
Abstract
The skin exhibits extraordinary plasticity, enabling it to adapt to mechanical changes in the environment. While transient deformations are accommodated without lasting structural effects, sustained mechanical stress induces durable tissue changes. To investigate if these responses are mediated by shifts in epidermal stem cell fate, we employed two-photon intravital imaging to visualize epidermal cells in live skin subjected to acute mechanical forces. Mechanical force triggered the formation of intracellular “stress” vesicles within epidermal stem cells that filled with extracellular fluid and progressively enlarged, deforming the nucleus. Lineage tracing analyses revealed that the extent of nuclear deformation can predict cell fate outcomes. Moreover, mechanical stress caused sustained elevation of intracellular calcium in basal epidermal stem cells, and conditional deletion of the mechanosensitive ion channel Piezo1 disrupted calcium dynamics and increased stress vesicle formation. Using human skin xenografts, we demonstrated that stress vesicles are conserved in mammalian skin. Together, these findings identify stress vesicles as key mediators linking mechanical stress, calcium signaling, and epidermal stem cell fate.
Similar content being viewed by others


Introduction
The mammalian skin serves as a critical barrier, protecting the body from external environmental stressors. To effectively fulfill this protective role, the skin possesses remarkable mechanical properties, accommodating continuous demands for flexibility and movement, while minimizing injury and maintaining functionality over time1. However, the cellular and molecular mechanisms enabling the skin to withstand and recover from acute mechanical stresses, while preserving its structural and functional integrity, remain incompletely understood2,3,4. Anatomically, skin comprises three distinct cellular layers: the epidermis, dermis and hypodermis. The dermis largely determines the mechanical properties of skin, primarily through its extracellular matrix, consisting of collagen and elastin fibers5,6,7,8,9. In contrast, relatively little is known of how epidermal keratinocytes, the primary cells forming the skin epithelial barrier, sense and transduce mechanical signals to regulate cellular activity and sustain epidermal homeostasis.
Previous studies have elucidated fundamental mechanisms by which individual cells perceive external mechanical forces and translate them into intracellular signaling cascades that propagate from the cell surface to the nucleus and ultimately the genome10,11,12,13,14,15. This fundamental process, known as mechanotransduction, enables cells to trigger a wide range of biological responses, including changes in cell fate16,17,18. Although mechanotransduction at the plasma membrane and the nucleus function as a mechanosensitive organelle are now well documented19,20,21,22,23,24,25,26,27, the detailed sequence of subcellular events connecting plasma membrane deformation, cytoskeletal rearrangements, and nuclear responses during acute mechanical stress in intact tissues such as the epidermis remains poorly understood. The epidermis exhibits rapid cellular turnover driven by basal layer keratinocytes, which balance proliferation and differentiation to sustain epidermal homeostasis28,29,30,31. Basal keratinocytes establish critical adhesions both with the basement membrane beneath and with their terminally differentiated progeny in the suprabasal epidermal layers. Given that basal keratinocytes uniquely retain the capacity to proliferate or undergo terminal differentiation, external mechanical cues capable of influencing their fate could profoundly impact overall tissue integrity. Therefore, the main goal of this study is to resolve how epidermal keratinocytes respond to mechanical stress in vivo and how these responses may regulate their cell fate decisions and skin homeostasis.
Changes in intracellular calcium dynamics represent one of the earliest cellular responses to mechanical stimuli, initiating downstream signaling pathways crucial for various biological processes32,33,34,35,36,37. Recent studies have substantially advanced our understanding of mechanosensitive calcium signaling across different cell types and tissues, both in vitro and in vivo25,35,38,39,40,41,42,43. However, calcium dynamics in epidermal keratinocytes specifically subjected to mechanical stress in their native environment remain unresolved. The mechanosensitive channel Piezo1 has emerged as a central mediator of calcium signaling, activated by membrane tension38,40,44,45,46,47,48, and implicated in diverse physiological processes, including vascular development39,49,50, erythrocyte volume maintenance51,52,53, cellular differentiation and proliferation23,54,55,56. Activation of Piezo1 by mechanical tension facilitates calcium flux across cell membranes23,57,58. Given the pivotal role of calcium signaling in keratinocyte differentiation and epidermal function, defining the specific contributions of Piezo1 to epidermal mechanobiology could significantly enhance our understanding of how mechanical stress regulates skin homeostasis and stem cell fate decisions.
Here, we employed intravital imaging of mouse skin to capture in real time the cellular events induced by physical force applied directly to the live tissue. We discovered previously unrecognized subcellular processes in basal layer epidermal keratinocytes as a direct result of acute mechanical stress. Using single-cell lineage tracing, we provide evidence that links these events to changes in cell fate. Furthermore, our data supports a role for Piezo 1 as a critical component of this molecular mechanism that translates physical cues to cellular behaviors via calcium signaling.
Results
Mechanical stress induces intracellular changes in basal epidermal cells
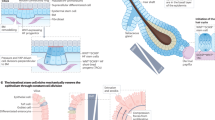
To investigate how keratinocytes sense and respond to mechanical stressors, we designed three independent assays to apply acute mechanical force onto the mouse skin epidermis. These included the application of negative (suction) or positive (compression) pressure in vivo, and lateral tension (stretching) ex vivo (Supplementary Fig. 1). These assays were integrated with two-photon intravital imaging, enabling us to visualize the morphological and structural responses of keratinocytes to mechanical stress within their native environment. To effectively track specific cellular compartments, we used mouse fluorescent reporter lines that label keratinocyte cell membranes (R26-mTom) and nuclei (K14H2BGFP) (Fig. 1a). Real-time intravital imaging, during or immediately following the application of mechanical stress to the mouse skin, revealed the rapid formation and growth of prominent intracellular vesicles (hereafter termed “stress vesicles”) (Fig. 1a–f, Supplementary Fig. 1a–c). These responses occurred predominantly in basal layer keratinocytes (Fig. 1g, h). Furthermore, we found that increasing the magnitude or duration of the applied mechanical force led to a higher number of cells with stress vesicles, a higher number of stress vesicles per cell and an increase in the average size of stress vesicles (Fig. 1a–d). These changes were accompanied by a proportional deformation of the nucleus in cells that exhibited stress vesicles (Fig. 1a, e, f, Supplementary Fig. 2a–c).

a Schematic representation of the experimental setup and representative live imaging snapshots showing the formation of stress vesicles (yellow arrows) in the adult mouse epidermis following the application of negative pressure. Scale bar: 10 µm. b Quantification of the percentage of epidermal cells with stress vesicles as a function of the duration of applied negative pressure. Each point represents an individual mouse. Data from N = 3 mice (0 min, control), N = 3 (1 min), N = 5 (3 min), N = 6 (5 min), and N = 3 (10 min). Overall group differences were assessed using the Kruskal-Wallis test (P = 0.0012). Pairwise comparisons were performed as follows: unpaired t-test with Welch’s correction between 0 min and 1 min (P = 0.0016); one-way ANOVA followed by Šídák’s multiple comparisons test between successive timepoints: 1 min vs 3 min (P = 0.0217), 3 min vs 5 min (P < 0.0001), and 5 min vs 10 min (P = 0.0003). Data are presented as mean ± SEM. c Quantification of stress vesicle size with increasing duration of applied negative pressure. Each data point represents a single vesicle. A total of 753 vesicles were analyzed across all groups. A Spearman’s rank correlation test revealed a strong positive correlation between stress duration and vesicle size (r = 0.7324, 95% CI: 0.6963-0.7647, P < 0.0001). d Quantification of the percentage of cells exhibiting multiple stress vesicles as a function of the duration of applied negative pressure. Each data point represents an individual mouse. Overall group differences were evaluated using the Kruskal-Wallis test (P = 0.0019). Pairwise comparisons were conducted as follows: an unpaired t-test with Welch’s correction between 0 min and 1 min (P = 0.0286); and one-way ANOVA followed by Šídák’s multiple comparisons test between sequential timepoints: 1 min vs 3 min (P = 0.5490, not significant), 3 min vs 5 min (P = 0.0015), and 5 min vs 10 min (P = 0.1702, not significant). Data are shown as mean ± SEM. e Quantification of the percentage of deformed cell nuclei under varying durations of negative pressure. Each data point represents an individual mouse. Group differences were assessed using the Kruskal-Wallis test (P = 0.0001). Pairwise comparisons were performed using an unpaired t-test with Welch’s correction between 0 and 1 min (P = 0.0079), and one-way ANOVA with Šídák’s multiple comparisons test between successive timepoints: 1 vs 3 min (P = 0.0051), 3 vs 5 min (P < 0.0001), and 5 vs 10 min (P < 0.0001). Data are presented as mean ± SEM. f Quantification of nuclear volume in deformed nuclei under different durations of negative pressure. Quantification of nuclear volumes was performed across stress durations of 0 min (control), 1 min, 3 min, 5 min, and 10 min. Each point represents an individual nucleus, with a total of 444 nuclei analyzed. Spearman’s rank correlation analysis revealed a moderate inverse relationship between stress duration and nuclear volume (r = −0.3326; 95% CI: −0.4152 to −0.2446; P < 0.0001). g Representative timeframes from real-time imaging of stress vesicle growth in the basal and suprabasal cell layers of the mouse epidermis. Scale bar: 10 µm. h Quantification of the percentage of epidermal cells with stress vesicles as a function of the duration of applied negative pressure. Data from N = 4 mice; n = 16 images analyzed for each group. Statistical analysis was performed using a two-tailed unpaired t-test. i Representative frames at 3-s intervals of stress vesicle growth. Similar results were obtained from at least three independent experiments. Scale bar: 2 µm.
We performed time-resolved live imaging to capture the dynamics of stress vesicle formation at high spatiotemporal resolution (Fig. 1i). Moreover, the use of dual reporter mice (K14H2BGFP; R26-mTom) enabled simultaneous visualization of basal keratinocyte membranes and nuclei. Thus, we visualized in real-time the formation and gradual expansion of stress vesicles as well as the deformation of the nucleus (Fig. 2a, b, Supplementary Movie 1). Ultrastructural analysis confirmed the vesicular nature of the stress-induced intracellular structures, without any detectable disruption of plasma membrane integrity, indicating that affected cells remained structurally intact (Fig. 2c). To test whether extracellular fluid influx contributes to stress vesicle formation, we administered fluorescently labeled dextran, a biologically inert tracer, via hypodermal injection before applying mechanical stress (Fig. 2d–f). Live imaging demonstrated dextran accumulation within newly formed stress vesicles, supporting our hypothesis that these vesicles form via extracellular fluid internalization (Fig. 2f). To further validate these observations, we utilized dual reporter mice to distinctly label the cell membrane (R26-mGFP) and cytoplasm (R26-cytTom). Imaging revealed that stress vesicles originated from the mGFP-labeled cell membrane and lacked cytTom fluorescence, confirming that vesicle contents originated from the extracellular environment rather than the cytoplasm (Fig. 2g).

a Representative live imaging frames illustrating the formation of stress vesicles during the application of positive pressure. Similar results were obtained from at least three independent experiments. Scale bar: 2 µm. See also Movie S1. b Three-dimensional renderings of basal epidermal cells before and after the application of positive pressure. The nucleus is shown in green, the plasma membrane in magenta, and stress vesicles in red. Scale bar: 2 µm. c High-resolution electron micrograph of basal epidermal cells in the mouse epidermis. A large cytoplasmic vesicle is visible in the stressed cell. The red dashed line marks the boundary between the epidermis and dermis. Similar results were obtained from two independent experiments. Scale bar: 1 µm. d Schematic of the experimental strategy used to track the internalization of extracellular fluid into epidermal cells following the application of negative pressure. e Quantification of epidermal cells with stress vesicles labeled with the extracellular fluid tracer Dextran Alexa-488. Data are from N = 4 mice, n = 16 areas measured. Statistical analysis: unpaired t-test. f Representative images of Dextran internalization in normal skin and skin subjected to 10 min of negative pressure. Scale bar: 10 µm. g Representative images of epidermal cells labeled with mGFP (membrane-bound) and cytTom (cytoplasmic) following negative force application. Stress vesicles are composed of plasma membrane, with lumens devoid of red signal, indicating the absence of cytoplasmic content. Scale bar: 5 µm. h Quantification of stress vesicle growth. Data are from N = 3 mice, n = 20 vesicles. i Representative images of stress vesicles filled containing Dextran-FITC dye, previously injected intradermally in a R26-mTom reporter mouse; Dextran-FITC labeled stress vesicles were processed through 3D rendering in Imaris. j Quantification of stress vesicle size measured by 3D volumetric analysis. Data are from N = 4 mice, n = 120 vesicles. k Representative images of stress vesicle disappearance after the removal of force. Scale bar: 5 µm. l Quantification of the decrease in vesicle numbers 1 h and 2 h after the removal of force loading. Data are from N = 5 mice for each group. Statistical analysis: two-tailed unpaired t-test. Data are presented as mean ± SEM Scale bars: 2 µm (c) and 5 µm (e).
Time-lapse intravital imaging also captured dynamic changes in stress vesicle morphology, characterized by a rapid increase in vesicle perimeter within the first 20 s of vesicle formation, followed by slower expansion thereafter (Fig. 2h, i). Three-dimensional volumetric image segmentation showed that stress vesicle volume averaged approximately 99 μm³, ranging from 17 μm³ to 265 μm³, and was associated with modest changes in both nuclear and overall cell volumes (Fig. 2j, Supplementary Fig. 2b, c). The cell nucleus exhibits intrinsic elasticity to mechanical stress14,25,27,59,60,61,62. Specifically, the nuclear lamina, composed predominantly of Lamin A/C proteins, is critical for nuclear structural stability and regulates chromatin organization and genome integrity. Previous studies have suggested an inverse correlation between nuclear compressibility and Lamin A/C expression, with cells experiencing higher nuclear deformation typically expressing lower levels of Lamin A/C63,64,65. In basal epidermal keratinocytes subjected to acute mechanical stress, we observed that the nuclear lamina, as indicated by Lamin A/C immunostaining, precisely mirrored the deformation of nuclear content labeled by H2B-GFP (Supplementary Fig. 2d). Upon removal of mechanical stress, vesicles gradually reduced in size, and the majority disappeared within two hours (Fig. 2k, l). To track extracellular fluid dynamics within stress vesicles, we performed hypodermal injections of fluorescent dextran and observed, via live imaging, that stress vesicles exhibited a sagging and wrinkled morphology but remained structurally intact upon resolution, without leaking dextran into the cytoplasm (Supplementary Fig. 2e–g). Moreover, nuclear deformation associated with stress vesicle formation reversed completely, as nuclei regained their original, rounded shape following force removal (Supplementary Fig. 3a–d).
To further characterize the molecular composition of stress vesicles and the mechanism of their formation, we performed histological analyses using markers specific for components of the cellular membrane, cytoskeleton, and cell adhesion apparatuses (Supplementary Fig. 4a–i). Our data show that stress vesicles are enclosed by a lipid bilayer, labeled by wheat germ agglutinin (Supplementary Fig. 4b). Moreover, we observed a strong enrichment of F-actin and Myosin at the vesicle periphery, indicating that the actomyosin activity may contribute to stress vesicle formation (Supplementary Fig. 4c, d). Enrichment for Vinculin but no E-cadherin was also detected at the periphery of stress vesicles (Supplementary Fig. 4e, f). Moreover, we found no evidence for a caveola-mediated mechanism of endocytosis (Supplementary Fig. 4g). Clathrin was also weakly detected in association with stress vesicles, indicating that clathrin-mediated endocytosis is likely not involved (Supplementary Fig. 4h). To investigate whether actomyosin activity is required for the formation of stress vesicles, we performed the stress-inducing in vivo assays using inhibitors against actin polymerization (Latrunculin-A)66 and non-muscle myosin (Blebbistatin)67 and documented their effect in basal keratinocytes by live imaging (Supplementary Fig. 5a, b). Under these conditions, treatment with either Latrunculin-A or Blebbistatin had a measurable inhibitory effect on the formation of stress vesicles (Supplementary Fig. 5c). Taken together, our data implicates the contractile actomyosin cytoskeleton in modulating the formation and/or growth of stress vesicles in epidermal stem cells (Supplementary Fig. 5d). Based on the size of stress vesicles, the rapid kinetics of their growth and the involvement of actomyosin dynamics suggest that the mechanism for formation of stress vesicles may be related to macropinocytosis; a process that has previously been described for the non-specific uptake of extracellular fluid.
Stress vesicles are associated with changes in stem cell fate
Epidermal homeostasis relies on a precise balance between proliferation and differentiation of basal layer keratinocytes. To investigate whether the formation of stress vesicles in epidermal stem cells influences their fate decisions, we implemented a lineage tracing approach by longitudinal live imaging. Specifically, we selectively labeled epidermal stem cells in the interfollicular epidermis using a photo-activatable nuclear reporter mouse line (K14H2BPAGFP), which generates a strong nuclear signal upon photoactivation, and which retains the long half-life of the histone H2B moiety (Fig. 3a). We initially marked a defined group of basal cells and applied acute mechanical stress to the skin, subsequently tracking these cells by re-imaging immediately afterward and at 24 h post-treatment (Fig. 3b–e). By quantifying changes in both the number and position of labeled cells over time, we inferred their relative tendencies to proliferate (increased cell numbers) or differentiate (delamination from the basal layer). In mechanically stressed epidermis, we observed that the labeled cell group increased in size, indicative of increased proliferation compared to unstressed controls (Fig. 3c). Moreover, a larger number of cells moved to the suprabasal epidermal layers within 24 h, consistent with increased differentiation following mechanical stress (Fig. 3d, Supplementary Fig. 6a, b). These observations suggest that stress vesicle formation correlates with changes in basal epidermal stem cell fate under acute mechanical stress.

a Schematic representation of the genetic alleles and strategy for lineage tracing of epidermal cells via in vivo photo-labeling. Prior to mechanical force application, equivalent groups of basal epidermal cells were scanned to activate the K14H2BPAGFP reporter. The same areas were re-imaged to track population changes over time. b Representative time-lapse images of photo-labeling and cell tracking following mechanical force application. Yellow arrows indicate the direction of cell movement as cells differentiate and move from the basal to the suprabasal layer. Scale bar: 10 µm. c, d Quantification of the relative fractions of cell proliferation and differentiation in tracked epidermal cells 1 day after mechanical force application. N = 3 mice, n = 12 images analyzed per condition. Statistical analysis: two-tailed unpaired t-test. e Experimental strategy for collecting mouse skin tissue immediately after applying negative pressure for panels F-I. f Representative immunofluorescence images showing stress vesicle formation in K10+, Ki67+, and K10-/Ki67- basal epidermal cells. Yellow arrows indicate basal layer cells with stress vesicles. A white dashed line marks the boundary between the epidermis and dermis. Scale bar: 5 µm. g Quantification of stress vesicle formation in three distinct groups of basal epidermal cells following mechanical force application. One-way ANOVA test (P < 0.0001) with Tukey’s mulitiple comparisons test: K10-; Ki67- vs Ki67+, P = 0.0089; K10-; Ki67- vs K10 + , P < 0.0001; Ki67+ vs K10+, P < 0.0001. Data are presented as mean ± SEM, N = 7 mice. h Representative whole-mount immunofluorescence images of stressed epidermis stained with Ki67 (nuclear signals) and Krt10 (membrane signals) antibodies. Scale bar: 10 µm. i Quantification of the ratio of stress vesicle formation in three different groups of basal epidermal cells from whole-mount-stained epidermis. One-way ANOVA test (P < 0.0001) with Tukey’s mulitiple comparisons test: K10-; Ki67- vs Ki67 + , P < 0.0001; K10-; Ki67- vs K10 + , P < 0.0001; Ki67+ vs K10 + , P < 0.0001. Data are presented as mean ± SEM, n = 16 images analyzed from 4 mice for each group. J Representative time-lapse images from live imaging lineage tracing, capturing varying levels of nuclear deformation and the subsequent fates of epidermal cells bearing nuclear deformation. Labeled epidermal cells were categorized into three groups based on their fates: undifferentiated cells (remaining in the basal layer), differentiated cells (delaminating and moving into the suprabasal layer), and proliferating cells (producing two daughter cells in the basal layer). Scale bar: 2 µm. k, l Quantification of nuclear deformation as a function of cell fate. Each data point represents a single nucleus. Statistical comparisons between groups were performed using One-way ANOVA with Tukey’s multiple comparisons test; p-values are indicated on the graph. Circularity: N = 4 mice, n = 59 (undifferentiated cells), n = 59 (differentiated cells), and n = 60 (proliferated cells). Statistical analysis: P < 0.0001. Angle: N = 4 mice, n = 93 (undifferentiated cells), n = 99 (differentiated cells), and n = 89 (proliferated cells). m, n Quantification of nuclear volume in differentiated cells (left graph) or proliferated cells (right graph) compared to undifferentiated cells. Data are from N = 3 mice, n = 40 undifferentiated cells, n = 23 differentiated cells, and n = 22 proliferated cells. Statistical analysis: two-tailed unpaired t-test. Data are presented as mean ± SEM.
In time-resolved intravital imaging experiments, stress vesicles gradually disappeared within approximately two to three hours after removal of the mechanical stressor (Fig. 2k, l, Supplementary Fig. 3a). To determine whether the increased proliferation observed after acute mechanical stress was indirectly driven by cell differentiation events, we isolated actively dividing (Ki67+) epidermal cells and performed cell-cycle analysis by flow cytometry. Our data revealed a clear shift in the cell-cycle phase, with an increase in cell progression from the G1 to the G2/M phase after mechanical stress, confirming activation of epidermal stem cell proliferation (Supplementary Fig. 6c–f). Next, we tested whether the observed changes in epidermal stem cell fate and overall cellular turnover were secondary effects of tissue damage or increased cell death caused by mechanical stress. Histological analysis showed no appreciable structural damage in mechanically stressed epidermis. Specifically, the epidermal barrier remained intact, including normal differentiated suprabasal layers and no noticeable alterations in the dermis (Supplementary Fig. 7a–c). Moreover, hair follicle epidermal cells neither formed stress vesicles nor showed changes in hair cycle dynamics in response to mechanical stress (Supplementary Fig. 7d, e). Consistent with these observations, there were no detectable changes in immune cell infiltration within the epidermis after acute mechanical stress (Supplementary Fig. 7f, g). Taken together, these findings strongly indicate that the observed epidermal stem cell responses are not secondary to tissue damage or inflammatory responses but rather represent specific adaptations to mechanical stress.
Our live imaging data revealed heterogeneity in the response of basal epidermal cells to mechanical stress, prompting us to investigate whether stress vesicle formation correlated with the differentiation status of epidermal stem cells. To address this, we categorized basal keratinocytes into three discrete groups based on proliferation and differentiation markers: 1) relatively quiescent stem cells (Krt10−/Ki67−), 2) actively cycling transit amplifying cells (Ki67+), and 3) committed differentiating cells (Krt10+). Our analysis showed that committed differentiating cells (Krt10+) exhibited a higher frequency of stress vesicle formation compared to either relatively quiescent or transit amplified cells (Fig. 3f–i). Collectively, these findings suggest that stress vesicle formation preferentially occurs in basal epidermal cells committed to differentiation, linking vesicle formation directly to the early stages of epidermal differentiation.
Stress-induced changes in nuclear architecture precede stem cell differentiation
Our data indicates that acute mechanical stress rapidly increases cellular turnover within the epidermis, likely driven by stem cell activation in the basal layer. To directly test this hypothesis, we analyzed the size of the stress vesicles and the degree of nuclear deformation for correlation to subsequent cell fate decisions. Using intravital imaging, we photo-labeled individual basal epidermal cells, applied mechanical stress, and traced their activity over time (Fig. 3j). By quantifying nuclear morphology parameters such as circularity, and nuclear deformation angle, we found that cells with substantial nuclear circularity changes exhibited a clear propensity to differentiate and delaminate from the basal layer. In contrast, cells that maintained intact nuclei and lacked stress vesicles were more likely to either proliferate or remain quiescent within the basal layer (Fig. 3k, l). To further elucidate the timing of these differentiation events, we performed sequential live imaging at 1, 6, 9, and 18 h following mechanical stress application. We observed that keratinocytes with pronounced nuclear deformation began to delaminate significantly earlier, within 6 to 9 h post-stress, compared to basal keratinocytes in unstressed conditions (Supplementary Fig. 6a, b). Together, these results indicate that stress vesicle formation and the associated nuclear deformation events are early predictive markers of epidermal stem cell commitment to differentiation.
Previous studies have shown that basal epidermal cells undergo major changes in cell shape, adhesiveness and cytoskeletal architecture throughout different phases of the cell cycle, and that these structural changes are closely associated with cellular responses to external mechanical stimuli68,69,70,71. Interestingly, the pro-differentiation phenotype of epidermal stem cells as a result of mechanical stress appears to be linked to the degree of nuclear deformation rather than to changes in nuclear volume (Fig. 3k–n). These findings indicate that stress vesicle formation and its associated influence on cell fate are likely to occur independently of changes in nuclear envelope stiffness, suggesting that alternative mechanisms mediate epidermal stem cell responses to mechanical stress.
Epidermal cells sense mechanical stress through persistent intracellular calcium signals
Calcium is widely recognized as a critical mediator of mechanotransduction34,37,38,43. Moreover, calcium is a major regulator of keratinocyte differentiation in vivo and in vitro36,70,72. Given our observation of stress vesicle formation in epidermal stem cells and their correlation with differentiation, we hypothesized that calcium dynamics might underpin these cellular responses. To directly test this possibility, we employed an intravital imaging approach using mice expressing a genetically encoded calcium reporter (GCaMP6s). This allowed real-time monitoring of calcium dynamics in vivo in the skin, under both normal and mechanically stressed conditions (Fig. 4a). In adult epidermis, keratinocytes are spatially organized into discrete layers corresponding to their differentiation states. This organization is accompanied by a calcium gradient, increasing progressively from the basal stem cell compartment toward the terminally differentiated suprabasal layers73,74. Consistent with previous reports70, our imaging confirmed the presence of this gradient in the epidermis and further revealed dynamic calcium fluctuations within basal keratinocytes at normal conditions (Fig. 4b).

a Schematic representation of calcium imaging in single epidermal cells during the application of compressive force applied on live mouse skin. b Representative time frames from in vivo calcium imaging of basal epidermal cells before (top panels) and after (bottom panels) application of positive pressure. Each frame is pseudo-colored and projected into the last frame (right). Four highlighted epidermal cells show initial intracellular calcium fluxes and persistent cytoplasmic calcium after compression. Graphs show relative calcium signal intensity as a function of time for each highlighted cell. Scale bar: 2 µm. c Quantification of calcium signals at rest and under positive pressure. Each line represents measurements from a single cell. N = 4 mice, n = 23 cells analyzed. d Quantification of calcium signal intensity at baseline and under stress. N = 4 mice, n = 24 cells (control) and n = 22 cells (stressed). Statistical analysis: two-tailed unpaired t-test. Data are presented as mean ± SEM. e Representative examples of calcium dynamics in cells with and without stress vesicles. Scale bar: 2 µm. f Quantification of relative calcium intensity over time for two representative cells. g Quantification of the number of cells with persistent high intracellular calcium versus calcium flux, which form stress vesicles. Data are from N = 5 mice, n = 25 cells analyzed. Statistical analysis: two-tailed unpaired t-test. Data are presented as mean ± SEM. h Schematic strategy for visualizing calcium dynamics during the recovery period after force removal. i Representative images showing high calcium signals persisting in stress vesicle-forming cells after force removal. Scale bar: 10 µm. j Quantification of the percentage of cells displaying varying calcium signal intensities along with the presence of stress vesicles. Data are from N = 5 mice, n = 20 cells analyzed per condition. Statistical analysis: two-tailed unpaired t-test. Data are presented as mean ± SEM.
To investigate how epidermal stem cells modulate intracellular calcium in response to acute mechanical stress, we applied compressive force to the mouse ear and directly visualized calcium dynamics in epidermal keratinocytes by intravital imaging. Immediately following mechanical force application, we observed a robust, tissue-wide increase in cytoplasmic calcium levels, particularly prominent in basal keratinocytes (Fig. 4b–d). However, upon prolonged compression, intracellular calcium gradually decreased. Importantly, stress vesicles preferentially persisted in cells that displayed constantly elevated cytoplasmic calcium levels, compared to cells showing transient calcium fluxes (Fig. 4e–g). This persistent association was further confirmed by tracking epidermal keratinocytes during their recovery phase after mechanical stress removal. Cells retaining stress vesicles consistently exhibited high cytoplasmic calcium signals. (Fig. 4h–j). Taken together, these findings establish a direct relationship between acute mechanical stress, persistent intracellular calcium signaling, and stress vesicle formation in epidermal stem cells. These data further suggest that sustained elevations in intracellular calcium and stress vesicle persistence are coordinated cellular mechanisms potentially driving epidermal stem cell differentiation.
Piezo1 deletion increases stress vesicle formation
Piezo1 is a mechanosensitive ion channel that enables cells in various tissues to perceive and respond to mechanical stimuli. Although Piezo1 functions broadly as a cation channel, it primarily influences cell responses by regulating calcium influx across membranes. Based on our findings that intracellular calcium dynamics and stress vesicle formation correlate with epidermal stem cell fate, we hypothesized that Piezo1 could participate in the molecular pathway governing these processes. To directly test this hypothesis, we conditionally deleted Piezo1 in adult epidermis using a specific driver for basal layer stem cells (p63CreER; Fig. 5a). Histological analysis confirmed Piezo1 expression in basal keratinocytes of wild-type mice and its effective deletion in Piezo1 conditional knockout (Piezo1 cKO; Supplementary Fig. 8a). Under physiological conditions, Piezo1 cKO mice showed no obvious differences in skin morphology compared to wild-type control. We then investigated whether epidermal cells with Piezo1 gene deletion differ in their response to mechanical stress. However, upon mechanical stress application, Piezo1-deficient epidermal cells exhibited a significant increase in stress vesicle formation compared to wild-type cells exposed to identical mechanical stress conditions (Fig. 5b, c). These data strongly suggest that Piezo1 plays a critical regulatory role in modulating stress vesicle formation in response to acute mechanical stimuli in epidermal keratinocytes.

a Schematic representation of the experimental design to conditionally ablate Piezo1 expression in basal epidermal cells and observe its effect on stress vesicle formation. b Representative images of basal epidermal cells from control and Piezo1 conditional knockout (cKO) mouse back skin, visualized before and immediately after mechanical force application. Scale bar: 10 µm. c Quantification of the fraction of basal epidermal cells with stress vesicles. Data are from N = 5 pairs of control and Piezo1 cKO mice. Statistical analysis: two-tailed unpaired t-test. d Schematic strategy to capture calcium dynamics in control and Piezo1 cKO mouse skin immediately after application of positive pressure. e Representative images of calcium signals at the basal layer of the skin from control and Piezo1 cKO mice, taken before (1st frame) and after the application of positive pressure. Scale bar: 20 µm. f Quantification of calcium signal intensity in basal epidermal cells from control and Piezo1 cKO mice after application of positive pressure, as a function of time. Each line shows representative measurements from individual cells. Data are from N = 8 control and 8 Piezo1 cKO mice, n = 15 and n = 15 image stacks analyzed. g Schematic strategy to visualize cell behaviors following mechanical stress applications in control and Piezo1 cKO mice. h Representative images of photo-activated basal epidermal cells and their tracked behaviors in control and Piezo1 cKO epidermis after application of negative pressure. Scale bar: 10 µm. i Quantification of the ratio of cell proliferation and differentiation in the epidermis of control and Piezo1 cKO mouse back skin. Data are from N = 5 control and 5 Piezo1 cKO mice, n = 25 and n = 29 image stacks analyzed. Statistical analysis: two-tailed unpaired t-test.
Acute mechanical force exerted on tissues is predicted to propagate across cellular membranes where Piezo1 channels are located, leading to channel activation and subsequent calcium influx into the cytoplasm from extracellular and/or intracellular stores. To test this prediction in epidermal keratinocytes, we bred mice expressing the calcium indicator GCaMP6s with or without conditional Piezo1 deletion. Under baseline conditions, both control and Piezo1 cKO mice displayed similar calcium flux patterns in basal epidermal keratinocytes (Supplementary Fig. 9a, b); however, upon acute mechanical stimulation, basal keratinocytes in both genotypes exhibited elevated intracellular calcium (Fig. 5d). However, the kinetics of calcium elevation was notably different, with calcium levels rising gradually in wild-type keratinocytes but increasing abruptly and rapidly in Piezo1-deficient cells (Fig. 5e, f). Moreover, unlike wild-type cells that exhibited transient calcium elevation, Piezo1-deficient keratinocytes maintained persistently high calcium levels long after mechanical stress was removed (Fig. 5e, f). These results demonstrate that basal epidermal cells respond to mechanical force by increasing intracellular calcium and that the kinetics of calcium influx are partially dependent on Piezo1 channel activity.
Next, we investigated whether the increased stress vesicle formation observed in Piezo1-deficient keratinocytes is associated with enhanced cell differentiation. To address this, we crossed the K14H2BPAGFP lineage-tracing-by-photolabeling mouse line with Piezo1 conditional knockout mice (Piezo1 cKO) and tracked basal keratinocyte proliferation and differentiation following mechanical stress application to the mouse dorsal skin. Consistent with increased vesicle formation, we observed greater cell differentiation, evidenced by an increased number of keratinocytes migrating into the suprabasal epidermal layers of Piezo1 cKO compared to control mice (Fig. 5g–i). These data strongly suggest that stress vesicle formation and sustained intracellular calcium elevation collectively promote epidermal keratinocyte differentiation. To further verify the role of Piezo1 in suppressing stress vesicle formation, we intradermally injected mice with the Piezo1-selective activator Yoda1 prior to mechanical stress application. Yoda1 treatment induced a pronounced increase in calcium influx (Supplementary Fig. 9c) and notably suppressed stress vesicle formation in vivo during mechanical stress (Supplementary Fig. 9d, e). In contrast, pretreatment with the general mechanosensitive calcium channel inhibitor GsMTx4 failed to prevent vesicle formation (Supplementary Fig. 9d, e). To test whether Piezo1 deletion impairs epidermal barrier integrity under mechanical stress, we applied sustained negative pressure to mouse dorsal skin in vivo. Strikingly, after just 10 min of continuous mechanical force, Piezo1 cKO mice exhibited extensive epidermal blistering, whereas control skin remained intact (Supplementary Fig. 8b, c). Collectively, these findings indicate that the mechanosensitive ion channel Piezo1 plays a protective and essential role in maintaining epidermal integrity during acute mechanical stress and highlight a specific regulatory function for Piezo1 channels in modulating stress vesicle formation and subsequent differentiation responses of epidermal keratinocytes.
To further elucidate the molecular pathways involved in epidermal stem cell responses to mechanical stress, we performed RNA sequencing on basal layer keratinocytes isolated from the interfollicular epidermis of wild-type and Piezo1 cKO mice. Gene expression analysis revealed downregulation of genes associated with epidermal barrier function and cell adhesion in Piezo1 cKO keratinocytes (Supplementary Fig. 8d, e). Consistent with these transcriptional changes, ultrastructural analysis also uncovered alterations in cell-cell junctions in Piezo1-deficient epidermis compared to controls. (Supplementary Fig. 8f). Interestingly, we identified upregulation of genes linked to immune activation pathways specifically in Piezo1 cKO keratinocytes (Supplementary Fig. 8d, e). Additionally, the differential gene expression signature prominently included developmental pathways, such as TGF-beta75, PKC76,77 and calcium-related genes, all previously implicated in mechanotransduction and epidermal differentiation.
Stress vesicles are a conserved phenomenon in mammalian skin
Despite molecular and functional similarities, the structural organization and mechanical properties of mouse skin are markedly different from human skin. We therefore investigated whether stress vesicle formation, as observed in the mouse epidermis, is also conserved in human keratinocytes in response to mechanical stress. Conventional cell culture assays inadequately recapitulate the biomechanical environment of intact skin. To overcome this limitation, we established a human skin xenograft model combined with intravital imaging, allowing us to directly visualize keratinocyte responses to acute mechanical force within a physiologically relevant environment (Fig. 6a). Human primary keratinocytes were engineered to express fluorescent reporter proteins highlighting nuclei, cell cortices, and plasma membranes. Following full stratification in organotypic 3D culture, engineered human skin was grafted onto immunodeficient mice, where it integrated stably and maintained viability long-term (Fig. 6b). Live imaging of the xenografted human skin confirmed successful labeling and revealed characteristic columns of differentiating keratinocytes (Fig. 6d). Histological analysis demonstrated that these engineered xenografts recapitulated essential features of human skin, including distinct epidermal morphology, ridge-like structures, and greater number of differentiated suprabasal layers compared to adjacent mouse epidermis (Fig. 6d). Importantly, applying the same mechanical stressors used previously resulted in consistent formation of stress vesicles in human keratinocytes within the xenografts, closely matching their morphology and abundance in mouse epidermis (Fig. 6e–h). Our results demonstrate that stress vesicle formation represents an evolutionarily conserved cellular mechanism through which mammalian epidermis adapts rapidly to acute mechanical stress, illuminating previously unrecognized aspects of skin mechanobiology.

a Schematic representation of the experimental design to generate a human skin xenograft for intravital imaging. b Representative examples of a stable human skin xenograft in a mouse. Magnified views show the mounting (top) and the wide-field epifluorescence image of the xenograft, acquired by live imaging. c Representative low- and high-magnification serial optical sections and corresponding orthogonal views of the human skin xenograft, obtained by two-photon microscopy. Note the ridge-like structures at the epidermal-dermal junction (Second Harmonic Generation in red), the epithelial clonal columns (K14-GFP), and the distinct multilayered organization recapitulating the human skin epidermis. Scale bars: 200 µm and 20 µm. d Hematoxylin and eosin (H&E) stained histological section of the entire length of the human skin xenograft. Scale bar: 100 µm. e, f Representative images showing nuclear deformation (e) and stress vesicle formation (f) in basal epidermal cells of the human skin xenograft. Note the nuclear deformation recapitulates what was observed in mouse skin. Similar results were obtained from four independent experiments. Scale bar: 10 µm. g, h Quantifications of the number of cells with nuclear deformation (g) and stress vesicle formation (h). Data are from N = 4 mice, n = 2500 and 2276 cells analyzed from the stressed area. Mann-Whitney test, p = 0.0286. Data are presented as mean ± SEM.
Discussion
The skin serves as a critical barrier that protects the body from external physical stressors, constantly adapting to mechanical forces to preserve tissue integrity. To maintain barrier function and counter cellular attrition under these conditions, the epidermis relies on continuous cellular turnover driven by stem cells in the basal layer. Although the skin often appears phenotypically unaffected following acute mechanical stress, the cellular mechanisms underlying this resilience remain poorly understood. Here, we developed three distinct mechanical stress assays designed to apply negative pressure (suction), positive pressure (compression), or lateral tension (stretching) to mouse skin, each capable of inducing distinct tissue deformations and cellular responses. Leveraging intravital imaging to directly visualize keratinocyte behavior at single-cell resolution within their native tissue environment, we identified a conserved sequence of intracellular events leading to the formation of “stress vesicles” in response to mechanical stress. Furthermore, we demonstrated that stress vesicles are closely associated with significant nuclear deformation. Crucially, the extent of nuclear deformation serves as a reliable predictor of whether keratinocytes subsequently commit to terminal differentiation, thereby directly implicating mechanical stress-induced nuclear remodeling in the regulation of cell fate within adult epidermis. Importantly, we established direct links between stress vesicle formation, intracellular calcium dynamics, and the mechanosensitive channel Piezo1. While the immediate physiological significance of these responses requires further exploration, our findings suggest several intriguing hypotheses regarding how mechanical cues integrate into cellular decision-making pathways to sustain epidermal homeostasis.
The adult skin is made up of three main layers: the epidermis, dermis, and subcutaneous tissue, all of which are perfused by extracellular fluid that mediates cellular interactions and supports tissue integrity78,79. Under normal conditions, extracellular fluid maintains a balanced hydrostatic pressure throughout the skin. Mechanical stress applied to the skin disrupts this equilibrium, resulting in rapid, localized increases in hydrostatic pressure. Such pressure elevations can disrupt cell-cell adhesions, allowing extracellular fluid to infiltrate intracellular spaces and force the inward invagination of plasma membranes. Similar phenomena involving fluid-driven membrane invaginations triggered by altered cell adhesions have previously been described both in vitro and during tissue development in vivo, strongly resembling the stress vesicles we observed80,81,82,83,84. Consistent with these studies, our experiments demonstrated direct uptake of extracellular fluid into stress vesicles. Importantly, each mechanical stress modality we applied—suction (negative pressure), compression (positive pressure), and stretch (lateral tension)—is expected to differently alter extracellular fluid dynamics: suction enhances local blood flow and tissue expansion; compression restricts blood flow and reduces swelling; and stretch elongates tissue architecture, altering fluid distribution patterns. While tissues can adapt gradually to these altered fluid dynamics, their limited capacity for rapid fluid redistribution leads to transient, localized increases in hydrostatic pressure. Our results further suggest that keratinocytes respond to these mechanical challenges by actively engaging their cytoskeletal machinery, forming stress vesicles as a protective mechanism to minimize cellular damage in response to acute mechanical stress.
Consistent with prior studies, our data suggest that multiple endocytic pathways may contribute to stress vesicle formation. The enrichment of acto-myosin around vesicles, together with their relatively large size, is consistent with macropinocytosis as the likely mechanism. This interpretation is supported by recent studies, which demonstrated that interstitial fluid can be cleared through macropinocytosis following larval zebrafish skin wound healing85. Moreover, we found evidence of clathrin enrichment in stress vesicles following mechanical perturbation to the skin. Whether clathrin localizes to vesicles to promote their formation or as part of a retrieval/remodeling process remains unresolved and will require further functional experiments using pharmacological or genetic perturbations to fully elucidate. Future work will be needed to determine how these pathways are integrated and how they couple to cytoskeletal dynamics and mechanosensory signaling to regulate vesicle turnover and function.
Epithelial cells have evolved sophisticated mechanisms to translate mechanical cues into biochemical signals. Central to this mechanotransduction machinery are adhesion complexes embedded in the plasma membrane, which relay physical forces from the external environment to the intracellular cytoskeleton and nucleus, ultimately influencing gene expression86,87,88,89. Additionally, mechanosensitive ion channels, such as transient receptor potential (TRP) channels and Piezo family members, act as potent sensors of mechanical forces. Upon activation by membrane tension, these channels facilitate calcium influx into the cytoplasm, initiating a signaling cascade that modulates cell adhesion dynamics and keratinocyte differentiation90,91. Piezo1, a well-characterized mechanosensitive cation channel expressed by epidermal keratinocytes, has previously been implicated in mechanical sensing and keratinocyte migration during wound repair91,92. Interestingly, Piezo1 signaling also appears to influence various intracellular processes by modulating cytoskeletal architecture and cell adhesion93. Consistent with these roles, our findings indicate that epidermal-specific Piezo1 deletion disrupts intracellular calcium homeostasis, perturbs the expression of adhesion-related and membrane dynamics genes, and markedly increases stress vesicle formation upon acute mechanical stress. Therefore, Piezo1 may function as a critical sensor or sentinel, priming the cytoskeletal and adhesive machinery of keratinocytes to rapidly adapt to changes in the mechanical environment. It is intriguing to consider that such Piezo1-mediated responses may underlie the observed regional variations in skin areas subject to different mechanical stress levels.
Stress vesicle formation associated with membrane deformation may likely also affect lipid raft organization and membrane fluidity. However, how these membrane structural changes influence membrane protein mobility and mechanosensitive ion channel activity remains unclear and warrants further study. It will also be essential to investigate how membrane remodeling during vesicle formation impacts intracellular signaling pathways linked to membrane trafficking and cellular repair mechanisms. Addressing these critical questions will enhance our understanding of mechanotransduction in epidermal biology and could facilitate innovative strategies for cell fate manipulation, tissue engineering, and targeted drug delivery.
The ultimate outcome of mechanotransduction is the regulation of gene expression, which controls the activity and fate decisions of cells. Multiple pathways have been proposed to translate mechanical cues into genomic responses, altering gene regulatory networks. Emerging evidence positions the nucleus itself as a central component of the mechanosensory apparatus. This suggests that mechanical forces exerted upon the nucleus can lead directly to changes of chromatin organization, thereby modulating transcriptional activity and cellular fate94.
Nuclear mechanotransduction is known to involve dynamic interactions among the cytoskeleton, nuclear lamina, and nuclear actin network25,95. Here, we demonstrate that acute mechanical stress applied to epidermal stem cells induces stress vesicle formation and associated nuclear deformation. Importantly, we find a strong correlation between the extent of nuclear deformation and subsequent cellular differentiation. These observations suggest that stress vesicles might represent a previously unrecognized mechanism of nuclear mechanotransduction, functioning as intermediates that relay external mechanical forces into nuclear shape alterations, thereby influencing stem cell fate decisions within epidermal tissue.
Throughout the cell cycle, epidermal keratinocytes undergo significant changes in their cytoskeletal architecture and cell-cell adhesion properties, potentially influencing their responsiveness to mechanical stressors. Our single-cell lineage tracing data reveal that keratinocyte responses to mechanical stress differ significantly depending on their differentiation status. Epidermal stem cells predominantly express Keratins 5/1496,97,98; however, one of the earliest molecular indicators of their commitment to terminal differentiation is the transition toward expressing Keratins 1 and 10. The keratin intermediate filament network is critical for maintaining the structural integrity of both the plasma membrane and nucleus96,99,100,101,102. Recent studies further suggest that intermediate filaments may directly influence cell fate decisions by mediating differentiation cues103. Our results specifically link the onset of Keratin 10 expression in basal cells to an increased propensity to form stress vesicles under acute mechanical stress. It is conceivable that the Keratin 1/10 intermediate filament network increases cellular stiffness, thereby sensitizing differentiating basal cells to mechanical forces. Future investigations will be essential to fully understand the mechanotransductive roles of intermediate filaments and their functional significance in controlling epidermal stem cell fate and differentiation.
Altogether, our findings demonstrate that mechanical stress triggers a conserved cascade of intracellular structural events in epidermal keratinocytes, initiated by the inward budding and expansion of stress vesicles and culminating in nuclear deformation and subsequent initiation of terminal differentiation. The mechanosensitive ion channel Piezo1 emerges as a central regulator in this process, linking intracellular calcium dynamics to cytoskeletal rearrangements, changes in cell-cell adhesion and stress vesicle formation. We propose that these coordinated cellular responses represent a robustness mechanism that safeguards epidermal tissue integrity and ensures homeostasis under acute mechanical stress conditions. Understanding these adaptive mechanisms could provide valuable insights into epidermal biology, tissue engineering, and therapeutic approaches for skin disorders characterized by impaired mechanotransduction.
Methods
Mice
All procedures involving animal subjects were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania., R26loxp-tdTom-stop-loxp-EGFP (R26-mTom in text), R26loxp-stop-loxp-tdTomato (R26-cytTom in text), K14CreER;R26loxp-tdTom-stop-loxp-EGFP (R26-mGFP in text), R26-CAG-GCaMP6s, Piezo1fl/fl mice were obtained from The Jackson Laboratory. K14H2BGFP mice were obtained from E. Fuchs (Rockefeller University). K14H2BPAGFP mice were generated by the Center for Animal Transgenesis and Germ Cell Research at the School of Veterinary Medicine of the University of Pennsylvania. p63CreER mice were created and kindly donated by J. Xu (Baylor College of Medicine). All mice that were used in this study were bred for multiple generations into a Crl:CD1(ICR) mixed background. For lineage tracing experiments and Piezo1 gene deletion experiments, Cre activation was induced with intraperitoneal injections of Tamoxifen in corn oil (0.1–2 mg per 20 g body weight). Mice were induced between 6–8 weeks old, and subsequent experiments were conducted at the indicated times after induction. Experiments included equal representation of males and females. There was no apparent difference in phenotype between genders. Mice were housed in a temperature and light-controlled environment and received food and water ad libitum.
Mechanical force assays
Mice were anesthetized with an intraperitoneal injection of Ketamine / Xylazine cocktail I PBS (0.1 mL / 20 g body weight: 87.5 mg /kg Ketamine, 12.5 mg / kg Xylazine). Mouse skin was shaved and treated with depilatory cream (Nair) prior to mechanical force application. Negative pressure: A modified cutaneous suction system with a glass vacuum chamber (10 mm in diameter) was applied to generate negative pressure on the back skin of anesthetized mice. Briefly, a vacuum of 80 kPa was used to generate a negative force (suction) on the mouse back skin. For monitoring the formation of stress vesicles, negative pressure was applied to the mouse back skin for 3 min before imaging (Supplementary Fig. 1a). For the mouse skin fragility study, the skin was carefully monitored for blistering formation during a 10 min application of suction force. The number of blisters was counted right after treatment. Positive force (Compression): The ear and back skin on each mouse was compressed by using a modified pinching method, in which a metal arm connected a cover slip that compressed the mounted skin. A pressure sensor was inserted beneath the mouse skin to monitor the pressure intensity. Three levels of compression force were applied respectively on different mouse skin: 1, 2 and 3 gf/mm2. Using this approach to generate compression on mouse skin, we can visualize the responses of epidermal cells to mechanical forces by live imaging in real time (Supplementary Fig. 1b). Mechanical stretching (lateral tension): Full-thickness skin (2 cm in width and 3.5 cm in length) was collected from the adult mouse back and put into PBS. Each skin sample was subsequently mounted onto a custom-designed tensile stretching device with a 37 °C heating element that was placed onto an imaging stage. The explant was then stretched to its original dimensions (3.5 cm) before applying tensile stress. During linear stretching, the explant was continually moisturized with PBS that was preheated to 37 C°. Individual skin samples were stretched to three different strain rates (low, medium, high) from 10% to 30% strain and that are quantified by the elongated length of the skin tissue after stretching (Supplementary Fig. 1c).
In vivo imaging
Imaging preparation and procedures were performed with the protocol previously described104. Mice were anaesthetized by intraperitoneal injection of a ketamine/xylazine cocktail in PBS (0.1 mL / 20 g body weight; 87.5 mg / kg Ketamine, 12.5 mg / kg Xylazine). A surgical plane of anesthesia was verified by the absence of pedal reflex responses following physical stimulation and was maintained during the imaging period with 1% vaporized isoflurane in oxygen and air delivered through a nose cone. The skin was mounted on a custom-made stage with a glass coverslip placed directly against it. To maintain body temperature, mice were placed on a heating pad throughout the experiment. Image acquisition was performed with an upright Olympus FV1200MPE microscope, equipped with a Chameleon Vision II Ti: Sapphire laser. The laser beam was focused through 10X, 20X or 25X objective lenses (Olympus UPLSAPO10X2, N.A. 0.40; UPLSAPO20X, N.A. 0.75; XLPLN25XWMP2, N.A. 1.05). Emitted fluorescence was collected by two multi-alkali and two gallium arsenide phosphide (GaAsP) non-descanned detectors (NDD). The following wavelengths were collected by each detector: NDD1 419–458 nm; NDD1 458–495 nm; GaAsP-NDD1 495–540 nm; GaAsP-NDD2 575–630 nm. GFP and Tomato reporters were excited at 930 nm, and their signal was collected by GaAsP-NDD1 and GaAsPNDD2, respectively. Second harmonic generation signal was generated using 930 nm excitation wavelength and detected in NDD2. Serial optical sections were acquired in 2.5–3 mm steps, starting from the surface of the epidermis and capturing the entire thickness of the epidermis and a partial section of the dermis (epidermis 30 mm, dermis 60–80 mm). Multi-day tracing experiments were performed by re-imaging the same field of view at the indicated times after the initial acquisition, with vasculature and micro-tattoos used as landmarks to identify the imaging region at low magnification and clusters of hair follicles used as landmarks at higher magnification. For all time-lapse videos, the live mouse remained anesthetized for the length of the experiment, and serial optical sections were captured at intervals of 10 s. After each imaging session, the mice were monitored and allowed to recover in a warm chamber before being returned to the housing facility.
In vivo photo-labeling
The procedures were performed with the protocol previously described105. The pre-activated form of the H2B-PAGFP fluorescent fusion protein was visualized by exciting with 850 nm wavelength, and the emission signal was collected in GaAsP-NDD1. Excitation with 930 nm verified that no signal is emitted by the reporters before activation. Photolabeling was achieved by scanning a defined region-of-interest (ROI) at the plane of the basal layer of the epidermis, with the laser tuned to 750 nm wavelength, for 5–10 s, using 5%–10% laser power. Immediately after photo-activation, a series of optical sections, with a range that includes the entire thickness of the skin, was acquired using the same acquisition settings as for GFP. Visualizing the signal of the activated form of PAGFP only within the ROI confirmed the successful photo-labeling of basal layer keratinocytes. Following the initial image acquisition immediately after photo-labeling, the same area of skin was re-imaged at the indicated times to evaluate the responses of the individual labeled basal keratinocytes or groups of labeled basal keratinocytes to mechanical forces compared to control cells or control groups.
Hypodermal drug injection
To label extracellular fluid, 10 kDa fluorescent dextran (Alexa Fluor 488 dextran Invitrogen #D22910) was injected subcutaneous into back skin prior to mechanical forces application. Briefly, a single anaesthetized mouse was placed on the heat pad, and fluorescent dye at a concentration of 20 mg/ml and 10 µl volume in total was injected into the skin dermal area. Mechanical force was applied right after fluorescent dye injection. Images were taken in the green channel immediately after mechanical force application. For the inhibitor experiments, mice were locally injected with Latrunculin-A (Abcam, ab144290, 50 µM), blebbistatin (Sigma, #B0560, 2 mg/kg per mouse) 1 h before mechanical force application, PBS was used as a vehicle control. For the activation of calcium influx experiment, 5 µM Yoda1 was injected locally in the back skin prior to mechanical force application, PBS diluted DMSO was used as a vehicle control. For the inhibitor of calcium dynamic experiments, GsMTx4 (R&D, #4912, 1 µM) was locally injected in the area prior to mechanical force application, and PBS was used as a vehicle control.
Whole-mount staining, Immunostaining, TUNEL assays, and RNAscope
To isolate epidermis for whole mount staining, dorsal skin was dissected right after mechanical force application, then was incubated in 5 mg/ml dispase II solution (Sigma, #4942078001) at 37 °C for 10 min. The epidermis was separated from the dermis using forceps. The epidermis was fixed in 4% paraformaldehyde in PBS for 45 min at room temperature, then washed in PBS, permeabilized and blocked for > 2 h (0.3% Triton-X, 5% normal goat serum and 1% BSA in PBS) and then incubated in primary antibodies overnight at 4 °C and secondary antibodies for 3 h at room temperature. For section staining, dorsal skins were dissected, laid flat and directly fixed with 4% paraformaldehyde in PBS for overnight at 4 C°. Fixed tissues were paraffin embedded and sectioned. Deparaffinized slides were incubated in a 1:50 dilution of Antigen Unmasking Solution (Vector Laboratories) and heated in a boiling bath for 10 min. Sections were permeabilized with 0.2% Tween 20 in PBS and incubated in blocking solution (10% normal goat or horse serum in 0.5% Tween 20 in PBS for 1 h. Samples were incubated with primary antibody diluted in staining solution (2% normal goat or horse serum 0.2% Tween 20 in PBS) overnight at 4 C°. After 2 h of washing in 0.2% Tween 20 in PBS, tissue sections were incubated with secondary antibody and DAPI diluted in staining solution for 1 h at room temperature, followed by washing three times with 0.2% Tween 20 in PBS for 5 min each. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed using the In Situ Cell Death Detection Kit (Roche) according to manufacturer’s instructions. In brief, following deparaffinization and slide rehydration, slides were treated with a 20 mg/ml Proteinase K (Denville Scientific) solution in PBS for 15 min at room temperature, washed in 0.2% Tween 20 PBS and incubated in TUNEL reaction mixture for 1 h at 37 C°, then washed prior to mounting. As a positive control for the reaction, we included a slide treated with DNase I for 15 min at room temperature prior to TUNEL reaction mixture incubation. For Hematoxylin and eosin (H&E) staining, sections were fixed in 4% paraformaldehyde for 10 min, washed in PBS and stained in hematoxylin for 8 min and eosin for 1 min. The following primary antibodies were used: Anti-Ki67 (1:100, 14-5698, eBioscience), anti-Keratin 14 (1:1000, 905301, BioLegend), anti-Keratin 10 (1:500, 905401, BioLegend), anti-BrdU (1:100, B-35128, Invitrogen), anti-Lamin A/C (1:100, ab133256, abcam), anti-Ecadherin (1:200, ab16505, abcam), anti-Vinculin (1:50, ab155120, abcam), anti-F-actin (1:100, MA1-80729, Invitrogen), anti-Clathrin (1:50, ab21679, abcam), anti-CD3 (1:200, ab5690, abcam), Wheat Germ Agglutinin (WGA) (1:1000, 29022-1, Biotium), and Non-muscle Myosin II A (1:100, 909802, BioLegend). The following secondary antibodies were used: Alexa Fluor 488 Goat anti-Mouse (1:500, A-11001, Invitrogen), Alexa Fluor 594 Goat anti-Rabbit (1:500, A-11012, Invitrogen), Alexa Fluor 488 Goat anti-chicken (1:500, A-110039, Invitrogen), Alexa Fluor 594 Donkey anti-goat (1:500, A-110058, Invitrogen) and Alexa Fluor 647 Donkey anti-goat (1:500, A-21447, Invitrogen). RNAscope experiment was performed by using the RNAscope Multiplex Fluorescent Reagent Kit, version 2 (#323100, Advanced Cell Diagnostic, Newark, CA) with Murine Piezo1 and K14 specific probe (Advanced Cell Diagnostics). All images were acquired using an Olympus BX51 equipped with a Hamamatsu Orca CCD camera or a Leica DM6 B equipped with a Leica DFC9000 GT fluorescent camera and a Leica DMC2900 brightfield camera.
Cell preparation for flow cytometry and sorting
The dorsal skin was dissected, and subcutaneous fat was removed from the skin. To separate epidermal and dermal layers, skin was cut into small pieces and floated, epidermis side up, on 0.25% Trypsin (GIBCO) in PBS for 1 h at 37 C°. Following the incubation, the epidermis was carefully scraped off and transferred to 10% FBS (fetal bovine serum) in DMEM (GIBCO. Single cell suspensions were obtained by pipetting gently and filtering through 70 mm cell strainers (VWR). Each sample was spun down and resuspended in 2% FBS in PBS. For cell staining, antibodies were directly added to the cell suspension and incubated for 10 min on ice. Afterward, cells were washed using 2% FBS PBS and resuspended for sorting in 5 mM EDTA PBS. The following antibodies were used for cell sorting experiments: anti-CD49f-APC (1:300, #313615, BioLegend), anti-CD49f-PercP/Cy5.5 (1:300, #313618, BioLegend), anti-CD34-Alexa Fluor 647 (1:50, #560230, BD PharMingen), anti-Sca1-Ly-6A/E-Violet 605 (1:1000, #108133, BioLegend), and DAPI (1:1000, Biotium). Epidermal cells for RNA-sequencing experiments were membrane GFP+, CD49f+, Sca1+, CD34- and DAPI-. Analysis of cell cycle: cells were fixed and permeabilized with BD Cytofix / Cytoperm buffer (BD Pharmingen) on ice after immunofluorescent staining of antibodies specific for cell surface markers; Afterward, resuspended cells were washed using 1X BD Perm/Wash buffer (BD Pharmingen), then cells were stained with intracellular antigen-specific antibodies, including Ki67 antibody (1:100, 14-5698, eBioscience); cells were incubated with DNase-free RNase A to digest doble stranded RNA; At last, to label the toal cellular DNA content for cell cycle analysis, cells were stained with propidium iodide (PI) (1:1000, P3566, Invitrogen). Stained cells were acquired on a flow cytometer. The following antibodies were used for cell cycle analysis: anti-CD49f-FITC (1:200, #313606, BioLegend), anti-Sca1-Ly-6A/E-Violet 605 (1:1000, #108133, BioLegend), anti-Ki67 (1:100, #14-5698, eBioscience), propidium iodide (PI) (1:1000, P3566, Invitrogen), Zombie Kit (1:1000, #423105, BioLegend), and DAPI (1:1000, Biotium). We used Flow cytometry was performed on a BD LSRII cytometer (BD Biosciences), sorting on a BD FACS Aria II sorter (BD Biosciences). Flow cytometry data was collected and exported using BD FACs Diva software (BD Biosciences) and analyzed and plotted using FlowJo software.
RNA sequencing and data analysis
p63CreER; Piezo1fl/fl; R26-mTmG (Piezo1 cKO) mice were administered tamoxifen injections (3 times, 20ug/dose) at P45-P52 to activate cre recombination and delete Piezo1 gene expression in basal layer keratinocytes. Basal epidermal cells were isolated from Piezo1 cKO and control mouse back skin using flow sorting. RNA was extracted using the RNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. The libraries for sequencing were prepared using the method described before 90. Briefly, RNA-seq libraries were prepared at the same time for all samples belonging to a single experimental cohort to reduce batch effects. All RNA-seq libraries were prepared using the NEBNext Poly(A) mRNA magnetic isolation module, followed by the NEBNext Ultra Directional RNA library preparation kit for Illumina. Library quality was checked by Agilent BioAnalyzer 2100, and libraries were quantified using the Library Quant Kit for Illumina. Libraries were then sequenced using a NextSeq500 platform [75–base pair (bp) single-end reads]. All RNA-seq was aligned using RNA STAR under default settings to Mus musculus GRCm38 fragments per kilobase per million mapped fragments, and differential expression analysis was performed using DESeq292. Statistical significance was obtained using an adjusted P value generated by DESeq2 of less than 0.05. Replicates were generated from 3 control and 3 Piezo1 cKO mice, isolated and sorted at different times. All GO analyses were performed using PANTHER at http://pantherdb.org/ to determine statistically overrepresented GO terms using Fisher’s exact test under the “biological process” category. P-values for GO terms are false discovery rate statistics. The top 12 plotted GO terms represent the GO terms with the highest fold enrichment under PANTHER’s default hierarchical clustering categorization. GO term figures are generated using ggplot2.
Human-engineered skin xenografts
Organotypic skin grafts containing human primary keratinocytes were performed as previously detailed methods106,107,108,109. All primary human keratinocytes were obtained from the University of Pennsylvania Skin Biology and Disease Research Center (SBDRC) STaR Core, and were isolated from deidentified discarded tissue and therefore considered to be IRB (Institutional Review Board) exempt by our institution and the National Institutes of Health (NIH). Primary human keratinocytes were cultured in a 1:1 mixture of Keratinocyte Growth Media (KGM) and Keratinocyte Media 50/50 (Gibco) containing 2% FBS, 1.2 mM calcium chloride, 100 nM Et-3 (endothelin 3), 10 ng/mL recombinant human stem cell factor, and 4.5 ng/mL recombinant basic fibroblast growth factor. Cells were transduced with CMV-H2BPAGFP and CMV-Lifeact-EGFP lentiviral vectors. Transduced human keratinocytes (10 × 105) were suspended in 80 µl mixture media, seeded onto the decellularized human dermis provided by the SBDRC STaR Core, and incubated at 37 C° for 4 days at the air-liquid interface to establish organotypic skin. Organotypic skin tissues were graft onto 5- to 7- week- old female ICR SCID mice (Taconic) according to an International Animal Care and Use Committee (IACUC) approved protocol at the University of Pennsylvania. Mice were anesthetized in an isoflurane chamber, and murine skin was removed from the upper dorsal region of the mouse. Organotypic human skin was trimmed to a uniform 11 mm by 11 mm square and grafted onto the back of the mouse with individual interrupted 6-0 nylon sutures. Mice were dressed in Bactroban ointment, Adaptic, Telfa pad, and Coban wrap. Dressings were removed 2 weeks after grafting. Mechanical treatments were performed 2 months after grafting.
Calcium imaging data analysis
To investigate the cellular response to mechanical forces, calcium dynamics were captured in individual epidermal cells within basal layer of epidermis in vivo using R26-CAG-GCaMP6s; R26-mTmG mice. At both rest and stressed condition, time lapses of one z plane (the basal layer of epidermis) were recorded. Time-lapse images were analyzed and plotted using the following steps. First, raw image stacks were imported into FIJI (ImageJ, NIH); To measure the calcium dynamics in the same cells in response to mechanical forces, region-of-interest (ROIs) on the cells that responded to stresses were chose and segmented manually based on tomato fluorescent labeled cell membrane. After setting multiple ROIs using ROI manager, the mean fluorescence intensity (GFP) of each segmented cell was measured and normalized to basal level for all segmented cells and frames.
Digital data analysis
Raw image files from two-photon imaging were imported into FIJI (Image J NIH) or Imaris (Bitplane) for analysis. Individual optical planes and sequential optical sections were used to assemble figures as previously described. 3-dimensional renderings were created using Imaris software. To quantify population clonal dynamics and high-resolution optical sections were obtained sequentially and used to construct 3-dimensional tiled views of the epidermis. The same mice were then re-imaged using identical acquisition parameters. For single-cell lineage tracing, individual high-magnification serial optical sections were obtained for each traced clone and 3-dimensional analysis was performed once the entire imaging time course was completed to analyze the state of basal and suprabasal cells at each time point. Departure of a cell from the basal layer and subsequent upward transit was scored as differentiation, while continuous increase in the basal cell number was scored as self-renewal. Clone measurements were performed manually. Images shown in figures typically represent maximum projections or single optical sections selected from the z stacks unless otherwise specified.
Electron microscopy
Trimmed skin samples were fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH7.4, overnight. Samples were submitted to and processed by the Electronic Microscopy Resource Lab (EMRL), a core facility at UPenn. Images were taken using Morada CCD and iTEM (Olympus) software.
Statistics and reproducibility
Data are expressed as column bar graphs and scatter plots. When comparing multiple groups in this manuscript, statistical analyses were conducted as follows: comparisons between the untreated group and each treated group (1 min, 3 min, and 5 min) were performed using the Mann-Whitney U test. Differences among all groups were assessed using the Kruskal-Wallis test. For pairwise comparisons among the treated groups (1 min vs. 3 min, 1 min vs. 5 min, and 3 min vs. 5 min), one-way ANOVA with Sidak’s multiple-comparisons correction was applied. Unpaired Student’s t-test or Mann-Whitney U-test was performed to analyze data sets with two groups. Statistical calculations were performed using the Prism software package (GraphPad). To evaluate the relationship between stress duration and vesicle size, a Spearman’s rank correlation analysis was performed using GraphPad Prism. This nonparametric test was chosen due to the non-normal distribution of vesicle size data. Correlation strength and significance were determined based on the correlation coefficient (r) and the corresponding p-value. No statistical method was used to predetermine sample size.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Reagents presented in this study are available from the corresponding author upon reasonable request. RNA sequencing data can be accessed through the Gene Expression Omnibus under accession number GSE217491. Source data are provided with this paper.
References
Joodaki, H. & Panzer, M. B. Skin mechanical properties and modeling: a review. Proc. Inst. Mech. Eng. Part H. J. Eng. Med. 232, 323–343 (2018).
Biggs, L. C., Kim, C. S., Miroshnikova, Y. A. & Wickström, S. A. Mechanical forces in the skin: roles in tissue architecture, stability, and function. J. Invest. Dermatol. 140, 284–290 (2019).
Filippo, R. E. D. & Atala, A. Stretch and growth: the molecular and physiologic influences of tissue expansion. Plast. Reconstr. Surg. 109, 2450–2462 (2002).
Tepole, A. B., Gosain, A. K. & Kuhl, E. Stretching skin: the physiological limit and beyond. Int J. Nonlinear Mech. 47, 938–949 (2012).
Yang, W. et al. On the tear resistance of skin. Nat. Commun. 6, 6649 (2015).
Oxlund, H., Manschot, J. & Viidik, A. The role of elastin in the mechanical properties of skin. J. Biomech. 21, 213–218 (1988).
Huang, X. et al. Matrix stiffness–induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Resp. Cell Mol. 47, 340–348 (2012).
Balaban, N. Q. et al. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 3, 466–472 (2001).
Plikus, M. V. et al. Fibroblasts: origins, definitions, and functions in health and disease. Cell 184, 3852–3872 (2021).
Le, H. Q. et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 18, 864–875 (2016).
Carley, E. et al. The LINC complex transmits integrin-dependent tension to the nuclear lamina and represses epidermal differentiation. eLife 10, e58541 (2021).
Lamaze, C. & Torrino, S. Caveolae and cancer: a new mechanical perspective. Biomed. J. 38, 367 (2015).
Swift, J. & Discher, D. E. The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J. Cell Sci. 127, 3005–3015 (2014).
Alisafaei, F., Jokhun, D. S., Shivashankar, G. V. & Shenoy, V. B. Regulation of nuclear architecture, mechanics, and nucleocytoplasmic shuttling of epigenetic factors by cell geometric constraints. Proc. Natl. Acad. Sci. 116, 13200–13209 (2019).
Pfeifer, C. R., Alvey, C. M., Irianto, J. & Discher, D. E. Genome variation across cancers scales with tissue stiffness – an invasion-mutation mechanism and implications for immune cell infiltration. Curr. Opin. Syst. Biol. 2, 102–113 (2017).
LeGoff, L. & Lecuit, T. Mechanical forces and growth in animal tissues. Csh Perspect. Biol. 8, a019232 (2015).
Matamoro-Vidal, A. & Levayer, R. Multiple influences of mechanical forces on cell competition. Curr. Biol. 29, R762–R774 (2019).
Jaalouk, D. E. & Lammerding, J. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Bio 10, 63–73 (2009).
Aureille, J., Belaadi, N. & Guilluy, C. Mechanotransduction via the nuclear envelope: a distant reflection of the cell surface. Curr. Opin. Cell Biol. 44, 59–67 (2017).
Stan, R. V. Structure of caveolae. Biochim. Biophys. Acta Bba - Mol. Cell Res 1746, 334–348 (2005).
Sinha, B. et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144, 402–413 (2011).
Joseph, J. G. & Liu, A. P. Mechanical regulation of endocytosis: new insights and recent advances. Adv. Biosyst. 4, 1900278 (2020).
Gudipaty, S. A. et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 543, 118–121 (2017).
Apodaca, G. Modulation of membrane traffic by mechanical stimuli. Am. J. Physiol.-Ren. 282, F179–F190 (2002).
Nava, M. M. et al. Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181, 800–817.e22 (2020).
Aragona, M. et al. Mechanisms of stretch-mediated skin expansion at single-cell resolution. Nature 584, 268–273 (2020).
Khilan, A. A., Al-Maslamani, N. A. & Horn, H. F. Cell stretchers and the LINC complex in mechanotransduction. Arch. Biochem Biophys. 702, 108829 (2021).
Senoo, M. Epidermal stem cells in homeostasis and wound repair of the skin. Adv. Wound Care 2, 273–282 (2013).
Kaur, P. Interfollicular epidermal stem cells: identification, challenges, potential. J. Invest Dermatol 126, 1450–1458 (2006).
Lim et al. Interfollicular epidermal stem cells self-renew via autocrine Wnt signaling. Sci. N. Y. N. Y 342, 1226–1230 (2013).
Dekoninck, S. & Blanpain, C. Stem cell dynamics, migration and plasticity during wound healing. Nat. Cell Biol. 21, 18–24 (2019).
Martino, F., Perestrelo, A. R., Vinarský, V., Pagliari, S. & Forte, G. Cellular mechanotransduction: from tension to function. Front Physiol. 9, 824 (2018).
Liu, Y.-S. & Lee, O. K. In search of the pivot point of mechanotransduction: mechanosensing of stem cells. Cell Transpl. 23, 1–11 (2012).
Gerard, J., Borst, G. & Helmchen, F. Calcium influx during an action potential. Methods Enzymol. 293, 352–371 (1998). [20].
Cho, C.-H., Woo, J. S., Perez, C. F. & Lee, E. H. A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 49, e378–e378 (2017).
Snoeck, H. Calcium regulation of stem cells. Embo Rep. 21, e50028 (2020).
Miroshnikova, Y. A. et al. Calcium signaling mediates a biphasic mechanoadaptive response of endothelial cells to cyclic mechanical stretch. Mol. Biol. Cell 32, 1724–1736 (2021).
Passini, F. S. et al. Shear-stress sensing by PIEZO1 regulates tendon stiffness in rodents and influences jumping performance in humans. Nat. Biomed. Eng. 5, 1457–1471 (2021).
Li, J. et al. Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282 (2014).
Friedrich, E. E. et al. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc. Natl. Acad. Sci. 116, 12980–12985 (2019).
Kraus, D. et al. Intracellular calcium dynamics - sparks of insight. J. Basic Clin. Physiol. Pharm. 11, 331–366 (2000).
Casteels, R. & Droogmans, G. Cell membrane responsiveness and excitation-contraction coupling in smooth muscle. J. Cardiovasc Pharm. 6, S304–S312 (1984).
Ji, G., Barsotti, R. J., Feldman, M. E. & Kotlikoff, M. I. Stretch-induced calcium release in smooth muscle. J. Gen. Physiol. 119, 533–543 (2002).
Retailleau, K. et al. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep. 13, 1161–1171 (2015).
Chen, X. et al. A feedforward mechanism mediated by mechanosensitive ion channel PIEZO1 and tissue mechanics promotes glioma aggression. Neuron 100, 799–815.e7 (2018).
Syeda, R. et al. Piezo1 channels are inherently mechanosensitive. Cell Rep. 17, 1739–1746 (2016).
Coste, B. et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60 (2010).
Gnanasambandam, R., Bae, C., Gottlieb, P. A. & Sachs, F. Ionic selectivity and permeation properties of human PIEZO1 Channels. PLoS ONE 10, e0125503 (2015).
Wang, S. et al. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Invest. 126, 4527–4536 (2016).
Liu, T. et al. Piezo1-mediated Ca2+ activities regulate brain vascular pathfinding during development. Neuron 108, 180–192.e5 (2020).
Ma, S. et al. A role of PIEZO1 in iron metabolism in mice and humans. Cell 184, 969–982.e13 (2021).
Cahalan, S. M. et al. Piezo1 links mechanical forces to red blood cell volume. Elife 4, e07370 (2015).
Murthy, S. E., Dubin, A. E. & Patapoutian, A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat. Rev. Mol. Cell Bio 18, 771–783 (2017).
Eid, E. S. & Kurban, M. S. A Piez-o the jigsaw: the Piezo1 channel in skin biology. Clin. Exp. Dermatol 47, 1036–1047 (2022).
Eisenhoffer, G. T. et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484, 546–549 (2012).
He, L., Si, G., Huang, J., Samuel, A. D. T. & Perrimon, N. Mechanical regulation of stem-cell differentiation by the stretch-activated Piezo channel. Nature 555, 103–106 (2018).
Cox, C. D. et al. Removal of the mechanoprotective influence of the cytoskeleton reveals that PIEZO1 is gated by bilayer tension. Nat. Commun. 7, 10366 (2016).
Lewis, A. H. & Grandl, J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. Elife 4, e12088 (2015).
Versaevel, M., Riaz, M., Grevesse, T. & Gabriele, S. Cell confinement: putting the squeeze on the nucleus. Soft Matter 9, 6665–6676 (2013).
Lele, T. P., Dickinson, R. B. & Gundersen, G. G. Mechanical principles of nuclear shaping and positioning. J. Cell Biol. 217, 3330–3342 (2018).
Kalukula, Y., Stephens, A. D., Lammerding, J. & Gabriele, S. Mechanics and functional consequences of nuclear deformations. Nat. Rev. Mol. Cell Bio. 23, 583–602 (2022).
Jones, M. L. et al. The elephant in the cell: nuclear mechanics and mechanobiology. J. Biomech. Eng. 144, 080802 (2022).
Amiad-Pavlov, D. et al. Live imaging of chromatin distribution reveals novel principles of nuclear architecture and chromatin compartmentalization. Sci. Adv. 7, eabf6251 (2021).
Srivastava, L. K., Ju, Z., Ghagre, A. & Ehrlicher, A. J. Spatial distribution of lamin A/C determines nuclear stiffness and stress-mediated deformation. J. Cell Sci. 134, jcs248559 (2021).
Goelzer, M., Goelzer, J., Ferguson, M. L., Neu, C. P. & Uzer, G. Nuclear envelope mechanobiology: linking the nuclear structure and function. Nucleus 12, 90–114 (2021).
Coué, M., Brenner, S. L., Spector, I. & Korn, E. D. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213, 316–318 (1987).
Liu, Z. et al. Blebbistatin inhibits contraction and accelerates migration in mouse hepatic stellate cells. Brit J. Pharm. 159, 304–315 (2010).
Li, Y. & Burridge, K. Cell-cycle-dependent regulation of cell adhesions: adhering to the schedule. Bioessays 41, 1800165 (2019).
Jones, M. C., Zha, J. & Humphries, M. J. Connections between the cell cycle, cell adhesion and the cytoskeleton. Philos. Trans. R. Soc. B 374, 20180227 (2019).
Moore, J. L. et al. Cell cycle controls long-range calcium signaling in the regenerating epidermis. J. Cell Biol. 222, e202302095 (2023).
Bayarmagnai, B. et al. Invadopodia-mediated ECM degradation is enhanced in the G1 phase of the cell cycle. J. Cell Sci. 132, jcs227116 (2019).
Munaron, L., Antoniotti, S. & Lovisolo, D. Intracellular calcium signals and control of cell proliferation: how many mechanisms?. J. Cell Mol. Med 8, 161–168 (2004).
Rinnerthaler, M., Streubel, M. K., Bischof, J. & Richter, K. Skin aging, gene expression and calcium. Exp. Gerontol. 68, 59–65 (2015).
Feingold, K. R., Schmuth, M. & Elias, P. M. The regulation of permeability barrier homeostasis. J. Invest Dermatol 127, 1574–1576 (2007).
Maeda, T. et al. Conversion of mechanical force into TGF-β-mediated biochemical signals. Curr. Biol. 21, 933–941 (2011).
Kröger, C. et al. Keratins control intercellular adhesion involving PKC-α–mediated desmoplakin phosphorylation. J. Cell Biol. 201, 681–692 (2013).
Jin, Y.-H. et al. Protein kinase C and calmodulin serve as calcium sensors for calcium-stimulated endocytosis at synapses. J. Neurosci. 39, 9478–9490 (2019).
Gonzalez-Molina, J. et al. Extracellular fluid viscosity enhances liver cancer cell mechanosensing and migration. Biomaterials 177, 113–124 (2018).
Choudhury, M. I., Benson, M. A. & Sun, S. X. Trans-epithelial fluid flow and mechanics of epithelial morphogenesis. Semin Cell Dev. Biol. 131, 146–159 (2022).
Dumortier, J. G. et al. Hydraulic fracturing and active coarsening position the lumen of the mouse blastocyst. Science 365, 465–468 (2019).
Mosaliganti, K. R. et al. Size control of the inner ear via hydraulic feedback. Elife 8, e39596 (2019).
Casares, L. et al. Hydraulic fracture during epithelial stretching. Nat. Mater. 14, 343–351 (2015).
Latorre, E. et al. Active superelasticity in three-dimensional epithelia of controlled shape. Nature 563, 203–208 (2018).
Morris, C. E., Wang, J. A. & Markin, V. S. The Invagination of Excess Surface Area by Shrinking Neurons. Biophys. J. 85, 223–235 (2003).
Kennard, A. S., Sathe, M., Labuz, E. C., Prinz, C. K. & Theriot, J. A. Post-injury hydraulic fracturing drives fissure formation in the zebrafish basal epidermal cell layer. Curr. Biol. 33, 2616 (2023).
Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006).
Humphrey, J. D., Dufresne, E. R. & Schwartz, M. A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Bio 15, 802–812 (2014).
Charras, G. & Yap, A. S. Tensile forces and mechanotransduction at cell–cell junctions. Curr. Biol. 28, R445–R457 (2018).
Kechagia, J. Z., Ivaska, J. & Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Bio 20, 457–473 (2019).
Elias, P. M. et al. Modulations in epidermal calcium regulate the expression of differentiation specific markers. J. Invest. Dermatol. 119, 1128–1136 (2002).
Wang, D., Chadha, G. K., Feygin, A. & Ivanov, A. I. F-actin binding protein, anillin, regulates integrity of intercellular junctions in human epithelial cells. Cell Mol. Life Sci. 72, 3185–3200 (2015).
Holt, J. R. et al. Spatiotemporal dynamics of PIEZO1 localization controls keratinocyte migration during wound healing. Elife 10, e65415 (2021).
Mikesell, A. R. et al. Keratinocyte PIEZO1 modulates cutaneous mechanosensation. Elife 11, e65987 (2022).
Dupont, S. & Wickström, S. A. Mechanical regulation of chromatin and transcription. Nat. Rev. Genet 23, 624–643 (2022).
Biedzinski, S. et al. Microtubules control nuclear shape and gene expression during early stages of hematopoietic differentiation. Embo J. 39, e103957 (2020).
Yoon, S. & Leube, R. E. Keratin intermediate filaments: intermediaries of epithelial cell migration. Essays Biochem 63, 521–533 (2019).
Santos, M., Paramio, J. M., Bravo, A., Ramirez, A. & Jorcano, J. L. The expression of keratin k10 in the basal layer of the epidermis inhibits cell proliferation and prevents skin tumorigenesis. J. Biol. Chem. 277, 19122–19130 (2002).
Ho, M. et al. Update of the keratin gene family: evolution, tissue-specific expression patterns, and relevance to clinical disorders. Hum. Genomics 16, 1 (2022).
Wang, F., Chen, S., Liu, H. B., Parent, C. A. & Coulombe, P. A. Keratin 6 regulates collective keratinocyte migration by altering cell–cell and cell–matrix adhesion. J. Cell Biol. 217, 4314–4330 (2018).
Russell, D., Andrews, P. D., James, J. & Lane, E. B. Mechanical stress induces profound remodelling of keratin filaments and cell junctions in epidermolysis bullosa simplex keratinocytes. J. Cell Sci. 117, 5233–5243 (2004).
Vassar, R., Coulombe, P. A., Degenstein, L., Albers, K. & Fuchs, E. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell 64, 365–380 (1991).
Jacob, J. T. et al. Keratin 17 regulates nuclear morphology and chromatin organization. J. Cell Sci. 133, jcs254094 (2020).
Redmond, C. J. & Coulombe, P. A. Intermediate filaments as effectors of differentiation. Curr. Opin. Cell Biol. 68, 155–162 (2021).
Huang, S. & Rompolas, P. Two-photon microscopy for intracutaneous imaging of stem cell activity in mice. Exp. Dermatol 26, 379–383 (2017).
Huang, S. et al. Lgr6 marks epidermal stem cells with a nerve-dependent role in wound re-epithelialization. Cell Stem Cell 28, 1582–1596.e6 (2021).
Ridky, T. W., Chow, J. M., Wong, D. J. & Khavari, P. A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med 16, 1450–1455 (2010).
McNeal, A. S. et al. CDKN2B loss promotes progression from benign melanocytic nevus to melanoma. Cancer Discov. 5, 1072–1085 (2015).
Natale, C. A. et al. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. Elife 7, e31770 (2018).
Doepner, M. et al. Endogenous DOPA inhibits melanoma through suppression of CHRM1 signaling. Sci. Adv. 8, eabn4007 (2022).
Acknowledgments
We thank George Cotsarelis, John Stanley and John Seykora for their invaluable advice that guided this study. We also thank Sara Wickström, Dennis E. Discher and Jean-Leon Maitre for insightful discussions. We are grateful to the University of Pennsylvania Skin Biology and Disease Research-based Center (SBDRC) for analysis of tissue sections (CPAT Core) and support with the establishment of human-engineered skin xenograft (STaR Core). We also acknowledge the support of the Institute for Regenerative Medicine and the entire stem cell community at Penn. S.H. was supported by an IRM postdoctoral fellowship. G.R. was supported by a training grant (T32GM007229) from NIH/NIGMS. P.K. was supported by the American Association for Cancer Research-John and Elizabeth Leonard Family Foundation Basic Cancer Research Fellowship. P.R. was supported by grants from NIH (R01EY036440) and from the American Cancer Society (RSG1803101DCC). Penn SBDRC was supported by a center core NIH/NIAMS grant (P30AR069589) and a shared instrumentation grant from the NIH Office of the Director (S10OD038384).
Author information
Authors and Affiliations
Contributions
S.H. and P.R. conceptualized the study, designed the experiments, and wrote the manuscript. S.H., P.K., A.B., M.M., G.R., and P.R. performed the experiments. J.Z. and B.C.C. assisted with the RNA sequencing and performed the bioinformatic analysis. P.K., M.D., and T.W.R. assisted with the establishment of human-engineered skin xenograft. T.Z. and S.P. assisted with the histological assays. J.D.B. assisted with the mechanobiology assays. All authors discussed results and participated in the manuscript preparation and editing. P.R. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jean-Léon Maître and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, S., Kuri, P., Zou, J. et al. Stress vesicles link epidermal mechanotransduction to stem cell differentiation. Nat Commun 17, 1578 (2026). https://doi.org/10.1038/s41467-026-68294-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68294-7
