMLP for electrolytes can accurately predict densities for a wide range of electrolytes and solvents.
Solvation structure
To examine the predicted solvation structures of electrolytes, the RDFs for LiTFSI/PC electrolytes at different concentrations are computed using AIMD, MLMD with a single-system MLP, and MLMD with the uMLP for comparison. As shown in Figure S8, the RDFs and coordination number for Li+ computed from MLMD with uMLP for electrolytes are almost the same as the results computed from MLMD with MLP for single system (labeled as MLMD* in Figure S8). As for the difference between the AIMD and MLMD, this is actually caused by the short time scale (about 50 ps) and small size (about 400 atoms) of AIMD simulations.
The solvation structures of Li+ were further analyzed. As illustrated in Fig. 3a, b, and d, for the low-concentration electrolyte LiFSI1.00DMC9.00, the first solvation shell of Li+ is primarily composed of DMC, with a relatively minor contribution from FSI−, compared to the high-concentration electrolyte LiFSI1.00DMC1.10. Additionally, upon the introduction of the BTFE diluent, the contribution of DMC to the first solvation shell of Li+ is reduced, approaching the levels observed in the highly concentrated LiFSI1.00DMC1.10 electrolyte (Fig. 3a). The contribution of FSI− to the first solvation shell of Li+ increases, reaching values similar to those in the high-concentration LiFSI1.00DMC1.10 electrolyte (Fig. 3d). Analysis of the contribution of oxygen atoms from BTFE to the first solvation shell of Li+ (Fig. 3c and d) reveals a coordination number of less than 0.1 in all electrolytes containing BTFE. By incorporating the coordination numbers into Fig. 3d, it is evident that the LHCE maintains a solvation structure similar to that of the high-concentration electrolyte (light blue region), exhibiting a clear distinction from the low-concentration counterpart(light pink region). This suggests a negligible contribution from BTFE to Li+ solvation and indicates that electrolytes containing BTFE form a locally concentrated solvation environment.

The radial distribution functions (RDF, g(r)) of (a) Li+ − ODMC, (b) \({{{{\rm{Li}}}}}^{+}-{{{{\rm{O}}}}}_{FS{I}^{-}}\), and c. Li+ − OBTFE, along with (d) the corresponding coordination numbers (CN) in diluted, concentrated LiFSI/DMC electrolytes and localized highly concentrated electrolytes. e The vibrational density of states (vDOS) of aqueous electrolytes with varying concentrations of LiTFSI, showing the blue shift of the O-H stretching mode from ∼3400cm−1 to ∼3600cm−1 with increasing concentration, indicating the isolation of water molecules by the ionic network formed by Li+ and TFSI−. f The vDOS of DME-based electrolytes with varying concentrations of LiFSI, showing a mode at ∼750cm−1 shifting to higher wavenumbers as the LiFSI concentration increases. The C, H, O, N, S, F and Li atoms are in gray, white, red, blue, yellow, green and pink, respectively.
Special solvation structures can be inferred from Raman spectra, including the O-H stretching mode of water molecules and the S-N-S bending mode of FSI−. To further validate the solvation structures calculated by MLMD using uMLP, the vibrational density of states (vDOS) of aqueous electrolytes with varying concentrations of LiTFSI were computed(Fig. 3e). The observed blue shift (from ∼3, 400cm−1 to ∼3600cm−1) of the O-H stretching mode with increasing LiTFSI concentration, which indicates that water molecules are progressively isolated by the ionic network of Li+ and TFSI−, is in excellent agreement with both the Raman spectra3,20 and previous MLMD calculations41. This further substantiates the capability of the MLMD method to accurately capture changes in solvation structure and corresponding spectroscopic features as a function of electrolyte concentration. Additionally, we computed the vDOS for DME-based electrolytes with varying concentrations of LiFSI. The results show that with increasing LiFSI concentration, the mode at ∼750cm−1 shifts to higher wavenumbers. Although there are slight blue shifts in the peak positions in comparison with experiments, this trend is consistent with Raman spectra reported in the literature84,85. Overall, the uMLP-based MLMD method provides accurate calculations of electrolyte solvation structures and establishes accurate computed vDOS and in comparison with Raman spectra, thereby facilitating the interpretation of spectroscopic data.
Viscosity
Viscosity is an important property in liquids, influencing ion transport and the wettability of electrolytes. Despite significant efforts to compute the viscosity of liquids86,87,88,89,90,91,92,93,94,95, accurately predicting liquid viscosity remains a challenge due to the need for ultra-long MD simulations89. In this study, we perform 3 ns MLMD simulations under the NVT ensemble using the uMLP method, with the Green-Kubo formula96,97 applied to compute the viscosity91. The densities of the electrolytes are obtained from 1 ns MLMD trajectories in the NpT ensemble. As shown in Figure S9, the viscosities of hybrid aqueous organic electrolytes are computed and compared with experimental data, with all data points closely aligning with the diagonal line. In a previous publication, 500 ps MLMD simulations were employed to calculate the viscosity of water at different temperatures. However, 500 ps MD simulations exhibit poor convergence in the low-frequency region of the power spectrum, which may significantly affect the accuracy of viscosity calculations. Therefore, it is crucial to note that long-timescale MD simulations are essential for obtaining accurate computed viscosity.
Ionic conductivity
Ionic conductivity is one of the most important physical properties of electrolytes, as it determines the internal resistance of batteries. To compute the ionic conductivity of the electrolytes, ∼10 ns MLMD trajectories are generated. The diffusion coefficients of cations and anions are calculated using TRAVIS98 and subsequently converted to ionic conductivity using the Nernst-Einstein equation. For validation, we selected three systems that have been experimentally investigated: LiTSI/FAN, LiFTSI/FAN, and LiTFSI/EC-EMC(3:7), to assess the accuracy of the computed ionic conductivity. As shown in Fig. 4a, the ionic conductivity of electrolytes with varying concentrations is computed using MLMD simulations (represented by crossed points). For comparison, the corresponding experimental values are also plotted (represented by circular points). The maximum ionic conductivity as a function of concentration is well reproduced by the MLMD simulation with uMLP. This indicates that the concentration of the electrolyte at which the highest ionic conductivity occurs can be predicted by MLMD simulations, consistent with experimental methods used for screening high-conductivity electrolytes16. Therefore, by utilizing MLMD simulations with uMLP, it becomes possible to compute ionic conductivity with high accuracy and efficiency, enabling the computational design of electrolytes.

a Ionic conductivity σ at different concentrations and b ionic conductivity of electrolytes at different temperatures from experiments and computed by MLMD simulations. The circular points (∘) represent experimental values, and the crossed points (×) are obtained through MLMD calculations.
Operating Temperature Range
To demonstrate the computation of the operating temperature ranges of electrolytes, the ionic conductivity is calculated using MLMD simulations across a range of temperatures. The primary objectives are twofold: firstly, to validate that MLMD based on uMLP can accurately computed ionic conductivity by comparing computed results with experiments, and secondly, to indicate that computing ionic conductivity of electrolyte at different temperatures can be used to screen electrolytes with wide operating temperature ranges. To evaluate the operating temperature ranges of electrolytes, ionic conductivity under different temperatures are computed by MLMD based on uMLP for electrolytes to determine the temperature intervals with high ionic conductivity. As shown in Fig. 4b, the trend of ionic conductivity variation with temperature can be accurately simulated. For the high-temperature range, the calculated values of ionic conductivity are close to the experimental values. However, in the low-temperature region, due to the slow dynamics at low temperatures, 10 ns MLMD simulation is insufficient to obtain converged diffusion coefficients. To obtain more accurate ionic conductivity, it is possible to reduce statistical errors by increasing the size of the electrolytes model and extending the simulation time scale of MLMD. In addition, the variation of ionic conductivity with temperature can be accurately predicted in comparison with experiments16, thereby determining the operating temperature range of the electrolyte. Therefore, through MLMD simulations based on uMLP for electrolytes, the operating temperature range of the electrolyte can be directly calculated, which will become an important step in the computational design of electrolytes.
Coordination dynamics
Although ionic conductivity and operating temperature range can be accurately computed through uMLP-based MLMD, the detailed correlation between Li+ coordination strength and these properties remains unclear. Weakly solvated Li+ is often associated with enhanced ionic mobility and higher conductivity due to reduced solvation barriers, while strongly solvated Li+ typically leads to sluggish ion transport as a result of stronger solvation shells hindering diffusion99,100,101. To bridge this gap, a quantitative analysis of Li+ coordination dynamics is essential, providing a clearer understanding of how solvation strength impacts macroscopic properties and enabling the targeted design of electrolytes optimized for ionic transport. Similar to the dynamics of hydrogen bonds in ionic liquids102,103, the autocorrelation function (ACF) for each potential coordination bonding pair is calculated from the MD trajectories. The integral over the ACF is then regarded as the lifetime (τ) of the coordination bond of Li+. In this work, the continuous ACF, in which pairs are not allowed to break and reform at a later time, is applied104. As illustrated in Figure S10, the lifetime of the solvation structure of Li+ is derived from the MLMD trajectories for three types of electrolytes with 1.2 M Li-salt. In 1.2 M LiFSI/FAN, the lifetimes of Li+-FAN and Li+ − FSI− are less than 15 ps, indicating rapid exchange of solvation structures of Li+. When LiFSI is replaced with LiTFSI, the corresponding lifetime of the Li+-anion pair increases to ∼50 ps, resulting in a significantly lower conductivity of the electrolyte. Conversely, in the case of 1.2 M LiPF6/EC-EMC(3:7), the lifetimes of Li+-anions are relatively short compared to the other two electrolytes. However, the lifetimes of Li+-solvent pairs are significantly longer, ∼40 ps. Note that, the coordination of Li+ is primarily formed by solvent molecules in 1.2 M LiPF6/EC-EMC(3:7), consequently resulting in an increased lifetime of solvents and lower ionic conductivity. Furthermore, temperature also impacts the lifetime of coordination bonding pairs in electrolytes.
As shown in Fig. 5a, with decreasing temperature, the lifetimes of Li+-FAN and Li+ − FSI− increase in the 1.3 M LiFSI/FAN electrolyte. At low temperatures, strong solvation bonding pairs exhibit significantly longer lifetimes, typically ranging from 200 to 1000 ps, as depicted in Figure S11, indicating that the strength of solvation can be quantitatively represented by the lifetime. In previous work99, two kind diffusion mechanism of Li+ in electrolyte is proposed, i.e. hopping and vehicle mechanism. However, quantitatively describing the diffusion mechanism remains challenging. Drawing inspiration from the lifetime of coordination bonding pairs, the averaged lifetime of solvation of Li+ is computed by \(\bar{\tau }=\frac{1}{{{{\rm{CN}}}}}\mathop{\sum }_{i}^{N}{{{{\rm{CN}}}}}_{i}\cdot {\tau }_{i}\), in which the τi is the lifetime of coordination pair i, CN is the total coordination number and \({{{{\rm{CN}}}}}_{i}\) is the coordination number of pair i. Then, the ionic conductivity (both from experiments and MLMD simulations) is plotted as a function of the corresponding averaged lifetime \(\bar{\tau }\), as illustrated in Fig. 5 b. The data points on Fig. 5b can be easily clustered into three parts: the rapid ligand exchange region (light blue region), the ligand retention region (light pink region), and the transition region (gray region).

a Coordination lifetime of Li+-FAN and Li+ − FSI− at various temperatures. The ionic conductivity from MLMD and experiments are plotted together for comparison. b The conductivities are plotted as a function of coordination lifetime (\(\bar{\tau }\)), illustrating the correlation between ion transport and the dynamics of the solvation shell. c Schematic showing the increase in \(\bar{\tau }\) from “weakly solvated" to “strongly solvated" Li+ structures and the corresponding decrease in ionic transport. The C, H, O, N, S, F and Li atoms are in gray, white, red, blue, yellow, green and pink, respectively.
As shown in Fig. 5c, the relationship between coordination lifetime (\(\bar{\tau }\)), solvation structure, and ionic transport is illustrated. The green arrow indicates a progression from “weakly solvated" (short \(\bar{\tau }\)) to “strongly solvated" (long \(\bar{\tau }\)), with representative Li+ solvation structures provided for different \(\bar{\tau }\) values. The lower blue arrow highlights the inverse relationship, where shorter \(\bar{\tau }\) values correspond to faster ionic transport, while longer \(\bar{\tau }\) values are associated with slower transport. This visualization underscores the importance of coordination dynamics in determining electrolyte properties and provides a conceptual basis for designing weakly solvated electrolytes to optimize ion transport and electrochemical performance. Coordination dynamics analysis offers significant insights into the concept of weak solvation, particularly by quantifying the dynamic characteristics of Li+ coordination. By calculating the coordination lifetime (τ), it provides a clear metric to evaluate weak solvation, where short lifetimes and frequent solvent exchange reflect weak interactions between Li+ and its solvation shell. This analysis elucidates the direct relationship between weak solvation and ion transport, demonstrating how reduced coordination strength enhances ionic conductivity. Moreover, it facilitates the design of weakly solvated electrolytes by enabling the systematic screening of solvent and salt combinations that optimize coordination dynamics for improved electrochemical performance. Importantly, coordination dynamics bridges experimental observations, such as high ionic conductivity and wide operating temperature range, with their underlying microscopic mechanisms, offering a powerful tool to validate hypotheses and optimize electrolyte efficiently.
In this study, we developed a uMLP for electrolytes using a concurrent learning scheme, enabling long-timescale MD simulations with ab initio accuracy. To ensure comprehensive coverage of the chemical space, an electrolyte generator was employed to create diverse systems with randomized compositions from a database of over 2300 solvents and 20 salts. This approach allowed the uMLP to accurately compute key properties, including density, solvation structures, ionic conductivity, and operating temperature ranges, all showing good agreement with experimental data across diverse electrolyte systems. The uMLP’s transferability often eliminates the need for retraining, making it broadly applicable to various electrolyte formulations. Leveraging MLMD trajectories, we quantitatively analyzed Li+ coordination dynamics by calculating the average coordination lifetime (\(\bar{\tau }\)). This analysis provides a direct and quantitative measure of solvation strength, linking \(\bar{\tau }\) to ionic conductivity and weak solvation, and offering a molecular-level understanding of ion transport mechanisms. The coordination dynamics framework supports the design of weakly solvated electrolytes by identifying solvent-salt combinations that enhance electrochemical performance. By integrating property computations with coordination dynamics analysis, the uMLP would significantly accelerate the preliminary screening of electrolyte candidates, minimizing time and computational resources. This comprehensive approach facilitates the rapid identification of promising electrolytes, bridges macroscopic properties with molecular mechanisms, and offers invaluable guidance for experimental development and optimization.
Methods
DFT calculations
DFT calculations are carried out using the freely available CP2K/QUICKSTEP package105. The density functional calculations were performed using a hybrid Gaussian plane wave approach. The cutoff energy for the plane wave density at the finest grid level was set to 900 Ry for electrolytes containing Li+, Na+, and K+, while higher cutoff energies were used for divalent ions: 2300 Ry for Mg2+, 1900 Ry for Ca2+, and 1600 Ry for Zn2+. During the iterative training, DFT calculations are performed with the Perdew-Burke-Ernzerhof (PBE) functional73 for exchange correlation approximation and Grimme D374 method for van der Waals correction (PBE-D3). The core electrons are treated with Goedecker-Teter-Hutter (GTH) pseudopotentials106,107. And the molecularly optimised basis set (BASIS_MOLOPT) with short-ranged double-ζ basis with a set of polarization functions (DZVP-MOLOPT-SR-GTH) together with GTH pseudopotentials is used with PBE-D3108.
Training MLP
Initial atomic configurations were generated using the PACKMOL software109, with corresponding OPLS/AA force field parameters110 derived via LigParGen111. input files for Classical Molecular Dynamics (CMD) were subsequently prepared using FFTOOL112. CMD simulations were then performed to obtain pre-equilibrated structures for the electrolyte systems. Following equilibration, a universal MLP (uMLP) was constructed using a concurrent learning scheme involving four iterative stages: training, exploration, screening, and labeling113. In the training phase, four DeepPot-SE models52,114 were initialized with different random seeds. The neural network architecture employed an embedding net of size [25, 50, 100] and a fitting net of [240, 240, 240]. A radial cutoff of 6.0 Å was applied, with a smoothing function introduced between 0.5 and 6.0 Å to ensure continuity. The models were trained for 4 × 105 steps, during which the learning rate underwent an exponential decay from an initial value of 1.0 × 10−3 to a final value of 1.8 × 10−8. For the exploration step, one of the four trained MLPs was utilized to drive the MD simulations. To enhance the sampling of configurational space, simulations were conducted under both NVT and NpT ensembles. The temperature range was set to 300–800 K, and for NpT simulations, the pressure ranged from 1 to 100 bar. Temperature and pressure were regulated using the Nose-Hoover thermostat and Parinello-Rahman barostat, respectively, with damping parameters set to 50 fs for temperature and 500 fs for pressure. During the screening phase, the maximum standard deviation of atomic forces predicted by the four models, denoted as the model deviation57, was calculated to assess prediction accuracy. Structures with a model deviation falling within the interval [0.40, 0.80] eV/Å were classified as “decent" candidates. These configurations were further filtered using clustering analysis to minimize redundancy. Finally, in the labeling step, the selected structures underwent DFT calculations to determine ground-truth potential energies, atomic forces, and virial tensors25,75,115,116, which were then added to the training dataset. Upon completion of the iterative cycles, the final production MLPs were trained on the comprehensive dataset for 4,000,000 steps.
Molecular dynamics
MLMD simulations are carried with the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) package117. NpT ensemble is applied to densities of electrolytes under different temperatures. The time step is set to 0.5 fs and the pressure is set to 1 bar. The NpT ensembles are used with the Nose-Hoover thermostat for temperature controlling and Parinello-Rahman barostat for pressure controlling, and the temperature and stress damping parameter are set to 50 fs and 500 fs, respectively. Then, over 5 ns MLMD simulations are run with NVT ensemble to compute viscosity, conductivity under different temperatures. The NVT ensembles are used with the Nose-Hoover thermostat for temperature controlling and the temperature damping parameter are set to 50 fs. On a single NVIDIA A100 GPU, our model achieves an efficient simulation rate of 240.142 katom-step/s (kilo-atom-steps per second), making it well-suited for large-scale MLMD simulations.
Data availability
The training datasets and universal machine learning potential generated in this study have been deposited in the AI4EC repository under accession code https://doi.org/10.12463/AI4EC/QZCYP1118. Source data are provided with this paper. For trial use of uMLP for electrolytes, please refer to the op-elyte emulator app at https://ai4ec.ac.cn/apps/op-elyte-emulator. In this web app, the structure, corresponding potential energies, atomic forces, and virial tensors are provided for each simulation, allowing for accuracy testing. Source data are provided with this paper.
Code availability
The machine learning molecular dynamics simulations and workflow management were performed using open-source software packages, including ai2-kit68, DeePMD-kit119, LAMMPS120 and CP2K105. The specific input configuration files, parameter sets, and scripts required to reproduce the reported results are available at reference121 under the GNU General Public License.
References
Tarascon, J.-M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. nature 414, 359–367 (2001).
Dunn, B., Kamath, H. & Tarascon, J.-M. Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011).
Suo, L. et al. "Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 350, 938–943 (2015).
Yamada, Y. & Yamada, A. Superconcentrated electrolytes for lithium batteries. J. Electrochem. Soc. 162, A2406 (2015).
Rodrigues, M.-T. F. et al. A materials perspective on Li-ion batteries at extreme temperatures. Nature Energy 2, 1–14 (2017).
Liu, Y., Zhu, Y. & Cui, Y. Challenges and opportunities towards fast-charging battery materials. Nat. Energy 4, 540–550 (2019).
Fan, X. & Wang, C. High-voltage liquid electrolytes for li batteries: progress and perspectives. Chem. Soc. Rev. 50, 10486–10566 (2021).
Yamada, Y. et al. Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries. J. Am. Chem. Soc. 136, 5039–5046 (2014).
Piao, Z., Gao, R., Liu, Y., Zhou, G. & Cheng, H.-M. A review on regulating Li+ solvation structures in carbonate electrolytes for lithium metal batteries. Adv. Mater. 35, 2206009 (2023).
Yuan, S. et al. Advanced nonflammable organic electrolyte promises safer Li-metal batteries: from solvation structure perspectives. Adv. Mater. 35, 2206228 (2023).
Logan, E. et al. A study of the physical properties of Li-ion battery electrolytes containing esters. J. Electrochem. Soc. 165, A21 (2018).
Liu, Y.-K. et al. Research progresses of liquid electrolytes in lithium-ion batteries. Small 19, 2205315 (2023).
Logan, E. & Dahn, J. Electrolyte design for fast-charging Li-ion batteries. Trends Chem. 2, 354–366 (2020).
Leng, F., Tan, C. M. & Pecht, M. Effect of temperature on the aging rate of Li ion battery operating above room temperature. Sci. Rep. 5, 12967 (2015).
Borodin, O. Challenges with prediction of battery electrolyte electrochemical stability window and guiding the electrode–electrolyte stabilization. Curr. Opin. Electrochem. 13, 86–93 (2019).
Lu, D. et al. Ligand-channel-enabled ultrafast Li-ion conduction. Nature 627,101–107 (2024).
Yao, Y.-X. et al. Regulating interfacial chemistry in lithium-ion batteries by a weakly solvating electrolyte. Angew. Chem. Int. Ed. 60, 4090–4097 (2021).
Yao, N. et al. Probing the origin of viscosity of liquid electrolytes for lithium batteries. Angew. Chem. Int. Ed. 62, e202305331 (2023).
Gao, Y.-C. et al. Data-driven insight into the reductive stability of ion–solvent complexes in lithium battery electrolytes. J. Am. Chem. Soc. 145, 23764–23770 (2023).
Yamada, Y. et al. Hydrate-melt electrolytes for high-energy-density aqueous batteries. Nat. Energy 1, 1–9 (2016).
Huang, J.-X., Csányi, G., Zhao, J.-B., Cheng, J. & Deringer, V. L. First-principles study of alkali-metal intercalation in disordered carbon anode materials. J. Mater. Chem. A 7, 19070–19080 (2019).
Mendez-Morales, T., Li, Z. & Salanne, M. Computational screening of the physical properties of water-in-salt electrolytes. Batteries Supercaps 4, 646–652 (2021).
Zhang, Y. et al. Water-in-salt LiTFSI aqueous electrolytes. 1. Liquid structure from combined molecular dynamics simulation and experimental studies. J. Phys. Chem. B 125, 4501–4513 (2021).
Zhang, Y. & Maginn, E. J. Water-in-salt LiTFSI aqueous electrolytes (2): transport properties and Li+ dynamics based on molecular dynamics simulations. J. Phys. Chem. B 125, 13246–13254 (2021).
Miyazaki, K. et al. First-principles study on the peculiar water environment in a hydrate-melt electrolyte. J. Phys. Chem. Lett. 10, 6301–6305 (2019).
Li, C.-Y. et al. Unconventional interfacial water structure of highly concentrated aqueous electrolytes at negative electrode polarizations. Nat. Commun. 13, 5330 (2022).
Zhou, M. et al. Correlating the potential-holding formation protocol of solid–electrolyte interphases with improving calendar aging on lithium metal anode. ACS Energy Lett. 8, 4702–4710 (2023).
Han, D. et al. A non-flammable hydrous organic electrolyte for sustainable zinc batteries. Nat. Sustain. 5, 205–213 (2022).
Feng, S., Yin, T., Bian, L., Liu, Y. & Cheng, T. Optimization of lithium metal anode performance: Investigating the interfacial dynamics and reductive mechanism of asymmetric sulfonylimide salts. Batteries 10, 180 (2024).
Bi, S. & Salanne, M. Cluster analysis as a tool for quantifying structure-transport properties in simulations of superconcentrated electrolyte. Chem. Sci. 15, 10908–10917 (2024).
House, R. A. et al. Delocalized electron holes on oxygen in a battery cathode. Nat. Energy 8, 351–360 (2023).
Ko, S. et al. Electrode potential influences the reversibility of lithium-metal anodes. Nat. Energy 7, 1217–1224 (2022).
Yao, N., Chen, X., Fu, Z.-H. & Zhang, Q. Applying classical, ab initio, and machine-learning molecular dynamics simulations to the liquid electrolyte for rechargeable batteries. Chem. Rev. 122, 10970–11021 (2022).
Salanne, M. et al. Best practices for simulations and calculations of nanomaterials for energy applications: avoiding “garbage in, garbage out”. ACS Nano 17, 6147–6149 (2023).
Smith, M. D., Rao, J. S., Segelken, E. & Cruz, L. Force-field induced bias in the structure of aβ21–30: A comparison of OPLS, Amber, CHARMM, and GROMOS force fields. J. Chem. Inf. modeling 55, 2587–2595 (2015).
Costanzo, F., Sulpizi, M., Valle, R. G. D. & Sprik, M. The oxidation of tyrosine and tryptophan studied by a molecular dynamics normal hydrogen electrode. J. Chem. Phys. 134, 244508 (2011).
Cheng, J., Sulpizi, M. & Sprik, M. Redox potentials and pKa for benzoquinone from density functional theory based molecular dynamics. J. Chem. Phys. 131, 154504 (2009).
Yu, J., Zhao, T.-S. & Pan, D. Tuning the performance of aqueous organic redox flow batteries via first-principles calculations. J. Phys. Chem. Lett. 11, 10433–10438 (2020).
Yang, X.-H., Papasizza, M., Cuesta, A. & Cheng, J. Water-in-salt environment reduces the overpotential for reduction of co2 to co2–in ionic liquid/water mixtures. ACS Catal. 12, 6770–6780 (2022).
Wang, F. & Cheng, J. Unraveling origin of reductive stability of super-concentrated electrolytes from first principles and unsupervised machine learning. Chem. Sci. 13, 11570–11576 (2022).
Wang, F., Sun, Y. & Cheng, J. Switching of redox levels leads to high reductive stability in water-in-salt electrolytes. J. Am. Chem. Soc. 145, 4056–4064 (2023).
Goloviznina, K., Serva, A. & Salanne, M. Formation of polymer-like nanochains with short lithium–lithium distances in a water-in-salt electrolyte. J. Am. Chem. Soc. 146, 8142–8148 (2024).
Blumberger, J., Tavernelli, I., Klein, M. L. & Sprik, M. Diabatic free energy curves and coordination fluctuations for the aqueous Ag+/Ag2+ redox couple: A biased born-oppenheimer molecular dynamics investigation. J. Chem. Phys. 124, 064507 (2006).
Sulpizi, M. & Sprik, M. Acidity constants from vertical energy gaps: density functional theory based molecular dynamics implementation. Phys. Chem. Chem. Phys. 10, 5238–5249 (2008).
Cheng, J., Liu, X., VandeVondele, J., Sulpizi, M. & Sprik, M. Redox potentials and acidity constants from density functional theory based molecular dynamics. Acc. Chem. Res. 47, 3522–3529 (2014).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Huan, T. D. et al. A universal strategy for the creation of machine learning-based atomistic force fields. npj Comput. Mater. 3, 1–8 (2017).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Behler, J. et al. Perspective: Machine learning potentials for atomistic simulations. J Chem. Phys. 145, 170901(2016).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Rupp, M., Tkatchenko, A., Müller, K.-R. & von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012).
Zhang, L., Han, J., Wang, H., Car, R. & Weinan, E. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Wang, H., Zhang, L., Han, J. & E, W. Deepmd-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 228, 178 – 184 (2018).
Chmiela, S. et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 3, e1603015 (2017).
Schütt, K. T., Arbabzadah, F., Chmiela, S., Müller, K. R. & Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Comm. 8, 6–13 (2017).
Zhang, Y. et al. DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models. Comput. Phys. Commun. 253, 107206 (2020).
Zhang, L., Lin, D.-Y., Wang, H., Car, R. & Weinan, E. Active learning of uniformly accurate interatomic potentials for materials simulation. Phys. Rev. Mater. 3, 023804 (2019).
Huang, J. et al. Deep potential generation scheme and simulation protocol for the li10gep2s12-type superionic conductors. J. Chem. Phys. 154, 094703 (2021).
Staacke, C. G., Huss, T., Margraf, J. T., Reuter, K. & Scheurer, C. Tackling structural complexity in Li2s-P2s5 solid-state electrolytes using machine learning potentials. Nanomaterials 12, 2950 (2022).
Wang, F. & Cheng, J. Understanding the solvation structures of glyme-based electrolytes by machine learning molecular dynamics. Chin. J. Struct. Chem. 42, 100061 (2023).
Zhu, D. et al. Investigation of the degradation of lipf6–in polar solvents through deep potential molecular dynamics. J. Phys. Chem. Lett. 15, 4024–4030 (2024).
Zhang, D. et al. Dpa-2: a large atomic model as a multi-task learner. npj Comput. Mater. 10, 293 (2024).
Batatia, I. et al. A foundation model for atomistic materials chemistry. J. Chem. Phys. 18, 163 (2025).
Zhang, S. et al. Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential. Nat. Chem. 16, 727–734 (2024).
Gong, S. et al. A predictive machine learning force-field framework for liquid electrolyte development. Nature Machine Intelligence 1–10 (2025).
Chen, J., Gao, Q., Huang, M. & Yu, K. Application of modern artificial intelligence techniques in the development of organic molecular force fields. Physical Chemistry Chemical Physics (2024).
Ju, S. et al. Application of pretrained universal machine-learning interatomic potential for physicochemical simulation of liquid electrolytes in Li-ion batteries. Digital Discovery 4, 1544–1559 (2025).
van der Oord, C., Sachs, M., Kovács, D. P., Ortner, C. & Csányi, G. Hyperactive learning for data-driven interatomic potentials. npj Comput. Mater. 9, 168 (2023).
Wang, F., Ma, Z. & Cheng, J. Accelerating computation of acidity constants and redox potentials for aqueous organic redox flow batteries by machine learning potential-based molecular dynamics. J. Am. Chem. Soc. 146, 14566–14575 (2024).
Goodwin, Z. A. et al. Transferability and accuracy of ionic liquid simulations with equivariant machine learning interatomic potentials. J. Phys. Chem. Lett. 15, 7539–7547 (2024).
Yang, J. H. et al. Room-temperature decomposition of the ethaline deep eutectic solvent. J. Phys. Chem. Lett. 16, 3039–3046 (2025).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Leung, K. & Budzien, J. L. Ab initio molecular dynamics simulations of the initial stages of solid–electrolyte interphase formation on lithium ion battery graphitic anodes. Phys. Chem. Chem. Phys. 12, 6583–6586 (2010).
Rana, R., Ali, S. M. & Maity, D. K. Structure and dynamics of the Li+ ion in water, methanol and acetonitrile solvents: ab initio molecular dynamics simulations. Phys. Chem. Chem. Phys. 25, 31382–31395 (2023).
Xu, L., Shao, W., Jin, H. & Wang, Q. Data efficient and stability indicated sampling for developing reactive machine learning potential to achieve ultralong simulation in lithium-metal batteries. J. Phys. Chem. C. 127, 24106–24117 (2023).
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013).
Zhang, L., Wang, H., Car, R. & Weinan, E. Phase diagram of a deep potential water model. Phys. Rev. Lett. 126, 236001 (2021).
Schuhmacher, J. et al. Extending the reach of quantum computing for materials science with machine learning potentials. AIP Adv. 12, 115321(2022).
Wengert, S., Csányi, G., Reuter, K. & Margraf, J. T. Data-efficient machine learning for molecular crystal structure prediction. Chem. Sci. 12, 4536–4546 (2021).
Niblett, S. P., Kourtis, P., Magdău, I.-B., Grey, C. P. & Csányi, G. Transferability of data sets between machine-learned interatomic potential algorithms. J. Chem. Theory Comput. 12, 6096–6112 (2025).
Zhou, P. et al. Rational lithium salt molecule tuning for fast charging/discharging lithium metal battery. Angew. Chem. 136, e202316717 (2024).
Pradhan, A. et al. Stabilization of lithium metal in concentrated electrolytes: effects of electrode potential and solid electrolyte interphase formation. Faraday Discussions 253, 314–328 (2024).
Yin, Y. et al. Coulombic condensation of liquefied gas electrolytes for li metal batteries at ambient pressure. Angewandte Chemie International Edition 64, e202420411 (2024).
Kuo, I.-F. W. et al. Liquid water from first principles: Investigation of different sampling approaches. J. Phys. Chem. B 108, 12990–12998 (2004).
Grossman, J. C., Schwegler, E., Draeger, E. W., Gygi, F. & Galli, G. Towards an assessment of the accuracy of density functional theory for first principles simulations of water. J. Chem. Phys. 120, 300–311 (2004).
Schwegler, E., Grossman, J. C., Gygi, F. & Galli, G. Towards an assessment of the accuracy of density functional theory for first principles simulations of water. ii. J. Chem. Phys. 121, 5400–5409 (2004).
LaCount, M. D. & Gygi, F. Ensemble first-principles molecular dynamics simulations of water using the scan meta-GGA density functional. J. Chem. Phys. 151, 164101 (2019).
Todorova, T., Seitsonen, A. P., Hutter, J., Kuo, I.-F. W. & Mundy, C. J. Molecular dynamics simulation of liquid water: hybrid density functionals. J. Phys. Chem. B 110, 3685–3691 (2006).
Malosso, C., Zhang, L., Car, R., Baroni, S. & Tisi, D. Viscosity in water from first-principles and deep-neural-network simulations. npj Comput. Mater. 8, 139 (2022).
Gillan, M. J., Alfe, D. & Michaelides, A. Perspective: how good is DFT for water? J. Chem. Phys. 144, 130901(2016).
Chen, M. et al. Ab Initio theory and modeling of water. Proc. Natl Acad. Sci. 114, 10846–10851 (2017).
Zheng, L. et al. Structural, electronic, and dynamical properties of liquid water by ab initio molecular dynamics based on scan functional within the canonical ensemble. J. Chem. Phys.148, 164505 (2018).
Kühne, T. D., Krack, M. & Parrinello, M. Static and dynamical properties of liquid water from first principles by a novel car- Parrinello-like approach. J. Chem. Theory Comput. 5, 235–241 (2009).
Green, M. S. Markoff random processes and the statistical mechanics of time-dependent phenomena. ii. Irreversible processes in fluids. J. Chem. Phys. 22, 398–413 (1954).
Kubo, R. Statistical-mechanical theory of irreversible processes. i. General theory and simple applications to magnetic and conduction problems. J. Phys. Soc. Jpn. 12, 570–586 (1957).
Brehm, M., Thomas, M., Gehrke, S. & Kirchner, B. Travis—a free analyzer for trajectories from molecular simulation. J. Chem. Phys.152,164105(2020).
Fan, Z. et al. Solvation structure dependent ion transport and desolvation mechanism for fast-charging Li-ion batteries. Chem. Sci. 15, 17161–17172 (2024).
Guo, J. et al. Weak solvation chemistry in fluorinated nonflammable electrolytes achieves stable cycling in high-voltage lithium metal batteries. ACS Applied Materials & Interfaces (2024).
Qin, M. et al. Microsolvating competition in Li+ solvation structure affording pc-based electrolyte with fast kinetics for lithium-ion batteries. Adv. Funct. Mater. 34, 2406357 (2024).
Brehm, M. & Sebastiani, D. Simulating structure and dynamics in small droplets of 1-ethyl-3-methylimidazolium acetate. J. Chem. Phys. 148,193802 (2018).
Thar, J., Brehm, M., Seitsonen, A. P. & Kirchner, B. Unexpected hydrogen bond dynamics in imidazolium-based ionic liquids. J. Phys. Chem. B 113, 15129–15132 (2009).
Rapaport, D. Hydrogen bonds in water: Network organization and lifetimes. Mol. Phys. 50, 1151–1162 (1983).
VandeVondele, J. et al. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 167, 103–128 (2005).
Hartwigsen, C., Goedecker, S. & Hutter, J. Relativistic separable dual-space gaussian pseudopotentials from h to rn. Phys. Rev. B 58, 3641–3662 (1998).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space gaussian pseudopotentials. Phys. Rev. B 54, 1703–1710 (1996).
VandeVondele, J. & Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (2007).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Robertson, M. J., Tirado-Rives, J. & Jorgensen, W. L. Improved peptide and protein torsional energetics with the OPLS-aa force field. J. Chem. theory Comput. 11, 3499–3509 (2015).
Dodda, L. S., Cabeza de Vaca, I., Tirado-Rives, J. & Jorgensen, W. L. Ligpargen web server: an automatic OPLS-aa parameter generator for organic ligands. Nucleic acids Res. 45, W331–W336 (2017).
Padua, A., Goloviznina, K. & Gong, Z. agiliopadua/fftool: XML force field files (v1.2.1). Zenodo. https://doi.org/10.5281/zenodo.4701065 (2021).
Guo, Y.-X., Zhuang, Y.-B., Shi, J. & Cheng, J. Checmate: a workflow package to automatically generate machine learning potentials and phase diagrams for semiconductor alloys. J. Chem. Phys. 159, 094801(2023).
Zhang, L. et al. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems. Adv. Neural Inf. Process. Syst. 31, 4441–4451(2018).
Leung, K. Predicting the voltage dependence of interfacial electrochemical processes at lithium-intercalated graphite edge planes. Phys. Chem. Chem. Phys. 17, 1637–1643 (2015).
Yamada, Y. et al. General observation of lithium intercalation into graphite in ethylene-carbonate-free superconcentrated electrolytes. ACS Appl Mater. Inter. 6, 10892–10899 (2014).
Thompson, A. P. et al. LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Computer Phys. Commun. 271, 108171 (2022).
Wang, F. et al. Domain oriented universal machine learning potential enables fast exploration of chemical space of battery electrolytes. OmniElectroData: DFT Dataset for Universal Machine Learning Potential (uMLP) for Electrolyte, https://doi.org/10.12463/AI4EC/QZCYP1 (2025).
Wang, H., Zhang, L., Han, J. & E, W. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Comm. 228, 178–184 (2018).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. https://doi.org/10.1016/j.cpc.2021.108171 (2022).
Feng, W. et al. Domain oriented universal machine learning potential enables fast exploration of chemical space of battery electrolytes. codes for umlp, https://doi.org/10.5281/zenodo.17826544 (2025).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 22225302, 92470201, 92461312, 22021001, 21991151, 21991150, 92161113 and 22411560277 to J.C.; Grant Nos. 22393901 and 22503037 to F.W.), the Fundamental Research Funds for the Central Universities (Grant Nos. 20720250005, 20720220009, and 20720230090 to J.C.), and the Laboratory of AI for Electrochemistry (AI4EC) and IKKEM (Grant Nos. RD2023100101 and RD2022070501 to J.C. and F.W.).
Author information
Authors and Affiliations
Contributions
J.C. and F.W. conceived and designed the project, analysed the results and wrote the manuscript. F.W. Y.H.T. Z.B.M. and Y.C.J. performed the MLMD simulations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, F., Tang, YH., Ma, ZB. et al. Domain oriented universal machine learning potential enables fast exploration of chemical space of battery electrolytes. Nat Commun 17, 1226 (2026). https://doi.org/10.1038/s41467-025-67982-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67982-0
