
Abstract
Thousands of short open reading frames (sORFs) are translated outside of annotated coding sequences. Recent studies have pioneered searching for sORF-encoded microproteins in mass spectrometry (MS)-based proteomics and peptidomics datasets. Here, we assessed literature-reported MS-based identifications of unannotated human proteins. We find that studies vary by three orders of magnitude in the number of unannotated proteins they report. Of nearly 10,000 reported sORF-encoded peptides, 96% were unique to a single study, and 12% mapped to annotated proteins or proteoforms. Manual curation of a benchmark dataset of 406 manually evaluated spectra from 204 sORF-encoded proteins revealed large variation in peptide-spectrum match (PSM) quality between studies, with immunopeptidomics studies generally reporting higher quality PSMs than conventional enzymatic digests of whole cell lysates. We estimate that 65% of predicted sORF-encoded protein detections in immunopeptidomics studies were supported by high-quality PSMs versus 7.8% in non-immunopeptidomics datasets. Our work stresses the need for standardized protocols and analysis workflows to guide future advancements in microprotein detection by MS towards uncovering how many human microproteins exist.
Similar content being viewed by others

Introduction
Ribosome profiling (Ribo-Seq) studies have demonstrated widespread translation of short open reading frames (sORFs) outside of annotated coding sequences in eukaryotic genomes1,2, suggesting that the proteome may be much larger than currently annotated in databases such as UniProtKB3,4,5,6. Several such individual sORF-encoded microproteins were experimentally found to be implicated in diverse biological processes across the tree of life such as muscle physiology and cancer7,8,9,10,11,12. Yet, these well-characterized cases represent only a small fraction of the microproteins that could be encoded by translated sORFs13. The translation products of many sORFs may be poorly conserved, of low abundance, or rapidly degraded, leading to uncertainty about their biological significance5,14,15. There is a need, therefore, to identify the sORF-encoded microproteins that exist in the cell and have the potential to perform biological activities.
One systematic approach to identify unannotated microproteins predicted by Ribo-Seq is to search for peptide-level evidence in mass spectrometry (MS)-based proteomics or peptidomics datasets16,17. In the typical case, a sequence database is constructed that consists of a curated protein sequence database (e.g. the UniProtKB human reference proteome18) joined together with a list of putative unannotated proteins (e.g. predicted products of translated sORFs cataloged by Ribo-Seq). This protein sequence database may then be used for analyzing conventional “shotgun” MS proteomics datasets, in which protein samples are digested using a protease, or for analyzing datasets generated by immunopeptidomics experiments, which attempt to identify peptides presented by human leukocyte antigens (HLAs) without requiring protease pretreatment19. In both conventional proteomics experiments and immunopeptidomics experiments, the collected spectra will be generated from peptides derived from both annotated and unannotated proteins in the sample. Confident inference of an unannotated protein detection requires that the peptide uniquely supports an unannotated protein; i.e., that one can exclude the possibility that it derives from a protein in a curated protein sequence database. Detection confidence is generally controlled using a target-decoy approach20, which enables the calculation of a false discovery rate (FDR). The FDR can be set at the level of peptide-spectrum matches (PSMs), peptides, or proteins. Peptides and their inferred proteins passing the thresholds, usually 1% FDR at the peptide/protein level, are reported as detected21. Protein-level MS evidence in a conventional proteomics experiment using trypsin or other proteases indicates that the protein existed in the cell. Immunopeptidomics can be used to validate Ribo-Seq predictions by confirming that an sORF was translated and the processed forms of its translation product was presented by HLA molecules, but cannot establish that the protein was stably present in the cell22.
Despite the promise of shotgun proteomics for rapid and large-scale microprotein identification, the small size, low abundance, atypical sequence characteristics and frequent transmembrane localization of microproteins pose major technical challenges for existing MS pipelines23,24,25,26. For example, it can be impossible to observe multiple unique supporting peptides for microproteins whose sequence is too short to hold multiple cleavage sites, or if only one peptide is within the mass-over-charge range of the spectrometer. Therefore, the guidelines established by the Human Proteome Project27 for MS detection of proteins are difficult to apply fully, and researchers use a variety of ad hoc strategies16. As the field develops and the number of reported microprotein detections grows, there is a need to assess which strategies are most effective for identifying genuine microproteins while minimizing false positives.
In this work, we brought together a group of experts to perform a systematic confidence assessment of previously reported unannotated protein MS detections. We find wide variation in the number of unannotated microproteins reported between different proteomic studies, with few microproteins reported in more than one study. Manual evaluation indicates a division between immunopeptidomics studies and studies using conventional tryptic proteomics: most microproteins reported in immunopeptidomics studies are supported by high-quality PSMs, while most microproteins reported in conventional proteomics studies are supported by only low-quality PSMs and may not represent genuine discoveries. Yet, a subset of microproteins is supported by strong evidence in conventional proteomics datasets, suggesting that more remain to be discovered. We outline advice for increasing confidence in proteomic detection of microproteins as this area of investigation continues to grow.
Results
Reported numbers of unannotated proteins vary greatly between studies
To evaluate the extent to which unannotated proteins can be detected in proteomics data, our group of microprotein researchers assembled in 2023 to conduct a literature search for all papers reporting human unannotated protein detections published between 2019 and 2022. We identified 12 such studies that were published in this time window (Table 1). Seven studies searched for unannotated proteins in conventional proteomics data, while two studies searched for peptides derived from unannotated proteins in immunopeptidomics data, and three studies searched both classes of proteomics data. From each study, we obtained a list of the unannotated proteins reported to be detected (of any length), together with the PSMs supporting these detections (Supplementary Data 1, Supplementary Table 1).
A key motivation for initiating this community effort was the large variation in the number of validated unannotated proteins reported between studies, ranging from 628 to 490329 (Fig. 1A, Table 1). The peptides reported in support of unannotated proteins in each study were largely distinct: of 9414 total reported peptides across the considered studies, only 326 (3.5%) were reported in more than one study. For 8 of 12 studies, fewer than 10% of the reported peptides were found in any of the other analyzed studies (Fig. 1B, Supplementary Data 2). The low rate of replication is despite some studies analyzing the same collections of mass spectra, albeit with not fully overlapping databases of sORF sequences (Table 1). We do not interpret the high variability between studies as indicating that most reported detections are false: this high variability among reported detected peptides likely reflects in part the high variability in the size and composition of the sORF databases tested (Table 1)16 and the quantity of proteomic data analyzed, as well as the diversity of cell types examined, MS techniques used, HLA allotypes among the immunopeptidomics studies, and search algorithms. Nevertheless, in the absence of robust replicability to establish confidence, a closer assessment of the strength of evidence provided in each study for their reported detected unannotated proteins is needed.

A The relation between the number of sORFs used to construct the protein database of each study and the number of sORF-encoded proteins reported detected by MS (Spearman correlation = 0.43, p = 0.2). Whether the sORF database was constructed using a curated list of known sORFs, all possible sORFs from three frame translation of a transcriptome, or a list of ORFs found to be translated using Ribo-Seq or RNC-seq data is indicated. B For each study, the proportion of reported peptides supporting an unannotated protein that are also found by another study in our analysis is shown. The numbers of peptides found in other studies out of the total reported in the study are indicated above the bars. C Proportion of peptides mapping to annotated proteins using the ProteoMapper tool, divided into categories depending on the number of common single nucleotide polymorphism (SNP) differences separating the peptide from the peptide present in the reference protein and whether the annotated peptide is tryptic; i.e., could be generated by cleavage after lysine or arginine. Semi-tryptic peptides (where only one peptide end is tryptic) are grouped with non-tryptic. Peptides from immunopeptidomics experiments were not generated by trypsin digestion and therefore are not classified as tryptic or non-tryptic. Peptides matching currently annotated proteins that were not annotated on UniProtKB/Swiss-Prot in 2016 (i.e., recently annotated proteins) are excluded. D For each study, the proportion of reported peptides supporting an unannotated protein that are also found by another study in our analysis, excluding peptides that match to annotated proteins according to the ProteoMapper tool. Note that most studies have focused on different biological systems, which can limit the overlap. Source data are provided as a Source Data file.
Do reported peptides uniquely support an unannotated protein?
We first assessed whether PSMs reported as evidence for the detection of an unannotated protein may also be attributed to an annotated protein. All the studies in our meta-analysis attempted to exclude potential annotated protein-matching peptides, but different analysis pipelines were implemented that might not have equally accounted for the full space of potential proteoforms of annotated proteins16.
To assess whether some peptides reported to derive from an unannotated protein could potentially be attributed to an annotated protein, we used the PeptideAtlas ProteoMapper30 tool. ProteoMapper takes neXtProt31 reported amino acid variants into account; i.e., it will find matches not just to the reference proteome but to proteins that differ from the reference by one or more variant amino acids. We restricted our analysis to peptides that differed from the reference sequence by at most one single amino acid variant. Given this restriction, 12% of peptides reported to support detection of an unannotated protein (1161 of 9732) also had a putative match to an annotated protein on ProteoMapper, with this rate varying from 0% to 96% across individual studies (Supplementary Data 1).
Recent updates in annotation could potentially explain why some reported peptides mapped to annotated proteins when we conducted this ProteoMapper search in 2023. To evaluate this possibility, we checked whether these annotated proteins were annotated in the 2016 version of UniProtKB/Swiss-Prot18, as all studies in our analysis used protein databases published after 2016 to define their annotated set (Table 1). Only eight distinct annotated proteins matching reported unannotated peptides in 2023 were absent from UniProtKB/Swiss-Prot in 2016, indicating that annotation updates are not a major explanation for peptides reported to support unannotated proteins mapping to annotated proteins.
Peptides reported to support unannotated proteins might also map to annotated proteins if the studies did not account for non-tryptic peptides or protein variants. We therefore divided the peptides mapping to annotated proteins by whether they were perfect matches to the UniProtKB/Swiss-Prot reference protein or differed by one single amino acid variant, and by whether they were predicted tryptic (i.e., peptides that could be generated by cleavage after arginine or lysine residues) or non-tryptic (including semi-tryptic) (Fig. 1C). We note that some peptides in Chong et al. 32. map to both unannotated proteins and common variants of annotated proteins, but since this study used customized databases of annotated proteins reflecting each patients’ sequenced genotypes, these common variants were shown to be absent in the patient samples. Without such a customized database, it is difficult to fully rule out an annotated protein source given the possibility of unknown variants of annotated proteins, especially in cell lines or cancer samples.
For two studies, Prensner et al. 33. and Duffy et al. 34., a substantial fraction of reported unannotated peptides (10% or more) were perfect matches to tryptic peptides in reference proteins. The relatively high rate of matching UniProtKB protein references in Prensner et al. 33. might be explained by either the use of the UCSC RefSeq database to define the set of annotated proteins rather than UniProtKB, which was used by most other studies (Table 1), or not preferentially allocating all shared peptides to the annotated set. For Duffy et al. 34., spectra searches were conducted against custom databases of both annotated and unannotated proteins inferred to be expressed in the specific type of brain tissue or cell based on Ribo-Seq data, while all other studies included the full set of human annotated proteins in their protein database. Likely, annotated proteins not detected by Ribo-Seq may still be present in the sample, leading to peptides from annotated proteins potentially being falsely assigned to unannotated proteins. For two other studies6,35, more than half of reported peptides that mapped to both unannotated and annotated proteins were non-tryptic (Fig. 1C). A peptide with a match to an annotated protein does not uniquely support an unannotated protein detection, even if the match is non-tryptic, as trypsin does not have perfect specificity and can vary in grade, cleavage could have been induced by other proteases (e.g. upon lysing cells and tissues), and protein processing in cells can yield non-tryptic peptides.
Overall, these results indicate a need to consider non-tryptic peptides and possible amino acid variants of annotated proteins to ensure that peptides uniquely map to an unannotated protein. Excluding potential hits to annotated proteins can be done with tools such as ProteoMapper30 or the neXtProt peptide uniqueness checker36, as suggested by the HUPO-HPP MS data interpretation guidelines27, or, ideally, using sample-specific customized protein sequence databases based on sequenced genotypes.
After excluding all reported peptides that mapped to annotated proteins according to ProteoMapper, the general trends we observed for the entire set of reported peptides supporting unannotated protein detections remained: for 8 of 12 studies, at least 90% of reported unannotated peptides were only reported in that study (Fig. 1D). Therefore, we next examined the level of support PSMs provided for claimed unannotated protein detections.
Assessing PSM quality by manual evaluation
To assess PSM quality among literature-reported peptides supporting detection of unannotated proteins, a random sample of PSMs from each study was manually evaluated by a panel of six expert evaluators. A total of 406 PSMs from 12 studies were evaluated (1.3% of total), corresponding to 307 peptides from 204 unannotated proteins. These PSMs were sampled after excluding peptides mapping to annotated proteins or proteoforms (Fig. 1C). Of these 406 PSMs, 155 were evaluated by two evaluators each to enable determination of the overall consistency between evaluators. Additionally, a common set of 10 negative control PSMs was included in each sample, consisting of high-scoring decoy-spectrum matches intended to mimic PSMs that perform relatively well according to algorithms. Each PSM was rated on a scale of 1-5. Full evaluation criteria along with example spectra and explanations of their rating are given in Appendix 1. The PSMs assigned to each evaluator were ordered randomly and the evaluators were not informed as to the source publication of each PSM (Supplementary Data 3).
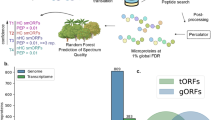
Agreement among evaluators was generally high. For the PSMs rated by two evaluators, ratings were well correlated (r = 0.82, p < 10-10) (Fig. 2A). Only 14 of 155 (9%) PSM scores differed by more than one point. The negative controls scored consistently poorly (average score of 1.5), as expected. Evaluator ratings were also well correlated (r = 0.74, p < 10−10) with the dot product between the observed spectra and the spectra predicted by MS2PIP (Supplementary Fig. 1)37. Among immunopeptidomics studies, PSMs with peptides that were predicted to bind to MHC molecules by NetMHC38 were rated more highly (n = 71, mean rating 3.94) than those with peptides not predicted to bind (n = 14, mean rating 3.29, p = 0.037 by two-sided permutation test, Supplementary Fig. 2, Supplementary Data 4), consistent with manual evaluation discriminating between true and false discoveries. To investigate consistency between manual ratings and machine learning methods for spectral prediction, we generated predicted spectral libraries for all evaluated PSMs under several models using Oktoberfest (see Methods)39. We observed a moderate correlation between the best spectral angle between the model-predicted and experimental spectra (a measure of spectral similarity) and evaluator rating (r = −0.56, p < 10−10, n = 274, Fig. 2B), suggesting both similarities and differences in how expert evaluators and this spectral prediction method assess PSM quality.

A Counts of each pair of ratings among the PSMs that were assessed by two evaluators (n = 155). The Pearson correlation between pairs of ratings is indicated. B For a set of manually evaluated PSMs (n = 274), the spectrum was also predicted using several machine learning models (see “Methods”). The spectral angle is an indicator of how different the observed PSM was from the closest predicted spectrum, with larger angles indicating a worse match. The best spectral angles are indicated among PSMs grouped by evaluator rating. The box in each boxplot indicates interquartile range between the first and third quartiles, while the center line indicates the median. The whiskers indicate minima and maxima within 1.5 times the interquartile range. C Mean ± standard error of ratings of PSMs sampled from each study, per each of six evaluators (n = 620 rated PSMs in total). Standard errors were corrected for finite population (total count of reported PSMs supporting unannotated proteins in the study). Ratings were given on a 1–5 scale. D Overall distribution of ratings for unannotated protein PSMs among all studies and evaluators (n = 620 PSMs). Bars indicate proportions ± standard errors. E Log Ribo-Seq read counts for ORFs expressing proteins in PSMs rated highly (rating > 3, n = 65 proteins) or lowly (rating < 3, n = 105 proteins). Reads are from a collection of human Ribo-Seq studies (see Methods). The box in each boxplot indicates interquartile range between the first and third quartiles, while the center line indicates the median. The whiskers indicate minima and maxima within 1.5 times the interquartile range. Differences between group means are tested using a two-sided permutation test. F Predicted lengths of proteins rated highly (>3,n = 65 proteins) or lowly (<3, n = 105 proteins). Box plot meaning is same as above. Differences between group means are tested using a two-sided permutation test. G Evaluated and extrapolated counts (±SEM) of HLA and non-HLA high-rated (rating of 4 or 5) protein detections. Extrapolated counts give the number of high-rated protein detections expected if the entire dataset had been evaluated. Source data are provided as a Source Data file.
There was also a general consistency between evaluators in average rating per study (Fig. 2C). The evaluated PSM quality varied across studies, with average rating ranging from 1.0 to 4.1 (Fig. 2C). Three studies had average PSM ratings that did not exceed the negative controls. For one of these studies, van Heesch et al. 6, the authors recognized the high FDR in their search results, which led them to develop a customized strategy for estimating a microprotein-specific FDR and to favor selected reaction monitoring (SRM) for their downstream analyses. We did not evaluate these SRM results but focused solely on the reported shotgun proteomics hits. For Douka et al. 40., the low ratings are understandable because, rather than using a 1% FDR threshold, this study used a 10% threshold in anticipation of the low abundance of microproteins. For Chothani et al. 4, unannotated protein PSMs were identified by searching hundreds of MS runs individually with a 1% FDR threshold after removing all matches to the annotated proteome, then assembling the hits into a master list. A likely explanation is that, since spectra matching annotated proteins were removed prior to searching for unannotated proteins, there were few genuine detections in the MS runs analyzed. Under conditions of few genuine detections, it is difficult to precisely estimate FDR, leading to potential false positives (Supplementary Fig. 3)41. Chothani et al. highlighted peptides found in multiple datasets; these peptides were not separately evaluated here.
The immunopeptidomics studies (Ouspenskaia et al. 29, Martinez et al. 42, and Chong et al. 32, and some peptides from Prensner et al. 33) reported substantially higher quality PSMs than most of the other studies (mean rating 3.8 vs. 2.3, n = 13, p = 0.024 for difference in mean by two-sided permutation test, Fig. 2C, D). The three studies that focused on HLA data have average scores above three, as do the HLA PSMs (but not non-HLA PSMs) from Prensner et al. 43. The only non-HLA studies with average scores of three or more were Cao et al. 44 and Bogaert et al. 28, which reported only 28 and 8 PSMs derived from unannotated proteins, respectively (Fig. 2C and Table 1). Overall, most (70%) evaluated PSMs supporting unannotated protein detections from HLA studies received a rating of at least 4, the threshold for convincing evidence of detection (See Appendix, Fig. 2D). In contrast, only 15% of ratings for reported matches in non-HLA data were in the 4-5 range. These results are consistent with a recent study, Deutsch et al. 45, where MS searches for peptide-level evidence supporting Ribo-Seq identified sORFs also found higher support in HLA than non-HLA datasets45.
Among 98 high-rated HLA peptides, 33 were reported in multiple studies, and 37 were validated by Deutsch et al. 45 (1 supporting an ORF in Tier 1A, 26 in Tier 1B, and 10 in Tier 2B, Supplementary Fig. 4). Of the 28 high-rated PSMs from non-HLA data, two involved peptides that were reported in multiple studies. Both peptides derive from the same sORF, located in the 5’ UTR of the MKKS locus. The protein encoded by this sORF (UniProt identifier Q9HB66 in UniProtKB/TrEMBL) has now accumulated enough peptide-level evidence to have become annotated as “core canonical” in PeptideAtlas in 2025, though it remains unannotated in UniProtKB/Swiss-Prot so far. Two high-rated non-HLA peptides were also identified as having strong evidence in Deutsch et al45. These peptides mapped to the sORFs c11riboseqorf4 in the Tier 1A class (the highest level of support that an ORF is protein-coding) and c12norep33 in the Tier 2A class (weaker support). These observations illustrate how searching multiple sources of MS data contributes towards a more comprehensive view of sORF-expressed proteins and improves annotations of the human proteome.
Higher rated PSMs are derived from more highly expressed sORFs
To assess whether our PSM ratings were influenced by the expression levels of the corresponding proteins, we compiled a large collection of human Ribo-Seq studies and analyzed translation levels harmoniously, using the iRibo program, for all the sORFs corresponding to evaluated PSMs for which genomic coordinates were provided by the original studies (191 sORFs; see “Methods”, Supplementary Data 5, 6)46. We found that reported unannotated proteins with corresponding PSMs rated 4 or 5 were more highly translated than those with corresponding PSMs rated 1 or 2 (difference in log Ribo-Seq read count per codon by two-sided permutation test, p = 0.005, Fig. 2E). This is consistent with more highly expressed proteins being more readily detectable by MS and thus generating higher quality PSMs47. Unexpectedly, high-rated proteins were also shorter on average by 37 amino acids than low-rated proteins (two-sided permutation test, p = 0.01, Fig. 2F). There was no significant correlation between log iRibo p-value, indicating level of confidence that the ORF is translated, and PSM rating (r = 0.098, p = 0.18).
Discovery of potential unannotated proteins
We next estimated the number of unannotated proteins we would expect to have strong MS support had we evaluated all reported detections. To do this, we extrapolated the number of unannotated protein detections that would be supported by high-scoring PSMs had we evaluated all PSMs among all studies, assuming the frequency of scores for each study would be the same as in the tested set (Fig. 2G). Among unannotated proteins reported in non-HLA data, 27 evaluated proteins were supported by at least one PSM rated 4 or 5. We predict 137 of 1749 (7.8%) would be supported by PSMs of this quality across the whole aggregated dataset. For HLA data, 94 evaluated proteins were supported by at least one PSM rated 4 or 5; we predict 3706 of 5705 (65%) would be found across the entire dataset. Other unannotated proteins are likely detectable in datasets outside our study scope. Thus, there is considerable potential for discovery even in the particularly challenging case of finding unannotated proteins in conventional enzymatically digested samples.
Discussion
Given the growing recognition of the importance of microproteins in human health48, there is an urgent need to prioritize sORF-encoded microproteins that are supported by MS evidence. Here, we reanalyzed twelve published studies that reported detection of unannotated microproteins with MS. While most reported PSMs (70%) in immunopeptidomics studies were of high quality, around 85% of non-HLA PSMs were evaluated by a panel of proteomics experts to be of too low quality to provide evidence of peptide detection. These results point to a need for caution in interpreting claimed unannotated protein detections reported in the literature and motivate technological improvements for the evaluation of microprotein evidence moving forward. Many unannotated protein detections do appear strong, and the microprotein literature has provided great value in expanding the protein universe with real discoveries of likely biological significance45. However, the idea that several hundreds to even thousands of unannotated proteins are genuinely detected in existing mass spectrometry datasets of conventional trypsin digests reflects an unrealistic expectation about the extent to which current shotgun proteomics can validate sORFs identified by Ribo-Seq.
Why do immunopeptidomics studies identify many high-quality PSMs supporting unannotated protein detections while studies using conventional enzymatic digests identify only few? Many unannotated sequences found to be translated by Ribo-Seq lack signatures of evolutionary conservation and may not encode proteins that provide any benefit to the organism5,15,49. It is plausible that many of these poorly conserved proteins are expressed but quickly degraded, and so can be found only as peptides bound to HLAs14,50. However, there are also technical explanations for why HLA-bound peptides derived from unannotated microproteins may be easier to detect. Immunopeptidomics concentrates peptides bound to HLAs, which decreases sample complexity and may thereby enrich for low abundance microproteins. HLA peptides also have physical and chemical properties different from tryptic peptides that may affect detectability. Most immunopeptidomics datasets are from cancer samples, and some proteins may be expressed in some cancers but not in normal physiological conditions. Furthermore, microproteins may preferentially reside in cellular compartments that are hard to sample through non-HLA MS, such as membranes26. Moreover, the laboratories that perform immunopeptidomics are often distinct from those that analyze non-HLA data and may differ in their sample preparation techniques, experimental setup, and analytical choices. Understanding which factors are most important to explaining the difference between immunopeptidomics and conventional shotgun proteomics may require the development of more sensitive proteomic techniques for identifying low-abundance and short-lived microproteins in the cell.
Why do several studies report low-quality spectra despite controlling FDR at 1%? Most of the studies we evaluated control only the proteome-wide FDR instead of controlling FDR for unannotated peptides or proteins specifically (Table 1)17,23,51. Since the proteome-wide FDR does not imply any particular FDR among unannotated proteins17,23, it does not imply high confidence in the unannotated list specifically. In a theoretical example experiment in which 1 million PSMs, 50,000 peptides and 10,000 proteins pass threshold, a 1% FDR corresponds to 10,000 incorrect PSMs, 500 incorrect peptides, or 100 incorrect proteins. If the analysis purports to detect 50 sORFs, the default assumption should be that these are mostly incorrect identifications until very carefully scrutinized. Studies that controlled FDR for unannotated proteins in a class-specific manner, such as Chong et al. 32. and Ouspenskaia et al. 29, scored high in our evaluations. We recommend that studies of the unannotated proteome report local or class-specific unannotated FDRs instead of, or in addition to, whole proteome FDRs, so that confidence in the list of reported unannotated proteins can itself be evaluated. To facilitate future work on the detection of unannotated microproteins by MS-based proteomics, we developed a set of guidelines based on our findings (brief advice in Box 1, detailed guidelines in Appendix 2). The guidelines in Appendix 2 are an extension of the Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.027. It is important to note that false positives can occur across the full range of PSM quality; a low-quality spectrum does not prove that a claimed detection is a false positive; nor is a high-quality spectrum conclusive evidence of detection. The gold standard for rigorous MS-based proteomics data validation requires demonstration that a synthetic peptide generates the observed spectrum and is retained on the liquid chromatography column to the same extent as the originally detected peptide, and that the endogenous spectrum is eliminated when the ORF is disabled genetically. Supporting evidence for the biological significance of a protein with inconclusive MS support can also come from outside proteomics, such as by demonstrating the evolutionary conservation of its amino acid sequence or reporting phenotypic impacts upon genetic perturbations23,45.
The thousands of sORFs identified by Ribo-Seq experiments suggest a massive potential for undiscovered microproteins of biomedical relevance, even at low proteomic validation rates. While our community assessment found relatively low proteomic support for these microproteins in the datasets generated by the pioneering studies we analyzed, this finding should not be interpreted to mean that only few sORF-encoded proteins are present in the cell. There are major technical limitations in the ability of proteomic experiments to find short and low-abundance proteins16,23,25, and the microproteins field is still in its infancy. The extent to which sORFs encode stable functional proteins thus remains an open question. To answer it, we will need to expand the limits of protein detectability through further methodological developments, including but not limited to improving the sensitivity of MS instruments. We hope the dataset of 406 manually curated PSMs generated here will prove useful for benchmarking much-needed new data analysis tools and pipelines for unannotated microprotein detection by MS (Supplementary Data 3).
Methods
Study selection
We conducted a search for all studies published in the 2019-2022 period that attempted to detect unannotated proteins using shotgun proteomics. For each study, we obtained information on the PSMs claimed to support each reported unannotated detection (Supplementary Data 1). For each PSM, we collected the information needed to construct a universal spectrum identifier (USI)52 so the PSM could be visualized. Where possible, we obtained the PSM data from the supplementary information provided with the study; otherwise, we attempted to obtain them from the study authors. The sources of data for each study are given in Supplementary Table 1. The authors of one study (Cai et al.)53 were unable to provide the necessary data so this study was not evaluated.
The set of “unannotated” proteins depends on the annotation database used; the proteins included in our analysis followed the definition used in each study. Unannotated proteoforms of annotated proteins were not included.
ProteoMapper analysis
All reported unannotated peptides were submitted to the ProteoMapper online tool30 in July 2023 using default settings. For each peptide, ProteoMapper returns a list of matches to known or predicted proteins, accounting for neXtProt31 amino acid variants. We determined whether each peptide mapped to a human annotated protein according to the 2023 build of the PeptideAtlas database54 and whether each peptide mapped to a protein present in the 2016 version of UniProtKB/Swiss-Prot18. Any peptide that mapped to a core canonical PeptideAtlas protein on ProteoMapper was not passed on for manual evaluation, even if it differed from the reference sequence by multiple neXtProt amino acid variants.
Manual evaluation of PSM quality
PSMs for each study were evaluated by a group of six expert evaluators. Each evaluator rated a random sample of PSMs from each study. A total of 424 PSMs from 12 studies were given for evaluation, out of which 406 were given ratings, as a few PSMs could not be displayed from the input USI. Out of the 406 PSMs evaluated, 155 were evaluated by two evaluators each to enable determination of the overall consistency between evaluators. Evaluations were done by visual inspection of the PSM using the ProteomeCentral USI web application (https://proteomecentral.proteomexchange.org/usi/) in May to June 2023. The evaluators were told to use no other information except the PSM as displayed on the USI application. A common set of 10 negative control PSMs was given to each evaluator; the evaluators were not informed of the existence of these controls. These negative controls consisted of high-scoring decoy-spectrum matches manually selected from among the strongest 30 decoy-spectrum matches in Duffy et al. 34. Each PSM was rated on a scale of 1-5; the rating scale is given in Appendix 1.
Comparing manual evaluations to spectral prediction machine learning methods
Spectra were predicted for each manually evaluated peptide sequence annotated to the set of experimental spectra using the open-source spectral library prediction pipeline Oktoberfest39. Multiple predicted spectra were generated for each peptide at various collision energies (CE = 25, 30, 35 and 40) and using 4 different intensity models (Prosit 2020 intensity HCD55, Prosit 2020 intensity CID, Prosit 2020 intensity TMT, AlphaPept ms2 generic)55,56,57,58. Only methionine oxidation, cysteine carbamidomethylation, and TMT6plex modifications were considered in the spectral predictions; peptides with other modifications were excluded for this analysis. MSP spectral library files output by Oktoberfest were then converted to MGF formatted spectra. Internal python scripts compared the experimental spectra vs. the predicted spectra by calculating spectral angles (SA) between each spectral pair. Similarity was ranked as being high if SA ≤ 20°, moderate if SA between 20°–45°, poor if SA between 45°–70°, and terrible if SA > 70°. The script further generated mirrored plots for each spectral pair and annotated peptide fragment ions. These spectral angles were then compared to the manual ratings for each PSM given by the evaluators.
Predicting HLA binding for immunopeptides supporting unannotated protein detections
For each evaluated immunopeptide from Ouspenskaia et al. 2021, Martinez et al. 42, or Chong et al. 32 used to support an unannotated protein detection, the HLA alleles for the cell type used in the experiment producing the peptide was found in the supplemental data of the study. NetMHC 4.0 was then used to predict binding of the peptide to the HLA-A, HLA-B, and HLA-C allele if the allele was available in NetMHC 4.0. A peptide was classified as being HLA-binding if it met the default criteria for being a weak (% rank <2%) or strong (% rank < 0.5%) binder in NetMHC 4.0.
Relating ORF properties to the probability of detection
The coordinates of each ORF with an evaluated peptide were taken from the supplementary data of each study and the ORF length determined. All ORF coordinates were converted to hg38 coordinates using LiftOver. ORFs from Chen et al. 35, Chong et al. 32., Cao et al. 44., and Lu et al. 59. were not considered because we were not able to identify the ORF coordinates from supplementary data files. To assess translation levels, we aggregated Ribo-Seq data from 109 studies (Supplementary Data 5-6) using the following procedure. Transcriptomes from MiTranscriptome60, FANTOM5 robust set60, CHESS61, RNA Atlas62, and Ensembl version 108 were merged using Stringtie63 version 2.2.1with Ensembl version 108 as the reference annotation (-G parameter). MiTranscriptome and FANTOM5 coordinates were lifted over from hg19 to hg38 prior to merging. Adapters in each ribo-seq run were removed with TrimGalore version 0.6.7 using default options. Trimmed Ribo-seq reads were then mapped to the merged transcriptome using STAR64,65 version STAR-2.7.10b using the parameters--outSAMtype BAM Unsorted --outFilterMismatchNmax 2 --outFilterMultimapNmax 1 --outSAMattributes Standard. The iRibo program46 was then used to aggregate the mapped reads from all studies and assign counts of ribosome P-sites to each position of each analyzed ORF.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data analyzed are available in a Figshare database (https://doi.org/10.6084/m9.figshare.30131869.v1). Source data are provided with this paper.
Code availability
All code required to reproduce the figures and data for analyses are available at: https://doi.org/10.6084/m9.figshare.30131869.v1.
References
Wright, B. W., Yi, Z., Weissman, J. S. & Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol. 32, 243–258 (2022).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Ingolia, N. T. et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 8, 1365–1379 (2014).
Chothani, S. P. et al. A high-resolution map of human RNA translation. Mol. Cell 82, 2885–2899.e8 (2022).
Wacholder, A. et al. A vast evolutionarily transient translatome contributes to phenotype and fitness. Cell Syst. 14, 363–381.e8 (2023).
van Heesch, S. et al. The translational landscape of the human heart. Cell 178, 242–260.e29 (2019).
Anderson, D. M. et al. A micropeptide encoded by a putative long noncoding rna regulates muscle performance. Cell 4, 595–606 (2015).
Jackson, R. et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 564, 434–438 (2018).
Brown, A. et al. Structures of the human mitochondrial ribosome in native states of assembly. Nat. Struct. Mol. Biol. 24, 866–869 (2017).
Andreev, D. E. et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife 4, e03971 (2015).
Merino-Valverde, I., Greco, E. & Abad, M. The microproteome of cancer: from invisibility to relevance. Exp. Cell Res. 392, 111997 (2020).
Hemm, M. R., Weaver, J. & Storz, G. Escherichia coli small proteome. EcoSal Plus 9, https://doi.org/10.1128/ecosalplus.ESP-0031-2019 (2020).
Mudge, J. M. et al. Standardized annotation of translated open reading frames. Nat. Biotechnol. 40, 994–999 (2022).
Kesner, J. S. et al. Noncoding translation mitigation. Nature 617, 395–402 (2023).
Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Cañas, J. L., Messeguer, X. & Albà, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nat. Ecol. Evol. 2, 890–896 (2018).
Prensner, J. R. et al. What can Ribo-seq, immunopeptidomics, and proteomics tell us about the non-canonical proteome? Mol. Cell. Proteomics 22,100631 (2023).
Nesvizhskii, A. I. Proteogenomics: concepts, applications and computational strategies. Nat. Methods 11, 1114–1125 (2014).
The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Chong, C., Coukos, G. & Bassani-Sternberg, M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol. 40, 175–188 (2022).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for mass spectrometry-based proteomics. in Proteome Bioinformatics (eds. Hubbard, S. J. & Jones, A. R.) 55–71. https://doi.org/10.1007/978-1-60761-444-9_5(Humana Press, 2010).
Aggarwal, S. & Yadav, A. K. False discovery rate estimation in proteomics. in Statistical Analysis in Proteomics (ed. Jung, K.) 119–128. https://doi.org/10.1007/978-1-4939-3106-4_7 (Springer, 2016).
Zhang, B. & Bassani-Sternberg, M. Current perspectives on mass spectrometry-based immunopeptidomics: the computational angle to tumor antigen discovery—PMC. J. Immunother. Cancer https://pmc.ncbi.nlm.nih.gov/articles/PMC10619091/(2023).
Wacholder, A. & Carvunis, A.-R. Biological factors and statistical limitations prevent detection of most noncanonical proteins by mass spectrometry. PLoS Biol. 21, e3002409 (2023).
Fijalkowski, I., Willems, P., Jonckheere, V., Simoens, L. & Van Damme, P. Hidden in plain sight: challenges in proteomics detection of small ORF-encoded polypeptides. microLife 3, uqac005 (2022).
Ahrens, C. H., Wade, J. T., Champion, M. M. & Langer, J. D. A practical guide to small protein discovery and characterization using mass spectrometry. J. Bacteriol. 204, e00353–21 (2022).
Makarewich, C. A. The hidden world of membrane microproteins. Exp. Cell Res. 388, 111853 (2020).
Deutsch, E. W. et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J. Proteome Res. 18, 4108–4116 (2019).
Bogaert, A. et al. Limited evidence for protein products of noncoding transcripts in the HEK293T cellular cytosol. Mol. Cell. Proteomics MCP 21, 100264 (2022).
Ouspenskaia, T. et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 40, 209–217 (2022).
Mendoza, L. et al. Flexible and fast mapping of peptides to a proteome with proteoMapper. J. Proteome Res. 17, 4337–4344 (2018).
Zahn-Zabal, M. et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res. 48, D328–D334 (2020).
Chong, C. et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 11, 1293 (2020).
Prensner, J. R. et al. Noncanonical open reading frames encode functional proteins essential for cancer cell survival. Nat. Biotechnol. 39, 697–704 (2021).
Duffy, E. E. et al. Developmental dynamics of RNA translation in the human brain. Nat. Neurosci. 25, 1353–1365 (2022).
Chen, J. et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020).
Schaeffer, M. et al. The neXtProt peptide uniqueness checker: a tool for the proteomics community. Bioinformatics 33, 3471–3472 (2017).
Degroeve, S. & Martens, L. MS2PIP: a tool for MS/MS peak intensity prediction. Bioinformatics 29, 3199–3203 (2013).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
Picciani, M. et al. Oktoberfest: open-source spectral library generation and rescoring pipeline based on Prosit. Proteomics 24, 2300112 (2024).
Douka, K. et al. Cytoplasmic long noncoding RNAs are differentially regulated and translated during human neuronal differentiation. RNA 27, 1082–1101 (2021).
Sticker, A., Martens, L. & Clement, L. Mass spectrometrists should search for all peptides, but assess only the ones they care about. Nat. Methods 14, 643–644 (2017).
Martinez, T. F. et al. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 16, 458–468 (2020).
Cao, X. et al. Comparative proteomic profiling of unannotated microproteins and alternative proteins in human cell lines. J. Proteome Res. 19, 3418–3426 (2020).
Cao, X. et al. Nascent alt-protein chemoproteomics reveals a pre-60S assembly checkpoint inhibitor. Nat. Chem. Biol. 18, 643–651 (2022).
Deutsch, E. W. et al. High-quality peptide evidence for annotating non-canonical open reading frames as human proteins. 2024.09.09.612016 Preprint at. https://doi.org/10.1101/2024.09.09.612016 (2024).
Turcan, A., Lee, J., Wacholder, A. & Carvunis, A.-R. Integrative detection of genome-wide translation using iRibo. STAR Protoc 5, 102826 (2024).
Ning, K., Fermin, D. & Nesvizhskii, A. I. Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-seq gene expression data. J. Proteome Res. 11, 2261–2271 (2012).
Hofman, D. A., Prensner, J. R. & Heesch, S. van. Microproteins in cancer: identification, biological functions, and clinical implications. Trends Genet. 41, 146–161 (2024).
Smith, C. et al. Pervasive translation in Mycobacterium tuberculosis. eLife 11, e73980 (2022).
Cuevas, M. V. R. et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 34, 108815 (2021).
Deutsch, E. W. et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 15, 3961–3970 (2016).
Deutsch, E. W. et al. Universal spectrum identifier for mass spectra. Nat. Methods 18, 768–770 (2021).
Cai, T. et al. LncRNA-encoded microproteins: a new form of cargo in cell culture-derived and circulating extracellular vesicles. J. Extracell. Vesicles 10, e12123 (2021).
Desiere, F. et al. The PeptideAtlas project. Nucleic Acids Res. 34, D655–D658 (2006).
Gessulat, S. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518 (2019).
Wilhelm, M. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 12, 3346 (2021).
Gabriel, W. et al. Prosit-TMT: deep learning boosts identification of TMT-labeled peptides. Anal. Chem. 94, 7181–7190 (2022).
Zeng, W.-F. et al. AlphaPeptDeep: a modular deep learning framework to predict peptide properties for proteomics. Nat. Commun. 13, 7238 (2022).
Lu, S. et al. A hidden human proteome encoded by ‘non-coding’ genes. Nucleic Acids Res. 47, 8111–8125 (2019).
Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015).
Pertea, M. et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 19, 208 (2018).
Lorenzi, L. et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 39, 1453–1465 (2021).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Kim, M.-S. et al. A draft map of the human proteome. Nature 509, 575–581 (2014).
Slany, A. et al. Contribution of human fibroblasts and endothelial cells to the hallmarks of inflammation as determined by proteome profiling. Mol. Cell. Proteomics 15, 1982–1997 (2016).
Shekari, F. et al. Proteome analysis of human embryonic stem cells organelles. J. Proteomics 162, 108–118 (2017).
Doll, S. et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 8, 1469 (2017).
Murillo, J. R. et al. Mass spectrometry evaluation of a neuroblastoma SH-SY5Y cell culture protocol. Anal. Biochem. 559, 51–54 (2018).
Brenig, K. et al. The Proteomic landscape of cysteine oxidation that underpins retinoic acid-induced neuronal differentiation. J. Proteome Res. 19, 1923–1940 (2020).
Sarkizova, S. et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 38, 199–209 (2020).
Shraibman, B. et al. Identification of tumor antigens among the HLA peptidomes of glioblastoma tumors and plasma. Mol. Cell. Proteomics MCP 18, 1255–1268 (2019).
Wen, B., Wang, X. & Zhang, B. PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res. 29, 485–493 (2019).
Declercq, A. et al. MS2Rescore: data-driven rescoring dramatically boosts immunopeptide identification rates. Mol. Cell. Proteomics 21, 100266 (2022).
Acknowledgements
This work was supported in part by a Research Grant from HSFP awarded to A.R-C: https://doi.org/10.52044/HFSP.RGP0042023.pc.gr.168590. M.B.-S. is supported by the Ludwig Institute for Cancer Research, by grants KFS-4680-02-2019 and KFS-5637-08-2022 from the Swiss Cancer Research Foundation (M.B.-S.), the Swiss National Science Foundation PRIMA grant PR00P3_193079 (M.B.-S.) and the Swiss Bridge Foundation Award (M.B.S). J.A.V. is supported by funding from Wellcome [grant number 223745/Z/21/Z], and from EMBL core funding. J.S.C. acknowledges funding from the Wellcome Trust [223745/Z/21/Z] and from the ICR core funding. M.A.B. is supported by a Junior 1 career award from the Fonds de Recherche du Quebec - Sante (FRQS). F.B. is supported by a FRQS scholarship. F.A.T. is supported by a FRQS scholarship. I.A. is supported by a FRQS scholarship. X.R. is supported by the Canadian Institutes for Health Research (CIHR) (Grant No. PJT-175322), and Canada Research Chair in Functional Proteomics and Discovery of Novel Proteins. J.M.M. is supported by the Wellcome Trust (108749/Z/15/Z), the National Human Genome Research Institute (NHGRI) of the U.S. National Institutes of Health (NIH) under award number (2U41HG007234), and the European Molecular Biology Laboratory (EMBL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Ensembl is a registered trademark of EMBL. K.J. is supported in part by a NIH Chemical Biology training grant (T32 GM149444). J.R.P. acknowledges funding from the National Institutes of Health / National Cancer Institute [K08-CA263552-01A1]; the V Foundation for Cancer Research [V2024-013]; Hyundai Hope on Wheels Foundation; the Yuvaan Tiwari Foundation; DIPG/DMG Research Funding Alliance; Tough2gether Foundation; CureSearch Foundation; Morgan Adams Foundation; ChadTough Defeat DIPG Foundation; Book for Hope Foundation; Curing Kids Cancer Foundation [20-3388093], and the Andrew McDonough B+ Foundation [1185689]. J.R.P. is the Ben and Catherine Ivy Foundation Clinical Investigator of the Damon Runyon Cancer Research Foundation [CI-127-24]. S.A.S. is supported by the Paul G. Allen Frontiers Group Distinguished Investigator Award. This work was funded in part by the National Institutes of Health grants R24 GM148372 (E.W.D.), R01 GM087221 (E.W.D., R.L.M.), S10 OD026936 (R.L.M.), and by National Science Foundation grants DBI-2324882 (E.W.D.) DBI-1933311 (E.W.D.), and MRI-1920268 (R.L.M.). N.H. was supported by a grant from the Leducq Foundation, an ERC Advanced Grant under the European Union Horizon 2020 Research and Innovation Program (AdG788970), a British Heart Foundation and a Deutsches Zentrum für Herz-Kreislauf-Forschung grant (BHF/DZHK: SP/19/1/34461), by German Research Foundation - DFG (CRC/SFB-1470 – B03), and in part by a grant from the Chan Zuckerberg Foundation (2019-202666). J.C.W. acknowledges the support of The Institute of Cancer Research and funding from Wellcome [grant numbers 208391/Z/17/Z, 223745/Z/21/Z]. S.L. is supported by Canadian Institutes for Health Research (CIHR) (Grant No. PJT-175322), and Canada Research Chair in Functional Proteomics and Discovery of Novel Proteins. P.V.B. is supported by Taighde Éireann – Research Ireland under Grant number [20/FFP-A/8929]. K.G. was supported by The Research Foundation—Flanders (FWO), project number G008018N. S.v.H. acknowledges funding from Fonds Cancers (FOCA, Belgium), Stichting Reggeborgh (the Netherlands), and Villa Joep. This publication is part of the project “Evolutionarily young microproteins in childhood brain cancer” (with project number VI.Vidi.223.022 of the research programme NWO talent programme Vidi, which is (partly) financed by the Dutch Research Council (NWO), awarded to S.v.H. Research reported in this publication was supported by Oncode Accelerator, a Dutch National Growth Fund project under grant number NGFOP2201, awarded to S.v.H. I.F-M. financial support was received from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 945405 (ARISE programme). S.C. is funded by Singapore Ministry of Health’s National Medical Research Council under OF-YIRG (OFYIRG23jan-0034). This work was supported in part by NIH/NIGMS grant R35GM157126 awarded to T.F.M. We are grateful for helpful feedback from Aviv Regev, Travis Law, Tamara Ouspenskaia, Karl Clauser, Susan Klaeger, Catherine J. Wu, Owen Rackham, Gong Zhang, Michelle Magrane, Erin Duffy, Brian Kalish, and Michael E. Greenberg.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.W., E.W.D., S.v.H., J.R.P., T.F.M., M.A.B., J.S., J.R-O., J.M.M., S.A.S., A-R.C. Methodology: A.W., A-R.C., E.W.D. Formal analysis: A.W., J.L., S.L., J.C.W., L.W.K., J.T.v.D Investigation: A.W., E.W.D., J.R.P., T.F.M., M.A.B., J.R-O., J.M.M., S.A.S. Resources: E.W.D. Data Curation: I.A., F.B., K.C., A.H.J., K.J., F-A.T., E.W.D. Writing - Original Draft: A.W. Writing - Review & Editing: S.v.H., L.W.K., J.T.v.D., I.F-M., E.W.D., M.B-S., S.C., J.A.V., J.S.C., M.A.B., X.R., J.M.M., J.R.P., P.V.B., J.R-O., N.H., S.A.S., T.F.M., A.B., D.F., K.G., R.L.M., A-R.C. Visualization: A.W. Project administration: A.W. and A-R.C. Supervision: A-R.C.
Corresponding author
Ethics declarations
Competing interests
J.R.P. has received research honoraria from Novartis Biosciences and Quantum-Si, and is a paid consultant for ProFound Therapeutics. P.V.B. is a cofounder and shareholder of EIRNA Bio. A.-R.C. is a member of the scientific advisory board for Flagship Labs 69, Inc. (ProFound Therapeutics). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wacholder, A., Deutsch, E.W., Kok, L.W. et al. Community benchmarking and evaluation of human unannotated microprotein detection by mass spectrometry based proteomics. Nat Commun 17, 1241 (2026). https://doi.org/10.1038/s41467-025-68002-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68002-x
