
Abstract
Behçet’s syndrome (BS), a chronic vasculitis, leads to recurrent oral ulcers, severely impacting quality of life. Here we report the results of a randomized, double-blind, placebo-controlled trial (NCT04065672) to evaluate the efficacy and safety of low-dose interleukin-2 (LD-IL-2) in BS patients. 60 participants with active oral ulcers are randomly assigned to receive LD-IL-2 or placebo. The primary endpoint is the oral ulcer count at week 12. The LD-IL-2 group has significantly fewer oral ulcers than the placebo group (0.69 ± 1.05 vs. 1.57 ± 0.90, P = 0.001) with greater improvements in ulcer pain, disease activity, and quality of life. No infections or severe adverse events are observed in either group. LD-IL-2 expands regulatory T (Treg) cells and decreased the ratio of effector T cell to Treg cells. Thus, LD-IL-2 therapy might be an effective and safe treatment in BS patients and is associated with the modulation of CD4 + T cell populations.
Similar content being viewed by others

Introduction
Behçet’s syndrome (BS) is a chronic and relapsing systemic vasculitis that causes recurrent oral aphthous ulcers, along with genital ulcers, skin lesions, and uveitis1. Patients may also present with arthralgia, arterial aneurysms, venous and arterial thrombosis, gastrointestinal tract and neurological symptoms may be involved as well2. Recurrent relapsing and remitting oral ulcers are the most common manifestations of BS, which have a substantial deleterious effect on quality of life3,4. Although colchicine is recommended as the first-line treatment for mucocutaneous lesions, the efficacy of colchicine has been debated5,6,7. Second-line immunosuppressive treatments for mucocutaneous involvement include azathioprine, thalidomide, interferon-α, and tumor necrosis factor (TNF-α) inhibitors5. Biologics, including TNF-α inhibitors and tocilizumab, play an important role in the treatment of other organ involvement in BS, such as vascular, CNS, and eyes1,2. Several randomized, controlled trials have been conducted to assess the efficacy of Apremilast (a small molecule selective inhibitor of the phosphodiesterase 4 enzyme) for the treatment of BS oral ulcers, and this agent has been approved by the US FDA and EMA for the BS indication8,9,10, however, adverse events such as gastrointestinal side effects often limit its further use11,12. In addition, some BS patients were refractory to colchicine and/or Apremilast. Thus, there is an unmet need for therapies with better efficacy and lower adverse effects for BS patients.
Interleukin-2 (IL-2) is a cytokine known to induce CD4 + T cell activation and regulation13. Regulatory T cells (Tregs) are one of the subsets of CD4 + T cells, which inhibit the production and function of effector T cells (Teff) (including T helper 1 [Th1], Th2, Th17, and T follicular helper [Tfh] cells)14. IL-2 regulates the development, proliferation, and survival of Tregs15. Different doses of IL-2 produce divergent effects on T cell subsets, and low-dose IL-2 (LD-IL-2) has been shown to selectively increase Tregs and suppress the differentiation of Tfh and Th17 subsets in various autoimmune diseases as well as inflammatory diseases16. In these diseases, LD-IL-2 helps to maintain immune tolerance, prevent autoimmunity, and inhibit inflammatory responses. Recently, LD-IL-2 has been extensively studied and used in the treatment of auto-inflammatory diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and inflammatory bowel disease (IBD)17,18,19,20. Previous studies have demonstrated that the dysregulation of T cells is functionally involved in the development of BS, and LD-IL-2 promoted Treg restoration in BS patients21,22. These findings have motivated the exploration of LD-IL-2 for treating BS, and three small studies have reported efficacy for LD-IL-2 in treating BS patients. One of the studies showed efficacy for LD-IL-2 in a BS patient with oral ulcers, genital ulcers, and erythema nodosum23, while the other study monitored circulating Treg cells but did not formally assess clinical response19, and a recent open-label study showed the LD-IL-2 treatment response in 5/8 patients24.
Here, we report the results of a randomized, double-blind, placebo-controlled phase 2 clinical trial demonstrating the efficacy and safety of LD-IL-2 in BS. Regarding the primary endpoint, the LD-IL-2 group has a significantly lower mean number of oral ulcers than the placebo group at week 12, with some patients achieving a complete response. Importantly, no infections or severe adverse events are observed in either group. The study also implicates CD4 + T cell subsets in the clinical effects of LD-IL-2 in BS patients. These findings support the pursuit of using LD-IL-2 as an intervention for different phenotypes of BS with expanded trials and might also inform future studies to treat patients with other autoimmune diseases.
Results
Patient disposition and baseline characteristics
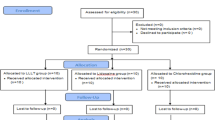
From October 2021 through July 2023, a total of 60 patients from Peking University People’s Hospital were deemed eligible and were randomly assigned to receive either LD-IL-2 (30 patients) or a placebo (30 patients). Of the 60 patients who underwent randomization, 56 completed the 12-week placebo-controlled period (26 in the LD-IL-2 group and 30 in the placebo group). Three patients in the LD-IL-2 group and two patients in the placebo group withdrew from the trial during the 12-week follow-up phase. Figure 1 shows the assignment of the patients to the trial groups and the reasons for discontinuation. In this trial, 23 females and 37 males were included, so the findings apply to both sexes. Patients were included regardless of sex or gender. There was no sex specific stratification implemented since no differences concerning efficacy and safety of LD-IL-2 in this regard were expected.

In a 1:1 ratio, 60 eligible patients were assigned to either the low-dose interleukin-2 (LD-IL-2) group or the placebo group by chance. The placebo-controlled treatment lasted for 12 weeks; 26 out of the LD-IL-2 group and 30 out of the placebo group completed the study. During the 12-week follow-up phase, three patients from the LD-IL-2 group and two from the placebo group withdrew from the trial.
The baseline demographic and disease characteristics of the patients, as well as their previous medications, were similar in the two trial groups (Table 1). Patients had a history of skin lesions (75%; including 27% erythema nodosum and 48% folliculitis at baseline), genital ulcers (40%), musculoskeletal involvement (20%), eye involvement (17%) (none of the patients had active uveitis at baseline, as judged by an ophthalmologist), and gastrointestinal involvement (12%). No patient had vascular or central nervous system (CNS) involvement. The concomitant medications in the two groups at baseline and week 12 are shown in Supplementary Tables 1, 2.
Primary outcome
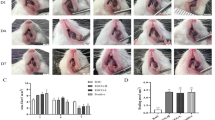
The mean number of oral ulcers per patient at baseline was 2.73 ± 2.20 (−95% CI, 1.84 to 3.62) in the LD-IL-2 group and 2.53 ± 2.08 (−95% CI, 1.75 to 3.31) in the placebo group. At week 12, the mean number of oral ulcers was significantly lower in the LD-IL-2 group than in the placebo group (0.69 ± 1.05, −95% CI, 0.27 to 1.12 vs.1.57 ± 0.90, −95% CI, 1.23 to 1.90, P = 0.001). The decrease in the number of oral ulcers was evident by week 4 in the LD-IL-2 group and was sustained throughout the full 12-week treatment phase (Fig. 2A). Percentage of patients who had a complete response to oral ulcers by week 12 was 61.5% in the LD-IL-2 group (16 of 26 patients) and only 16.7% in the placebo group (5 of 30 patients) (P < 0.001). The AUC of the total mean (±SE) number of oral ulcers during the 12-week placebo-controlled period was 85.6 ± 70.6 in the LD-IL-2 group, as compared with 147.9 ± 85.7 in the placebo group (least-squares mean difference, −62.3; 95% CI, −104.8 to −19.8, P = 0.005). The results regarding the primary and secondary endpoints are presented in Table 2 and Supplementary Table 5.

The clinical manifestation change from baseline in the Low-dose interleukin-2 (LD-IL-2) group(n = 26) and placebo group(n = 30) was shown and compared using a T-test with two-sided P values(degrees of freedom = 54, confidence intervals = 95%). No adjustments made for multiple comparisons. No replication. The treatment period is 0 to 12 weeks, and the follow-up period is 12 to 24 weeks. a The changes in the number of oral ulcers. Week 4 t = −2.59, P = 0.012. Week8 t = −4.40, P < 0.001. Week 12 t = −3.34, P = 0.002. b The changes in the number of genital ulcers. Week 4 t = −3.29, P = 0.002. Week 12 t = −2.45, P = 0.018. Week 24 t = −12.06, P < 0.001. c The changes in Behcet’s disease current activity form scores(BDCAF). Week 12 t = −3.00, P = 0.004. d The changes in visual-analog scale scores(VAS). Week 12 t = −2.22, P = 0.030. Data points represent mean values, and error bars indicate the standard error of the mean (SEM). *p < 0.05(LD-IL-2 vs. placebo). Source data are provided as a Source Data file.
Secondary clinical outcomes
A total of ten patients in the LD-IL-2 group and 13 patients in the placebo group had genital ulcers at baseline. The mean number of genital ulcers per patient at baseline was 0.69 ± 1.09 (−95% CI, 0.25 to 1.13) in the LD-IL-2 group and 0.67 ± 0.99 (−95% CI, 0.30 to 1.04) in the placebo group. The mean number of genital ulcers at week 12 was significantly lower in the LD-IL-2 group than in the placebo group (0 ± 0, −95% CI, 0 to 0 vs. 0.17 ± 0.38, −95% CI, 0.025 to 0.31, P = 0.023) (Fig. 2B). The percentage of patients who were free from genital ulcers at week 12 was 100% in the LD-IL-2 group (26 of 26 patients) and 83.3% in the placebo group (25 of 30 patients) (P = 0.055). A total of 51 patients completed week 24 of the trial. The prespecified analyses of the number of oral ulcers and genital ulcers at week 24 are shown in Fig. 2A, B, respectively.
At week 12, significant reductions from baseline in the pain from oral ulcers (as assessed on a 100-mm VAS) were observed for the LD-IL-2 group compared to the placebo group (LD-IL-2 group: −3.46, − 95% CI, −4.39to −2.53; placebo group: −1.67, − 95% CI, −2.48 to −0.85; P = 0.004). The mean change from baseline in the BSAS at week 12 was −20.12 (−95% CI, −24.54 to −15.69) in the LD-IL-2 group, which was significantly greater than for the placebo group (−6.47, −95%CI, −10.17 to −2.76) (P < 0.001). At week 12, LD-IL-2 group patients had more improvements in BDCAF value compared to the placebo group (LD-IL-2 group: −0.73, −95%CI, −1.10 to −0.36; placebo group: −0.47, −95% CI, −0.83 to −0.10; P = 0.303). The mean change of BD-QOL from baseline to week 12 was significantly greater in the LD-IL-2 group than in the placebo group (LD-IL-2 group: −3.42, −95%CI, −4.14 to −2.70; placebo group: −1.23, −95% CI, −1.57 to −0.89; P < 0.001).
During the follow-up period, three patients in the LD-IL-2 group developed new skin lesions, presenting as folliculitis. In the placebo group, there were two cases of new folliculitis, three cases of new arthralgia, and one case of new uveitis during the follow-up.
Safety
No infections were observed in either group during the treatment or follow-up periods. There were injection site reactions in four of the LD-IL-2 group patients (13.3%), and one patient (3.3%) discontinued the trial due to a severe injection reaction. There was one injection site reaction (3.3%) in the placebo group. Whereas there were no fevers for the placebo group, two patients (6.7%) in the LD-IL-2 group had fevers after the IL-2 injection. However, the fever abated spontaneously within 24 hours without any medical treatment (Supplementary Table 4).
Analysis of CD4 + T cell subsets in peripheral blood
Immunological analysis from patient peripheral blood samples included flow-cytometry-based enumeration of Tregs and Teff subsets (Th17 and Tfh cells). Briefly, the LD-IL-2 group displayed a significant expansion of Treg cells as a proportion of total CD4 + T cells (from 9.23% at baseline to 15.66% at week 12, P < 0.001) (Supplementary Fig. 1A), whereas no differences from baseline in the proportions of any examined cells were detected for the placebo group (Supplementary Fig. 1A–E). Moreover, the ratio of Teff (Tfh and Th17) cells to Tregs decreased rapidly following each LD-IL-2 administration (Supplementary Fig. 1D, E and Table 6).
Discussion
Management of Behçet’s syndrome is challenging due to the heterogeneous nature of the disease2. Different phenotypes of BS are thought to be governed by diversified mechanisms and may benefit from tailored therapeutic approaches25,26,27. In this double-blind, placebo-controlled study, we showed that LD-IL-2 effectively alleviates mucosal ulcers and associated pain in BS patients, with over 60% of patients achieving complete remission after 12 weeks. LD-IL-2 therapy significantly improved disease activity and quality of life (as indicated by reduced BSAS, BDCAF, and BD-QOL scores). It enabled substantial tapering of corticosteroids, with over one-third of corticosteroid users able to discontinue their use.
While there was a significant difference in the number of oral and genital ulcers between the two groups, the difference between changes in ulcers in each group was not highly significant (oral ulcers: p = 0.047, genital ulcers: p = 0.488). One potential explanation could be the small sample size of the study. Another explanation is that the relatively low number of ulcers at baseline (2.73 in this trial vs. 3.90 in the RELIEF trial19) which may have an impact on the statistical analysis. Therefore, clinical stratification in a larger sample size with more active BS patients would improve our understanding of the particular clinical features which likely to benefit from LD-IL-2 therapy.
LD-IL-2 has also demonstrated impressive clinical improvements in various skin disorders beyond BS, such as graft-versus-host disease (GVHD), HCV-associated vasculitis, and chronic urticaria28,29,30, mainly through modulation of tissue-resident regulatory Tregs in the skin30. Tregs help suppress local inflammation and autoimmunity in the skin, leading to improved skin health and symptom reduction. Our study observed a marked increase in Tregs and a decrease in Th17 cells in the LD-IL-2 group, which correlates with the clinical improvements seen in cutaneous manifestations. This shift in the immune cell balance is crucial for reducing inflammation and prom2506pt?>At week 12, the proportions of these patients for whom BDCAF decreased were 92% (23/25) in patients with skin lesions, 71% (5/7) with musculoskeletal involvement, 50% (2/4) with eye involvement and 100% (1/1) with gastrointestinal involvement. Analysis of organ-specific responses revealed that mucocutaneous and musculoskeletal involvement patients responded particularly well to LD-IL-2. However, the effect on other domains (ocular, gastrointestinal, vascular, and CNS) requires further investigation, as our trial did not include vascular or CNS involvement patients.
There were no serious adverse events observed in this study. Common adverse reactions such as injection site reactions and transient fever were mild and self-limiting. Importantly, there were no infections during the study period, aligning with previous findings that LD-IL-2 enhances the number and function of CD8 + T cells and NK cells31,32,33,34, which are required to build immune responses to infections.
In this study, we selected a dose of 1 million IU IL-2 administered every other day for 3 months. Our chosen dose falls within the range commonly employed in recent trials for conditions such as HCV-induced vasculitis (1.5–3 million IU)29, graft-versus-host disease (0.3–3 million IU per square meter of body-surface area)28, and type I diabetes (0.33–3 million IU)32. Within this range, LD-IL-2 has been shown to induce a dose-dependent increase in Tregs, while remaining associated primarily with mild, non-serious adverse events. The rationale for our dosing regimen was based on prior evidence of efficacy and safety in immune modulation and practical considerations for clinical application in patients with active disease35,36. Compared to other regimens, such as the protocol by ref. 19, which used 1 million IU daily for the first 5 days followed by 1 million IU every 2 weeks for 6 months to evaluate long-term immunomodulation, our approach features more frequent administration over a shorter period. This schedule may be preferable for the acute and intensive management of active BS, providing the potential for rapid symptom improvement and timely assessment of clinical response.
Interestingly, sustained improvement in disease activity scores persisted up to week 24, even after discontinuation of LD-IL-2 at week 12, despite a decline in expanded Tregs. This prolonged benefit may be explained by improved Treg suppressive function31,33 and a lingering reduction in pro-inflammatory cytokines37,38, effects that can outlast the active dosing period. Further immunological studies are needed to delineate these long-term effects and to optimize dosing regimens.
Different mechanisms have been investigated to clarify the effect of LD-IL-2 on autoimmune disease. In our previous trial of primary Sjögren Syndrome (pSS), the immunological analysis revealed that LD-IL-2 induced expansion of CD24highCD27+B cells, which is the key regulator in B cells31. NK cells are important both in keeping the autoimmune balance and protecting against viral infections. The CD56bright NK subset was preferentially expanded by LD-IL-2 in SLE patients33. In this study, although a mild increase in NK cell proportions and a decrease in B cell proportions were observed in the LD-IL-2 group after 12 weeks of treatment (Supplementary Table 7), these changes were not statistically significant. Further studies with larger sample sizes and detailed immunophenotyping are warranted to clarify the potential immunoregulatory mechanisms of LD-IL-2 in BS.
While LD-IL-2 demonstrated clear benefits in our study, several limitations remain to be addressed. First, excluding patients with vascular and central nervous system involvement restricts the generalizability of our findings. Second, notable improvements observed in the placebo group may be attributed to lingering effects of concurrent immunosuppressive therapies, such as thalidomide and colchicine, emphasizing a need for adequate washout periods and stratification by treatment history in future trials. Third, this was a single-center study with a relatively small cohort. Future larger sample sizes and multicentric studies with details of all background and previous therapies will be needed to gain insights into the therapeutic effects of LD-IL-2 in BS. Additional methodological concerns, including potential measurement bias and random confounding, should also be addressed in future research. Furthermore, the optimal dosing regimen for LD-IL-2 and its ability to sustain long-term remission in BS remain to be determined.
Importantly, our current data do not definitively establish whether LD-IL-2 therapy is superior to conventional treatments. Comparative studies directly evaluating the efficacy and safety of LD-IL-2 versus standard agents, such as colchicine, apremilast, and anti-TNF therapies, are required to clearly delineate its relative benefits and to determine its place in long-term disease management.
In summary, LD-IL-2 had a significant effect on BS patients by reducing oral ulcers and genital ulcers. The clinical benefits observed might result from the rise of Tregs and reduction of Teff cells, which may contribute to disease remission in BS.
Methods
Trial design and oversight
This was a Phase 2, randomized, double-blind, placebo-controlled, parallel-group, superiority design trial to evaluate the efficacy and safety of LD-IL-2 in Behçet’s syndrome patients with active oral ulcers or genital ulcers that did not respond to previous topical glucocorticoid or systemic treatment. The trial was conducted from Oct 2021 to Jul 2023, at Peking University People’s Hospital. Trial reporting conformed to the Consolidated Standards of Reporting Trials (CONSORT) reporting guidelines. The trial design is shown in Fig. 1. The protocol was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice guidelines and Chinese law for research involving human participants and was approved by the Peking University People’s Hospital Ethics Committee. All participants provided written informed consent. Full details of the trial can be found in the protocol.
Participants
Eligible patients were aged 18 to 70 years and fulfilled the criteria of the 1990 International Study Group for Behçet’s Disease39. All patients had at least one oral ulcer within 28 days before screening, and had at least two oral ulcers at the time of randomization, despite the previous use of at least one non-biologic medication, such as (but not limited to) topical or systemic glucocorticoids, non-steroidal anti-inflammatory drugs, colchicine, thalidomide, or immunosuppressants.
Patients were excluded if they had BS-related active major organ involvement, such as uveitis requiring systemic treatment or vascular or central nervous system involvement during the 12 months preceding trial entry or had a history of biologics usage, severe comorbidities, allergies to relevant reagents, active or chronic infection, or malignant neoplasm (see the protocol in the Supplement for details).
Sex was not considered in the study design, and it was determined based on self-report.
Randomization and masking
A simple randomization method was used to randomly group the participants, based on computer-generated random numbers prepared by a statistician who had no involvement in conducting the trial. Eligible participants were randomly assigned at a 1:1 ratio to receive either recombinant human IL-2 or a placebo in a blinded manner. Randomization was assigned by the order in which patients qualified for treatment. The investigators and the study participants were masked to the allocation sequence and the intervention (study drug containing LD-IL-2 or placebo). The study drug was packaged, labeled, and randomly assigned by an independent third party (Beijing Stemexel Technology Co). The packaging and appearance of the placebo were identical to those of the active drug. At the study site, the study drug was matched to the independent randomization schedule and then distributed to each randomized study participant.
Procedures
After a screening period that lasted up to 4 weeks, patients were randomized to receive IL-2 (recombinant human IL-2Ala125 [Beijing SL Pharma]) at a dose of 1 million IU or placebo (sterile water for injection containing the same adjuvant as LD-IL-2) subcutaneously every other day. After the initiation of the therapy, patients could continue with concurrent medication but were prohibited from changing or adding immunosuppression therapy during the study. After a 12-week placebo-controlled treatment period and a 12-week observational treatment-free follow-up, patients were assessed for clinical symptoms, and both routine laboratory tests and the peripheral blood lymphocyte subsets were assessed at each visit (weeks 0, 4, 8, 12, and 24).
End points and assessments
The primary efficacy endpoint was the mean number of oral ulcers at week 12. Secondary efficacy endpoints for the placebo-controlled phase included the change in pain from oral ulcers from baseline to week 12, as measured on a 100-mm visual-analog scale (VAS, with 0 representing no pain and 100 the worst pain ever experienced)40 and the change in disease activity from baseline to week 12. Disease activity was evaluated using the Behçet’s Syndrome Activity Score (BSAS, a scale ranging from 0 to 100, with higher scores indicating more active disease)41 and the Behcet’s Disease Current Activity Index score (BDCAF, ranging from 0 to 12, with higher scores indicating more active disease)42. Quality of life was evaluated at baseline and week 12 with the use of the Behçet’s Disease Quality of Life scale (BD-QOL, on which scores range from 0 to 30, with higher scores indicating greater impairment of quality of life)43.
Secondary endpoints for the placebo-controlled period included the mean number of genital ulcers at week 12, the proportion of patients with a complete response to oral ulcers (defined as the proportion of patients who had no oral ulcers at week 12), and the percentage of patients with genital ulcers at baseline who were ulcer-free at week 12. Secondary efficacy endpoints for the follow-up period were the mean number of oral ulcers and genital ulcers at week 24. The oral ulcers and genital ulcers were evaluated by a physician.
At each trial visit, the safety endpoints were assessed, including discontinuations, incidence, and severity of adverse events, serious adverse events, the relationship of such events to LD-IL-2, and pre-established events of special interest. Adverse events were coded using the Medical Dictionary for Regulatory Activities version 18.0.
Immunological analysis
Protocol-specific immunophenotypic analysis of peripheral blood lymphocyte subsets was performed at baseline and every 4 weeks until week 12 (weeks 0, 4, 8, and 12). Relative proportions of Treg, Tfh, and Th17 cell subsets were analyzed by flow cytometry using a FACSAria III instrument (BD) and FlowJo software (Tree Star). Tregs were defined as CD3+CD4+CD25highCD127low, Tfh cells as CD3+CD4+CD45RA-CXCR5highCCR7low. IL-17 was stained intracellularly by fluorophore-conjugated monoclonal antibodies to isolate Th17 from CD3+CD4+lymphocytes. (Supplementary Fig. 2 and Table 8). The Treg/Th17 and Treg/Tfh ratios were calculated based on these subsets.
For flow-cytometry analysis and sorting, single-cell suspensions were obtained and stained with the following monoclonal antibodies: CD3(SP34-2, dilution 1/100), CD4(RPA-T4, dilution 1/100), CD25(BC96, dilution 1/100), CD127(HIL-7R-M21, dilution 1/100), CD3 (SK7, dilution 1/100), CCR7 (2-L1-A, dilution 1/100), CXCR5 (RF8B2, dilution 1/100), PD-1 (EH12.1, dilution 1/100), CD45RA(HI100, dilution 1/100), CD4 (RPA-T4, dilution 1/100), IL-17A (N49-653, dilution 1/100).
Statistical analysis
The sample size of 26 patients per treatment group was chosen to provide 80% power to detect a treatment difference of 0.80 in the mean number of oral ulcers per patient between the placebo group and the LD-IL-2 group at week 12. This power calculation adopted a fixed superiority margin of 10%. Allowing for a 15% dropout rate, we aimed to recruit 30 participants for each group of the study.
Efficacy and safety outcomes were analyzed using the intention-to-treat (ITT) principle. Data for multiple comparisons and non-normally distributed data are expressed as medians [interquartile range (IQR)]. For normally distributed data, the results are presented as the mean ± standard deviation (SD). S-W test or K–S test was used to assess the Normality assumption. Differences between any two groups were analyzed using the Student’s t-test or the Mann–Whitney U-test as appropriate. Levene’s test was used to assess the homogeneity of variance. Quantitative outcomes were assessed with an analysis of the covariance (ANCOVA) model. Differences among the two groups were analyzed using the Kruskal–Wallis test followed by Dunn’s post-hoc test with Bonferroni correction. Correlations were analyzed with Spearman’s rank order test. A nominal significance level of 0.05 (two-sided) was applied to all of the statistical analyses, which were carried out using SPSS (version 20.0, IBM) or Graph Pad Prism (Version 5.0, Graph Pad Software).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available on request from the corresponding authors, Tian Liu and Jing He. The raw data were protected and are not publicly available due to data privacy laws. Access to the data is restricted to rheumatology specialists, with a response timeframe of 2 weeks and a validity period of three months. The datasets will be distributed by email upon reasonable request. The data used to generate all tables and figures in this study are provided in the Source Data file. Source data are provided with this paper.
References
Saadoun, D., Bodaghi, B. & &Cacoub, P. Behçet’s syndrome. N. Engl. J.Med. 390, 640–651 (2024).
Emmi, G. et al. Behçet’s syndrome. Lancet 403, 1093–1108 (2024).
Yazici, H. et al. Behçet syndrome: a contemporary view. Nat. Rev. Rheumatol. 14, 119 (2018).
Yazici, Y. et al. Behçet syndrome. Nat. Rev. Dis. Primers. 7, 67 (2021).
Hatemi, G. et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann. Rheum. Dis. 77, 808–818 (2018).
Leccese, P. et al. Management of skin, mucosa and joint involvement of Behçet’s syndrome: a systematic review for update of the EULAR recommendations for the management of Behçet’s syndrome. Semin. Arthritis. Rheum. 48, 752–762 (2019).
Alibaz-Oner, F. & Direskeneli, H. Advances in the treatment of Behcet’s disease. Curr. Rheumatol. Rep. 23, 47 (2021).
Hatemi, G. et al. Apremilast for Behçet’s syndrome-a phase 2, placebo-controlled study. N. Engl. J. Med. 372, 1510–1528 (2015).
Hatemi, G. et al. Trial of apremilast for oral ulcers in Behçet’s syndrome. N. Engl. J. Med. 381, 1918–1928 (2019).
Hatemi, G. et al. Apremilast for oral ulcers associated with active Behçet’s syndrome over 68 weeks: long-term results from a phase 3 randomised clinical trial. Clin. Exp. Rheumatol. 132, 80–87 (2021).
Lopalco, G. et al. Real-world effectiveness of apremilast in multirefractory mucosal involvement of Behçet’s disease. Ann. Rheum. Dis. 78, 1736–1737 (2021).
Takeno, M. et al. Apremilast in a Japanese subgroup with Behçet’s syndrome: results from a Phase 3, randomised, double-blind, placebo-controlled study. Mod. Rheumatol. 32, 413–421 (2022).
Klatzmann, D. & Abbas, A. K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Rheumatol. 15, 283–294 (2015).
Dominguez-Villar, M. & Hafler, D. A. Regulatory T cells in autoimmune disease. Nat. Immunol. 19, 665–673 (2018).
Boyman, O. & Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Rheumatol. 12, 180–190 (2012).
Liao, W., Lin, J. X. & Leonard, W. J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 38, 13–25 (2013).
He, J. et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 22, 991–993 (2016).
Zhang, X. et al. Efficacy and safety of low-dose interleukin-2 in combination with methotrexate in patients with active rheumatoid arthritis: a randomized, double-blind, placebo-controlled phase 2 trial. Signal Transduct. Target Ther. 7, 67 (2022).
Rosenzwajg, M. et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann. Rheum. Dis. 78, 209–217 (2019).
Cheng, J. et al. Low-dose interleukin-2 for the treatment of systemic lupus erythematosus with recurrent rash. Rheumatol. Autoimmun. 4, 265–266 (2024).
Liu, X. et al. Low-dose IL-2 effectively restored decreased regulatory T cells in patients with Behçet’s disease. Clin. Exp. Rheumatol. 39, 746–752 (2021).
Geri, G. et al. Critical role of IL-21 in modulating TH17 and regulatory T cells in Behçet disease. J. Allergy. Clin. Immunol. 128, 655–664 (2011).
Zhou, W. et al. Refractory Behçet’s disease treated with low-dose interleukin-2: a case report. Medicine 101, e31173 (2011).
Lorenzon, R. et al. The universal effects of low-dose interleukin-2 across 13 autoimmune diseases in a basket clinical trial. J. Autoimmun. 144, 103172 (2024).
Bettiol, A. et al. Treating the different phenotypes of Behçet’s syndrome. Front. Immunol. 10, 2830 (2019).
Soejima, Y. et al. Changes in the proportion of clinical clusters contribute to the phenotypic evolution of Behçet’s disease in Japan. Ann. Rheum. Dis. 23, 49 (2021).
Zhong, H. et al. Efficacy and safety of tocilizumab in Behçet’s syndrome with refractory arterial lesions: a single-centre observational cohort study in China. Rheumatology 61, 2923–2930 (2022).
Koreth, J. et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 365, 2055–2066 (2011).
Saadoun, D. et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N. Engl. J. Med. 365, 2067–2077 (2011).
Wang, J. Q. et al. Consecutive injections of low-dose interleukin-2 improve symptoms and disease control in patients with chronic spontaneous urticaria. Clin. Immunol. 247, 109247 (2023).
He, J. et al. Efficacy and safety of low-dose interleukin 2 for primary Sjögren syndrome: a randomized clinical trial. JAMA Netw. Open 5, e2241451 (2022).
Hartemann, A. et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet. Diabetes Endocrinol. 1, 295–305 (2022).
He, J. et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 79, 141–149 (2022).
Miao, M. et al. Short-term and low-dose IL-2 therapy restores the Th17/Treg balance in the peripheral blood of patients with primary Sjögren’s syndrome. Ann. Rheum. Dis. 77, 1838–1840 (2018).
Miao, M. et al. Treatment of active idiopathic inflammatory myopathies by low-dose interleukin-2: a prospective cohort pilot study. Rheumatol. Ther. 8, 835–847 (2021).
Zhufeng, Y. et al. Modification of intestinal microbiota dysbiosis by low-dose interleukin-2 in dermatomyositis: a post hoc analysis from a clinical trial study. Front. Cell Infect.Microbiol. 12, 757099 (2022).
Kolios, A. G. A. et al. Interleukin-2 and regulatoryT cells in rheumatic diseases. Nat. Rev. Rheumatol. 17, 749–766 (2021).
Tchitchek, N. et al. Low-dose IL-2 shapes a tolerogenic gut microbiota that improves autoimmunity and gut inflammation. JCI Insight 7, e159406 (2022).
International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 335, 1078–1080 (1990).
Yilmaz, S. et al. Patient-driven assessment of disease activity in Behçet’s syndrome: cross-cultural adaptation, reliability and validity of the Turkish version of the Behçet’s syndrome activity score. Clin. Exp. Rheumatol. 31, 77–83 (2013).
Hatemi, G. et al. Core set of domains for outcome measures in Behçet’s syndrome. Arthritis. Care Res. 74, 691–699 (2022).
Bhakta, B. B. et al. Behçet’s disease: evaluation of a new instrument to measure clinical activity. Rheumatology 38, 728–733 (1999).
Gilworth, G. et al. Development of the BD-QoL: a quality of life measure specific to Behçet’s disease. J. Rheumatol. 31, 931–937 (2014).
Acknowledgements
We express our gratitude to all patients who participated in this trial. The authors thank Dr. John Hugh Snyder for providing medical writing support. This study was supported by grants from the National Natural Science Foundation of China (92374202 and 32141004 to Z.L., 32441099 to F.H., 82202003 to J.C.), National Key R&D Program of China (2022YFE0131700 to J.H.), Shenzhen Medical Research Fund (C2404002 to Z.L.), Bethune-Puai Medical Research Fund (PAYJ-023 to T.L.), Beijing High-Level Innovation and Entrepreneurship Talent Program (G202522095 to X.S.) and Beijing Nova Program (20220484206 to J.H.).
Author information
Authors and Affiliations
Contributions
T.L., J.H., and Z.L. designed the research; T.L., W.Z., Y.Z., J.C., X.X., B.H., R.F., H.Y., S.C., X.Z., M.S., C.L., and Y.J. conducted the clinical trial, enrollment and managed the patients. X.L., X.Z., Y.Z., and R.L. performed the analysis of peripheral blood lymphocyte subsets. X.Z., Y.S., H.L., J.L., M.M., and Y.J. analysed and interpreted the data. T.L., W.Z., Y.Z., and W.X. wrote the paper; F.H., J.G., X.S., J.H. and Z.L. revised the paper. The decision to submit the paper for publication was made jointly by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Vinod Ravindran, Michelle Rosenzwajg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, T., Zhou, W., Zhu, Y. et al. Efficacy and safety of low-dose interleukin 2 for Behçet’s syndrome: a randomized, placebo-controlled, double-blind, phase 2 clinical trial. Nat Commun 17, 1344 (2026). https://doi.org/10.1038/s41467-025-68100-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68100-w
This article is cited by
-
Mundschleimhauterkrankungen im Kindesalter
hautnah (2026)
