
Abstract
The preS1 domain present on hepatitis B and D virus particles is an intrinsically disordered peptide (IDP) essential for recognizing the host receptor, sodium taurocholate co-transporting polypeptide (NTCP). However, it remains poorly understood how the disordered preS1 peptide achieves high-affinity binding through its conformational change. Here, we reveal a stepwise binding process through which multiple regions within preS1 cooperate to establish the stable binding to NTCP. By combining structure-based molecular simulations with virological assays, we show that Asn9preS1, Gly12preS1, and His17preS1 primarily mediate the NTCP binding by forming a tandem loop core structure that docks into the bile acid tunnel of NTCP, creating an extensive binding interface. Another interaction via Trp41preS1 stabilizes the flexible preS1 33–48 aa stretch embedded onto the NTCP outerface, particularly involving Asn87NTCP and Tyr146NTCP. Mutation in these key residues abolishes virion infectivity in cell culture and mouse models, demonstrating the biological significance of this binding maturation. These findings propose a multistep mechanism for viral IDP-mediated receptor recognition that ensures high virion infectivity and strict host specificity.
Similar content being viewed by others


Introduction
Hepatitis B and D viruses (HBV, HDV) exhibit a narrow host range, specifically infecting hepatocytes of limited animal species, including humans and chimpanzees1,2,3,4. This restricted tissue and species tropism is largely attributed to the highly specific viral entry process, which is initiated by high-affinity interaction of the viral surface protein to host cell receptor5,6,7,8. The infectious HBV virion displays three surface proteins, small, middle, and large surface proteins (SHBs, MHBs, LHBs). HDV, a satellite virus of HBV, relies on HBV-derived surface proteins for virion production and infectivity. LHBs, particularly its N-terminal region known as the preS1 region, plays an essential role in binding to the host receptor9,10. Specifically, the N-terminally myristoylated 2–48 aa of preS1 (myr-2–48preS1), located at the outermost part of the virions, is responsible for binding to the host receptor, sodium taurocholate co-transporting polypeptide (NTCP)11,12,13. NTCP is a bile acid transporter specifically expressed in hepatocytes7,8. The preS1-NTCP interaction is crucial in determining key virological features of HBV/HDV, including virion infectivity, transmission efficiency, pathogenesis, and host tropism. The preS1 region is structurally an intrinsically disordered peptide (IDP) with conformational flexibility, distinguishing it from surface proteins of many other viruses (env, spike, hemagglutinin, and capsid, etc.), which require stably structured proteins to mediate receptor binding14,15,16. Despite its fundamental importance, the mechanism by which the intrinsically disordered preS1 peptide establishes a high-affinity interaction with NTCP remains unclear. Elucidating this mechanism could provide valuable insights into the unique infection process of HBV and HDV, potentially crucial for the development of novel therapeutic and preventive strategies.
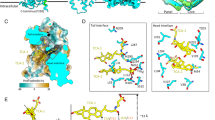
We and other groups have solved the cryo-electron microscopy (cryo-EM) structure of NTCP17,18,19,20. NTCP is a nine transmembrane (TM) protein with two functional domains, the panel domain (consisting of TM1, 5, and 6) and the core domain (consisting of TM2, 3, 4, 7, 8, and 9), with forming a bile acid tunnel between the two domains. Recently, we and another group solved the cryo-EM structure of myr-2–48preS1 in complex with NTCP21,22. PreS1 adopts a lasso-like structure, intricately folded and producing four loops in its N-terminal 31 amino acids (myr-2–32preS1), which is densely packed in the bile acid tunnel of NTCP (Fig. 1a, b). The C-terminal stretch (33–48preS1) appears in contact with the extracellular surface of NTCP. This preS1 structure creates numerous contact sites with a large interface area with NTCP to achieve a high-affinity interaction. However, the process involved in forming this intricate conformation remains enigmatic.

a Schematic representation of the structure of preS1-NTCP complex. NTCP is shown by cylinder (left) and surface representation (right). N-terminally myristoylated preS1 (shown in ribbon model) of the 2–19 aa (myr-2–19preS1, salmon) and 20–32 aa (20–32preS1, pink) buried in the bile acid tunnel of NTCP, followed by the 33–48 aa stretch (33–48preS1, light pink), contacting to the extracellular surface of NTCP. b Close-up view of the bile acid tunnel. Myr-2–19preS1 and 20–32preS1 form four loops V7–G12 (loop 1), F13–Q18 (loop 2), L19–K24 (loop 3), and A25–P30 (loop 4). c NTCP binding activity of each preS1 region. HepG2-NTCP or HepG2 cells were treated with TAMRA-labeled preS1 peptide consisting of the indicated regions (myr-2–48preS1, myr-2–19preS1, 20–32preS1, 33–48preS1, 2–48preS1, or 2–19preS1) at the indicated concentrations (40, 100, 250, and 625 nM) for 4 °C for 30 min. Cell-bound TAMRA fluorescence (red) as well as the nuclei (blue) were detected using fluorescence microscopy. Myrcludex-B (myr-B), an HBV-cell binding inhibitor, was used to show the background signal levels. Fluorescence intensities of the red signal were quantified and are shown as the percentages relative to the signals for myr-2–48preS1 for each concentration (right graph). Scale bar, 100 μm. d NTCP binding assay of the wild type and the alanine mutants at the 7–19 preS1 residues of myr-2–48preS1. Red and blue signals indicate the cell-bound TAMRA fluorescence and the nuclei, respectively. The right graph shows the percentages of the TAMRA fluorescence intensity relative to the wild type of myr-2–48preS1 for each concentration tested. e Competition capacity of the indicated myr-2–48preS1 peptide in HDV infection assay. HepG2-NTCP cells were inoculated with HDV in the presence of the indicated preS1 peptide at the indicated concentrations (1, 5, 25, 125, and 625 nM). Intracellular HDV RNA levels were quantified by real-time RT-PCR at six days post-inoculation. The graph shows the percentages of single inhibition compared to the signal without a peptide. 50% inhibitory concentrations (IC50s) are shown at the bottom of the graph. Bars showed the means of three independent experiments (n = 3) and error bars represent standard deviation (SD). Source data are provided as a Source Data file. myr-B, myrcludex-B.
In this study, by combining virological assays with cryo-EM structure-based molecular simulation and statistical mechanical analyses, we identify multiple preS1 amino acids in distinct regions that formed substructures crucial for tight binding to NTCP. Our analysis suggests that the N-terminal myristoylated 2–19 aa region, especially Asn9preS1, Gly12preS1, and His17preS1, forms the core structure that produces the tandem loop, fitting in the bile acid tunnel for de novo NTCP binding. PreS1 undergoes another interaction involving Trp41preS1 to the extracellular surface loops of NTCP, restraining the 33–48 aa region for dynamic fluctuation and stabilizing the tight preS1-NTCP binding. This multistep binding maturation process, driven by an intrinsically disordered preS1 peptide, would secure the high-affinity receptor interaction.
Results
The myristoylated 2–19 aa region of preS1 primarily mediates NTCP binding
The solved structure of the myr-preS1(2–48)-NTCP complex reveals that the 2–32 aa region of preS1 forms a lasso-like structure with four loops, which is buried in the bile acid tunnel of NTCP, whereas the following 33–48 aa region crawls along with the extracellular surface of NTCP (Fig. 1a)21. The myristoyl group at the N-terminus is anchored to the lipids in the outer leaflet of the membrane22. PreS1 contacts both the bile acid tunnel of NTCP and its extracellular surface, while forming multiple intramolecular interactions that create a four-loop structure (V7–G12 for loop 1, F13–Q18 for loop 2, L19–K24 for loop 3, and A25–P30 for loop 4) (Fig. 1b). The 2–19 aa region, comprising loops 1 and 2, is completely buried in the tunnel between TM1, 5, and 8b, whereas the 20–32 aa residues, forming loops 3 and 4, occupy a more outer area of the tunnel along with TM1 (Fig. 1b).
To examine which region in myr-preS1(2–48) serves for initiating the NTCP binding, we divided it into three segments, myr-2–19 aa, 20–32 aa and 33–48 aa to evaluate their binding capacity to NTCP-overexpressing cells. HepG2-NTCP cells were treated with TAMRA fluorescence labeled-preS1 peptides consisting of these regions (myr-2–19preS1, 20–32preS1 and 33–48preS1) as well as myr-2–48 aa (myr-2–48preS1) at varying concentrations (40–625 nM) for 30 min, followed by washing to detect cell-bound TAMRA fluorescence as well as nuclear staining with DAPI (Fig. 1c)23,24. The TAMRA intensities were quantified and presented as relative values to that for myr-2–48preS1 in the right graph. As a positive control, myr-2–48preS1 showed fluorescence binding to NTCP-expressing cells, which was completely abolished by treatment with an HBV attachment inhibitor, myrcludex-B (myr-B) (Fig. 1c, panels 1–5)25. These signals were not observed in HepG2 cells lacking NTCP expression at any tested concentrations (Fig. 1c, panels 29–32), conforming the specificity of the observed fluorescence signals for NTCP binding. The myr-2–19preS1 peptide exhibited cell-bound fluorescence in a concentration-dependent manner, although the signals were lower than those observed with myr-2–48preS1 especially at low preS1 peptide concentrations (Fig. 1c, panels 6–10). In contrast, neither 20–32preS1 nor 33–48preS1 showed any cell-binding signals at any concentrations (Fig. 1c, panels 11–20). Moreover, non-myristoylated 2–19preS1 and 2–48preS1 failed to produce cell-bound fluorescence signals (Fig. 1c, panels 21–28), indicating that 2–19preS1-mediated NTCP binding requires the N-terminal myristoyl group.
These data were supported by peptide competition assays. HepG2-NTCP cells were treated with TAMRA labeled-myr-2–48preS1 peptide together with non-labeled preS1 peptides (myr-2–19preS1, 20–32preS1, 33–48preS1, or myr-2–48preS1) at varying concentrations (40–625 nM) as competitors (Supplementary Fig. 1a). The myr-2–19preS1 peptide inhibited the cell binding of myr-2–48preS1 in a concentration-dependent manner (Supplementary Fig. 1a, panels f–i), whereas 20–32preS1and 33–48preS1 had no inhibitory effect at any concentrations (Supplementary Fig. 1a, panels j–q). Consistently, in the peptide competition in the HDV infection assay, myr-2–19preS1, but not 20–32preS1 or 33–48preS1, competed with HDV infection to HepG2-NTCP cells in a dose-dependent manner (Supplementary Fig. 1b). The 50% inhibitory concentration (IC50) for myr-2–19preS1 was 56.2 nM, which was 15-fold lower potency compared to that for myr-2–48preS1 (IC50 = 3.8 nM) (Supplementary Fig. 1b). These results suggest that the myristoylated 2–19 aa region of preS1 mediates the primary NTCP binding, with the help of the N-terminal myristoyl group. In contrast, C-terminal regions (20–48 aa) lack the binding ability to NTCP by itself, although the 33–48preS1 region was later shown to enhance the binding capacity of preS1 that is already bound to NTCP.
Asn9, Gly12, and His17 in preS1 played a major role in NTCP binding
Within the 2–19 aa region of preS1, amino acids after position 7 enter deeply into the bile acid tunnel of NTCP (Fig. 1b). To elucidate the specific amino acids within the 7–19 aa region having a major contribution to NTCP binding, we introduced amino acid substitutions in the preS1 peptide. We synthesized TAMRA-labeled myr-2–48preS1 peptides, each carrying an alanine substitution at individual positions within the 7–19 region and examined their NTCP binding capacity using the method described in Fig. 1c. Among these modified myr-2–48preS1 peptide, substitutions at positions 9, 12, and 17 almost completely abolished NTCP binding, even at the highest concentration of TAMRA-preS1 treatment (Fig. 1d, N9A, G12A, and H17A). Substitutions at F13 and Q18 significantly but moderately reduced NTCP binding, whereas other substitutions had no or a lesser effect.
Peptide competition assays also showed that myr-2–48preS1 peptides with alanine residue at position 9, 12 or 17 exhibited lower ability to compete with wild type preS1 for binding to NTCP (Supplementary Fig. 1c). Consistently, these modified myr-2–48preS1 peptides showed 24–39 fold lower potency in inhibiting HDV infection compared to the wild type peptide (IC50 = 122.5 nM for N9A, 172.0 nM for G12A, 193.9 nM for H17A v.s. 5.1 nM for the wild type) (Fig. 1e). These data collectively suggest that N9, G12, and H17 in preS1 play a crucial role in establishing the efficient NTCP binding. Notably, these residues are positioned at the N-terminus (N9), center (G12), and C-terminus (H17) of the region constituting the loops 1 (V7–G12) and 2 (F13–Q18). They closely contact each other (within 3.5 Å) to form a “core” structure to bundle the tandem loops (loops 1 and 2) that create many contact sites to NTCP amino acids in the bile acid tunnel (Fig. 2a).

a Left, the core structure in preS1 involving N9preS1 (yellow), G12preS1 (red), and H17preS1 (pink) located in the bile acid tunnel of NTCP (green) by cryo-EM. Middle, schematic representation for the core structure (pink) between TM1 and TM8 of NTCP (green). Right, Close-up structure views of the binding interface for N9preS1, G12preS1, and H17preS1. NTCP (green, surface model) and preS1 (pink, ribbon model) are shown. Yellow highlights the side chain of N9preS1, G12preS1, and H17preS1. Dark green in NTCP indicates the amino acids within 4.5 Å from preS1, including L27, S28, L31, V32, L35, V263, and Q264. b PreS1 binding capacity of the wild type and mutant NTCP. HepG2 cells expressing the wild type and mutant NTCP were incubated with TAMRA-labeled myr-2–48preS1 peptide at the indicated concentration (2.6, 6.4, 16, and 40 nM). Quantified TAMRA intensities are shown in the right graph. c Virus infection susceptibility of the wild type and mutant NTCP. HDV infection was evaluated using HepG2 cells overexpressing the wild type or the indicated mutants of NTCP by quantifying HDV RNA in the cells at six days post-inoculation of HDV. d Expression levels of the wild type and mutant NTCP. NTCP in the cell surface (upper) and total cell lysate (middle) as well as actin in total cell lysate as an internal control (lower) were detected by immunoblotting as shown in Methods. The gel data are shown from one representative experiment. Uncropped gels are shown in the Source data. e Transporter activity. HepG2 cells expressing the wild type and mutant NTCP were incubated with [3H]-taurocholic acid as a substrate either in the presence (gray) or absence (white) of sodium. Intracellular radioactivity was measured at 15 min after incubation. Bars showed the means of three independent experiments (n = 3), and error bars represent standard deviation (SD). Source data are provided as a Source Data file. WT, wild type; myr-B, myrcludex-B.
Leu31 and Gln264 in NTCP are essential counterparts of the preS1 core, supporting preS1 binding
To identify the counterpart amino acids in the NTCP side responsible for the preS1 core recognition, we comprehensively selected the amino acid residues in NTCP that are located within a 4.5 Å distance from either N9preS1, G12preS1, or H17preS1 (Supplementary Table 1), and identified L27, S28, L31, V32, and L35 in TM1, and V263 and Q264 in TM8b of NTCP as potential candidates (Fig. 2a). We examined the role of these amino acids in supporting preS1 binding by mutation analysis: The wild type NTCP or variants carrying alanine substitutions at each candidate position were overexpressed in HepG2 cells, which were then subjected to a preS1 binding assay, HDV infection assay, bile acid transport assay, and cell surface protein expression analysis, as described previously24. Whereas the cells expressing L27A, S28A, V32A, L35A, and V263A mutants exhibited preS1 binding levels comparable to those expressing the wild type NTCP (Fig. 2b, panels 5–12, 17–28), L31A and Q264A mutants demonstrated a drastic reduction in the capacity to support preS1 binding, especially at low concentration ranges of preS1 peptide (Fig. 2b, panels 13–16, 29–32). Consistently, HDV infection assay revealed a reduced susceptibility to infection of cells overexpressing L31A and Q264A NTCP mutants (Fig. 2c). In these experiments, cell surface expression levels of NTCP variants, including L31A and Q264A, were variable but were not markedly impaired compared to the wild type NTCP (Fig. 2d). In bile acid transport assay, L31A and Q264A mutants showed reduced (48–53%) but apparently retained the activity for Na+-dependent taurocholic acid uptake (Fig. 2e), indicating that the abolished preS1 binding and HDV infection in L31ANTCP- and Q264ANTCP-producing cells were not due to the lack of cell surface NTCP expression or loss of general protein functions by deleterious mutations, but rather to specific functional defects as a viral receptor. Structural information indicates that L31 in TM1 and Q264 in TM8b are the only amino acids in NTCP that are within 4.5 Å distance to all three amino acids, N9preS1, G12preS1, and H17preS1 (Supplementary Table 1). Thus, the preS1 core is tightly clamped by L31 on the TM1 side and Q264 on the TM8b side of NTCP, highlighting the critical nature of these residues in inducing and/or stabilizing the preS1 core structure that generates large NTCP contact sites in the loops 1 and 2.
Molecular dynamics simulation showing the role of the preS1 core in forming the stable complex with NTCP
To investigate how N9preS1, G12preS1, and H17preS1 contribute to NTCP binding, we performed all-atom molecular dynamics (MD) simulations: Three independent 1 μs MD simulations were performed embedded in a membrane-water system (see Methods in detail) for NTCP-complexed 2–19preS1 of the wild type (WT), the N9A, G12A, and H17A mutants, and the L11A and F14A mutants as negative controls (Supplementary Fig. 2a). Structures of 2–19preS1 during the MD simulations were analyzed by a principal component analysis (PCA), in which snapshot structures of 2–19preS1 during MD simulations were projected onto the pc1-pc2 surface (pc1 and pc2 correspond to elongation and twisting motions, respectively) as a frequency heat map (Fig. 3a). The structural distribution of WTpreS1(2–19) showed a single elongated basin formed by two connected basins, one near the initial cryo-EM structure (black square) and another (denoted as WT_rep) at the positive region in pc1 appeared the most frequently (Fig. 3a, WT). In contrast, the structural distributions of the N9A, G12A, and H17A mutants showed different profiles: N9A and G12A mutants distributed in a broader area with splitting at three basins (N9A_repA, N9A_repB, N9A_repC, and G12A_repA, G12A_repB, G12A_repC), two of which appeared far from the cryo-EM structure or WT_rep (Fig. 3a, N9A and G12A). H17A mutant showed one basin (H17A_repA), which appeared, however, far from the cryo-EM structure and outside the area that WT covered (WT_area, shown by green) (Fig. 3a, H17A). In contrast, L11A and F14A mutants, which retained the NTCP binding capacity, showed apparently stable distributions close to the initial cryo-EM structure and quite a similar distribution pattern to WT (Fig. 3a, L11A and F14A). These results indicate that the loss of binding to NTCP is related with less stable retention of the binding pose for the complex with NTCP, compared with WT.

a PCA analysis showing the structural distributions of myr-2–19preS1 (WTpreS1) and its mutants carrying the N9A, G12A, H17A, L11A, and F14A substitutions during MD simulations. Frequencies are shown by color (Brown to white, high to low). The initial cryo-EM structure is shown by black dots. The area covering the whole distribution for WTpreS1 is highlighted by a green box (WT_area). The points showing the most frequent distribution are shown by x (_rep or _repA). WT_rep is shown by a black triangle in the mutant panels. At lower right, PC vectors for pc1 (magenta, elongation) and pc2 (blue, twisting) on the structure after an energy minimization step of NTCP (green as ribbons) and 2–19preS1 (pink as cartoon) are shown. b, c Frequency of the distances between the Cα atoms for position 9preS1 and position 12preS1 (left), those for position 12preS1 and position 17preS1 (middle), and those for position 9preS1 and position 17preS1 (right), in the MD simulations for myr-2–19preS1 WT (salmon) and its mutants, N9A (yellow), G12A (red), H17A (magenta), L11A (light orange), and F14A (purple). The distances of the initial cryo-EM structure are shown by black vertical lines. d Contact frequency of L31NTCP with either 9preS1 (yellow), 12preS1 (red), or 17preS1 (magenta) (left) and that of Q264NTCP with either 9preS1, 12preS1, or 17preS1 (right) in the MD simulations using myr-2–19preS1 (lanes 1–3, 13–15) or its N9ApreS1 (lanes 4–6, 16–18), G12ApreS1 (lanes 7–9, 19–21), or H17ApreS1 (lanes 10–12, 22–24) mutants. e Schematic representation for the impact of alanine mutations at N9preS1, G12preS1, and H17preS1 on the intra- and inter-molecular contacts for the preS1 core structure formation.
To evaluate the local structure for maintaining the core (N9preS1-G12preS1-H17preS1 structure), we quantified Cα atoms for the three distances, position 9preS1-position 12preS1, position 12preS1-position 17preS1, and position 9preS1-posirtion 17preS1 (Fig. 3b, c and Supplementary Fig. 2a). All the three distances for WT were maintained near their initial values, ranging stably within 4–6, 4–7, and 5–7 Å (peaked at 5, 5, and 6 Å), respectively (Fig. 3b, c, salmon); whereas these distances were more diverse and larger, especially for the position 12preS1-position 17preS1 (ranging 4–11 Å, peaked at 6, 6, and 8 Å for N9A, G12A, and H17A, respectively) and position 9preS1-position 17preS1 (ranging 5–12 Å, peaked at 6, 10, and 8 Å for N9A, G12A, and H17A, respectively) distances in N9A, G12A, and H17A mutants (Fig. 3b, yellow, red, and pink); in contrast, all the three distances for L11A and F14A were in good agreement with those for WT (Fig. 3c, light orange and purple). The representative structures at WT_rep and repA for each mutant, showing the most frequent basin in the PCA in Fig. 3a, were shown in Supplementary Fig. 2a. These data suggest that the core structure was well-maintained in WT and the L11 and F14 mutants, but was destabilized by mutation at position 9, 12, or 17.
We further analyzed the reasons why the core was structurally destabilized by mutation at position 9, 12, or 17. Destabilization of the core structure was associated with the loss of hydrogen bonds by alanine substitution at positions 9 and 17, and a steric hindrance by alanine substitution at position 12, as described below: The preS1 residues forming hydrogen bonds with the side chains of N9preS1 and H17preS1 during simulations are listed in Supplementary Table 2. The N9preS1 side chain primarily forms hydrogen bonds with the main chain of L11preS1, but they were completely disrupted by N9A mutation and thus the distance between A9 and L11 (between Cβ atom at position 9 and N atom at position 11) was not fixed, showing a higher diversity (Supplementary Fig. 2b, yellow). Similarly, although the H17 side chain formed hydrogen bonds with the main chains of F13preS1 and F14preS1 in WTpreS1, the H17A mutation disrupted the hydrogen bonds, resulted in more diverse and increased distance between the H17 side chain and the main chains of F13 and F14 (Supplementary Fig. 2c, pink). In the G12A substitution, the presence of the methyl group side chain induced a steric hindrance to the close contact of the H17 side chain to the position 12 amino acid, resulted in divergent and larger distance between Cα atom at position 12 and Cβ atom at position 17 (Supplementary Fig. 2d, red). In addition to intramolecular interactions, these preS1 amino acids contribute to the intermolecular interactions with L31 in TM1 and Q264 in TM8b of NTCP, quantified by contacts between these residues and L31NTCP or Q264NTCP (Fig. 3d). The N9A substitution drastically reduced the contact frequency of N9ApreS1–L31NTCP (Fig. 3d, lane 4), the G12A substitution reduced the contact of H17preS1–L31NTCP (Fig. 3d, lane 9), and the H17A substitution reduced the multiple contacts of H17ApreS1–L31NTCP and H17ApreS1–Q264NTCP (Fig. 3d, lanes 12 and 24). These analyses suggest that N9preS1, G12preS1, and H17ApreS1 maintain both the stable intramolecular interaction and the intermolecular interaction with NTCP (Fig. 3e as a summary).
We confirmed that the conformation of the N-terminal myristoyl chain or its contact patterns to the membrane-bound NTCP were not essentially affected by the mutations in preS1 (Supplementary Fig. 3a): During the MD simulations for the NTCP/2–19preS1 complex, the myristoyl chain was extended with ~ 15 Å of the distance between the N- and C-terminal edges (C1 and C14 atoms) (Supplementary Fig. 3b), and with nearly 180° of the angle defined by C1, C7, and C14 atoms (Supplementary Fig. 3c). The myristoyl chain moved closer and farther apart (Supplementary Fig. 3d) but kept in contact with membrane-bound NTCP during almost the whole duration of the simulations (Supplementary Fig. 3e) via TM5 (Supplementary Fig. 3f, cyan showing the NTCP residues with high contact frequencies, mostly at TM5). These behaviors of the myristoyl chain described above were essentially similar in WT and the mutants (N9A, G12A, and H17A) (Supplementary Fig. 3b–f).
N9preS1, G12preS1, and H17preS1 are essential for rendering virion infectivity to cell and mouse models
To investigate the biological significance of the N9preS1, G12preS1, and H17preS1- induced stable core formation in acquiring the virion infectivity, we generated HDV variants carrying the LHBs bearing substitutions at either N9, G12, or H17 in preS1. HDV virions were produced by co-transfecting Huh-7 cells with an HDV-encoding plasmid and a plasmid expressing LHBs with respective mutations (Fig. 4a)26. These mutations did not affect the virion production itself, as shown by the equivalent levels of HDV RNA in the immunoprecipitates from the culture supernatant of the producing cells that captured HBs-packaged HDV among each variant (Fig. 4a, anti-HBs).

a Production of HDV carrying the N9A, G12A, or H17A mutations in LHBs. HDV was produced from Huh-7 cells by cotransfecting with the plasmid encoding the HDV genome and that encoding LHBs or its mutant (N9A, G12A, or H17A). Culture supernatants were purified by immunoprecipitation with either anti-HBs antibody (black) or normal mouse IgG as a negative control to show the background signal (white), or without immunoprecipitation as a total fraction (gray), followed by quantification of HDV RNAs to evaluate the production of HBs-packaged HDV. b Infectivity of HDV to HepG2-NTCP cells. HepG2-NTCP cells were inoculated with HDV produced in (A) at 5, 25, or 125 GEq/cell, followed by detection of intracellular HDV RNA (graph, for 5 and 25 GEq/cell HDV) and HDAg (pictures, for 125 GEq/cell HDV) at six days post-inoculation. Myr-B was used as a control to inhibit HDV infection (white). c Infectivity of HDV in vivo. Nine-day-old hNTCP transgenic mice were inoculated with wild type or mutant HDVs produced in (A) at 1.3 × 107 copies/mouse, and intrahepatic HDV RNAs were quantified six-days post-inoculation. Myr-B was used to inhibit HDV infection. Bars showed the means of three independent experiments (n = 3) (a, b) or five independent samples (c) and error bars represent standard deviation. Statistical significance of p-values is indicated as follows: **p < 0.05. Source data are provided in a Source Data file. WT, wild type; myr-B, myrcludex-B.
We then examined the infectivity of these HDV variants both in cell culture and in vivo models. In cell culture assay, HepG2-NTCP cells were inoculated with each HDV variant at 5, 25, and 125 genome equivalent (GEq)/cell, followed by detection of HDV RNA and HDAg in the cells at six days post-inoculation (Fig. 4b). Whereas wild type HDV showed dose-dependent detection of HDV RNA and HDAg in inoculated cells, HDV variants carrying mutations at N9, G12, or H17 exhibited negligible signals at any inoculation doses (Fig. 4b). For in vivo assay, we inoculated human NTCP-transgenic (hNTCP Tg) mice with wild type or mutant HDVs. Intrahepatic viral RNA was detected six days post-infection of the wild-type virus, as reported previously (Fig. 4c)27, and was reduced by administration of myr-B prior to virus infection. In sharp contrast to the wild-type HDV, HDV RNA was under detectable levels following inoculation of HDV variants carrying the N9, G12, or H17 mutants. Overall, these data clearly indicate that the N9preS1, G12preS1, and H17preS1-involved core formation in preS1 plays a fundamental role in rendering virion infectivity.
33–48preS1 interaction with cell-surface of NTCP enhances the preS1-NTCP binding
While the above data show that N9, G12, and H17-produced core structure for the tandem loop is essential for NTCP binding and infectivity, the lower NTCP binding capacity of myr-2–19preS1 compared to myr-2–48preS1 (Fig. 1) suggests an additional role of the 20–48preS1 region in achieving maximal NTCP binding. To identify the regions that maximize the binding capacity of myr-2–48preS1, we analyzed the contact regions between NTCP and myr-2–48preS1 during the MD simulations and divided it into myr-2–19preS1, 20–32preS1, and 33–48preS1 contributions. The contact regions and frequency are depicted in Supplementary Fig. 3g (red to blue, high to low contact frequency in Supplementary Fig. 3g). The high-frequency contact area with myr-2–48preS1, shown in red, encompassed TM1, TM2–3 loop, TM3b, TM4–5 loop, TM5, TM8b, TM8–9 loop, and TM9 (Supplementary Fig. 3g). Whereas most of these areas, including TM1, TM3b, TM4-5 loop, TM5, TM8b, and TM9, were covered by myr-2–19preS1, the remaining TM2–3 loop, TM4–5 loop, and TM8–9 loops were exclusively covered by 33–48preS1, with no contribution of 20–32preS1 to acquisition of additional contact areas (Supplementary Fig. 3g). We therefore focused on 33–48preS1 for potential supportive function in NTCP binding. The top amino acids in 33–48preS1 having the highest frequency to NTCP contact during MD simulations, include W41preS1, D43preS1, A44preS1, N45preS1, and K46preS1, which contacted the loops located at either TM2–3, TM4–5, or TM8–9 in NTCP (Supplementary Table 3). Although most these preS1 amino acids contacted a single loop, mainly TM2–3 loop, W41preS1 especially contacted to multiple NTCP loops involving N87, Y146, and F274 reportedly important for preS1 binding or HBV infection (Fig. 5a)3,8,27,28,29.

a Close-up view of the interface between preS1 amino acid (W41preS1, D43preS1, A44preS1, N45preS1, and K46preS1 in orange, yellow and brown) and the counterpart NTCP regions (K86, N87, and I88 in TM2–3 loop, Y146 in TM4–5 loop, and F274 in TM8–9 loop, light blue). b NTCP binding of TAMRA-labeled myr-2–48preS1 or its mutants (W41A/D43A/A44R, N45A/K46A, W41A) was examined as shown in Fig. 1c. c, d PreS1 binding capacity and HDV infection susceptibility for the cells expressing NTCP and its mutant (N87A/Y146A) were examined as shown in Fig. 2b and c. e, f Production and infectivity of HDV carrying mutations in preS1. HDV having the indicated mutations in preS1 was produced and inoculated to HepG2-NTCP cells to evaluate infectivity as described in Fig. 4. Bars showed the means of three independent experiments (n = 3) and error bars represent standard deviation (SD). Source data are provided as a Source Data file. WT wild type; myr-B myrcludex-B.
We then experimentally validated the role of these amino acids in promoting the NTCP-preS1 binding. As shown in Fig. 5b, substitutions W41A/D43A/A44R and N45A/K46A significantly reduced the NTCP binding capacity (Fig. 5b). W41A single mutation demonstrated the strongest reduction in NTCP binding, although still retaining the low NTCP binding (Fig. 5b), which suggests a major role of W41preS1 within the 33–48preS1 region in enhancing the NTCP binding. In support of this, alanine mutation at the NTCP counterparts of W41preS1, N87 (TM2–3 loop), and Y146 (TM4–5 loop) on the extracellular NTCP surface showed an impairment of preS1 binding capacity especially at lower preS1 concentrations, although showing low preS1 binding at higher preS1 concentrations (Fig. 5c). Loss of function by the N87A/Y146ANTCP substitution was more prominent in HDV infection assay (Fig. 5d). Furthermore, HDV carrying the preS1 with the W41A substitution on virion surface completely abolished the infectivity to HepG2-NTCP cells, although it did not affect the production of HDV virion itself (Fig. 5e, f). These data indicate the crucial role of W41preS1, and the counterpart N87A/Y146ANTCP, in rendering the preS1-NTCP binding and the virion infectivity.
Thus, although 33–48preS1 did not have the capacity for initiating the binding to NTCP by itself (Fig. 1b and Supplementary Fig. 1a and b), this region potentiated the NTCP binding that was already established by myr-2–19preS1.
The statistical mechanical model predicts that 2–48preS1 folding is initiated by tandem loop structure
These experimental data raise the possibility that the formation of the core structure for tandem loop in myr-2–19preS1 initiates the de novo binding to NTCP, and 33–48preS1-mediated interaction follows to stabilize the complex with the NTCP extracellular surface. To further analyze the order of the folding and the binding to NTCP, we employed a coarse-grained statistical mechanical model called the WSME-L model30. The model uses the final folded structure of protein as input and systematically evaluates the free energies of diverse conformations accessible along the folding pathway, thereby enabling quantitative prediction of the folding processes. In this study, we applied the NTCP-bound structure of 2–48preS1 to predict the folding process of the intrinsically disordered preS1 peptide into its bound conformation. The model showed a gradual increase in the free energy along with the elevated folding (order parameter) of 2–48preS1 (Fig. 6a), indicating that 2–48preS1 is difficult to fold and is largely unfolded, consistent with preS1 being an IDP. Nevertheless, calculations of the folding degree of each residue along the order parameter with the WSME-L model reveal residue-specific folding propensities, allowing the inference of the overall sequence of folding steps that would occur if the peptide were to adopt a folded conformation upon binding to NTCP. In the model wherein preS1 folds by itself, along with the increase in the order parameter, shifting to the high degree of folding (shown by yellow to red) started at the N-terminal 7–19 aa region, and was followed by at other regions, with the slowest folding state at the C-terminal 39–48 aa (Fig. 6b); e.g., when setting 0.5 as order parameter, (Fig. 6b, white broken line), high folding state (above 0.25) was shown at 7–15 and 17–19 aa but not any other regions in 2–48preS1 (Fig. 6c), indicating that 7–19 aa region in preS1 that corresponds to the tandem loop (loops 1 and 2) folds first, and the region 39–48 aa shows the slowest folding. Figure 6d depicts the predicted structure of preS1 during the early process for folding, showing the folded structure at 7–15 and 17–19 aa, but not at any other regions (Fig. 6d). We further calculated the two-dimensional free energy landscape for preS1 folding by artificially enhancing intramolecular contacts to monitor its folding process in detail (Fig. 6e): The landscape indicates that the pathway from the fully disordered state (x = 0, y = 0) to the fully structured state (cryo-EM structure of preS1) (x = 1, y = 1) undergoing along the lowest free energy areas (shown by gray line) follows the increase in the folding in 2–19preS1 (rightward shift) at first and then the increase in the 20–48preS1 folding afterwards (upward shift), implicating that the 2–19preS1 region undergoes robust folding (up to 0.75), followed by the late 20–48preS1 folding (Fig. 6e). Similar predictions were reproduced for the two preS1 structures bound to NTCP (PDB IDs: 8HRX and 8RQF) (Fig. 6; Supplementary Fig. 4). These results, together with the experimental results and MD simulations, support the stepwise binding mechanism with the coupled folding reaction of preS1: myr-2–19preS1 initiates the folding to fit to the bile acid tunnel, which is followed by the 33–48preS1-mediated interaction with the extracellular face to stabilize the complex with NTCP.

a One-dimensional free energy landscape of 2-48preS1 predicted using the WSME-L model. The horizontal axis is an order parameter indicating the degree of folding reaction, where 0 and 1 represent the unfolded state and the structured form (NTCP-bound form), respectively. b Degree of structure formation (referred to as Φ-value) for each residue of 2–48preS1 (horizontal axis) along the varying folding state (vertical axis). Φ-values are represented by the color (red to purple, folded to unfolded). The white broken line indicates 0.5 as the order parameter, used for the analysis in (c). c Degree of structure formation (Φ-value) of each residue in 2–48preS1 for the order parameter of 50%. d Structure of 2–48preS1 during folding. Residues with Φ-values greater than 0.25 (7–15 and 17–19 aa) are shown by spheres. e Two-dimensional free energy landscape of 2–48preS1 predicted using the WSME-L model by artificially enhancing intramolecular contacts. The gray line indicates the pathway traveling along the lowest free energy from fully unfolded (x = 0, y = 0) to fully structured state (x = 1, y = 1). These analyses were based on the 2–48preS1 structure in PDB ID: 8HRX. f Schematic hypothesis for preS1 binding to NTCP. Upper, (i) Membrane proximation: The N-terminal myristoyl group initiates transient interaction with the target cell membrane. (ii) Core formation: Asn9preS1/Gly12preS1/His17preS1 in 2–19preS1 binds to Leu31NTCP and Gln264NTCP in the narrow bile acid tunnel and form the tandem loop core structure that creates a wide interface to NTCP. (iii) Outerface interaction: The myr-2–19preS1-bile acid tunnel interaction enables the secondary interaction of 33–48preS1, including Trp41preS1, to the NTCP extracellular loops to stabilize the binding state of the preS1/NTCP complex. Lower, as another hypothesis, (ii) Outerface interaction: 33–48preS1 interacts with the extracellular loops of NTCP, which facilitates (iii) Core formation: 2–19preS1 forms the tandem loop structure in the NTCP bile acid tunnel. This stepwise induced-fit mechanism of preS1 binding acquires a high affinity to NTCP, leading to the productive viral infection to hepatocytes.
Discussion
PreS1 is a multi-functional protein that is essential for receptor binding during cell entry and for HBV virion assembly by interacting with viral and cellular factors7,8,14,31. The structural feature of preS1 as an IDP is likely to enable to interact with multiple viral and cellular counterparts at different steps of the viral life cycle, probably by forming distinct protein conformations14,15,16. In contrast to the typical virus-receptor interaction that involves firmly-structured viral surface proteins (e.g., envelope, spike, hemagglutinin, and capsid), preS1 follows dynamic conformational change to fit to the target protein and establishes high-affinity binding. The structural flexibility of preS1 allows itself to be inserted into the narrow bile acid tunnel of NTCP, although its precise mechanism for forming a lasso-structure remains elusive21. The present study shows that the multiple subregions, myristoylated 2–19 and 33–48 in preS1, coordinately interact with NTCP. Interestingly, these distinct subregions are likely to function sequentially, rather than simultaneously or redundantly. Our data show that N9preS1, G12preS1, and H17preS1 interact intramolecularly to form a core structure of the tandem loop (Fig. 2a), producing a large interface for binding to the NTCP bile acid tunnel. This core formation would be induced, at least in part, by L31 in NTCP TM1 and Q264 in TM8b. Consistent with the general role of myristoyl group in inserting into the lipid membranes in a non-specific and low-affinity manner to recruit the proteins onto cellular membranes32, our chemical crosslink preS1-cell binding assay (for examination of low-affinity binding) showed that myr-2–48preS1 interacted with NTCP-negative HepG2 cells, in a myristoyl group-dependent but N9preS1, G12preS1, and H17preS1-independent manner. Loss of myristoyl group abolished detectable affinity of preS1 peptide to NTCP-positive cells (Supplementary Fig. 5). These data suggest that 2–48preS1-NTCP binding follows the myristoyl group-mediated cell membrane proximity, consistent with the general role of N-terminal myristoyl group in inducing the primary and transient target membrane proximation33 that triggers secondary more specific and high affinity protein-protein interaction. 33–48preS1 by itself did not show affinity to NTCP but enhanced the preS1 binding to NTCP that was already established, likely used as a scaffold. These experimental results were well supported by a statistical mechanical model explaining that the robust folding within 2–19preS1 precedes the 20–48preS1-mediated NTCP interaction (Fig. 6a–e). However, the possibility is not fully excluded that 33–48preS1 interaction with the outer NTCP surface precedes the 2–19preS1 docking to the bile acid tunnel. Based on the evidence so far, the possible mode of preS1 binding to NTCP through the multiple subregions is proposed (Fig. 6f). This is a hypothetical model, rather than a definitive conclusion, based on the collective results obtained from our experimental data and theoretical modeling.
(i) The N-terminal myristoyl group initiates transient and non-specific interaction with the target cell membrane (Membrane proximation). This is followed by the peptide region-mediated NTCP interactions that has two possible orders: (ii) 2–19 aa peptide folds to form a core with N9preS1, G12preS1, and H17preS1 that bundles the tandem loop to create a large interface with the amino acids in the bile acid tunnel of NTCP (Core formation), and then (iii) The 33–48 aa stretch including W41preS1 interacts with the extracellular loops of the NTCP surface, including N87NTCP and Y146NTCP, to stabilize its dynamic fluctuation stably embedded onto the NTCP surface (Outerface interaction): Alternatively, it is possible that (ii) the 33–48 aa stretch-mediated outerface interaction precedes, which then facilitates (iii) 2–19 aa region-involved core formation and docking to the bile acid tunnel. Detailed order of the binding to NTCP has not been fully demonstrated, which should be further characterized in future subjects. However, this model involving the multiple preS1 subregions in receptor binding is consistent with the previous evidences reporting the role of multiple regions in preS1 (essential role of 9–15 aa and promoting role of 2–9, 16–20, and 34–48 aa) in viral infection25, and the involvement of TM1, TM8b, 84–87 aa in the TM2–3 loop, and 146 aa in the TM4–5 loop of NTCP for preS1 binding3,8,19,21,24,27,28,30. This “binding maturation” mechanism for receptor recognition would be significant in that multiple functional sites are coordinately involved to secure a high-affinity receptor interaction.
Within the whole HBV genome, the preS1-coding region shows the highest sequence diversity among HBV strains/genotypes and heterogeneity in infected patients34,35,36. This region is under strong positive selection, which is a sign for adaptive evolution under selective pressure to improve infection efficiency as well as to escape from immune response. Despite this overall diversity, the 9–17 aa region is highly conserved, suggesting its indispensable function for virus spread. In particular, N9preS1, G12preS1, and H17preS1 are conserved in 99.8% (13919/13952), 99.9% (13942/13952), and 99.9% (13933/13952) respectively, among the HBV genomic sequences publicly registered in the Hepatitis B Virus Database (HBVdb, https://hbvdb.lyon.inserm.fr/HBVdb/HBVdbDataset) (Supplementary Fig. 6a). These three amino acids are also highly conserved in HBV-related hepadnaviruses infecting in primates, including wooly monkey, capuchin monkey, and great apes (Supplementary Fig. 6b)37. It is notable that although the C-terminal region after aa 20 is more diverse in sequence, W41preS1 is highly conserved in 99.7% (13912/13952) of HBV strains, and also in primate hepadnaviruses (Supplementary Fig. 6). This suggests that the mode of receptor recognition mediated by these four key amino acids is conserved among the majority of HBV strains and primate hepadnaviruses.
In conclusion, we propose a model of preS1 recognition of its receptor, wherein the narrow transporter tunnel is primarily used for induced-fit folding for de novo binding, and the extracellular surface is secondarily used for stabilization of the binding. This mechanism features a high-affinity virus-receptor interaction by a viral IDP, explaining the host selection by HBV/HDV and providing insights for the development of antiviral agents targeting this dynamic process.
Methods
Cell culture
HepG2 and HepG2-hNTCP-C4 cells were cultured with Dulbecco’s modified Eagle’s medium (D-MEM)/F-12 + GlutaMax (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Nichirei), 10 mM HEPES (pH 7.4, Nacalai Tesque), 100 units/mL penicillin, 100 μg/mL streptomycin (Invitrogen), and 5 μg/mL insulin (Wako) in the presence (HepG2-hNTCP-C4 cells) or absence (HepG2 cells) of 0.4 mg/mL G418 (Nacalai Tesque) at 37 °C in 5% CO223. Huh-7 cells were cultured in D-MEM supplemented with 10% FBS, 10 mM HEPES, 100 units/mL penicillin, 100 mg/mL streptomycin, 100 mM non-essential amino acid, 1 mM sodium pyruvate (Invitrogen) at 37 °C in 5% CO2.
Generation of NTCP-expressing cells
The plasmids encoding the gene for human NTCP (NM_003049.3 [https://www.ncbi.nlm.nih.gov/nuccore/NM_003049.3/]) tagged with c-myc at the C-terminus was described previously38. The plasmids encoding a mutant NTCP were constructed by oligonucleotide-directed mutagenesis using the above plasmid as a template, as described21.
HepG2 cells were transfected with these expression plasmids with ViaFect transfection reagent (Promega) according to the manufacturer’s protocol and were used for the assays at 72 h post-transfection. The transfection efficiency was roughly 30–40%.
PreS1 binding assay
The binding between preS1 and NTCP-expressing cells was examined essentially as described previously39. HepG2 cells transfected with the expression plasmid for either the wild type or the mutant NTCP or the corresponding empty vector were incubated with the indicated concentrations (40, 100, 250, and 625 nM in Figs. 1c, 1d, 2b, 3a, and 2.56, 6.4, 16, and 40 nM in Fig. 5b) of C-terminally 6-carboxytetramethylrhodamine (TAMRA)-labeled preS1 peptide consisting of 2–48 aa (2–48preS1), its N-terminally myristoylated form (myr-2–48preS1), 2–19 aa (2–19preS1), its N-terminally myristoylated form (myr-2–19preS1), 20–32 aa(20–32preS1), 33–48 (33–48preS1), or 2–48 preS1 carrying a point mutation at the indicated amino acid to Ala at 4 °C for 30 min. After washing free peptide, the cells were fixed with 4% paraformaldehyde and stained with 0.02% DAPI to observe the cell-bound preS1 fluorescence (red) and the nucleus (blue) with a fluorescence microscope, BZ-X810 (Keyence, Osaka, Japan). The fluorescence intensities were quantified with BZ-H4A and BZ-H4C (Keyence). Co-treatment with myrcludex-B, a preS1-NTCP binding inhibitor40, at 2 μM together with the fluorescence peptide at 625 nM was used to show the background level of the assay. Treatment of HepG2 cells with myr-2–48preS1 also showed the background level.
In the peptide competition assay, the cells were treated with the indicated non-fluorescence-labeled preS1 peptide at the indicated concentrations (40, 100, 250, and 625 nM) together with TAMRA-labeled myr-2–48preS1 at 40 nM to detect cell-bound fluorescence as shown above.
Transient and low-affinity binding to target cells was assessed under chemical crosslinking as follows: HepG2-hNTCP-C4 and HepG2 cells were treated with the respective TAMRA-labeled preS1 peptide at 625 nM in the presence of 2.5 mM ethylene glycol bis (sulfosuccinimidyl succinate) as a chemical crosslinker at 4 °C for 30 min, followed by the same method for detection of TAMRA-fluorescence and DAPI-stained nuclei.
HDV preparation and infection assay
To examine the role of the preS1 amino acids in virus infection, we employed the HDV infection assay carrying the mutants in the surface protein, as described previously9,26 (validation of mutated HBV infectivity by a trans complementation strategy remains for future study). The wild type HDV and the variants carrying a point mutation in its LHBs on the virion were recovered from the culture supernatant of Huh-7 cells transfected with the expression plasmid for HDV genome (pSVLD3, kindly provided by Dr. John Taylor at Fox Chase Cancer Center) and that for the wild type HBV surface antigen (pT7HB2.7) or its corresponding mutants, which were then concentrated with 10% PEG8000 and 2.3% NaCl26. To examine HDV production level, the recovered culture supernatant was immunoprecipitated with anti-HBs antibody (Abcam, ab9193) or normal IgG (Dako) as a negative control for 1 h, followed by incubation with protein G-Sepharose (Cytiva) for 30 min and extensive washes. HDV RNA titer in the precipitates was extracted with RNeasy Mini Kit (Qiagen) and virus particles were quantified by real-time RT-PCR analysis as described41. The culture supernatant without immunoprecipitation was also detected as the total fraction.
For the HDV infection assay, the above culture supernatant was incubated to HepG2-hNTCP-C4 cells or HepG2 cells overexpressing the indicated NTCP variants or the wild type at 5, 25, or 125 genome equivalent (GEq)/cell in supplement with 5% PEG8000 for 16 h. After washing out the free virus, the cells were cultured for another 6 days. Viral infection was evaluated by quantifying the intracellular HDV RNA by real-time RT-PCR analysis or detecting the intracellular HDAg by immunofluorescence analysis as described previously41. In the peptide competition assay, the indicated preS1 peptide at varying concentrations (1, 5, 25, 125, and 625 nM) were added during HDV inoculation.
Detection of HDV RNA
HDV RNA was extracted by RNeasy Mini Kit (Qiagen) and was then quantified by one-step RT-qPCR kit (THUNDERBIRD Probe One-step RT-qPCR kit, TOYOBO) by using 5’-GGACCCCTTCAGCGAACA-3’ and 5’-CCTAGCATCTCCTCCTATCGCTAT-3’ as primers and 5’-AGGCGCTTCGAGCGGTAGGAGTAAGA-3’ as a probe as described previously42.
Cell surface biotinylation assay
HepG2 cells transfected with the expression plasmid for either the wild type or mutant NTCP were washed out with PBS and incubated with EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) for 30 min. After washing and lyzing the cells, the supernatants were pulled down with streptavidin beads (Cytiva) for 2 h. The precipitates were detected by immunoblotting to examine the cell surface expression of NTCP. The samples without pull-down were also analyzed to examine the expression level in the whole cells.
Immunoblot analysis
Recovered cell lysates were treated with 250 U peptide-N-glycosidase F to digest N-linked oligosaccharides of NTCP before loading on SDS-PAGE24. Proteins were detected with anti-c-myc (Santa Cruz Biotechnology, 9E10) (1:3000 dilution) and anti-actin (Sigma, A5441) (1:10000 dilution) antibodies as primary antibodies38.
NTCP transporter assay
The cells were incubated with [3H]-taurocholic acid as a substrate in the buffer (4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgCl2, 1.3 mM CaCl2, 2.6 mM D-glucose, 25 mM HEPES, 10 μM taurocholate, pH 7.4) either in the presence or absence of 125 mM sodium at 37 °C for 15 min39. After washing and lysing the cells, the intracellular radioactivity was measured with a liquid scintillation counter (Perkin Elmer, Waltham, USA). Bile acid uptake into the cells in the presence of sodium presents the NTCP-dependent transporter activity.
Infection assay in hNTCP-transgenic mice
HDV infection to hNTCP transgenic mice (JAX stock #030535)43 was performed essentially as described44. Nine-day-old mice were subcutaneously injected with dexamethasone at 50 mg/kg body weight, and were intraperitoneally challenged with HDV at 1.32 × 107 copies/mice. Six days post-inoculation, the livers were harvested, and total RNA was extracted with ISOGEN (NIPPON GENE) according to the manufacturer’s instructions. For validation of hNTCP-dependent viral entry, 2 mg/kg body weight of myr-B was injected subcutaneously 1 h prior to viral inoculation and once per day for the next two days post-inoculation. The animal experiment was approved by the ethical of the National Institute of Infectious Diseases (NIID-124096) and conducted in accordance with its guidelines regarding the care and use of laboratory animals.
Molecular dynamics simulations
The NTCP-2–19preS1 structure was prepared by cutting the 20–48preS1 moiety from the NTCP-2–48preS1 structure (PDB: 8HRY), and was used as an initial structure of MD simulations. The membrane-water systems were prepared using Membrane Builder module implemented in CHARMM-GUI45,46,47. During setup in CHARMM-GUI, missing hydrogen atoms were added, according to the protonation states of residues determined by their pKa values estimated by PROPKA implemented in PDB2PQR48, using the initial NTCP-2–19preS1 (WT) structure at pH 7 condition: E257NTCP was set to a protonated glutamic acid, and D16preS1 was set to a protonated aspartic acid. All histidine residues in NTCP-2–19preS1 were set to neutral histidine, and all the histidine residues except for H57NTCP and H17preS1 were protonated at the Nδ atom only (HSD in the CHARMM force field), and H57NTCP and H17preS1 were protonated only NƐ atom (HSE in the CHARMM force field). The N- and C-termini in NTCP were set to NH3+ and COO–, respectively. In preS1, the myristoylation of the N-terminus (GLYM in the CHARMM force field) was added, and the terminal capping CONH2 (CT2 in the CHARMM force field) was added to the C-terminus. N9A, G12A, H17A, L11A, and F14A substitutions in preS1 were performed in the setup process, i.e., the substitution was added to the initial NTCP-2–19preS1 structure. The protonation states for the mutants were set to the same states determined in WT described above. The complex was embedded in a 70 Å × 70 Å 1-palmitoyl-2-oleoyl-glycero-3-phosphocholin (POPC) bilayer in the x–y plane in the center of the rectangular unit cell. The orientation of NTCP relative to the membrane was set the same as that of NTCP deposited in the Orientations of Proteins in Membranes database (PDB: 7FCI). After filling the unit cell with water molecules (TIP3water model), sodium and chloride counterions (NaCl) were added with 150 mM ion concentration. The constructed system is illustrated in Supplementary Fig. 3a. and details of the system setup were summarized in Supplementary Table 4.
All-atom conventional MD simulations were performed using the MD program package GROMACS ver. 2021.449 under periodic boundary conditions. The CHARMM36m force field50,51,52, which is often used for membrane-protein systems, was used. Energy minimization and equilibration runs were performed according to the default setting in Membrane Builder; detailed conditions were the same as those of our previous simulations53. Then, three independent 1 μs production runs with different initial velocities and initial configurations were performed; each independent MD was performed from the energy minimization process. The NPT ensemble was used, and temperature and pressure were set at 300 K and 1 atm, respectively. The type of pressure coupling was semi-isotropic. The thermostat and barostat used were the Nose-Hoover scheme and the Parrinello–Rahman approach, respectively. The particle mesh Ewald method was used for evaluating electrostatic interactions, and the van der Waals interactions were smoothly truncated using the switching function within a 10–12 Å range. Bond length involving hydrogen atoms was constrained by the P-LINCS algorithm, and a 2-fs timestep was used. MD simulations for NTCP/2–19preS1 (mutants) were performed using the same procedure and conditions as described above. The system settings and MD simulations for NTCP-2–48preS1(WT) were performed similarly using the same protocols for NTCP/2–19preS1 systems as described above. Here, three independent 1-μs production runs were performed per each system, i.e., NTCP-2–19preS1(WT, N9A, G12A, H17A, L11A, and F14A) and NTCP-2–48preS1(WT) systems in this study. Snapshots were saved every 1 ns from a 1 μs MD simulation and used for trajectory analyses. After structural alignment of snapshots to the initial structure using Cα atoms for the 2–19 residues of preS1, RMSD values of Cα atoms for the 2–19 residues of preS1 were calculated in three independent simulations. Principal component analysis (PCA) of the binding pose of 2–19preS1 was performed using Cα atoms for the 2–19 residues of preS1 after a structural alignment of the snapshots using Cα atoms for the 2–19preS1, and the PC axes were determined using all the MD simulations of NTCP/2–19preS1 complex for WT and the five mutants (N9ApreS1, G12ApreS1, H17ApreS1, L11ApreS1, and F14ApreS1) to ensure that full conformational variability was captured. Contributions of PC1 and PC2 against the total degree of freedom were 25% and 16.8%, respectively. The free energy landscapes shown as color maps were evaluated by –lnP, where P is the existence probability calculated on the normalized histogram of 0.5 Å x 0.5 Å pixels on the PC1-PC2 surface. The representative structure of the basin on the FEL was selected by the central structure of the cluster with the most members after clustering the structures at 1 pixel corresponding to the minimum of the basin, in which the hierarchical clustering of Cα and Cβ atoms was performed, using the MMTSB tool. To analyze the contact frequency of preS1-NTCP, contacts were defined as distances < 5 Å between nonhydrogen atoms of NTCP and preS1. Hydrogen bonds analyses were performed using the HBonds Plugin implemented in the Visual Molecular Dynamics (VMD) ver.1.9.3, and the donor-acceptor distance and the angle cutoff were set to 3.5 Å and 30 degrees, respectively.
The statistical mechanical model
The free energy landscapes of preS1 were calculated by a coarse-grained statistical mechanical model called the WSME-L model using the native three-dimensional structure as input30. The details on the free-energy landscape calculations based on the WSME-L model are provided in the Supplementary Notes. Briefly, in the WSME-L model, each residue is assigned a two-state variable mk (mk = 1 for the folded conformation and mk = 0 for other conformations). For a protein of N residues, the conformational state of a protein defined by the set of mk values \(\left\{m\right\}=\{{m}_{1},\,{m}_{2},\cdots,{m}_{N}\}\) is associated with the following energy:
where \({\varepsilon }_{i,j}\) denotes the contact energy between residues i and j, and \({m}_{i,j}\) is defined as:
The order parameter representing the progress of the folding reaction is defined as follows:
The partition function at order parameter n is given by:
where kB denotes the Boltzmann constant, T the temperature, and Si the entropy reduction associated with residue i adopting the folded conformation. Trn represents the sum over all states at the given value of the order parameter n.
Next, to account for non-local interactions, we consider a situation in which residues u and v are brought into proximity by a virtual linker. The corresponding energy is given as follows:
where \({E}^{(u,v)}={\varepsilon }_{u,v}+{\varepsilon }_{u+1,v}+{\varepsilon }_{u-1,v}+{\varepsilon }_{u,v+1}+{\varepsilon }_{u,v-1}\). \({\varepsilon }_{i,j}^{\left(u,v\right)}\) is defined to be zero when \(\left(i,j\right)=\left(u,v\right),\,\left(u\pm 1,v\right)\), and \(\left(u,v\pm 1\right)\), and otherwise takes the same value as \({\varepsilon }_{i,j}\). \({m}_{i,j}^{(u,v)}\) is defined as follows:
The partition function corresponding to this energy is expressed as follows:
Taken together, the partition function of the WSME-L model is defined as the ensemble of partition functions obtained by introducing a virtual linker on a per-contact basis, as follows:
where \({S}^{{\prime} \left(u,v\right)}\left(n\right)\) represents the entropy cost associated with the reduction in the number of accessible states due to the presence of a virtual linker. The cost is calculated by treating the protein chain as a Gaussian chain30. The two-dimensional free energy landscape was obtained by computing the partition function using order parameters corresponding to the N-terminal (residues 2–19) and C-terminal (residues 20–48) regions of 2–48preS1. In addition, the degree of folding for each residue was evaluated through theoretical Φ-value analysis. The Φ-value ranges from 0 to 1, where 0 indicates that the residue is unfolded and 1 indicates that it is folded. For each residue, the theoretical Φ-value was obtained by perturbing its contact energy, calculating the resulting free energy difference at order parameter n, and normalizing with respect to the stability change of the folded state.
The WSME-L model was applied to 2–48preS1 as follows. First, the three-dimensional structure of the NTCP-preS1 complex (PDB ID: 8HRX or 8RQF) was energy minimized using AMBER24/ff14SB with restraints for the backbone atoms54. The energy-minimized structure was used to calculate the AMBER-derived contact energy by the MMPBSA.py module for implicit solvents55. The AMBER-derived contact energy was scaled to be consistent with the experimentally obtained dissociation constant for the NTCP-preS1 interaction (67 nM)40. Then, the intramolecular contact energy of preS1, \({\varepsilon }_{i,j}\), was used to calculate the one- and two-dimensional free energy landscapes of the folding of an isolated preS1 and the residue-specific structure formation (theoretical Φ-value analysis) of preS1 during folding. In the calculations, the contacts with neighboring residues (j ≤ i + 2) were set to \({\varepsilon }_{i,j}=0\), and the virtual linkers were introduced only for the contacts between residues i and j with an AMBER-derived contact energy less than –0.6 kcal/mol. The entropic cost of the main chain of the i-th residue was set to Si = –3.5 cal/(mol·K), and the temperature was set to T = 298 K. The free energy landscapes and Φ-values were drawn using Mathematica 14.1 (Wolfram, Champaign, IL, USA). When artificially enhancing the intramolecular contacts of preS1, the contact energies of preS1 were enhanced ~ 2-fold to make the free energies of the folded and unfolded states similar.
Figure preparation
The figures of the structure models were prepared by PyMOL. Graphs were prepared by GraphPad Prism9. The HBV genomes were aligned by Clustal Omega online software56,57. The figures of alignment were prepared by ESPript58.
Statistics
Bar graphs show the means ± standard deviation from triplicated experiments. One-way ANOVA was performed by GraphPad Prism9.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The input structure, MD input files, and all the trajectory data from MD simulations generated in this study have been deposited in the Biological Structure Model Archive (BSM-Arc) under accession code BSM-ID BSM00094 [https://bsma.pdbj.org/entry/94]. The protein structures used in the statistical mechanical model in this study are available in PDB under accession codes: 8HRX [https://doi.org/10.2210/pdb8HRX/pdb] and 8RQF [https://doi.org/10.2210/pdb8RQF/pdb]. Source data are provided in this paper.
Code availability
No custom code was developed for this study. The analyses conducted in the MD analyses did not involve any non-standard algorithms or specialized computational procedures. All reported results can be fully reproduced from the trajectory data deposited in the BSM-Arc database (BSM-ID BSM00094) using commonly available MDs analysis tools.
All codes for the statistical mechanical calculations with the WSME-L model have been previously published and are available at https://github.com/ut-arailab/WSME-L_model. These codes are also archived in Zenodo with the identifier https://doi.org/10.5281/zenodo.8280372.
References
Burwitz, B. J. et al. Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat. Commun. 8, 2146 (2017).
Lempp, F. A. et al. Sodium taurocholate cotransporting polypeptide is the limiting host factor of hepatitis B virus infection in macaque and pig hepatocytes. Hepatology 66, 703–716 (2017).
Liu, Y. et al. Targeted viral adaptation generates a simian-tropic hepatitis B virus that infects marmoset cells. Nat. Commun. 14, 3582 (2023).
Liu, Y., Maya, S. & Ploss, A. Animal models of hepatitis B virus infection-success, challenges, and future directions. Viruses 13, https://doi.org/10.3390/v13050777 (2021).
Guidotti, L. G., Matzke, B., Schaller, H. & Chisari, F. V. High-level hepatitis B virus replication in transgenic mice. J. Virol. 69, 6158–6169 (1995).
Lucifora, J. et al. Hepatitis B virus replication in primary macaque hepatocytes: crossing the species barrier toward a new small primate model. Hepatology 51, 1954–1960 (2010).
Yan, H. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1, e00049 (2012).
Ni, Y. et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146, 1070–1083.e1076 (2014).
Sureau, C., Guerra, B. & Lanford, R. E. Role of the large hepatitis B virus envelope protein in infectivity of the hepatitis delta virion. J. Virol. 67, 366–372 (1993).
Le Seyec, J., Chouteau, P., Cannie, I., Guguen-Guillouzo, C. & Gripon, P. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J. Virol. 73, 2052–2057 (1999).
Glebe, D. et al. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 129, 234–245 (2005).
Gripon, P., Cannie, I. & Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 79, 1613–1622 (2005).
Barrera, A., Guerra, B., Notvall, L. & Lanford, R. E. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J. Virol. 79, 9786–9798 (2005).
Jürgens, M. C. et al. The hepatitis B virus preS1 domain hijacks host trafficking proteins by motif mimicry. Nat. Chem. Biol. 9, 540–547 (2013).
Watashi, K., Shionoya, K., Kobayashi, C. & Morita, T. Hepatitis B and D virus entry. Nat. Rev. Microbiol.23, 318–331. (2024).
Chi, S.-W., Kim, D.-H., Lee, S.-H., Chang, I. & Han, K.-H. Pre-structured motifs in the natively unstructured preS1 surface antigen of hepatitis B virus. Protein Sci. 16, 2108–2117 (2007).
Goutam, K., Ielasi, F. S., Pardon, E., Steyaert, J. & Reyes, N. Structural basis of sodium-dependent bile salt uptake into the liver. Nature 606, 1015–1020 (2022).
Asami, J. et al. Structure of the bile acid transporter and HBV receptor NTCP. Nature 606, 1021–1026 (2022).
Park, J. H. et al. Structural insights into the HBV receptor and bile acid transporter NTCP. Nature 606, 1027–1031 (2022).
Liu, H. et al. Structure of human NTCP reveals the basis of recognition and sodium-driven transport of bile salts into the liver. Cell Res. 32, 773–776 (2022).
Asami, J. et al. Structural basis of hepatitis B virus receptor binding. Nat. Struct. Mol. Biol.31, 447–454 (2024).
Liu, H. et al. Structure of antiviral drug bulevirtide bound to hepatitis B and D virus receptor protein NTCP. Nat. Commun. 15, 2476 (2024).
Iwamoto, M. et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 443, 808–813 (2014).
Shionoya, K. et al. Structural basis for hepatitis B virus restriction by a viral receptor homologue. Nat. Commun. 15, 9241 (2024).
Schulze, A., Schieck, A., Ni, Y., Mier, W. & Urban, S. Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol. 84, 1989–2000 (2010).
Le Duff, Y., Blanchet, M. & Sureau, C. The pre-S1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J. Virol. 83, 12443–12451 (2009).
He, W. et al. Modification of three amino acids in sodium taurocholate cotransporting polypeptide renders mice susceptible to infection with hepatitis D virus in vivo. J. Virol. 90, 8866–8874 (2016).
Yan, H. et al. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J. Virol. 87, 7977–7991 (2013).
Zakrzewicz, D. et al. Tyrosine 146 of the human Na(+)/taurocholate cotransporting polypeptide (NTCP) is essential for its hepatitis B virus (HBV) receptor function and HBV entry into hepatocytes. Viruses 14, https://doi.org/10.3390/v14061259 (2022).
Ooka, K. & Arai, M. Accurate prediction of protein folding mechanisms by simple structure-based statistical mechanical models. Nat. Commun. 14, 6338 (2023).
Bruss, V. Envelopment of the hepatitis B virus nucleocapsid. Virus Res. 106, 199–209 (2004).
Chen, B., Sun, Y., Niu, J., Jarugumilli, G. K. & Wu, X. Protein lipidation in cell signaling and diseases: function, regulation, and therapeutic opportunities. Cell Chem. Biol. 25, 817–831 (2018).
Resh, M. D. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451, 1–16 (1999).
Li, S., Wang, Z., Li, Y. & Ding, G. Adaptive evolution of proteins in hepatitis B virus during divergence of genotypes. Sci. Rep. 7, 1990 (2017).
Chen, B. F. et al. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology 130, 1153–1168 (2006).
Chen, B. F. Hepatitis B virus pre-S/S variants in liver diseases. World J. Gastroenterol. 24, 1507–1520 (2018).
Littlejohn, M., Locarnini, S. & Yuen, L. Origins and evolution of hepatitis B virus and hepatitis D virus. Cold Spring Harb. Perspect. Med. 6, a021360 (2016).
Takeuchi, J. S. et al. A single adaptive mutation in sodium taurocholate cotransporting polypeptide induced by hepadnaviruses determines virus species specificity. J. Virol. 93, https://doi.org/10.1128/jvi.01432-18 (2019).
Kobayashi, C. et al. Fungal secondary metabolite exophillic acid selectively inhibits the entry of hepatitis B and D viruses. Viruses 14, https://doi.org/10.3390/v14040764 (2022).
Meier, A., Mehrle, S., Weiss, T. S., Mier, W. & Urban, S. Myristoylated PreS1-domain of the hepatitis B virus L-protein mediates specific binding to differentiated hepatocytes. Hepatology 58, 31–42 (2013).
Iwamoto, M. et al. Identification of novel avian and mammalian deltaviruses provides new insights into deltavirus evolution. Virus Evol. 7, veab003 (2021).
Kaneko, M. et al. A novel tricyclic polyketide, vanitaracin A, specifically inhibits the entry of hepatitis B and D viruses by targeting sodium taurocholate cotransporting polypeptide. J. Virol. 89, 11945–11953 (2015).
Winer, B. Y. et al. Preclinical assessment of antiviral combination therapy in a genetically humanized mouse model for hepatitis delta virus infection. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aap9328 (2018).
He, W. et al. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLOS Pathog. 11, e1004840 (2015).
Jo, S., Kim, T. & Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLOS ONE 2, e880 (2007).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Lee, J. et al. CHARMM-GUI Input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
MacKerell, A. D. Jr., Feig, M. & Brooks, C. L. Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc. 126, 698–699 (2004).
Klauda, J. B. et al. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 114, 7830–7843 (2010).
Muraoka, T. et al. A synthetic ion channel with anisotropic ligand response. Nat. Commun. 11, 2924 (2020).
Case, D. A. et al. Amber 2024 (University of California, San Francisco, 2024).
Miller, B. R. et al. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012).
Sievers, F. & Higgins, D. G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145 (2018).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
The HDV-expression plasmid, pSVLD3, was kindly provided by Dr. John Taylor at Fox Chase Cancer Center. We appreciate for a discussion with Drs Diter Glebe and Joachim Geyer at Justus-Liebig University Giessen for a back to back submission of the related paper to different journals. This work was supported by Japan Agency for Medical Research and Development (AMED) under grand numbers JP25fk0310525 (K. Wakae., S-Y.P., M.Ikeguchi., K.Watashi.), JP25fk0310533 (K.Watashi.), JP25fk0310547 (K.Watashi.), JP23ama121007 (N.N., K.Watashi.), JP25ak0101271 (K.Watashi.), JP23fk0310517 (M.Ikeguchi., M.M.), JP23ama121023 (M.Ikeguchi.); JSPS/MEXT under KAKENHI grant numbers JP25KJ2119 (C.K.), JP24K02290 (K.Watashi.), JP23H02724 (N.N., K.Watashi.), JP25K21767 (N.N., K.Watashi.), JP25K21782 (K.Watashi.), JP24K18078 (K.O.), JP25H01280 (M.A., K.O.), JP25H00976 (S-Y.P.); J.S.T. MIRAI program grant number JPMJMI22G1 (K.Watashi.); J.S.T. SPRING, Grant Number JPMJSP2151 (C.K.); The Takeda Science Foundation (K.Watashi., N.N.); the Chemo-Sero-Therapeutic Research Institute (Kaketsuken) (S.-Y.P.); the grant for 2021-2023 Strategic Research Promotion (No. SK202202 to M.Ikeguchi.) of Yokohama City University; Joint Usage/Research Center Program of the Institute for Life and Medical Sciences at Kyoto University (N.N.).
Author information
Authors and Affiliations
Contributions
The experimental design was developed by K.Watashi. Virological and biochemical experiments were performed by C.K., J.M., and K.M. MD simulations were performed by T.E. and M.Ikeguchi. Statistical mechanical calculations were performed by K.O. and M.A. Original draft was written by C.K., T.E., M.A., M.Ikeguchi., and K.Watashi. with critical discussion by K.O., J.M., K.M., L.Q., Y.L., A.K., T.M., K.S., M.N., K.Wakae., M.Isogawa., M.M., U.O., N.N., S-Y.P., and C.S. Fundings were acquired by C.K., K.O., M.M., N.N., S-Y.P., M.A., M.Ikeguchi., and K.Watashi. The project was supervised by K.Watashi.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kobayashi, C., Ekimoto, T., Ooka, K. et al. Multistep receptor binding of the hepatitis B virus preS1 domain. Nat Commun 17, 102 (2026). https://doi.org/10.1038/s41467-025-68062-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68062-z
