
Abstract
Patients with recurrent high-grade glioblastoma have a median survival of 6-8 months, with limited therapeutic options. In recent years, interest has grown in applying chimeric antigen receptor T (CAR-T) cells to solid cancers, including advanced gliomas. Here we generated off-the-shelf CRISPR-Cas9–edited IL-13Rα2-specific allogeneic universal CAR-T cells (MT026) by disrupting the endogenous TCR to prevent graft-versus-host disease and knocking out HLA class I molecules to mitigate the host-versus-graft response, and observed minimal NK-cell–mediated rejection in preclinical studies. In a first-in-human, single-center, open-label investigator-initiated trial (ChiCTR2000028801) in patients with high-grade glioma with prior therapy failure and short life expectancy, intrathecal injection of MT026 via lumbar puncture (1.0-3.0×10^7 cells per dose) demonstrated favorable tolerability and safety (primary outcome), pharmacokinetic characteristics, and preliminary clinical activity (secondary outcomes). Among the five patients enrolled, one achieved a complete response and three achieved partial responses. No grade ≥3 adverse events were observed; the predominant treatment-related toxicities were grade 1-2 pyrexia, hypoxia, and vomiting. Trial enrolment was halted after enrolment of the first five patients, however these preliminary clinical data support the potential benefit of locally administered allogeneic universal CAR-T cell therapy for recurrent glioblastoma.
Similar content being viewed by others

Introduction
Glioblastoma accounts for over 49.1% of newly diagnosed primary brain malignancies each year1. The current standard of care for glioblastoma includes maximal safe surgical resection followed by radiation, chemotherapy with temozolomide, and tumor-treating fields2,3. However, nearly all glioblastomas relapse after standard-of-care treatment, and patients survive only 25–30 weeks4. For recurrent disease, standard-of-care options are suboptimal, and effective therapeutic approaches remain to be established. Therefore, patient participation in clinical trials is recommended per the NCCN guideline2.
Administration of chimeric antigen receptor T (CAR-T) cells into the CSF allows bypassing the blood–brain barrier and may help overcome the immunosuppressive glioma microenvironment by delivering a preponderant supply of tumor-specific T cells to immunologically “cold” gliomas, which kill tumor cells by homing to tumor-associated antigens5.
IL-13Rα2, expressed in over 75 % of glioblastomas and associated with increased tumor invasiveness and poor prognosis, is a well-recognized CAR-T target5,6,7,8. Intrathecal administration of autologous CAR-T cells targeting IL-13Rα2 for glioblastoma has been reported with favorable tolerance and tumor regression in a single patient9. However, autologous T cell immunotherapy for glioblastoma may face obstacles due to suboptimal T cell quality and quantity. The patient-specific nature and complex manufacturing process make autologous cell generation costly and time-consuming. The fact that patients with recurrent high-grade glioma have survival measured in months in the absence of effective therapies10,11 calls for more efficient CAR-T production. Developing off-the-shelf, on-demand allogeneic CAR-T to overcome exhaustion and preserve effector function and persistence is desirable to achieve complete clinical potential.
Nevertheless, intrinsic TCRs on allogeneic T cells can recognize recipient alloantigens, which may result in graft-versus-host disease (GvHD). In addition, expression of HLA molecules on the surface of allogeneic T cells can trigger prompt rejection by the host immune system through host-versus-graft reaction (HvGR)12.
Recent advances in genome-editing technologies have greatly expanded the ability to modify genomic sequences as desired13,14,15. The Major Histocompatibility complex class I (MHC-I), known as human leukocyte antigen class I (HLA-I), includes the classical HLA-A, HLA-B, and HLA-C molecules. HLA matching affects clinical outcomes of hematopoietic stem cell transplantation16. Numerous studies have aimed to generate hypoimmunogenic induced pluripotent stem cells or to refine hematopoietic stem cell transplantation (HSCT) to minimize immune rejection caused by HLA-type mismatches17,18,19,20.
To modulate immune responses mediated by T cells, NK cells, and macrophages against transferred cells, researchers have tested knocking out beta 2-microglobulin (B2M) to eliminate HLA class I surface expression, knocking out CIITA to inhibit MHC class II expression, and overexpressing HLA-E and CD47 to reduce NK- and macrophage-mediated rejection17,19,20,21. The primary challenges in employing allogeneic T cells are GvHD and HvGR. The former can be mitigated by removing the TCR, typically achieved through knockout (KO) of the constant domain in either the α or β chain, or by substituting specific TCR components to impair antigen recognition22. Although disruption of the common subunit β2-microglobulin (B2M) completely prevents HLA-I surface expression, it may render the cell susceptible to NK-cell lysis23. However, knocking out specific subunits of HLA-I molecules alone still carries the risk of host T cell attacks against the remaining HLA molecules. Novel approaches to bypass surveillance by the host immune system of allogeneic universal CAR-T (UCAR-T)cells have been intensively investigated12,19,24,25.
Here, we successfully generated MT026 by knocking out TRAC and disrupting HLA-I α chains, enabling control of GvHD and mitigation of HvGR. We present an alternative research approach and show that disruption of most HLA class I molecules enhances the persistence of IL-13Rα2-specific allogeneic CAR-T cells in patients with recurrent glioblastoma. Data from a first-in-human, single-center, open-label investigator-initiated trial (IIT; ChiCTR2000028801) using CRISPR-Cas9–edited universal CAR-T therapy, delivered intrathecally to tumor sites, demonstrated favorable tolerability, safety, pharmacokinetic (PK) characteristics, and preliminary efficacy of IL-13Rα2-targeted allogeneic UCAR-T therapy.
Results
Preparation and characterization of allogeneic UCAR-T cells
We initially generated allogeneic UCAR-T cells by transducing a lentiviral vector encoding an anti-IL-13Rα2 CAR into CD3-positive T cells. To minimize both GvHD and HvGR in recipients of allogeneic UCAR-T cells, we designed four sgRNAs targeting the first exon of the TRAC gene and multiple sgRNAs for the HLA-I α chain, rather than the B2M gene, in PBMCs from an HLA-A2–positive donor. Cas9/sgRNA RNP complexes were electroporated, and we screened for the single sgRNA that produced the highest disruption efficiency. Finally, multiple batches suitable for clinical use were stably manufactured after process development and optimization (Supplementary Table 1). Vector copy number (VCN) analysis showed an average of 1.13 ± 0.38 integrations in UCAR-T cells (Supplementary Fig. 1a–d). Surface expression levels of TCR and HLA-I, as well as the phenotype, were confirmed by flow cytometry to be marginal. The percentage of CAR-positive cells was 45.3%, and the percentages of TRAC knockout (by CD3-negative staining) and HLA-I knockout (by HLA-ABC staining) were 99.52 % and 95.23 %, respectively (Fig. 1a; and Supplementary Fig. 1c). Moreover, expression of the early activation markers CD69 and CD137 was assessed in vitro following co-culture with U251 target cells to determine the activation level of allogeneic UCAR-T cells. The flow cytometry data indicated that double knockout of TCR and HLA-I enabled activation by target cells (Fig. 1b).

a Expression of TCR and HLA-I T cells in double-knockout T cells. b Evaluation of T cell activities in double-knockout CAR-T cells. c NK cells were mixed with K562 cells or UCAR-T cells or WT T cells at a 2:1 ratio for 24 h. Data are from 1 of 3 similar experiments. d GvHD modeling assay to validate the HLA-TRAC dual-editing strategy: MT026 cells (TCR-knockout UCAR-T) showed no proliferation when co-cultured with allogeneic PBMCs for 120 h, contrasting with significant expansion in conventional CAR-T (TCR-intact) and WT T cells. e Cell count of CAR-T cells with different knockout strategies co-cultured with allogeneic PBMCs (NK-cell depleted) for 12 days was detected. f Cell count of cells with distinct knockout strategies co-cultured with allogeneic NK cells for 24 h was measured. Representative data from a single donor are shown. Each sample was set with 3 technical replicates, and the experiment was performed at least in 3 biologically independent repeats. Data from all three donors are provided in Supplementary Table 1. Source data are provided as a Source Data file.
Off-target activity assessment
To address concerns about off-target specificity and chromosomal stability of the dual TRAC-HLA CRISPR-Cas9-edited MT026 product, we used orthogonal genome-wide profiling methods (AID-seq, PEM-seq, GUIDE-seq, and iGUIDE-seq) to systematically characterize genetic perturbations.
AID-seq identified 465 (Sg1/TRAC) and 1,491 (Sg2/HLA) putative off-target sites (Supplementary Table 2), with no overlap across sgRNA-specific loci. GUIDE-seq and iGUIDE-seq detected minimal off-target editing in dual-edited samples (sample 1: 56–62 sites; sample 2: 41–56 sites), with only one coding-region hit (MUC4; < 0.1% clone fraction) indicating negligible oncogenic risk. Single-gene–edited controls (TRAC: 106–129 sites; HLA: 42–48 sites) showed comparable off-target profiles, confirming specificity that is independent of sgRNA.
PEM-seq revealed low-frequency translocations at on-target loci (TRAC: 10.62%, HLA: 1.79 %). No individual translocation event exceeded a mutation frequency of 5 %, and none involved cancer-related genes (Supplementary Table 3). A notable 4.4 % translocation frequency at the CD8 locus was excluded from analysis due to junctional criteria but warrants clinical monitoring for functional impact.
Amplicon sequencing with CRISPResso2 analysis (20-bp quantification window ± 10 bp from cut sites) confirmed 100 % on-target editing specificity in dual-edited MT026 cells (samples 1 and 2), with no detectable off-target modifications in flanking regions (Supplementary Table 4). Collectively, these multilayered assessments demonstrate: 1) sgRNA-specific off-target profiles with limited coding region impact, 2) low structural variation risk at therapeutic thresholds, and 3) precise on-target editing, supporting the safety and clinical translatability of this dual-edited UCAR-T product.
Evaluation of the effects of HLA-TRAC dual-editing strategy on TCR and HLA-I functions
To validate the efficacy of the HLA–TRAC dual-editing strategy in mitigating GvHD and HvGR risks, we systematically evaluated the functional consequences of TCR and HLA-I disruption.
To verify the protective role of the HLA-I gene-editing strategy, we used allogeneic NK cells with IL-2 to enhance NK function. UCAR-T cells were co-cultured with NK cells for 24 h. Only 20–30 % of UCAR-T cells were lysed by NK cells, whereas the positive-control cell line K562, which lacks HLA-I expression, exhibited ~60 % lysis. As expected, WT T cells with HLA-I expression resisted killing (Fig. 1c). In order to further evaluate whether the dual knockout allogeneic UCAR-T cells would induce GvHD on recipients as well as HvGR, which were the common issues of allogeneic UCAR-T cells, UCAR-T generated by different knockout strategies were challenged by allogeneic PBMCs. MT026 cells (TCR-knockout UCAR-T) showed no proliferation when co-cultured with allogeneic PBMCs, in stark contrast to conventional CAR-T cells (TCR-intact) and WT T cells, which exhibited significant activation and expansion (Fig. 1d).
For HvGR evaluation, we conducted co-culture experiments using cells with different knockout strategies—HLA-I KO (α-chain knockout), TCR KO, TCR–B2M dKO, and MT026 (TCR and HLA-I dKO), with allogeneic PBMCs (NK cell-depleted) or purified NK cells. Knocking out HLA-I effectively prevented rejection by allogeneic T cells (Fig. 1e). However, these cells still did not evade rejection by NK cells, with effects similar to those observed for the established B2M knockout strategy (Fig. 1f). These findings highlight the need to integrate HLA-I α-chain knockout with NK-cell suppression strategies or to develop multi-targeted engineering approaches to mitigate residual NK cell–mediated rejection, thereby supporting the development of safer and more effective allogeneic CAR-T therapies.
IL-13Rα2 allogeneic UCAR-T cells potently inhibit glioma growth in vitro and in vivo
To determine whether CRISPR/Cas9-mediated knockout of TCR and HLA-I affects the effector function of IL-13Rα2 allogeneic UCAR-T cells relative to conventional CAR-T cells expressed using a viral system, we first performed in vitro cytotoxicity assays against U251 target cells (Supplementary Fig. 1f; Fig. 2a). The results indicate that the killing activity of IL-13Rα2 allogeneic UCAR-T cells with HLA-I knockout (MT026) was equivalent to that of allogeneic UCAR-T cells with intact HLA-I.

a The U251 tumor cells were co-cultured with MT026 (Anti-IL-13Rα2 allogeneic UCAR-T) cells from two different donors or CAR-T cells, and the specific killing of tumor cells was measured by flow cytometry over 24 h, with 3 biologically independent experiments for each sample. Error bars represent mean ± SD. n = 3 for CAR-T and MT026 at effector:target ratios of 0.5:1 (CAR-T mean = 38.75, MT026 mean = 39.81), 1:1 (CAR-T mean = 51.28, MT026 mean = 52.19), 2:1 (CAR-T mean = 60.78, MT026 mean = 62.09), 5:1 (CAR-T mean = 70.83, MT026 mean = 68.94), and 10:1 (CAR-T mean = 79.86, MT026 mean = 79.03). Differences are not significant (ns), calculated by two-way ANOVA. b Fluorescence images of the GFP-labeled organoids co-cultured with IL-13Rα2 CAR-T cells at various E: T ratios for 0, 12, 24, 48, and 72 h. Scale bar, 3000 μm; c Quantification of the relative fluorescence intensity of GFP-labeled organoids in panel B was performed using Image J software, with 4 biologically independent samples for each sample. Error bars represent ± SDs. ****p < 0.0001 calculated by two-way ANOVA. d Schematic illustration of the in vivo experiment; e Kaplan-Meier survival curves of the Patient-Derived Orthotopic Xenograft (PDOX) mouse models, which were intratumorally injected with IL-13Rα2 CAR-T cells in (d), n = 8 mice per group. Significance of difference was determined by two-tailed log rank test. ****p < 0.0001. f Representative bioluminescent images (BLI) of the Patient-Derived Orthotopic Xenograft (PDOX) mouse models, which were intratumorally injected with IL-13Rα2 CAR-T cells in (d), n = 8 mice per group. g Tumor burden measured by BLI in mice of each treatment group in (d), n = 8 mice per group. Error bars represent ± SDs. **p = 0.0031, ****p < 0.0001 calculated by two-way ANOVA. Source data are provided as a Source Data file.
We next investigated optimal E:T ratios by co-culturing glioma organoids expressing a GFP reporter in a time-dependent manner (Fig. 2b). Relative GFP fluorescence intensities of glioma organoids were quantified. A visible reduction in GFP fluorescence after > 48 h indicated progressive elimination of glioma organoids (Fig. 2c). As the E:T ratio increased to 5:1, the cytotoxicity of IL-13Rα2 allogeneic UCAR-T cells was maximized, consistent with the cytotoxicity assays (Fig. 2a).
Given the pronounced efficacy of IL-13Rα2 allogeneic UCAR-T cells in vitro, we further assessed antitumor effects in a patient-derived orthotopic xenograft (PDOX) model using female 6-week-old BALB/c-nu mice. Ten fully formed organoids (1 mm diameter) were transplanted into the brain. Glioma organoid cells were transduced to express a luciferase reporter, enabling quantification throughout the study by bioluminescence imaging (BLI; Fig. 2d).
The survival curve of the PDOX model indicated that IL-13Rα2 allogeneic UCAR-T therapy suppressed tumor progression in vivo and improved survival, whereas mice in the no-treatment control (NT) group exhibited continuous tumor proliferation, resulting in death from day 30 onward (Fig. 2e). Brain tumor growth was significantly suppressed for 7 days after intratumoral injection of IL-13Rα2 allogeneic UCAR-T cells; however, slight tumor recurrence was observed since 14th day after injection (day 29 after model initiation) (Fig. 2f). Quantitative analysis of BLI in PDOX mice was consistent with survival results: the control group showed a substantial increase in bioluminescence, whereas the experimental group exhibited only a minimal increase (Fig. 2g). Taken together, the in vitro and in vivo data indicate that IL-13Rα2 allogeneic UCAR-T therapy effectively inhibits tumor growth and confers sustained tumor suppression in model animals.
Clinical trial - patients and treatment overview
The key objective was to assess the clinical feasibility, tolerability, and safety of intrathecal administration of CRISPR-Cas9–edited allogeneic IL-13Rα2 UCAR-T cells for treatment of recurrent high-grade glioma (WHO grades 3 and 4) after standard tumor resection with chemoradiotherapy. The study, which ran from August 2020 to July 2022, faced operational challenges due to COVID-19 restrictions. As a result, enrollment, dose-escalation schedules, and assessment schedules were impacted. Five eligible patients with recurrent IL-13Rα2-overexpressing glioblastoma out of a planned twelve were enrolled and received study treatment and assessments. When visits were missed due to pandemic restrictions, follow-up calls were performed to monitor patient safety and survival status. All patients were followed until death upon the development of disease progression. The derived data were valuable for understanding preliminary safety and therapeutic response to CRISPR-Cas9–edited allogeneic IL-13Rα2 UCAR-T cells delivered intrathecally in a patient context.
All five patients were Chinese, with a mean age of 52 years (range, 45–63), and four were women. Four patients had KPS < 70, and one had KPS 90; the mean KPS was 60. Four of five had medium IL-13Rα2 expression, and one (MT026-02) had high expression before study entry (Table 1). All patients shared the same prognostic genetic profile: IDH-wildtype and MGMT promoter–unmethylated. Each patient received at least one round of conventional therapy, including surgical resection and systemic chemotherapy (temozolomide) and radiation, before study entry.
Patients received more than four intrathecal injections of CAR-T cells, totaling 1.0 × 107 to 3.0 × 107 cells over 3.4 months of exposure, with the longest course comprising eight injections over up to 7.9 months (MT026-02) before disease progression (Supplementary Fig. 2). At the PI’s discretion, patient MT026-02 continued with two additional study-drug administrations after concurrent surgical treatment with chemoradiation at progression (Supplementary Fig. 2b, c).
After disease progression in patient MT026-003, a tumor biopsy was performed, and other investigational targeted cell therapies—EGFR-targeted or B7H3-targeted cells—were administered based on new targets identified by immunohistochemistry (Supplementary Fig. 2d).
Clinical bioactivity of IL-13Rα2 allogeneic UCAR-T cell administration via an intrathecal route
Of five patients, CSF CAR DNA copies were consistently elevated 1 day after each of the first five study-drug administrations, peaked 2–4 days after infusion, and remained detectable between consecutive injections with repeated intrathecal dosing in patient MT026-005. CSF CAR DNA copies reached maximal levels from baseline after the fourth intrathecal injection in patients MT026-004 and MT026-005. (Fig. 3a, b and Supplementary Fig. 3). No CAR DNA copies were detected in peripheral blood at any time point (data not shown). While the observed high CAR DNA peaks suggest that cell expansion may have occurred following intrathecal injection, we cannot definitively confirm expansion of the cells in vivo.

a, b CAR DNA copies and IL-6 concentrations in cerebrospinal fluid of MT026 before and 1 day after each administration. Baseline value is defaulted to zero. The administration interval is ~1 month. c Change of multiple cytokines in CSF before and after the 4th MT026 administration in Patient 005. d IL-6 concentrations in cerebrospinal fluid and blood of MT026 after different administration. Error bars represent ± SDs. Biological replicates: for CSF, n = 3 per group for 1st-4th, n = 1 for 5th; for PB, n = 2 per group for 1st-4th, n = 1 for 5th. e Change of blood lymphocyte count with treatment of MT026 in the 5 patients. f Change of neutrophils to lymphocytes ratio in blood with treatment of MT026 in the 5 patients. Source data are provided as a Source Data file.
Effector cytokines, including TNFα, IFNγ, IL-2, IL-6, IL-8, and IL-10, and chemokines, including CCL2 and CXCL10, increased in CSF after intrathecal delivery of the study drug in patient MT026-005 (Fig. 3c). These changes, exemplified by CSF IL-6 elevation, appeared temporally associated with increases in CSF CAR DNA copies, as observed in patients MT026-004 and MT026-005.
Peripheral blood IL-6 concentrations were elevated after each CAR-T administration; however, levels were lower than in CSF, with CSF concentrations 79- to 647-fold higher than blood. These data suggest a more localized effector response at tumor proximity (patients MT026-004 and MT026-005), Fig. 3d).
In Patient MT026-005, the blood lymphocyte count was low at baseline and increased with treatment. In this patient, the blood neutrophil-to-lymphocyte ratio was high at baseline and decreased in response to treatment (Fig. 3e, f). These results are consistent with the notion that early NLR reduction is significantly associated with better prognosis in mCRC patients treated with immunotherapy26.
Safety of repeated exposure of IL-13Rα2 allogeneic UCAR-T cell administration via an intrathecal route
During the study, study drug–related adverse events included fever (4/5, 80 %), hypoxia (2/5, 40 %), vomiting (2/5, 40 %), and headache (1/5, 20 %) (Table 2). These AEs were grade 1 or 2 and resolved or stabilized spontaneously or with optimal management. Reported CSF laboratory abnormalities were increased interleukin-6 (5/5, 100 %), increased total protein (4/5, 80 %), and increased lactate dehydrogenase (3/5, 60 %). Serum laboratory abnormalities included elevated IL-6 in two of five patients (2/5, 40 %) and elevated C-reactive protein in three of five patients (3/5, 60 %). These elevated cytokine and proinflammatory markers were most likely part of downstream signaling stimulated by CAR-T immunotherapy delivered into the CSF space and circulating in the blood, as described above.
No grade ≥3 AEs, SAEs, immune effector cell–associated neurotoxicity syndrome (ICANS), GvHD, or infections were reported during the study. Only Grade 1 AEs were observed, including myelosuppression (decreased lymphocytes and leukocytes), leukocytosis, and Grade 1 cytokine release syndrome (CRS) manifesting as fever (Table 3). In summary, repetitive dosing of up to 8 infusions of IL-13Rα2-specific CAR-T cell clones, with doses ranging from 1.0 × 107 to 3.0 × 107 cells administered into the CSF space via an intrathecal route, exhibited an acceptable safety profile with limited, transient, low-grade adverse events.
Evidence of preliminary therapeutic benefit of IL-13Rα2 allogeneic UCAR-T cell intrathecal administration in a patient context
Although therapeutic benefit could not be established in this small cohort, we report individual patient–level data on the preliminary efficacy of this CRISPR-Cas9–edited allogeneic IL-13Rα2–specific UCAR-T product administered intrathecally in refractory high-grade glioma.
Antitumor effect was assessed using the Immunotherapy Response Assessment in Neuro-Oncology (iRANO) criteria. All five patients showed a decrease in tumor size after treatment (Fig. 4a–e; and Supplementary Fig. 4). Among the five patients, the objective response rate (ORR) was 80 % (Fig. 4d, f; and Table 1), comprising one complete response, three partial responses, and one case of stable disease (Fig. 4f). The median duration of response was 3.8 months, with a maximum of 7.4 months (Fig. 4f; and Table 3). Recognizing the limitations of cross-study comparisons, the 80% overall response observed with MT026 appears numerically higher than reported rates with bevacizumab, 25.9 % (95 % CI, 17.0 %, 36.1 %) and 19.6% (95 % CI, 10.9 %, 31.3 %) in two studies27. An exceptionally high response rate (4/5) was observed in this difficult-to-treat population (Table 4).

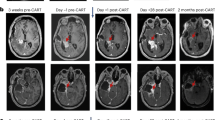
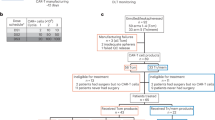
a CONSORT Flow diagram. b Study Schema. c Disease development over time in the five patients. d The ratio of cross-sectional area of the target lesion relative to baseline at each assessment point as the rate of tumor growth. Black, red, orange, and purple triangles indicate IL-13Rα2-, B7-H3-, HER2-, and EGFR-targeted CAR-T therapies, respectively. e MRI assessment of tumor size at baseline and best overall response after intra-thecal, via lumbar puncture injection of MT026 in the five patients. The number above the MRI image represents the exact scanning time, the characters on the right side of the image indicate the cycle count, the best evaluation result, and the number of days after the first administration. C represents cycle, CR complete response, PR partial response, SD stable disease. f The best percent change from baseline in tumor size in the five patients, where the data cutoff date was July 6, 2022 (median follow-up, 8.4 months). Source data are provided as a Source Data file.
Patients MT026-001 to MT026-005 survived 7.6, 17.2, 8.3, 10.5 and 5.7 months, respectively, after the first MT026 infusion, with a median OS of 8.3 months (95 % CI, 6.7-9.8), a maximum survival of 17.2 months, and a 12-month OS rate of 20.0% (95 % CI, 3.5-100 %). When survival was calculated from the time of recurrence, the same patients survived 12.8, 33.2, 9.2, 13.7, and 13.1 months, respectively, yielding a median overall survival of 13.1 months (95 % CI, 12.6-13.6) and a 12-month overall survival rate of 80% (95 % CI, 51.6-100 %). The longest survival was 33.2 months for Patient MT026-002 (Table 1). Survival in these high-grade patients with glioma appeared numerically better than the expected poor median overall survival of ~6–8 months following recurrence4, although comparisons across studies were limited. Mean progression-free survival was 5.6 months (95 % CI: 3.1-8.2), with a maximum of 7.9 months, and the 6-month progression-free survival rate was 40.0 % (95 % CI: 11.8-76.9 %). Two of the five patients (MT026-002 and MT026-003) subsequently received additional investigational CAR-T cell therapies as part of subsequent treatment, as indicated in Fig. 4c. Patient MT026-002 received two administrations of B7H3 targeted CAR-T cells and one administration of HER2 targeted CAR-T cells, whereas Patient MT026-003 received one administration each of B7H3 and EGFR targeted CAR-T cells.
In summary, this study of five subjects with recurrent high-grade glioma demonstrated the feasibility of treating these subjects with engineered allogeneic CAR-T cells via intrathecal delivery to tumor sites. The absence of therapy-related serious adverse events and the potential clinical benefit warrant further investigation.
Discussion
Recent studies have investigated CAR T cell therapies for recurrent glioblastoma and other high-grade gliomas, reporting both promising signals and ongoing challenges. Choi et al. (NEJM, 2024) reported rapid, transient tumor regression using CARv3-TEAM-E cells targeting EGFRvIII and wild-type EGFR via intraventricular infusion28. Bagley et al. (Nat Med, 2024) evaluated intrathecally delivered bivalent CAR T cells targeting EGFR and IL-13Rα2, demonstrating preliminary safety and bioactivity with early radiographic changes but no confirmed objective responses29. Brown et al. (Nat Med, 2024) conducted a phase 1 trial of locoregional IL-13Rα2–targeted CAR T cells in 65 patients, showing acceptable safety and clinical activity, with stable disease or better in 50% of patients but limited median overall survival30. This dual-editing strategy in MT026 eliminates TCR-mediated GvHD and reduces HvGR, providing a potential off-the-shelf therapeutic option (Fig. 5). Although these cells, similar to those generated by the existing B2M knockout strategy, still could not evade rejection by NK cells, this approach demonstrated antitumor activity with reduced alloreactivity in both in vitro and in vivo tumor mouse models via multiplex CRISPR technology. Consistent with the preclinical data, clinical data from this first-in-human, single-center, open-label IIT validated antitumor activity in the study population.

This figure summarizes the genome editing strategy for engineering universal chimeric antigen receptor (CAR)-T cells, alongside the functional goals of these modifications. Central Schematic (Edited T Cell): The cartoon depicts a T cell modified via CRISPR/Cas9 (scissors symbols at TRAC and HLA-I loci). Key edits include: TRAC Knockout (Label ①): Disruption of the TRAC locus (encoding TCR α-chain) to eliminate endogenous T cell receptor (TCR) expression. HLA-I Knockout (Label ②): Inactivation of HLA-I α-chain (HLA-A/B/C) to ablate HLA-I surface presentation. CAR Insertion (Label ③): Introduction of a CAR to equip the cell with tumor-targeting specificity. Adjacent Panels: Our Editing Strategy lists the three core manipulations: TCR (TRAC) knockout, HLA-I α-chain knockout, and CAR insertion. Main Objective of Edit outlines the strategy’s functions: preventing graft-versus-host disease (GvHD; via TCR knockout), mitigating T/NK-mediated rejection (via HLA-I knockout), and directing tumor killing (via CAR).
Despite the advent of bevacizumab as a treatment option, recurrent glioblastoma remains a largely unmet medical need, highlighting the need for novel and effective therapies. Among emerging therapeutic strategies, CAR-T cells offer potential options for patients refractory to standard therapy. To date, two clinical studies investigating autologous IL-13Rα2–specific CAR-T cells suggest that IL-13Rα2–specific CAR-T cells may be a beneficial therapeutic approach for recurrent glioblastoma, and the intrathecal route has been broadly applied in the clinic because of reduced peripheral organ biodistribution and biased delivery to the brain27. However, the time-consuming, complex manufacturing process for autologous CAR-T cells has limited their broader application in candidates for T-cell immunotherapy9,31.
The study design incorporated the following features to overcome barriers and improve patient benefit: (1) use of “off-the-shelf” allogeneic CAR-T cells to avoid long generation time and to circumvent failed autologous T-cell generation from patient donors with secondary T-cell exhaustion; 2) use of the intrathecal route via lumbar puncture instead of intracavity injection to allow exposure of CAR-T cells to newly seeding lesions, given that multifocal gliomas account for 17.2% of all gliomas32; (3) use of intrathecal delivery via lumbar puncture instead of the intraventricular route to improve ease of administration, as drug distribution in CSF via intraventricular or intrathecal routes is similar 4-6 h after administration33; 4) use of monthly rather than weekly dosing to reduce patient burden in clinic settings; and (5) elimination of lymphodepletion. Preconditioning with lymphodepleting agents may create a “favorable” environment for expansion and survival of CAR-T cells in the body by eliminating regulatory T cells; however, this step is used primarily in intravenous CAR-T cell therapy. There is still controversy over whether CAR-T will produce different effects after lymphodepleting chemotherapy in solid tumors. The pre-conditioning with lymphodepleting chemotherapy is not necessary for a local route of administration; it remains uncertain whether lymphodepleting chemotherapy is necessary34,35.
The clinical data showed favorable tolerability and safety profiles, with no grade 3 or higher adverse events, no typical or severe cytokine release syndrome, and no severe neurotoxicity. These findings are consistent with other clinical studies involving IL-13Rα2–specific CAR-T cells9,31 using local product delivery. A plausible explanation is that lower tumor burden and lack of prior lymphodepletion contribute to the absence of cytokine release syndrome and neurotoxicity, even when cell doses in these studies were comparable to those administered to patients with B-cell malignancies36,37,38. However, another study reported more severe adverse events at higher cell doses31, suggesting a dose-dependent safety pattern. In published studies of GD2 CAR-T cells in patients with H3K27M-mutated diffuse midline gliomas, neurotoxicity was observed in patients treated with ICV. No significant neurotoxicity was observed in this study, and blood IL-6 levels were significantly lower than those in CSF, possibly related to the CAR-T cell construct and the total number of cells injected into CSF39, suggesting that intrathecal administration via lumbar puncture may limit systemic effects.
Notably, MT026 did not result in any adverse events related to graft-versus-host disease40, which supports the approach of constructing allogeneic universal CAR-T cells by knocking out T-cell receptor α chains with CRISPR/Cas9 technology to eliminate this specific toxicity.
The study population was more advanced, with worse disease status (i.e., lower KPS) and faster disease progression as indicated by baseline prognostic markers (MGMT, IDH1/IDH2), compared with participants in other pivotal studies. Baseline characteristics included KPS ≥ 40 with a mean of 60, whereas the published cohort required KPS ≥ 7027. In addition, methylated MGMT status is the strongest predictor of prolonged survival in GBM, and gliomas with mutated IDH1 or IDH2 have improved prognosis compared with gliomas with wild-type IDH1/IDH241. All five patients in this study had unmethylated MGMT and wild-type IDH1/2, whereas published cohorts included both wild-type and mutant IDH1/2 and a mix of methylated and unmethylated MGMT promoters (Table 5)41,42,43,44,45,46. It is encouraging that the overall response rate of MT026 was numerically higher than that reported in bevacizumab studies that enrolled populations with higher KPS (90 − 100 for 45 % of patients and 70 − 80 for 55 % of patients in one study; 90-100 for 68 % of patients in another), although other differences between MT026 and bevacizumab study designs limit direct comparison.
Several published studies have shown that long-term remissions of hematologic malignancies are associated with expansion and sustained persistence of CAR-T cells47. Based on preliminary data, we hypothesize that CAR-T cell expansion and persistence in brain tumor areas, delivered via lumbar infusion, may have contributed to the immune response and the remissions observed in patients with glioma treated with MT026 CAR-T cells.
MRI does not necessarily distinguish between tumor, edema, and radiation necrosis. We expect that allogeneic UCAR-T cells will continuously infiltrate, proliferate, and elicit inflammation within tumor tissue40,48. A study of CAR-T cell therapy for H3K27M-mutated diffuse midline gliomas demonstrated that pseudo-progression may occur within days after therapy and lasts only several days39. A more intensive radiographic assessment schedule should be implemented to further characterize pseudo-progression related to MT026.
As previously reported, when survival was calculated from the first MT026 infusion, the OS of the five enrolled patients ranged from 5.7 to 17.2 months, with a 12-month OS rate of 20.0 % (95 % CI, 3.5-100 %). When calculated from the time of recurrence, the same patients survived 9.2 to 33.2 months, with a 12-month overall survival rate of 80 % (95 % CI, 51.6-100 %). Although these values appear numerically superior to the epidemiologically reported median OS of 6-8 months for recurrent high-grade glioma, interpretation of the potential survival benefit attributable to MT026 is limited by several factors. First, most observed responses were short-lived: Patient MT026-003 exhibited loss of IL-13Rα2 expression after re-biopsy, suggesting immune escape due to target loss; Patient MT026-004 discontinued MT026 after only four infusions for personal reasons; and Patient MT026-005 experienced highly aggressive tumor growth, with progression occurring within 33 days post-surgery, likely reflecting extreme tumor kinetics and suboptimal dosing frequency. Second, the limited sample size and the fact that two of the five patients received additional investigational UCAR-T therapies targeting other antigens further confound interpretation. Despite these limitations, the clinical data provide preliminary efficacy signals, including tumor responses and potential survival benefit, supporting the need for larger-scale, randomized controlled trials to more definitively determine the therapeutic impact of MT026 in this patient population.
In summary, this study report an approach using allogeneic IL-13Rα2–specific CAR-T cells designed to enhance the persistence of IL-13Rα2–specific allogeneic CAR-T cells, with a tolerable safety profile and potential clinical benefits in recurrent high-grade glioma. These findings support further clinical investigation in this patient population.
Moving forward, we will incorporate a prespecified translational framework into future cohorts to strengthen mechanistic understanding and enhance platform generalizability. Specifically, we plan to perform serial CSF-based TCR sequencing and immune phenotyping to dynamically monitor CAR-T cell clonal expansion and persistence, utilize single-cell cytokine profiling to comprehensively characterize immune activation, and include NK-activity readouts to evaluate innate immune responses. Moreover, considering that allogeneic CAR-T cells remain vulnerable to NK cell–mediated clearance, we will explore mitigation strategies such as HLA-E overexpression to engage inhibitory NK receptors, CIITA knockout to reduce HLA class II expression, and CD47 engineering to deliver “don’t eat me” signals. Integration of these translational approaches will provide key insights into mechanisms of therapeutic resistance, optimize CAR-T cell persistence, and facilitate broader applicability of this platform-based strategy.
Methods
Production of lentiviral vectors
HEK 293 T cells (ATCC, # CRL-3216) were cultured in 10-cm dishes and transfected at a density of 6 × 105 cells/mL. Transfection used PEI complexes containing VSV-G (3.5 µg), GagPol (5.5 µg), Rev (2.5 µg), and the transfer vector MK005-A (10 µg; encoding the CARs). Lentiviral supernatants were collected 48 h after transfection, passed through a 0.45 µm filter, and concentrated by centrifugation for 2 h at 50,000 g. Lentiviral vector titers (TU/mL) were determined by serial dilution, and the percentage of CAR-positive cells was measured by flow cytometry 7 days post-transduction.
Manufacturing of cell products
The IL-13Rα2 CAR sequence (WO/2014/072888, Pfizer INC.) was cloned into the lentiviral vector backbone. T cells collected from healthy donors were enriched by magnetic separation using anti-CD4 and anti-CD8 microbeads (Miltenyi Biotec), and activated with T Cell TransAct (Miltenyi Biotec). At 24 h after activation, lentivirus was mixed with T cells at a final MOI of 3 in 6-well round-bottom plates. For each well, 5 × 106 T cells were combined with medium containing lentivirus (1 × 109 TU/mL) in 15 μL and then spinfected at 800 × g for 60 min. Cells from the same condition were pooled and seeded at 5×105 cells/mL in culture flasks. Two days post-transduction, cells were collected and pelleted for nucleofection of Cas9 ribonucleoproteins (RNPs) to generate TCR and HLA-I knockouts. sgRNA for the HLA-I α chain: 5′-CTGACCATGAAGCCACCCTG-3′; sgRNA for TRAC: 5-AGAGTCTCTCAGCTGGTACA-3′; sgRNA for B2M: 5-GAGTAGCGCGAGCACAGCTA-3′. RNPs for each target locus were complexed separately at a 2.5:1 sgRNA (GenScript Biotech): SpCas9 (Thermo) ratio. The two RNPs were then combined to 42 pmol TRAC RNP and 42 pmol HLA-I RNP per million cells. The RNP mixture was diluted in P3 solution (Lonza) and mixed with the cell pellet, then nucleofected using the EO115 program (Lonza). Nucleofected cells were seeded at 1 × 106 cells/ mL. Magnetic separation using anti-CD3 microbeads (Miltenyi Biotec) was performed to remove CD3+ cells and improve the purity of allogeneic universal CAR-T cells. T cells were cultured in X-VIVO 15 medium (Lonza) supplemented with 5% CTS Immune Cell Serum Replacement (Gibco), recombinant human IL-2 (200 U/mL), IL-7 (10 ng/mL), and IL-15 (5 ng/mL) for 14 days. Cell products were stored in cryoprotectant after harvest.
Off-target analysis
To assess CRISPR-Cas9 off-target activity, multiple sequencing methods were employed. In AID-seq, genomic DNA from unedited T cells was fragmented, ligated to hairpin adapters, and digested with exonucleases to reduce false positives. Cleavage by Cas9 ribonucleoprotein exposed ends for biotinylated adapter ligation, followed by streptavidin enrichment, nested PCR, and alignment to identify on- and off-target sites. GUIDE-seq and iGUIDE-seq utilized dsODN tags integrated via NHEJ to mark cleavage sites in living T cells. iGUIDE-seq employed a longer dsODN for improved precision. Tagged DNA was fragmented, adapter-ligated, and PCR-amplified before sequencing and alignment. PEM-seq involved shearing DNA, PCR extension with biotinylated primers, and dual barcoding before high-throughput sequencing and analysis via PEM-Q. For editing efficiency, amplicon sequencing of target regions was performed, followed by CRISPResso analysis for quality control, alignment, and indel characterization. All methods enabled comprehensive profiling of CRISPR-induced on- and off-target edits.
Primary natural killer cell (NK Cell) rejection assay
Isolate natural killer cells (NK cells) from peripheral blood mononuclear cells (PBMCs) by positive selection with CD56 magnetic beads, label them with Far Red fluorescent dye (Thermo Fisher Scientific, Cat# C34572), co-culture them with CTV-labeled UCAR-T cells or K562 cells (Cat# 1101HUM-PUMC000039) which were purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China) at ratios of 2:1, and assess UCAR-T cell survival at 48 h. PBMCs used for NK cell isolation were obtained from healthy donors who provided written informed consent, and the procedure was approved by the Medical Ethics Committee of Ningbo University Affiliated People’s Hospital (approval number: 2024-013).
Graft-versus-host reaction (GVHR) assay
Label universal chimeric antigen receptor T cells (UCAR-T cells) from different groups with Carboxyfluorescein Diacetate Succinimidyl Ester (CTV) (Thermo Fisher Scientific, Cat# C34557), co-culture them with allogeneic peripheral blood mononuclear cells (PBMCs) treated with mitomycin at a ratio of 2:1 for 5 days (120 h), and assess the fluorescence shift of CTV in UCAR-T cells relative to the group containing only UCAR-T cells (Only group).
Mixed lymphocyte reaction (MLR) assay
After isolating natural killer cells (NK cells) from PBMCs using CD56 magnetic beads (Miltenyi Biotec, Cat# 130-050-401), co-culture them with CTV-labeled UCAR-T cells at a ratio of 10:1 for 12 days. Replace the culture medium every 3 days, stain with CD3 and a Fixable Viability Dye by flow cytometry, and analyze longitudinal changes in the numbers of UCAR-T cells and allogeneic T cells.
In vitro CAR T Cell Killing assay
UCAR-T cells and control cells (Mock-T) were co-cultured at different ratios with the U251 cell lines (Cat# BFN608006387) which were purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China) for 24 h. The U251 cell lines were pre-labeled with CellTrace™ Violet, and post-co-culture cell viability was assessed by PI staining. Flow cytometry was performed to quantify dead target cells, and the percentages of specific CAR-T killing were calculated using the formula: (Dead target cells% -Spontaneous dead target cells %) / (100- Spontaneous dead target cells %) x 100.
Co-culture of CAR-T cells with glioma organoids
All patient-derived organoids used in this experiment were obtained with written informed consent from the donors, and the use of these specimens was approved by the Medical Ethics Committee of the Fourth Affiliated Hospital of Soochow University (Dushu Lake Hospital) (approval number: 190009). According to the neural tissue dissociation kit instructions, dissociate approximately five organoids measuring 1–1.5 mm in diameter. Perform manual cell counts in triplicate to determine the average cell number within the 1-1.5 mm range. Pre-recover CAR-T cells and assess viability by trypan blue exclusion. Transfer individual organoids into separate wells of a 96-well plate and add CAR-T cells at the desired effector-to-target ratios. Bring the volume to 250 µL with T-cell culture medium. Incubate at 37 °C, 5 % CO2, and 90 % relative humidity, and co-culture on a shaker at 120 rpm. At specified time points, collect supernatant for cytokine analysis, perform immunofluorescence imaging of organoids, or fix organoids for immunohistochemical analysis. The glioma organoid samples are preserved at the Fourth Affiliated Hospital of Soochow University for further analyses. Requests for access to these samples should be directed to the corresponding author.
Patient-derived orthotopic xenograft (PDOX) models
All animal experiments complied with relevant ethical regulations and were approved by the Animal Ethics Committee of the Fourth Affiliated Hospital of Soochow University (Dushu Lake Hospital) (approval number: 190009). Female BALB/c-nu nude mice (6 weeks old) were used in this study. Mice were housed under standard conditions with a 12-h light/dark cycle, an ambient temperature of 22 ± 2 °C, and relative humidity of 50 ± 10 %. The choice of sex was based on experimental consistency and previous literature showing comparable tumor growth kinetics in female nude mice; therefore, sex was not considered as a biological variable in data analysis.
We ground the tips of the spinal needles and sterilized them at high temperature in advance. Anesthetize the mice with isoflurane and secure each mouse’s head in a stereotaxic frame. Maintain continuous anesthesia via a face mask. Using long forceps, pick up organoids individually and load them into the proximal end of the lumbar puncture needle. Fix the needle in the syringe holder of the stereotaxic frame. Disinfect the scalp with 75 % ethanol. Make a longitudinal incision of approximately 0.7 cm slightly posterior to the line connecting the eyes. Using a 2.0 mm electric grinder bit, create a small hole 2 mm posterior to the right side of the fontanelle. With the stereotaxic instrument, position the needle tip in the hole, ensuring contact with the brain surface. Slowly lower the needle by 2.5 mm and wait for 2 min. Retract the needle by 0.5 mm and slowly inject the organoids. Leave the needle in place for 2 min, then remove it slowly. Close the cranial window with bone wax and suture the skin.
After tumor formation, immobilize the mice and sterilize the scalp as described above. Lower a Hamilton syringe 2.5 mm beneath the skull surface and inject 5 µL of concentrated CAR-T cells (total CAR-T cell number five times the initially implanted organoid cell count). Withdraw the Hamilton syringe after 2 min and suture the skin. In vivo bioluminescence imaging was performed at regular intervals to monitor tumor progression and therapeutic response. Euthanasia was performed when predefined humane endpoints were reached, including: > 20% body weight loss compared with baseline, persistent weight loss > 15% over 48 h, severe neurological deficits (e.g., seizures, circling, loss of righting reflex, or inability to ambulate), marked reduction in activity or inability to access food and water, severe distress or pain unrelieved by analgesics, or rapid tumor-associated neurological deterioration. Mice meeting any of these criteria were euthanized using CO₂ inhalation followed by cervical dislocation in accordance with institutional ethical regulations.
Flow Cytometry Analysis
For flow cytometry, the antibodies used included HLA-ABC-APC (clone W6/32, Thermo Fisher Scientific, Cat# 17-9986-42), TCR-APC (clone IP26, Thermo Fisher Scientific, Cat# 17-9986-42), CD137-BV421 (clone 4B4-1, BD Biosciences, Cat# 564091), CD69-FITC (clone EN50, BioLegend, Cat# 310904), IL-13Rα2-PE (clone 47, BioLegend, Cat# 360306), CD62L-PerCP-eFluor710 (clone DREG-56, Thermo Fisher Scientific, Cat# 46-0629-42), and CD45RO-FITC (clone UCHL1, BioLegend, Cat# 304242). Detailed information of all antibodies, including clones, manufacturers and catalog numbers, and dilutions ratio were summarized in Supplemental Table 5. For flow cytometry analysis, cells were sampled and washed with FACS buffer (DPBS with 0.5 % BSA), incubated with antibodies in FACS buffer on ice in darkness for 30 min, washed again with FACS buffer twice. Samples were acquired on a CytoFLEX flow cytometer (Beckman Coulter Life Sciences) and analyzed using FlowJo software (v10.1, Tree Star).
Healthy donor
Donors were initially healthy volunteers who were screened using a questionnaire addressing risk factors for transmissible and hematologic diseases. Donors were required to be <30 years of age, have a body mass index <30 kg/m2, test negative for cytomegalovirus, and be homozygous for HLA-A*02. Donor screening and eligibility were determined per 21 Code of Federal Regulations 1271 criteria. Donors were screened for hepatitis B, hepatitis C, human immunodeficiency virus 1/2, human T-lymphotropic virus I/II, cytomegalovirus, syphilis, West Nile virus, Chagas (Trypanosoma cruzi), human herpesvirus 6/7/8, BK virus, Epstein–Barr virus, and human parvovirus B19. ABO and Rh typing were performed for all donors. HLA confirmatory typing was completed during screening. Additional information on healthy donors is provided in the Methods in the Supplementary Table 1.
Patients
This study complied with all relevant ethical regulations and was approved by the Ethics Committee of the First Affiliated Hospital of Soochow University (approval number: 2020001). The study was conducted in accordance with the principles of the Declaration of Helsinki. All patients enrolled met the ethical requirements for tumor burden, with actual tumor volume not exceeding the maximum limit approved by the ethics committee, defined as ≤ 5 cm × 4 cm × 3 cm (~30 mL) on MRI.
We enrolled adults with histologically or cytologically confirmed recurrent glioblastoma with IL-13Rα2 antigen expression ≥ 50% in tumor tissue. Patients were required to have received prior standard-of-care treatment for glioblastoma and to have relapsed with measurable tumor lesions according to the Immunotherapy Response Assessment in Neuro-Oncology (iRANO) criteria of the Response Assessment in Neuro-Oncology Working Group, and to have a Karnofsky Performance Score (KPS) ≥ 40 (on a 100-point scale, with lower numbers indicating greater disability). This study was preregistered in the Chinese Clinical Trial Registry (ChiCTR2000028801), with the first posting date on January 4, 2020 and the study initiation date on July 1, 2020. The last patient was enrolled on January 14, 2022. The study registration can be accessed at (https://www.chictr.org.cn/showproj.html?proj=47867). The originally planned enrollment of 12 patients was reduced to 5 due to delays caused by the COVID-19 pandemic. The study protocol and endpoints reported in this manuscript remain consistent with the preregistered version. Consent to publish clinical information potentially identifying individual participants was obtained from all enrolled patients.
Detection of IL-13Rα2 Expression in Tumor Tissues
IL-13Rα2 expression in tumor tissues was assessed by immunohistochemistry (IHC) based on the principle of specific antigen–antibody binding. Formalin-fixed, paraffin-embedded tumor sections were incubated with a rabbit anti-human IL-13Rα2/CD213a2 monoclonal antibody (clone E7U7B, Cat. #85677, Cell Signaling Technology, USA), followed by horseradish peroxidase (HRP)-conjugated secondary antibody (Cat. #7074, CST). Visualization was achieved using 3,3′-diaminobenzidine (DAB; Cat.#8059, CST) as the chromogenic substrate, and sections were counterstained with hematoxylin. IL-13Rα2 antigen–positive cells exhibited membranous or cytoplasmic staining ranging from light yellow to dark brown. For scoring, tumor samples were classified as IL-13Rα2-positive when ≥50 % of tumor cells displayed staining, consistent with the study’s enrollment criteria. Expression levels were further categorized as follows: medium expression was defined as 50–69 % of tumor cells with weak-to-moderate staining, whereas high expression was defined as ≥70 % of tumor cells with moderate-to-strong staining.
Study design and treatment
In this single-center, open-label, single-arm, exploratory trial, patients received MT026, an IL-13Rα2–specific allogeneic universal CAR-T cell preparation (IL-13Rα2 allogeneic UCAR-T cells; target dose, 2.5 × 107 cells, actual doses, 1.0 − 3.0 × 107 cells), administered by intrathecal lumbar puncture once monthly until disease progression, unacceptable toxicity, withdrawal of consent, death, loss to follow-up, or discontinuation for the benefit of patients at the discretion of the investigator. Dose selection for each patient was guided by clinical tolerance and safety considerations: if a patient developed fever up to 39 °C (corresponding to Grade 2 CRS), dose escalation was halted. For example, Patient MT026-002 had already achieved a complete response after treatment, and further dose escalation was not pursued to minimize risk of severe complications such as cerebral edema. Accordingly, the principal investigator chose to continue treatment at intermediate doses that had demonstrated both safety and early signs of efficacy. Dose adjustment was permitted in the event of severe toxic effects or inadequate therapeutic effect (Supplementary Fig. 2).
Assessment
Efficacy was assessed by brain magnetic resonance imaging (MRI; gadolinium-enhanced T1 and T2/FLAIR sequences), as well as new lesions, corticosteroid use, and clinical status according to iRANO criteria for CNS tumors. MRI was performed at baseline and then monthly until the end of treatment, and images were evaluated by radiologists. Two radiologists (Y.Z. and A.J.D.; 20 and 6 years of experience in neuroradiology, respectively) independently interpreted the images and conducted two-dimensional tumor measurements while blinded to clinical outcomes. Data from Y.Z. were used for analysis, and data from A.J.D. were used to test repeatability. The intraclass correlation coefficient (ICC) for tumor size between the two radiologists was 0.916 (95 % CI: 0.819–0.963, P < 0.001). Disease progression and survival status were assessed and recorded every 3 months. Baseline IL-13Rα2 expression in archival or newly obtained formalin-fixed tumor samples was assessed at a local laboratory using an investigational immunohistochemistry assay developed by T-MAXIMUM.
Adverse events were monitored during treatment and for 30 days after the end of treatment (90 days for serious adverse events). Events of interest specific to allogeneic universal CAR-T cells included cytokine release syndrome, immune effector cell–associated neurotoxicity syndrome, and graft-versus-host disease.
CAR DNA copies in CSF before and 1 day after each administration were quantified using an investigational assay developed by T-MAXIMUM. Routine CSF and blood tests and genetic testing were performed at a local laboratory. Cerebrospinal fluid and blood IL-6 concentrations were assessed by another local laboratory before and 1 day after each administration.
End points
The primary endpoint was the occurrence and severity of adverse events among all patients who received at least one dose of MT026. Secondary end points included overall survival, progression-free survival, objective response rate, duration of response, disease control rate, and pharmacokinetic parameters. Exploratory endpoints were PK characteristics (CAR DNA copies), changes in pharmacodynamic (PD) parameters (cytokines, lymphocyte count, and neutrophil-to-lymphocyte ratio), and relationships between demographic stratification variables (sex, KPS, IL-13Rα2 antigen expression level, IDH1/2 mutation, and MGMT promoter methylation) and response. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0 (NCI CTCAE 5.0). All efficacy endpoints were evaluated according to iRANO criteria for CNS tumors.
Statistical analysis
A sample size of 12 patients was predicated on the assumption that it would be sufficient to assess the initial acute and severe toxicity of MT026 at the tested dose in humans. Adverse event frequency and severity were tabulated by preferred terms and summarized using descriptive statistics. Rates of objective response, progression-free survival, and overall survival were summarized as percentages and estimated using Kaplan–Meier methods, with curves presented. Overall survival, progression-free survival, duration of response, and time to response were summarized as medians in months. Ninety-five percent confidence intervals for these parameters were calculated using the Clopper–Pearson exact method.
Data gathered and stored
During data collection, respondents were clearly informed of the study purpose and data confidentiality, and patient information was collected only after consent was obtained. During data analysis, researchers evaluated the treatment process, while MRI images and laboratory test data were processed by specialists. These procedures were implemented to ensure scientific rigor and data accuracy.
Study oversight
This investigator-initiated study was approved by the institutional review boards of The First Affiliated Hospital of Soochow University and The Fourth Affiliated Hospital of Soochow University. All patients provided written informed consent before enrollment, in accordance with the principles of the Declaration of Helsinki. All healthy donors provided written informed consent before T-cell donation. The authors vouch for the completeness and accuracy of the data and the fidelity of the study to the protocol.
Sex and gender considerations
This study enrolled both male and female patients with recurrent glioblastoma. The inclusion criteria were based solely on clinical and molecular characteristics, regardless of sex or gender. Given the small sample size (n = 5), no sex- or gender-based statistical analyses were performed, as the study was not powered to detect sex- or gender-related differences. Sex of participants was determined based on self-report during enrollment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The off-target detection datasets—comprising GUIDE-seq, PEM-seq, AID-seq, and amplicon sequencing—have been deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation, under accession code HRA015321. To protect donor privacy concerning HLA genetic sequences, these data are under controlled access for two years following publication. During this two-year controlled-access period, qualified researchers may request access through the GSA controlled-access system or by contacting the corresponding author, Yulun Huang (Y.H.), for legitimate scientific purposes. Additionally, since the sequencing primers used in the data analysis methods implicate donor HLA privacy, all inquiries concerning data analysis methods must be addressed directly to the corresponding author. A copy of the study protocol is available in the Supplementary Information file. Individual de-identified participant data, including clinical information, imaging data, and treatment responses, are provided in the manuscript and/or the Supplementary Information. Additional de-identified participant-level data may be made available for academic, non-commercial research purposes upon reasonable request to the corresponding author Yulun Huang (Y.H.), subject to institutional approvals and a data-sharing agreement. All other data supporting the findings of this study are available in the article, its supplementary files and source data and/or from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. 20, iv1–iv86 (2018).
Horbinski, C. et al. NCCN guidelines(R) insights: central nervous system cancers, Version 2.2022. J. Natl. Compr. Canc Netw. 21, 12–20 (2023).
Stupp, R. et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318, 2306–2316 (2017).
Walbert, T. & Mikkelsen, T. Recurrent high-grade glioma: a diagnostic and therapeutic challenge. Expert Rev. Neurother. 11, 509–518 (2011).
Bagley, S. J. et al. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 20, 1429–1438 (2018).
Lin, Y. J., Mashouf, L. A. & Lim, M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front Immunol. 13, 817296 (2022).
Taraseviciute, A. et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discov. 8, 750–763 (2018).
Xu, S. et al. Immunotherapy for glioma: current management and future application. Cancer Lett. 476, 1–12 (2020).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med 375, 2561–2569 (2016).
Lombardi, G. et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 20, 110–119 (2019).
Stupp, R. et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur. J. Cancer 48, 2192–2202 (2012).
Braud, V. M. et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 391, 795–799 (1998).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Cox, D. B., Platt, R. J. & Zhang, F. Therapeutic genome editing: prospects and challenges. Nat. Med 21, 121–131 (2015).
Choo, S. Y. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med J. 48, 11–23 (2007).
Xu, H. et al. Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 24, 566–578 e7 (2019).
Kitano, Y. et al. Generation of hypoimmunogenic induced pluripotent stem cells by CRISPR-Cas9 system and detailed evaluation for clinical application. Mol. Ther. Methods Clin. Dev. 26, 15–25 (2022).
Torikai, H. et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 122, 1341–1349 (2013).
Torikai, H. et al. Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci. Rep. 6, 21757 (2016).
Ren, J. et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res 23, 2255–2266 (2017).
Depil, S. et al. Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 19, 185–199 (2020).
Duygu, B. et al. HLA Class I molecules as immune checkpoints for NK cell alloreactivity and anti-viral immunity in kidney transplantation. Front Immunol. 12, 680480 (2021).
Sun, W. et al. Universal chimeric antigen receptor T cell therapy - The future of cell therapy: A review providing clinical evidence. Cancer Treat. Res Commun. 33, 100638 (2022).
Meril, S. et al. Targeting glycosylated antigens on cancer cells using siglec-7/9-based CAR T-cells. Mol. Carcinog. 59, 713–723 (2020).
Ouyang, H. et al. Baseline and early changes in the neutrophil-lymphocyte ratio (NLR) predict survival outcomes in advanced colorectal cancer patients treated with immunotherapy. Int Immunopharmacol. 123, 110703 (2023).
Rui, Y. & Green, J. J. Overcoming delivery barriers in immunotherapy for glioblastoma. Drug Deliv. Transl. Res 11, 2302–2316 (2021).
Choi, B. D. et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N. Engl. J. Med 390, 1290–1298 (2024).
Bagley, S. J. et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Ralpha2 in recurrent glioblastoma: phase 1 trial interim results. Nat. Med 30, 1320–1329 (2024).
Brown, C. E. et al. Locoregional delivery of IL-13Ralpha2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat. Med 30, 1001–1012 (2024).
Brown, C. E. et al. Bioactivity and safety of IL13Ralpha2-Redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res 21, 4062–4072 (2015).
Haque, W. et al. Patterns of management and outcomes of unifocal versus multifocal glioblastoma. J. Clin. Neurosci. 74, 155–159 (2020).
Chamberlain, M. C. et al. Pharmacokinetics of intralumbar DTC-101 for the treatment of leptomeningeal metastases. Arch. Neurol. 52, 912–917 (1995).
Mackensen, A. et al. CLDN6-specific CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: the phase 1 BNT211-01 trial. Nat. Med 29, 2844–2853 (2023).
Kalbasi, A. et al. Potentiating adoptive cell therapy using synthetic IL-9 receptors. Nature 607, 360–365 (2022).
O’Rourke, D. M., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017).
Brown, M. P., Ebert, L. M. & Gargett, T. Erratum: clinical chimeric antigen receptor T-cell therapy: a new and promising treatment modality for glioblastoma. Clin. Transl. Immunol. 10, e1331 (2021).
Davila, M. L. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med 6, 224ra225 (2014).
Majzner, R. G. et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603, 934–941 (2022).
Waxman, E. S. & Gerber, D. L. Pseudoprogression and immunotherapy phenomena. J. Adv. Pr. Oncol. 11, 723–731 (2020).
Brandes, A. A. et al. Temozolomide in patients with glioblastoma at second relapse after first line nitrosourea-procarbazine failure: a phase II study. Oncology 63, 38–41 (2002).
Chang, S. M. et al. Temozolomide in the treatment of recurrent malignant glioma. Cancer 100, 605–611 (2004).
Brada, M. et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann. Oncol. 12, 259–266 (2001).
Yung, W. K. et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 83, 588–593 (2000).
Moen, M. D. Bevacizumab: in previously treated glioblastoma. Drugs 70, 181–189 (2010).
Kim, M. M., Umemura, Y. & Leung, D. Bevacizumab and glioblastoma: past, present, and future directions. Cancer J. 24, 180–186 (2018).
Chen, Z., Hu, Y. & Mei, H. Advances in CAR-engineered immune cell generation: engineering approaches and sourcing strategies. Adv. Sci. (Weinh.) 10, e2303215 (2023).
Martinez, M. & Moon, E. K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 10, 128 (2019).
Acknowledgements
This work was supported by foundation of Chongqing International Institute for Immunology (2021YZH01 to X-Y. S), National Natural Science Foundation of China (No. 82173279, 82472981 to Y-L.H), and National Science and Technology Resource Sharing Service Platform (YCZYPT[2020]06-1 to Y-L.H), Suzhou Medical and Health Innovation Project (CXYJ2024A05 to Y-L.H), Suzhou Industrial Park Healthcare Talent Support Initiative (2024-54 to Y-L.H), Gusu Talent Program(2024-105 to Y-L.H), and the National Natural Science Foundation of China (82350112 to H-B.L.). These funders provided support for study design, data collection and analysis, and manuscript preparation. We thank the patients who participated in this trial and their families for making this trial possible; the study team, caregivers, and other personnel for their individual professional assistance; and the staff of the department of technology, T-MAXIMUM Pharmaceuticals (Suzhou) Co., Ltd., for technical support. We also extend our gratitude to Yijin Li and Xiu Zhao from T-MAXIMUM Pharmaceuticals for their valuable insights and fruitful discussions that significantly contributed to the development of this study.
Author information
Authors and Affiliations
Contributions
Y.H., X.S., and Y.W. conceived the clinical trial concept. ZhongW., X.L, ZhiminW., J. L., X. Y., H.Z. and Z.X. enrolled patients in the clinical trial, X.Z., X.R., Y.L., X.L., J.S., Z.B., and L.H. evaluated toxicity and participated in critical discussions, as well as manuscript writing and editing. W.G. performed the pathology assessments. H.L. and L.W. performed the immunological assays. X.J., YangZ., and J.C. conducted formal analysis, data visualization, writing—original draft and writing—review and editing. X.S., YuZ., J.G., X.M., and Y.Wang obtained resources, managed data, performed project administration, and performed writing—review and editing. Y.W. and Y.H. contributed to writing—original draft, writing—review and editing, funding acquisition, formal analysis, and data curation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Shang, X., Liu, J. et al. Intrathecal CRISPR-edited allogeneic IL-13Rα2 CAR T Cells for recurrent high-grade Glioma: preclinical characterization and phase I trial. Nat Commun 17, 1362 (2026). https://doi.org/10.1038/s41467-025-68112-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68112-6
