
Abstract
Nitrite (NO2⁻) is a high-value chemical pivotal to agriculture and pharmaceuticals, yet its conventional via the Ostwald process is energy-intensive and polluting. Electrochemical urea oxidation reaction (UOR) offers a sustainable NO2⁻ synthesis pathway with concurrent energy-saving hydrogen (H2) production, but suffers from non-selective N2/CO2 pathways. Here, we report Cr3+ Lewis acid sites in Ni3S2 that act as hydroxyl (OH⁻) pumps, dynamically spilling OH⁻ to adjacent Ni sites via a Lewis acid-base interaction. This triggers a urea-to-NO2⁻ pathway, achieving a NO2⁻ yield of 120.98 mg h-1 cm-2 (600 mA cm-2). The OH⁻ spillover accelerates C-N cleavage while suppressing N-N coupling, enabling energy-saving H2 production (3.7 kWh m-3 at 500 mA cm-2) and Zn-urea-air batteries (charging potential 288 mV lower than Zn-air). Techno-economic analysis reveals $1,210.5 per ton of urea processed at 400 mA cm-2. This work establishes OH⁻ spillover as a universal design principle for selective electrocatalysis.
Similar content being viewed by others

Introduction
Nitrogen oxides (NO2⁻/NO3⁻) are vital industrial chemicals used in food preservation, pharmaceuticals, and synthetic materials, with global demand exceeding millions of tons annually1,2,3. Current production predominantly relies on the energy-intensive Ostwald process, which generates significant harmful gases emissions (NO, NO2, etc.)4,5. Electrochemical ammonia oxidation reaction (AOR) offers a sustainable alternative but suffers from a high operating potential (>1.5 VRHE at 10 mA cm−2) and slow kinetics6,7,8,9,10. Urea, a stable nitrogen-rich compound abundant in wastewater (constituting 70-80% of nitrogen pollutants)11,12, presents an opportunity for simultaneous wastewater remediation and nitrogen oxide synthesis via the urea oxidation reaction (UOR). In addition, the anode UOR kinetics (−0.166 VSHE) is significantly lower than the oxygen evolution reaction (OER, 1.23 VSHE), which can assist the green H2 production at low energy consumption or support Zn-air batteries (ZAB) through electrochemical energy conversion13,14,15. Furthermore, among numerous small-molecule oxidation reactions, the UOR offers a cost-effective route for producing high-value products (nitrites)16,17, while utilizing a readily available feedstock and thereby eliminating the need for complex purification.
Under alkaline conditions, UOR can follow two pathways: (i) the traditional 6e⁻ pathway to N2/CO2, or (ii) hydroxyl (OH⁻)-mediated overoxidation to NO2⁻/NO3⁻ when hydroxide concentration exceeds the N-N coupling rate16,18. Current UOR research predominantly focuses on the urea-to-N2/CO2 pathway to reduce the hydrogen production overpotential19,20. But obviously, the latter pathway, which mainly involves C-N bond cleavage and amino group dehydrogenation-oxygenation, typically achieved by OH⁻ nucleophilic attack, is more valuable21,22,23. Consequently, enriching OH⁻ can significantly accelerate UOR kinetics, promoting the selective oxidation of urea to NO2⁻. Nickel-based catalysts dominate UOR research due to their oxyphilic nature24, but their unselective multi-coordination sites often lead to mixed products14. Therefore, improving the oxyphilic termination of Ni-based catalysts and designing catalysts capable of quickly capturing active OH⁻ groups are crucial for enhancing the selectivity of nitrogen oxyanions.
Lewis acid sites serve as an ideal platform for promoting OH⁻ enrichment process: as electron-deficient centers, they rapidly and efficiently capture OH⁻ via Lewis acid-base interactions25,26,27. Recently, Lu et al. engineered a Ni/ZnO catalyst with optimal Lewis acidity, effectively balancing water dissociation kinetics and *OH* binding energetics, thus enhancing neutral HER performance28. Also, Han et al. developed Al-doped Cu electrocatalysts for CO2 reduction, where Lewis acidic Al sites enhanced the adsorption strength of surface oxygen species and stabilized key intermediates, resulting in a high Faradaic efficiencies (FEs) for C2+ alcohols29. Notably, chromium ions (Cr3+), as a hard Lewis acid (pKa = 2.05), exhibit strong adsorption affinity for OH⁻ ions. Qiao et al. have demonstrated that chromium oxide (Cr2O3) layers enhanced OH⁻ capture during seawater oxidation, resulting in a localized alkaline environment that promoted reaction kinetics and rejected Cl- adsorption25. Therefore, the strategic integration of Cr3+ may offer an effective approach to modulate the reaction pathways of Ni-based catalysts. The spillover process, a key phenomenon in heterogeneous catalysis, involves the migration of activated species (e.g., OH⁻) from their initial adsorption site to an adjacent, distinct active site30. This process enables sites that lack intrinsic activity to participate in reactions by utilizing these migrated species, thereby achieving synergistic catalysis and potentially altering the reaction pathway, as demonstrated in various systems31. The strong Lewis acid-base effects at Cr sites may initiate OH− spillover. This spillover subsequently promotes the selective oxidation of urea to nitrite/nitrate, ultimately facilitating the efficient electrochemical synthesis of nitrogen oxyanions. However, electrochemical UOR lacks systematic research on producing high-value-added liquid products, especially nitrogen oxyanions, and controlling their selectivity. The lack of relevant research substantially impedes the understanding of the molecular-level reaction mechanism underlying urea electrooxidation.
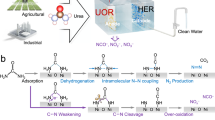
Herein, we leverage Lewis acid-base interactions to steer UOR pathways. A Cr-doped Ni3S2 (Cr-Ni3S2) as a multifunctional electrocatalyst is reported, which achieves high selectivity and activity for NO2⁻ synthesis via UOR while enabling efficient hydrogen production. Unlike conventional UOR pathways involving N-N coupling toward N2, we demonstrate that the Cr-induced Lewis acid-base interaction (Cr- OH⁻) triggers dynamic OH⁻ spillover. This optimizes reaction pathways for favorable energetics, while enriched OH⁻ groups promote urea-to-NO2⁻ conversion and enhance catalytic kinetics, thereby boosting overall activity (Fig. 1). This phenomenon stems from the adsorption energy difference of OH⁻ between Cr and Ni sites, along with electronic structure modulation via doping. The NO2⁻ production performance at an industrial current density compares favorably with reported values for similar systems. Significantly, Cr-Ni3S2 can be used as a multifunctional anode material.

Schematic diagram of different reaction pathways for Cr-Ni3S2 and Ni3S2 in UOR.
Results
Synthesis and characterizations of catalysts
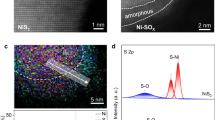
Cr-Ni3S2 (Fig. 2a) was synthesized through a two-step hydrothermal method, and a series of control electrodes was fabricated by adjusting the metal and sulfur source feed ratios. As shown in Fig. 2b and Supplementary Fig. 1, X-ray diffraction (XRD) patterns reveal that the characteristic peaks of CrNi-LDH and Ni3S2 correspond to JCPDS No. 38-0715 and JCPDS No. 44-1418, respectively, confirming their successful synthesis. As shown in Supplementary Fig. 2, the characteristic peak at 2θ ≈ 31.6° of Cr-Ni3S2 exhibits a peak shift toward the higher angles compared to that of Ni3S2, which can be attributed to lattice contraction induced by the Cr doping with a smaller ionic radius32. Scanning electron microscopy (SEM) images indicate the lamellar structure of CrNi-LDH and Ni3S2 (Supplementary Figs. 3, 4). As illustrated in Supplementary Fig. 4, Cr-Ni3S2 exhibits a block-like morphology composed of stacked lamellar layers. Notably, the Cr doping induces a significant reduction in nanosheet dimensions, as confirmed by SEM analysis, which can facilitate the exposure of additional active sites33. Moreover, the lamellar structure of Cr-Ni3S2 is also confirmed by transmission electron microscopy (TEM) image (Fig. 2c). Figure 2d and Supplementary Fig. 5 displays the HADDF-STEM image and the EDX mapping image, which indicate that the elements Ni, Cr, and S are uniformly distributed throughout the catalyst without obvious phase separation (with weaker S, O signals). Further, since different Cr incorporation modes may affect catalytic performance differently, we used aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) to determine the doping mode of Cr in Cr-doped Ni3S2. The results reveal that each bright spot in the HAADF-STEM image corresponds to the superposition of many atoms, presented as an atomic column. Due to the similar atomic numbers of Cr (Z = 24) and Ni (Z = 28), distinguishing individual Cr atom positions is challenging as their positions overlap within atomic columns34. As shown in Fig. 2e, f and Supplementary Fig. 6, it can be found that Cr atoms may be the displacement atoms rather than the interstitial or surface adsorbed atoms in the lattice, which displays the darker spots in the HAADF-STEM images (circled by the yellow dotted circle). Further, this result is also verified by the presence of Cr and Ni signals in the electron energy loss spectroscopy (EELS) spectra for region A in Fig. 2e (Fig. 2g). The substitution of larger Ni2+ by smaller Cr3+ induces lattice contraction and a decrease in interplanar spacing. Specifically, the (202) and (015) crystal facets of Cr-Ni3S2 exhibit slight decreases in interplanar spacing to 0.19 and 0.13 nm, respectively, compared to those of Ni3S2 (0.20 and 0.14 nm), which aligns with the findings from XRD analysis (Fig. 2e, Supplementary Fig. 7)35. Besides, inductively coupled plasma optical emission spectroscopy (ICP-OES) results exhibit the specific contents of 3.28 wt% Cr in Cr-Ni3S2, further confirming the successful incorporation of Cr (Supplementary Table 1)36.

a Schematic illustration of the synthesis of Cr-Ni3S2. b XRD patterns of Cr-Ni3S2 and Ni3S2. c TEM image of Cr-Ni3S2. d HADDF-STEM image and TEM mapping images of Cr-Ni3S2. e AC HAADF-STEM image, f False-color HAADF-STEM image and g corresponding EELS spectra in region I of (e) for Cr-Ni3S2. Source data are provided as a Source Data file.
The surface element composition, element valence and bonding information of Cr-Ni3S2 and Ni3S2 were analyzed by X-ray photoelectron spectroscopy (XPS). Supplementary Fig. 8a displays the coexistence of elements Ni, Cr, S and O, consistent with the above results. As indicated in Supplementary Fig. 8b, the high-resolution XPS spectra of Cr 2p shows the peaks at 576.9 and 586.5 eV, corresponding to Cr3+ 2p3/2 and Cr3+ 2p1/2, respectively, while the peaks at 578.1 and 587.8 eV are ascribed to the 2p3/2 and 2p1/2 orbitals of Cr-OH, verifying the successful introduction of Cr37. In addition, compared to Ni3S2, the peaks for Ni0 (853.0 eV), Ni2+ (855.8 eV), and Ni3+ (856.9 eV) in Cr-Ni3S2 shift by 0.1 eV toward the higher binding energies due to Cr doping (Supplementary Fig. 8c), whereas the peaks for S 2p3/2 (at 162.2 eV) and S 2p1/2 (at 163.4 eV) shift by 0.2 eV toward the lower binding energies (Supplementary Fig. 8d), respectively38,39. These findings indicate that the doping of Cr regulates the electronic structures of Ni3S2 and facilitates the electron transfer from Ni to S. To further investigate the electronic configuration and coordination environment of Cr and Ni sites in Cr-Ni3S2, synchrotron radiation X-ray absorption fine structure (XAFS) spectroscopy was employed for structural characterization. As shown in Fig. 3a, the Ni K-edge XANES spectra reveal that the absorption edge position of Cr-Ni3S2 lies between those of NiO and Ni2O3, confirming a mixed +2/+3 oxidation state for Ni in Cr-Ni3S240. It can be found that the Ni valence state has increased compared to that of pristine Ni3S2 (Fig. 3b), which is consistent with the electron delocalization effect induced by Cr doping, as evidenced by the above XPS analysis. Fourier-transform extended X-ray absorption fine structure (EXAFS) analysis (Fig. 3c, Supplementary Figs. 9, 10a, and Supplementary Table 2) exhibits an obvious peak at ~1.6 Å, ascribed to the Ni-S coordination in the first shell for Cr-Ni3S2, showing a 0.3 Å compression compared to pristine Ni3S2 (~1.9 Å) accompanied by a reduced coordination number of 3.8 (Cr-Ni3S2) vs. 4.0 (Ni3S2), indicative of Cr-induced Ni-S bond length shortening41. This structural modulation is also evidenced by the wavelet transform (WT)-EXAFS analysis (Fig. 3g, h, and Supplementary Fig. 11), which exhibits attenuated Ni-S scattering intensity in R-space compared to Ni3S2, being advantageous to facilitating electron transfer and reaction kinetics owing to the shortened Ni-S bond length42. Cr K-edge XANES analysis (Fig. 3d, e) indicates that the oxidation state of Cr in Cr-Ni3S2 is close to +3, being similar to that of Cr2O3. Furthermore, FT-EXAFS profiles (Fig. 3f, Supplementary Fig. 10b and 12) further verify that Cr incorporates into the Ni3S2 lattice as dopants, with no detectable Cr2O3 or metallic Cr phases, ruling out the formation of secondary phases (the main peak position in the Cr-Ni3S2 catalyst lies between those typical of Cr-O and Cr-S bonds, suggesting the complex chemical environment where Cr coordinates with both S and O (e.g., surface Cr-OH). The clear evidence for bulk Cr-S bonding, provided by high-resolution XPS (S 2p) and XRD, supports the assignment of the dominant EXAFS peak in Cr-Ni3S2 to primarily Cr-S/O bonding, with a minor Cr-O component. This result is consistent with the HAADF-STEM analysis. Besides, WT-EXAFS studies also confirm the uniform dispersion of Cr dopants within the Cr-Ni3S2 lattice, excluding local clustering or phase separation (Fig. 3i, j, and Supplementary Fig. 11).

a Ni K-edge XANES spectra of Ni foil, NiO, Cr-Ni3S2, Ni3S2, and Ni2O3. The inset image is a magnified region of the Ni K-edge XANES. b The oxidation states of Ni obtained from Ni K-edge XANES for the NiO, Ni2O3, Ni3S2, and Cr-Ni3S2. c Fourier transform of the EXAFS spectra of NiO, Cr-Ni3S2, and Ni3S2. d Cr K-edge XANES spectra, e The oxidation state of Cr obtained from Cr K-edge XANES, and f Fourier transform of the EXAFS spectra of Cr2S3, Cr2O3, and Cr-Ni3S2. WT-EXAFS contour plots of g Cr-Ni3S2, h Ni3S2, i Cr-Ni3S2, and j Cr2O3, respectively. Source data are provided as a Source Data file.
Electrocatalytic UOR performances
The electrochemical OER and UOR performance was respectively tested in 1 M KOH and 1 M KOH + 0.5 M urea solutions using standard three-electrode systems. Initially, the UOR activity of the control electrodes with various Cr: Ni molar ratios (Cr1-Ni3S2, Cr0.5-Ni3S2, Cr0.3-Ni3S2, Cr0.25-Ni3S2) and the different feed ratios of sulfur source were studied. The results demonstrated that Cr0.5-Ni3S2-0.35 displayed the optimal UOR activity (Supplementary Fig. 13), and the following electrochemical investigations were all based on the optimal sample Cr0.5-Ni3S2-0.35. As shown in Fig. 4a, for the OER performance, Cr-Ni3S2 only required 1.51 VRHE to achieve 100 mA cm-2 in 1 M KOH, lower than that of Ni3S2 (1.56 VRHE). When urea was added into the electrolyte, the potentials of both Cr-Ni3S2 and Ni3S2 significantly decreased. Cr-Ni3S2 only required 1.30, 1.43, and 1.57 VRHE to achieve 100, 500, and 1000 mA cm−2, respectively, while Ni3S2 exhibited severe current passivation at the potentials higher than 1.45 VRHE, which is likely because Ni3S2 tends to undergo OER rather than UOR at high potentials43. For Cr-Ni3S2, with the increasing potentials, although the enrichment of more active OH⁻ groups was potentially beneficial to OER, it exhibited better UOR activity (the specific reasons will be discussed below). Its UOR activity also outperformed industrial benchmark catalysts (Pt/C and RuO2, Supplementary Fig. 14). The catalytic activity of Cr-Ni3S2 for the alkaline UOR is comparable to other reported catalysts, even those based on precious metals (Supplementary Table 3). As evidenced by the Tafel slopes in Supplementary Fig. 15, Cr-doped Ni3S2 showcased faster UOR kinetics (27.67 mV·dec−1) compared to OER (114.32 mV·dec−1). This enhancement is particularly notable when compared to pristine Ni3S2 (28.32 vs. 169.76 mV·dec−1 for UOR and OER, respectively), highlighting the catalytic advantage of Cr incorporation in Ni3S2. Besides, Supplementary Fig. 16 shows that Cr-Ni3S2 possesses the smallest charge transfer resistance (Rct), and a significantly enhanced electrochemically active surface area (ECSA), as reflected in cyclic voltammetry (CV) scanning (Supplementary Figs. 17, 18). These results aligned with the quantification of active Ni sites generated on Cr-Ni3S2/NF and Ni3S2/NF determined by CV curves, as illustrated in Supplementary Fig. 19. In order to further evaluate the intrinsic activity of the catalysts, the ECSA-normalized current density and turnover frequency (TOF) of Cr-Ni3S2 and Ni3S2 were calculated (Supplementary Figs. 20, 21). It demonstrated that Cr-Ni3S2 still exhibited the lower onset potential as well as the higher TOF value than those of Ni3S2, indicating that the Cr doping improves the intrinsic activity of Ni3S2.

a LSV curves of Cr-Ni3S2 and Ni3S2 in 1 M KOH with and without urea with 100% iR compensation (the electrode area is 1 cm × 0.5 cm, and the resistance is 1.15 ± 0.5 Ω). FEs of different products in the UOR process at different current densities for b Cr-Ni3S2 and c Ni3S2 in 1 M KOH + 0.5 M urea solution. d Nitrite yields of Cr-Ni3S2 and Ni3S2 in the UOR process at different current densities. Error bars correspond to the standard deviations of three independent measurements, and the center value for the error bars is the average of three independent measurements. e LSV curves of different Lewis acids-doped Ni3S2 and Ni3S2, and Cr-doped Cu2S and Cu2S with 100% iR compensation (the electrode area is 1 cm × 0.5 cm, and the resistance is 1.51 ± 0.8 Ω). f FE\(\scriptsize {{{\rm{NO}}}}_{2}^{-}\) and FEN2 for Cr-Ni3S2, Sn-Ni3S2, Ti-Ni3S2, Ni3S2, Cr-Cu2S and Cu2S at the optimal current density of 400 mA cm−2. g Radar chart of some key indicators of UOR performance for Cr-Ni3S2 and Ni3S2. h Chronoamperometric testing of Cr-Ni3S2 at a current density of 750 mA cm−2 in 1 M KOH + 0.5 M urea, along with the FE of NO2- and Cr leaching concentration at each electrolyte replacement without iR compensation. All the measurements were tested in 1.0 M KOH with and without 0.5 M urea solution as the electrolyte (pH = 13.97 ± 0.02) at room temperature. Source data are provided as a Source Data file.
As shown in Fig. 4b, c and Supplementary Fig. 22, Supplementary Table 4, we systematically analyzed the FEs of NO3⁻, NO2⁻, N2, and O2 generated during the UOR process for Cr-Ni3S2 and Ni3S2 catalysts, respectively. Notably, the detected O2 originated from the competing OER at the anode, a parasitic process that increased energy consumption and reduced UOR activity. Thus, monitoring O2 production was critical for accurate UOR catalyst evaluation. Figure 4b, c reveal that NO2⁻ is the dominant UOR product for both Cr-Ni3S2 and Ni3S2, which suggests that the UOR pathway predominantly follows the OH⁻-mediated urea-to-NO2⁻ electrooxidation mechanism (CO(NH2)2 + 8 OH⁻ → NO2⁻ + NCO- + 6H2O + 6e-) rather than the N-N coupling pathway44. Strikingly, Cr-Ni3S2 demonstrated a significantly higher FE for NO2⁻ (85.71 ± 3.52%) compared to Ni3S2 (63.79 ± 1.89%) at 400 mA cm−2. Furthermore, the Cr-Ni3S2 catalyst achieved a NO2⁻ production rate of 120.98 ± 5.06 mg h−1 cm−2 at 600 mA cm−2 (Fig. 4d), a performance that compares favorably with documented values in the literature (Supplementary Table 5). Besides, Cr-Ni3S2 exhibited a consistently low FE for O2 evolution (< 1.5%) across the entire tested potential range, markedly inferior to pristine Ni3S2. These results indicated that Cr doping in Cr-Ni3S2 simultaneously enhanced urea-to-NO2⁻ conversion to boost NO2⁻ generation and suppressed the competitive OER, thereby synergistically reducing UOR potential while improving catalytic performance. Furthermore, as shown in Fig. 4e, f and Supplementary Figs. 23, 24, the use of other Lewis acids (Ti and Sn) as the dopants also improved the UOR activity and FE of NO2⁻ for Ni3S2. The UOR activity followed the order of Cr-Ni3S2 > Sn-Ni3S2 > Ti-Ni3S2 > Ni3S2, which was consistent with the trend in their Lewis acid strength (Supplementary Fig. 25 and Supplementary Table 6). Besides, the strategy of introducing Lewis acids as the dopants was also applicable to other catalyst matrix, for which Cr-doped Cu2S (Cr-Cu2S) also showcased the enhanced UOR activity over Cu2S, indicating the universality of this strategy. Figure 4g illustrates the comparisons of some key indicators of UOR performance between Cr-Ni3S2 and Ni3S2, including the potential at large current density, Tafel slope, active surface area, and TOF value, highlighting the promoting effect of Cr doping in Cr-Ni3S2 on UOR performance. Further, the stability and durability of a catalyst is a critical factors in assessing catalytic performance. As shown in Supplementary Fig. 26, after 3000 CV cycles, the Cr-Ni3S2 catalyst retained its UOR activity at 500 mA cm-2 with only a 1% potential decrease. More importantly, as illustrated in Fig. 4h and Supplementary Table 7, the Cr-Ni3S2 electrode maintained continuous operation for over 360 h at a high current density of approximately 750 mA cm−2. Throughout this period, the FE\(\scriptsize {{{\rm{NO}}}}_{2}^{-}\) remained stable, fluctuating narrowly between 64% and 69%, while metal leaching was negligible, with only 0.134 ppm of Cr detected after 360 h. These results collectively confirm the stability and durability of the Cr-Ni3S2 catalyst. To evaluate the structural and morphological stability of Cr-Ni3S2 after the UOR stability test, XRD, SEM, and XPS analyses were performed. As shown in Supplementary Fig. 27, the XRD patterns confirmed that the crystalline structure of Cr-Ni3S2 maintained intact after the UOR test. SEM image after UOR stability test, shown in Supplementary Fig. 28 exhibited no significant morphological changes. However, the nanosheet-stacked blocks showed a looser arrangement, and the nanosheets slightly decreased in size, indicating that the underlying structure of Cr-Ni3S2 was largely maintained. Supplementary Fig. 29 shows the XPS analysis of Cr-Ni3S2 before and after the UOR stability test, which uncovers the surface chemical states of Ni 2p and Cr 2p. It can be found that the characteristic peaks of Ni 2p3/2 and Cr 2p3/2 were both positively shifted after UOR stability testing, which were consistent with the electron loss process in the anodic oxidation reaction45. It is noteworthy that after UOR stability test, the peak area of Cr-OH increased, which can be attributed to the more Cr-OH bonding formation induced by the substantial capture of OH⁻ by Cr sites during the UOR process. This explains the increased UOR activity of Cr-Ni3S2. As a hard Lewis acid, Cr enhanced OH⁻ spillover by capturing and transferring it to adjacent nickel sites. This not only promoted the generation of UOR-critical Ni3+ active sites but also maintained a steady OH⁻ supply during catalysis, synergistically boosting UOR activity through dual mechanisms: active site engineering and reactant availability.
UOR mechanism study of Cr-Ni3S2
The electrosynthesis of nitrite depends critically on rapid C–N bond cleavage in urea, requiring a kinetic preference for the urea-to-NO2⁻ pathway over N-N coupling14. To prioritize this reaction selectivity, urea molecules must be preferentially adsorbed onto catalytic sites and undergo OH⁻ binding interactions rather than participating in intra-/intermolecular N-N bond coupling. Consequently, effective electrochemical nitrite synthesis necessitates catalysts that simultaneously optimize two critical functionalities: (1) robust urea adsorption capability to ensure adequate reactant supply, and; (2) enhanced OH⁻ affinity to facilitate C-N bond cleavage, and suppress competing N-N coupling reactions, and drive urea-to-NO2⁻ kinetics. Given the electrochemical NO2⁻ synthesis activity of Cr-Ni3S2, we conducted LSV tests in varying KOH concentrations to investigate the kinetic interactions between Cr-Ni3S2 and OH⁻. As shown in Supplementary Fig. 30, the reaction order of Cr-Ni3S2 (1.44) is higher than that of Ni3S2 (1.40), suggesting that both catalysts exhibit OH⁻-dependent UOR kinetics, with Cr-Ni3S2 demonstrating a stronger dependence on OH⁻ concentration during the reaction process. Further, to probe the generation and utilization of hydroxyl radicals (OH) during the UOR process, we conducted liquid-phase electron paramagnetic resonance (EPR) measurements using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the OH⁻ trapping agent in 1 M KOH with 0.5 M urea. As shown in Supplementary Fig. 31, under open-circuit potential (OCP), both Cr-Ni3S2 and Ni3S2 exhibited DMPO-OH adduct signals, with Cr-Ni3S2 demonstrating a markedly stronger intensity, reflecting the enhanced OH⁻ adsorption capability compared to Ni3S2. Notably, at an applied potential of 1.3 VRHE, the DMPO-OH signal for Cr-Ni3S2 vanished completely, whereas a faint yet discernible signal persisted for Ni3S2. These results suggested that under electrochemical bias, Cr-Ni3S2 efficiently consumed OH⁻ to drive urea-to-NO2⁻, while Ni3S2 exhibited limited OH⁻ utilization, leading to residual OH⁻ detection. To validate this speculation, we utilized a dynamic light scattering analysis-based instrument to examine the surface zeta potential difference between Cr-Ni3S2 and Ni3S2. As shown in Supplementary Fig. 32, the measured zeta potential of Cr-Ni3S2 was -1.38 mV, significantly lower than that of Ni3S2 (-0.85 mV). This difference can be attributed to the strong OH⁻ adsorption of Cr-Ni3S2, which enhanced the negative charge density on the surface of Cr-Ni3S2, thereby reducing the surface electrostatic potential.
Generally, Ni sites are widely recognized as the key catalytic active sites for UOR46. Therefore, the role of Cr sites in Cr-Ni3S2 requires further clarification. As shown in Supplementary Fig. 33, the UOR activity of a Cr-S catalyst without Ni species was first evaluated, which exhibited negligible activity, suggesting that Cr was unlikely to be the primary active site for UOR. Further quasi-in situ XPS analysis indicated that during the initial stages of the UOR process (1.2 ~ 1.25 VRHE) (Supplementary Fig. 34), the ratio of the deconvolved Cr-OH peak area to the entire peak area remained consistently high with negligible change. However, as the reaction progressed (at the higher potentials of 1.3 ~ 1.4 VRHE), the peak ratio decreased, which can be attributed to the substantial consumption of OH⁻ adsorbed on Cr sites during the UOR process. Thus, we concluded that during the UOR process, Cr sites played a crucial role in capturing abundant OH⁻ through Lewis acid-base interactions. These OH⁻ species subsequently overflowed to the neighboring Ni sites (the primary catalytic active sites), where they drove the urea-to-NO2⁻ process. This highly efficient bimetallic site tandem catalysis can significantly enhance the catalytic reaction kinetics. Furthermore, we employed in situ Raman spectroscopy to investigate the effect of different OH⁻ abundance on Ni sites during the UOR process. As illustrated in Supplementary Fig. 35, the structural evolution of Cr-Ni3S2 and Ni3S2 was monitored under an applied voltage range of 1.2 ~ 1.5 VRHE during UOR. As the reaction progressed, for Cr-Ni3S2, new peaks corresponding to δNiⅢ-O (486 cm−1) and νNiⅢ-O (547 cm−1) emerged at 1.3 VRHE, indicating that Ni sites were attacked by OH⁻ during UOR, inducing the in-situ reconstruction of Ni2+ and Ni3+ into δNiⅢ-O and νNiⅢ-O, which has been reported as key active sites for UOR47. More importantly, compared to Cr-Ni3S2, the reconstruction process of Ni3S2 was significantly delayed, occurring at 1.4 VRHE. These results suggested that the strong OH⁻ adsorption capability of Cr-Ni3S2 lowered the initial potential for reconstruction, enabling the formation of active sites at an earlier stage, thereby effectively enhancing catalytic activity. Post-UOR TEM characterization further corroborated these findings. As shown in Supplementary Fig. 36, the HADDF-STEM image of Cr-Ni3S2 after UOR test revealed the formation of an amorphous Cr-NiOOH layer on Cr-Ni3S2 surface. This structural evolution was consistent with the analysis of increased Ni3+ content in XPS and EELS after stability testing. Further, to demonstrate the dynamic migration and transformation of OH⁻ between the Cr and Ni sites during the UOR process of Cr-Ni3S2, we performed another Raman spectroscopy test in an 18O-labeled electrolyte by using H218O. As shown in Fig. 5a, Cr-Ni3S2 exhibited a Cr-18OH peak (304 and 352 cm−1) that intensified initially due to enhanced OH⁻ adsorption on Cr sites48. Upon increasing potential, this Cr-18OH peak disappeared while δNiⅢ-18O (469 cm−1) and νNiⅢ-18O (546 cm−1) peaks significantly enhanced (the NiⅢ-18O peak exhibits a redshift relative to the NiⅢ-16O peak). It can clearly confirm the OH⁻ spillover from Cr to Ni sites for promoting NiOOH generation. However, in the same 18O-labeled electrolyte, Ni3S2 exhibited a higher reconstruction potential and lower peaks for δNiⅢ-18O and νNiⅢ-18O, consistent with the results in Supplementary Fig. 3549. To exclude electronic effects of Cr-doping, Na2SiO3 was injected in the electrolyte at 1.4 VRHE to passivate Cr3+ sites. SiO32- can selectively bind Cr3+, blocking OH⁻ adsorption while negligibly affecting Ni sites50,51. As revealed in Fig. 5b, Supplementary Fig. 37, upon SiO32- injection, for Cr-Ni3S2, the Cr-18OH signal significantly decreased. Subsequently, during the reconstruction process, the peak intensities of δNiⅢ-18O and νNiⅢ-18O lost their original competitiveness, becoming comparable to that of Ni3S2. This confirmed the passivation of Cr sites, and the poisoned Cr sites hindered OH⁻ enrichment on Cr-Ni3S2, thereby delaying Ni site reconstruction. Collectively, the above results definitively confirm the Cr-triggered OH⁻ spillover from Cr to Ni in this reported Cr-Ni3S2 catalyst during UOR.

In-situ Raman spectra ranging from OCP to 1.5 VRHE (without iR correction) of a Cr-Ni3S2 and Ni3S2 in an 18O-labeled electrolyte; b Cr-Ni3S2 and Ni3S2 in an 18O-labeled electrolyte containing SiO32- added during the test. In-situ FTIR spectra of c, e Cr-Ni3S2 and d, f Ni3S2 under applied potentials from 0.8 to 1.5 VRHE (without iR correction). In situ DEMS patterns of g Cr-Ni3S2 and h Ni3S2 during UOR process. Source data are provided as a Source Data file.
To gain deeper insight into the reaction mechanism of Cr-Ni3S2 during the urea-to-NO2⁻ process, we conducted in-situ Fourier transform infrared spectroscopy (in-situ FTIR) analysis (Supplementary Fig. 38). As shown in Fig. 5c, d, the characteristic peak at 1121 cm−1 can be attributed to the N-H stretching vibration. Compared to Ni3S2, Cr-Ni3S2 exhibited a more pronounced absorption peak of N-H band with increasing potentials, indicating an enhanced adsorption of Cr-Ni3S2 to urea molecules/reaction intermediates of UOR52. This can be attributed to the Cr doping in Ni3S2, which effectively modulated the electronic structure of the catalyst, improved its ability to adsorb reactants, and accelerated reaction kinetics. A similar conclusion is drawn from an evaluation of intermediate adsorption behavior in the Helmholtz layer during UOR (Supplementary Fig. 39). An OCP test was performed in 1 M KOH for Cr-Ni3S2 and Ni3S2, respectively, where, after an electrolysis for 300 s, urea was injected for the following electrolysis. The results showed that the potential reduction for Cr-Ni3S2 (51 mV) was significantly higher than that for Ni3S2 (24 mV), also confirming that more urea molecules were adsorbed in the Helmholtz layer of Cr-Ni3S2 than that of Ni3S2. Moreover, as displayed in Fig. 5e, f, the characteristic peak at 3618 cm−1 corresponded to the OH⁻ absorption band53. With increasing potentials, Cr-Ni3S2 exhibited a more pronounced OH⁻ variation band compared to Ni3S2. More importantly, compared to Ni3S2, Cr-Ni3S2 exhibited an enhanced characteristic peak at 3747 cm−1 in its in-situ FTIR spectra, assigned to the Cr/Ni-OH vibration band, verifying the effective OH⁻ capture and utilization by Cr sites54. Nevertheless, the intensified C-N band in urea molecules at 1440 cm−1 under increasing potentials resulted from continuous attack by abundant OH⁻55. It is noteworthy that the spectra of Ni3S2 displays a vibration peak at 1506 cm−1, which can be attributed to the N-H vibration of the amide (-CONNH) group, ascribed to the intramolecular N-N coupling during the UOR process, which is a typical intermediate in the UOR-N2 pathway56. However, no corresponding characteristic peaks were observed in the spectra of Cr-Ni3S2, which demonstrated that Cr-Ni3S2 followed a more definite urea-to-NO2⁻ reaction pathway compared to Ni3S2 during the UOR process. More importantly, the presence of the absorption bands of -NH2OH (1050 cm−1), -NO2 (1348 cm−1), and -NOH (1501 cm−1) were the direct evidence for the occurrence of the dominant urea-to-NO2⁻ pathway, and the generation of NO2⁻ ions in the UOR process57. This result was also confirmed by the mass spectrometry analysis of the generated products after UOR electrolysis for Cr-Ni3S2 (Supplementary Fig. 40). Additionally, to verify the UOR pathway on Cr-Ni3S2, in-situ differential electrochemical mass spectrometry (DEMS) was also employed. As shown in Fig. 5g and h, the key intermediate *NHCONH (59) for the UOR-to-N2 pathway was detected on both Cr-Ni3S2 and pristine Ni3S2. However, its signal intensity on Cr-Ni3S2 was markedly weaker than those of the dehydroxylation intermediates essential for the UOR-to-NO2⁻ pathway, such as *NH2OH (33), *NHO (31), *NO (30), *NO2 (46), and *NO3 (62). In contrast, the intensity difference between these two groups of intermediates was far less pronounced on pristine Ni3S2. These results clearly demonstrate the effective selectivity of Cr-Ni3S2 toward the UOR-to-NO2⁻ pathway, agreeing well with the in situ FTIR findings.
Theoretical calculation analysis
To elucidate the influence of the Lewis acid-base interaction on the adsorption of active *OH in the urea-to-NO2⁻ pathway during the UOR, density functional theory (DFT) calculations and molecular dynamics (MD) simulations were employed to investigate *OH adsorption, diffusion, and the overall UOR mechanism. As shown in the Supplementary Fig. 41a, theoretical structural models of Cr-Ni3S2, Ni3S2, and their corresponding reconstructed species (Cr-NiOOH, NiOOH) were established (The modeling process was guided by EXAFS results and model optimization, Supplementary Data 1). First, the adsorption energies of *OH on different metal sites of Cr-Ni3S2 and Ni3S2 were calculated, respectively. As shown in Supplementary Fig. 41b, 42, the adsorption energy of *OH on the Cr site in Cr-Ni3S2 is −1.08 eV, which is significantly lower than that on the Ni site in Cr-Ni3S2 (−0.77 eV), and the Ni site in Ni3S2 (−0.54 eV), suggesting the Cr site in Cr-Ni3S2 exhibits the strongest adsorption ability to *OH. Besides, as shown in Fig. 6a and Supplementary Fig. 43, the overflow energy of *OH from Cr site to Ni site in Cr-Ni3S2 is 2.49 eV, which is significantly lower than the deprotonation energy of *OH → *O (4.53 eV) on the Cr site. This indicates that the *OH captured by the Cr site is more inclined to overflow to the neighboring Ni site rather than undergoing deprotonation at the Cr site31, as further supported by MD simulation. As depicted in Fig. 6b and Supplementary Video 1, the diffusion energy barrier of *OH on Cr-Ni3S2 is approximately 1.44 eV, remarkably lower than that on Ni3S2 (3.93 eV), which also indicates that *OH diffuses more easily on Cr-Ni3S2 than on Ni3S2.

a The deprotonation energy of *OH absorbed on the Cr site in Cr-Ni3S2. b The spillover energy barrier of *OH on Cr-Ni3S2 and Ni3S2, respectively. c Illustration of Ni 3d, Cr 3d and O 2p band centers obtained by theoretical calculations for catalyst models Cr-Ni3S2 and Ni3S2, respectively. d Free energy diagrams of the optimal UOR pathways for Cr-NiOOH and Ni-OOH in the urea-to-NO2⁻ pathway (solid line) and urea-to-N2 pathway (dashed line). e Schematic diagram of the UOR pathway for Cr-Ni3S2. Source data are provided as a Source Data file.
For Ni-based catalysts, in the typical UOR process, Ni2+ sites usually undergo reconstruction under the attack of *OH, transforming into NiOOH active sites, while some NiOOH sites are spontaneously reduced back into Ni2+ sites again by urea, for which has also been confirmed in our previous studies58. This reaction mechanism effectively prevents excessive adsorption of reaction intermediates on Ni3+ sites, thereby accelerating the UOR reaction kinetics. In this study, we systematically investigated the impact of Cr doping on the surface reconstruction of Ni3S2 and the corresponding Gibbs free energies for the reduction processes of Ni-OOH and Cr-Ni-OOH. As illustrated in Supplementary Fig. 44, Cr doping lowers the free energy barrier for the interconversion between Ni2+ and Ni3+ through electronic structure modulation, thereby significantly enhancing the redox activity of Ni sites. To confirm this, a series of electrochemical tests and analyses was further conducted. As shown in Supplementary Fig. 45a, b, in 1 M KOH, the peak current density (j1/2) corresponding to the transformation of Ni2+ to Ni3+ in Cr-Ni3S2 increased linearly with increasing urea concentrations. This phenomenon is attributed to the spontaneous reduction of Ni3+ to Ni2+ by urea, resulting in a higher current density. The redox index of Cr-Ni3S2 during the UOR process reached 2466, significantly higher than that of Ni3S2 (1570), indicating that Cr-Ni3S2 exhibited faster Ni3+/Ni2+ conversion kinetics than Ni3S2 (Supplementary Fig. 45c, d), which was further confirmed by periodic electrochemical measurements. As exhibited in Supplementary Fig. 46, for Cr-Ni3S2, in 1 M KOH electrolyte with urea or without urea conditions, a fixed potential of 1.45 VRHE was initially applied, which generated oxidation currents corresponding to the formation of the reconstructed NiOOH species. Under 1 M KOH electrolysis conditions, a pronounced reduction current emerged at 1.05 VRHE, corresponding to the conversion of Cr-Ni3S2-derived Cr-NiOOH to Cr-Ni(OH)2. However, in the third electrochemical stage, no significant reduction current response was observed at 1.05 VRHE, regardless of whether initial urea addition was present or under OCP test conditions. This further verified that urea can spontaneously reduce the reconstructed NiOOH to Ni2+, highlighting the enhanced catalytic activity of Cr-Ni3S2 in the UOR process. The Cr doping in Cr-Ni3S2 effectively promoted the oxidation process of Ni2+ and inhibited the occurrence of the competitive OER at high current densities. These findings can also be confirmed by the in situ Raman spectroscopy results from the urea injection experiments (Supplementary Fig. 47). This also explained why Ni3S2 tended to proceed OER with increasing potentials, while Cr-Ni3S2 showed high UOR activity.
Moreover, the broadening between the d-band of the metal site (Cr-d or Ni-d) in Cr-Ni3S2 and Ni3S2 and the p-band of the adsorbed *OH (O-p) confirms the enhanced *OH adsorption capability of Cr sites in Cr-Ni3S2. As demonstrated in Fig. 6c, the broadening between Cr-d in Cr-Ni3S2 and O-p bands is only 0.59 eV, significantly lower than that between Ni-d in Cr-Ni3S2 and O-p bands (0.79 eV), and between Ni-d in Ni3S2 and O-p bands (1.12 eV). These results indicate that the Lewis acid-base interaction between Cr sites and *OH effectively lowers their binding energy barrier59, being agreement well with the experimental findings. Further, Gibbs free energy calculations were conducted to investigate the possible reaction pathways and free energy differences (ΔG) of Cr-NiOOH and NiOOH during the UOR process. As depicted in Fig. 6d and Supplementary Fig. 48, Cr-NiOOH and NiOOH favor the urea-to-NO2⁻ pathway rather than intramolecular coupling of *NH2CONH → *NHCONH or intermolecular coupling *NH → *NH-NH2CONH in the UOR process (Supplementary Data 1). Furthermore, for both Cr-NiOOH and NiOOH, the rate-determining step (RDS) in the urea-to-NO2⁻ pathway is *NH2CONH → *NH2 + NCO-. Notably, the ΔG for the RDS on Cr-NiOOH is 0.98 eV, significantly lower than that of NiOOH (1.41 eV). Notably, the correlation coefficient between the pH value and the onset potential of Cr-Ni3S2 is calculated to be 70.04 mV pH−1 (Supplementary Fig. 49), which reveals a significant Nernst-type dependence of the onset potential on pH value, suggesting that the rate-determining step (RDS) of Cr-Ni3S2 likely involves a proton-coupled electron transfer. In contrast, the RDS for most reported Ni-based catalysts is the CO2 desorption step in the urea-to-N2 conversion13. These findings indicate that Cr doping effectively reduces the energy barrier of RDS for urea-to-NO2⁻, facilitates the reaction kinetics, thereby boosting the UOR activity.
In general, based on the above electrochemical tests and a series of in-situ and ex-situ characterizations, the OH⁻-mediated urea-to-NO2⁻ reaction mechanism for Cr-Ni3S2 was determined (Fig. 6e). The Cr sites facilitated the capture and thereafter adsorption of abundant OH⁻, which continuously overflowed to the neighboring Ni sites in Cr-Ni3S2, thereby promoting the in-situ reconstruction of Ni2+ into NiOOH. Since NiOOH species are the active sites in the UOR process, their prompt and abundant generation is conducive to boosting the UOR performance. Moreover, the doping of Cr effectively modulated the electronic structure of the catalyst, enhancing the catalytic reaction kinetics, which was also confirmed in XANES. The OH⁻-mediated urea-to-NO2⁻ reaction pathway in the UOR process is specified as follows: first, urea molecules are adsorbed on the surface of Cr-Ni3S2. The abundant OH⁻ ions, acting as nucleophilic species, are easily captured by Cr, which ceaselessly attack the C-N bonds in urea molecules, resulting in their cleavage. Thereafter, the remaining -NH2 groups undergo dehydrogenation and oxidation under the action of a large amount of OH⁻, generating NO2⁻ products but preventing N-N coupling. This selectivity stems from OH⁻ spillover, which keeps the energy barrier for urea-to-NO2⁻ conversion lower than that for nitrogen-containing intermediates (*NH, *NHCONH2) combining into N2, making NO2⁻ formation kinetically favorable over N2 release (as supported by DFT calculations).
Urea-assisted water splitting and Zn-urea-air battery
Since the catalytic activity of Cr-Ni3S2 in the UOR process, we constructed a urea-assisted water splitting cell using Cr-Ni3S2 as the anodic catalyst and Ni2P/CoP (reported previously) as the cathodic HER catalyst. The XRD tests shown in Supplementary Fig. 50 confirm the successful synthesis of Ni2P/CoP60, and its HER activity is also verified (Supplementary Fig. 51, HER overpotential = 151 mV at 100 mA cm−2). As exhibited in Fig. 7a and Supplementary Fig. 52, the Cr-Ni3S2| |Ni2P/CoP urea-assisted overall water splitting system required only 1.55 V to drive a current density of 500 mA cm−2, which was 310 mV lower than that of the conventional overall water splitting system, demonstrating the significant energy efficiency enhancement for H2 production through the combination of UOR and HER over the combination of OER and HER. Notably, even at ampere-level current density of 1 A cm−2, the system operated at a voltage as low as 1.59 V. Importantly, as indicated in Fig. 7b, the Cr-Ni3S2| |Ni2P/CoP urea-assisted overall water splitting flow cell system exhibited sustained operation for over 240 h at a current density of 700 mA cm-2, demonstrating its electrochemical stability under high-current conditions. The performance of the Cr-Ni3S2| |Ni2P/CoP system in urea-assisted water splitting was comparable to that of other advanced systems reported in recent studies (Fig. 7c, Supplementary Table 8). Moreover, to demonstrate the advantages of the urea-assisted H2 production system over traditional water electrolysis, we calculated the electricity cost per m3 of H2 for both UOR| |HER and OER| |HER configurations based on the LSV curves in Fig. 7a. As shown in Fig. 7d, the findings revealed that within a broad current density range (200 ~ 1000 mA cm−2), UOR| |HER consumed less power to yield one m3 of H2. Further, to assess the economic viability of the Cr-Ni3S2| |Ni2P/CoP catalyst for nitrite production and concomitant H2 generation via urea oxidation, a techno-economic analysis (TEA) was conducted (Fig. 7e, f, Supplementary Fig. 53). The TEA results indicated that operating at industrially relevant current densities (400 mA cm−2), the urea-to-nitrite conversion coupled with hydrogen evolution using Cr-Ni3S2| |Ni2P/CoP generated substantial economic returns of approximately $1,210.5 per ton of processed urea. This enhanced profitability primarily stemmed from the high-value nitrite co-production alongside hydrogen, contrasting sharply with conventional electrolysis, where oxygen evolution adds no economic value. Moreover, in a simulated wastewater environment, Cr-Ni3S2 also demonstrated higher UOR activity than Ni3S2, due to its enhanced OH⁻ adsorption capability. This property effectively limited the adsorption of competing anions on the catalyst surface (Supplementary Fig. 54, Supplementary Table 9).

a LSV curves of Cr-Ni3S2| |Ni2P/CoP for urea-assisted water splitting for H2 production, and conventional overall water splitting for H2 production in 1 M KOH with and without 0.5 M urea with 100% iR compensation (the scan rate is 5 mV s−1, and the resistance is 3.29 ± 0.58 Ω), respectively. b Chronoamperometry curves of Cr-Ni3S2| |Ni2P/CoP in 1 M KOH + 0.5 M urea at large current density of 700 mA cm−2 (Inset: optical picture of the flow cell). c Comparison of catalytic activity between Cr-Ni3S2| |Ni2P/CoP and the other most advanced catalysts reported toward urea-assisted water splitting for H2 production at 500 mA cm−2 in literature (relevant references can be found in Supplementary Information). All the measurements were tested in 1.0 M KOH with and witout 0.5 M urea solution as the electrolyte (pH = 13.97 ± 0.02) at room temperature. d The corresponding electricity consumption of Cr-Ni3S2| |Ni2P/CoP for UOR| |HER and OER| |HER at different current densities, respectively. e Cost and revenue analysis of the Cr-Ni3S2| |Ni2P/CoP urea-assisted overall water splitting system at 400 mA cm−2. f TEA results of Cr-Ni3S2| |Ni2P/CoP for nitrite production at 400 mA cm−2. g Electrochemical measurement for Zn-air battery and Zn-urea-air battery in 6 M KOH with and without urea (at current density of 10 mA cm−2). h Energy efficiency of Zn-urea-air battery and Zn-air battery in 6 M KOH with and without urea. Source data are provided as a Source Data file.
Further, a Zn-urea-air battery was fabricated by replacing the conventional OER electrode with Cr-Ni3S2 catalyst (using Fe-N-C catalysts for the oxidation-reduction reaction). The Zn-air and Zn-urea-air batteries using the Cr-Ni3S2/Fe-N-C catalyst exhibited nearly the same open-circuit voltage (Supplementary Fig. 55). By replacing the OER with the UOR during the charging process, this system achieved a significantly lower charging voltage in both 1 M and 6 M KOH. In the 6 M KOH electrolyte with 0.5 M urea, the required voltage was 1.79 V, a performance that is competitive with that of many reported Zn-air battery (Supplementary Fig. 56 and Table 10). This result was consistent with the findings from XAFS and quasi-in situ XPS analyses, which confirmed that Cr doping in Ni3S2 facilitated electron transfer and accelerated reaction kinetics. Furthermore, at the different tested current densities (1, 2, 5, 10, 15 and 20 mA cm−2), the charging potential of the Zn-urea-air battery remained stable at each specific current density for every two hours (Supplementary Fig. 57). Constant-current charge-discharge measurements revealed that the Zn-urea-air battery exhibited a 213 mV reduction in charging potential relative to traditional Zn-air battery, while maintaining stable performance for over 100 h (Fig. 7g, Supplementary Fig. 58). Significantly, Fig. 7h shows that the Zn-urea-air battery remains high and stable energy efficiency throughout this operational period.
Discussion
In conclusion, we have developed a Lewis acid Cr site-doped Ni3S2 electrocatalyst (Cr-Ni3S2) to realize low-energy saving H2 production accompanied by selective nitrite electrosynthesis via urea-assisted water splitting. According to in situ characterization and theoretical calculations, the Cr3+ sites can act as OH⁻ pump, enabling dynamic OH⁻ spillover to adjacent Ni sites. This process facilitated in situ generation of active NiOOH species while maintaining a sustained OH⁻ supply for C-N bond cleavage, thereby suppressing N-N coupling and steering nitrite selectivity via the urea-to-NO2⁻ pathway. Importantly, this approach can be extended to other Lewis acid sites (Sn, Ti) and matrix (Cu2S). Befitting from electronic structure modulation and optimized Ni redox cycling, the Cr-Ni3S2 achieved a NO2⁻ yield of 120.98 mg h-1 cm-2 at industrial-scale current densities and showed operational stability over 300 h at 750 mA cm−2. This approach enables efficient H2 conversion (3.7 kWh m−3 H2), substantial economic returns ($1,210.5 per ton of processed urea) and high-efficiency Zn-urea-air batteries. Our work clarifies Lewis acid triggering the OH⁻ spillover mechanism and provides a universal strategy for the design of advanced electrocatalysts affording green hydrogen production, coupling valuable chemical synthesis.
Methods
Synthesis of CrNi-LDH/NF, Cr-Ni3S2, Ni3S2, Ti-Ni3S2, Sn-Ni3S2, Cr-Cu2S, Cr-S
0.24 g Cr(NO3)3·9H2O (General-Reagent, AR), 0.35 g Ni(NO3)2·6H2O (Tianjin Fengchuan Chemical Reagent Co, Ltd, 99%) and 0.31 g urea (Tianjin Fengchuan Chemical Reagent Co, Ltd, 99%) were dissolved in 30 mL deionized water, which the mixture was stirred until completely dissolved and then poured into a 50 mL Teflon reactor. A piece of pretreated nickel foam (NF, 2 cm * 3 cm) was vertically placed into the Teflon reactor and heated at 120 °C for 10 h. Then, the obtained product was rinsed with deionized water several times and then put into a vacuum drying oven at 60 °C for drying, which the product being CrNi-LDH/NF. 0.35 g thiourea (Macklin, 99%) was dissolved in 30 mL deionized water, which was stirred thoroughly and then poured into a 50 mL reactor. The above-obtained CrNi-LDH/NF was put vertically into the reactor and heated at 120 °C for 24 h. After that, the product obtained was rinsed with deionized water several times and then put into a vacuum drying oven at 60 °C for drying, where the product being Cr-Ni3S2/NF. The synthesis of Ni3S2 is similar to that of Cr-Ni3S2, except that Cr(NO3)3·9H2O is not added. The synthesis of Ti-Ni3S2 and Sn-Ni3S2 follows the same procedure as that of Cr-Ni3S2, but with Ti(SO4)2·9H2O (Macklin, 96%), SnCl4·5H2O (Tianjin Damiao Chemical Reagent Factory, AR) replacing Cr(NO3)3·9H2O. Similarly, Cr-Cu2S is synthesized analogously to Cr-Ni3S2, using Cu(NO3)2·3H2O (Tianjin Fengchuan Chemical Reagent Co, Ltd, 99%) in place of Ni(NO3)2·6H2O. The synthesis process of Cr-S catalysts is similar to that of Cr-Ni3S2, except without the addition of Ni(NO3)2·6H2O. The carbon cloth is employed as the substrate.
Characterization
XPS measurements were carried out using a VG Scientific ESCALAB Mark II spectrometer equipped with two ultra-high vacuum chambers. The spectra were calibrated using the C-C peak of the C 1 s orbital at 284.8 eV. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) characterizations were performed using a JEOL JEM-ARM200F and Gatan GIF Quantum ER. Scanning electron microscopy (SEM) measurements were conducted using a QUANTA 430 FE-SEM. XRD analysis was performed with Cu Kα radiation (λ = 1.5418 Å), at a scanning rate of 0.013° and in the range of Bragg angles from 5° to 80°. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was measured by a Thermo ICAP PRO. Ultraviolet-visible (UV-Vis) spectral data were obtained using a UV-Vis-NIR spectrometer (Cary 5000) equipped with an integrating sphere unit and an automatic reflectance measurement unit. The Zeta potential was tested by a Nano-ZS900 instrument. Specifically, a certain amount of the catalyst was dispersed in a 1 M KOH solution (with a concentration of 0.1%), and then ultrasonicated for 5 minutes. Subsequently, a certain volume of the solution was injected into a U-shaped sample cell for the test. Using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the *OH trapping agent, electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMX Plus spectrometer. In-situ Fourier transform infrared (FTIR) spectra were obtained at different potentials applied on a CHI660E electrochemical workstation using a Nicolet iS50 Fourier transform infrared spectrometer with a resolution of 4 cm−1, and the infrared transmission window was a Ge crystal. Surface-enhanced Raman spectroscopy (SERS) was performed in situ using a LABRAM ARAMIS laser Raman spectrometer (Horiba, France). Prior to analysis, the catalyst was dispersed in a 1: 9 (V/V) mixture of naphthol reagent and ethanol (1 mL total volume) to form an ink, which was uniformly coated onto gold nanoplates and air-dried. Measurements were conducted using a 50 × long-working-distance objective lens with 532 nm laser excitation at 4.8 mW power. The spectrometer was configured with a 600 grooves/mm diffraction grating and 2-second integration time, scanning the spectral range of 1000 ~ 1800 cm⁻¹. For in situ electrochemical measurements, the modified gold nanoplates served as working electrodes, with Raman signals acquired after maintaining each applied potential for 300 seconds to ensure stable measurement conditions. Online DEMS measurements were performed on the CHI660E electrochemical workstation, combined with a tandem mass spectrometer (Linglu QAS 100) to identify key intermediates and products in the UOR process. The X-ray absorption fine structure spectra (Ni K-edge and Cr K-edge) were collected at BL14W beamline in Shanghai Synchrotron Radiation Facility (SSRF). The storage rings of SSRF were operated at 3.5 GeV with a stable current of 200 mA. Using Si (111) double-crystal monochromator, the data collection was carried out in transmission mode using a Lytle detector. All spectra were collected in ambient conditions.
Electrochemical measurement
Electrochemical measurements were conducted using a CHI660E electrochemical workstation and a Coster CS2350M workstation in a two-electrode or three-electrode system at room temperature (~25 oC) in 1 M KOH with and without 0.5 M urea. The electrolyte was prepared by dissolving a certain amount of KOH (56.11 g) and urea (30.03 g) into ultrapure water. All solutions were freshly prepared prior to each experiment and used directly. The final pH was adjusted to 13.97 ± 0.02 (three measurements). They were stored in sealed glass vials at room temperature (~25 oC) and protected from ambient CO2 to prevent the formation of carbonate species. A carbon rod was used as the counter electrode, the reference electrode was Hg/HgO. The Hg/HgO reference electrode was calibrated in a sealed three-electrode configuration using a Pt foil as both the working and counter electrode. Prior to calibration, the electrolyte was saturated with high-purity hydrogen for at least 30 minutes. CV test was conducted at a scan rate of 1 mV/s, revealing the reversible hydrogen electrode (RHE) potential to be -0.923 V. Accordingly, all measured potentials were converted to the RHE scale using the equation: \({{{{\rm{E}}}}}_{{{{\rm{RHE}}}}}\,=\,{{{{\rm{E}}}}}_{{{{\rm{Hg}}}}/{{{\rm{HgO}}}}}\,+\,0.923{{{\rm{V}}}}\). The measured offset (0.923 V) agrees closely with the theoretical value of 0.926 V in 1 M KOH at 25 °C, confirming the reliability of the reference electrode throughout the experimental study. The working electrode was the electrocatalyst (1 cm × 0.5 cm; the self-supported electrodes were cut to a fixed geometric area of 0.5 cm2 for all electrochemical tests. Due to the chemical transformation of the nickel foam surface during the in-situ growth process, the exact catalyst mass loading could not be measured gravimetrically. Therefore, all current densities are normalized to the geometric area of the electrode to ensure consistent comparison of electrocatalytic performance. All electrochemical tests were conducted at room temperature (~25 oC) and atmospheric pressure. Linear sweep voltammetry (LSV) polarization curves for UOR and OER were obtained in 1 M KOH with and without 0.5 M urea at a scan rate of 5 mV s-1 within a potential range of 0.923 ~ 1.723 VRHE (100% iR compensation). Three thousand accelerated durability tests (ADT) were carried out in 1 M KOH + 0.5 M urea, and the tested potential range was 0.923 ~ 1.723 VRHE.
Electrochemical impedance spectroscopy (EIS) analysis for UOR was performed at a potential of 1.30 VRHE, with a frequency range from 10 kHz to 0.1 Hz. The electrochemical surface area (ECSA) was estimated via the electrochemical double-layer capacitance (Cdl). Cyclic voltammetry (CV) was performed in a non-Faradaic potential window (0.372–0.472 VRHE) at various scan rates (20, 40, 60, 80, and 100 mV s-1). The Cdl was determined by plotting the charging current density difference at a fixed potential within the window against the scan rate. The slope of this linear plot is equal to twice the Cdl. The ECSA was then calculated using the equation: ECSA = Cdl/Cs, where Cs is the specific capacitance. A standard value of Cs = 0.040 mF cmECSA−2 in alkaline media was used for the estimation. The current density-time curve (i-t) was measured in 1 M KOH + 0.5 M urea using a flow cell. In this test, the electrolyte was replaced every 24 h to ensure a constant urea concentration. Simultaneously, the FE\(\scriptsize {{{\rm{NO}}}}_{2}^{-}\)(the main target UOR product) and the possibly leached metal ion concentrations (Cr, Ni) in the electrolyte were monitored. The two-electrode measurements were conducted using an H-type electrolytic cell, and an anion exchange membrane (FAB-PK-75, Fumatech) was used in the electrochemical cell. The membrane has a thickness of 60 μm. Prior to use, it was pretreated by soaking in a 3 M NaCl solution for 12 h.
Product quantification
Determination of NO3 ⁻
5 mL electrolyte was removed from the electrolytic cell, and 5.1 mL 1 M HCl and 0.01 mL 0.8 wt % sulfamic acid solution were added to the above solution. Absorption spectra were detected using a UV-Vis spectrophotometer, and absorbance at wavelengths of 220 and 275 nm was recorded, respectively. The final absorbance value was calculated by the formula:
The concentration-absorbance curve was calibrated using a series of potassium nitrate standard solutions.
Determination of NO2 ⁻
The amount of NO2⁻ was analyzed spectrophotometrically using Griess reagent. Griess reagent is prepared by dissolving 0.01 g N-(1-naphthyl)ethyldiamine dihydrochloride, 0.1 g sulfonamide, and 0.294 mL H3PO4 in H2O to 5 mL. In a typical colorimetric assay, Griess reagent (1.0 mL) is added to the sample solution (1.0 mL) and H2O (2.0 mL), and the absorbance at 540 nm is recorded by UV-Vis absorption spectroscopy.
Determination of N2 and O2
The gas products (N2, O2) formed during UOR were analyzed every 20 minutes by online gas chromatograph with GC7920 instrumental detection. The GC was equipped with an HP-PLOT Q and CP-Molsieve 5 Å PT column, together with thermal conductivity (TCD) and flame ionization (FID) detectors.
The FE of the generated product was calculated as follows:
In which e is the number of transferred electrons per product, F is the Faraday constant, Q is the charge, I is the applied current density, t is the reaction time, and n is the total molar number of the products. To exclude any possible errors, measurements of all products were repeated at least three times at each applied potential.
Calculation details
The density functional theory (DFT) calculations were carried out with the VASP code61. The Perdew-Burke-Ernzerhof (PBE) functional within generalized gradient approximation (GGA) was used to process the exchange-correlation62, while the projector-augmented-wave pseudopotential (PAW) was applied with a kinetic energy cut-off of 450 eV63, which was utilized to describe the expansion of the electronic eigenfunctions. The Brillouin-zone integration was sampled by a Γ-centered 5 × 5 × 1 Monkhorst-Pack k-point. All atomic positions were fully relaxed until energy and force reached a tolerance of 1 × 10-6 eV and 0.01 eV/Å, respectively. The dispersion corrected DFT-D method was employed to consider the long-range interactions64. The model was established using Materials Studio software. The entire system uses VASP’s machine learning force field. During the molecular dynamics simulation, the NVT ensemble is used to relax the system, with a time step set to 1 ps and a total duration of 1000 ps.
The Gibbs free energy change (ΔG) was calculated by the computational hydrogen electrode (CHE) model as follows:
where ΔE is the reaction energy obtained by the total energy difference between the reactant and product molecules absorbed on the catalyst surface, and ΔS is the change in entropy for each reaction, ΔZPE is the zero-point energy correction to the Gibbs free energy.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Canfield, D. onaldE. et al. The evolution and future of earth’s nitrogen cycle. Science 330, 192–196 (2010).
Wang, Y. et al. Electrochemical synthesis of nitric acid from air and ammonia through waste utilization. Natl. Sci. Rev. 6, 730–738 (2019).
Krebs, M. L. et al. Electrochemical Synthesis of nitrite and nitrate via cathodic oxygen activation in liquefied ammonia. J. Am. Chem. Soc. 146, 30753–30757 (2024).
Rouwenhorst, K. H. R. et al. From the Birkeland-Eyde process towards energy-efficient plasma-based NOX synthesis: a techno-economic analysis. Energy Environ. Sci. 14, 2520–2534 (2021).
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, 6611 (2018).
Zhu, K. et al. Simultaneous electrosynthesis of nitrate and hydrogen by integrating ammonia oxidation and water reduction. Chin. J. Catal. 55, 216–226 (2023).
Tian, Y. et al. Highly stabilized and selective ammonia electro-oxidation over CuNi metallic glass nanoarray. Carbon Energy 7, e667 (2024).
Mao, Z. et al. Modulation of charge distribution enabling CuNi nano-alloys for efficient ammonia oxidation reaction to nitrite production. Chem. Eng. J. 484, 149570 (2024).
Vu, T. M. et al. High-Rate, High-selectivity electrochemical oxidation of ammonia to nitrite with a silver-based catalyst. ACS Catal. 14, 10974–10986 (2024).
Wang, H. et al. Unraveling the mechanism of ammonia electrooxidation by coupled differential electrochemical mass spectrometry and surface-enhanced infrared absorption spectroscopic studies. J. Am. Chem. Soc. 146, 15926–15940 (2024).
Chen, Y. et al. The nitrogen removal performance and mechanisms for urea wastewater by simultaneous urea hydrolysis, partial nitritation and anammox in one reactor. J. Clean. Prod. 332, 130124 (2022).
Huang, S. M. et al. Structural insights into the mechanisms of urea permeation and distinct inhibition modes of urea transporters. Nat. Commun. 15, 10226 (2024).
Wang, P. et al. Directed urea-to-nitrite electrooxidation via tuning intermediate adsorption on Co, Ge co-doped Ni sites. Adv. Funct. Mater. 33, 2300687 (2023).
Li, J. et al. Deciphering and suppressing over-oxidized nitrogen in nickel-catalyzed urea electrolysis. Angew. Chem. Int. Ed. 60, 26656–26662 (2021).
Zhang, J. et al. Regulating reconstruction-engineered active sites for accelerated electrocatalytic conversion of urea. Angew. Chem. Int. Ed. 63, e202407038 (2024).
Gao, X. et al. Urea catalytic oxidation for energy and environmental applications. Chem. Soc. Rev. 53, 1552–1591 (2024).
Wang, T. et al. Progress in hydrogen production coupled with electrochemical oxidation of small molecules. Angew. Chem. Int. Ed. 61, e202213328 (2022).
Tatarchuk, S. W. et al. Nickel-catalyzed urea electrolysis: From nitrite and cyanate as major products to nitrogen evolution. Angew. Chem. Int. Ed. 61, e202209839 (2022).
Gao, X. et al. Membrane-free water electrolysis for hydrogen generation with low cost. Angew. Chem. Int. Ed. 64, e202417987 (2024).
Zhan, G. et al. Highly selective urea electrooxidation coupled with efficient hydrogen evolution. Nat. Commun. 15, 5918 (2024).
Shen, Q. L. et al. Lewis acid acceleration of C-N bond-forming reductive elimination from heteroarylpalladium complexes and catalytic amidation of heteroaryl bromides. J. Am. Chem. Soc. 129, 7734–7735 (2007).
Jin, H. et al. Identifying the bifunctional mechanism in alkaline water electrolysis by Lewis pairs at the single-atom scale. J. Am. Chem. Soc. 147, 3874–3884 (2025).
Cao, X. et al. Regulation of oxide pathway mechanism for sustainable acidic water oxidation. J. Am. Chem. Soc. 146, 32049–32058 (2024).
Geng, S. K. et al. Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst. Nat. Energy 6, 904–912 (2021).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Qin, H. et al. Regulating the electrochemical microenvironment of Ni(OH)2 by Cr doping for highly efficient methanol electrooxidation. ACS Catal. 14, 16234–16244 (2024).
Pei, Z. et al. Regulating the local reaction microenvironment at chromium metal-organic frameworks for efficient H2O2 electrosynthesis in neutral electrolytes. Adv. Mater. 37, 2500274 (2025).
Wang, L. L. et al. Tailoring Lewis acidity of metal oxides on nickel to boost electrocatalytic hydrogen evolution in neutral electrolyte. J. Am. Chem. Soc. 147, 7555–7563 (2025).
Zhang, L. et al. Oxophilicity-controlled CO2 electroreduction to C2+ alcohols over Lewis acid metal-doped Cuδ+ catalysts. J. Am. Chem. Soc. 145, 21945–21954 (2023).
Gu, K. et al. Sustained hydrogen spillover on Pt/Cu(111) single-atom alloy: dynamic insights into gas-induced chemical processes. Angew. Chem. Int. Ed. 62, e202312796 (2023).
Shen, Y. et al. Cr dopant mediates hydroxyl spillover on RuO2 for high-efficiency proton exchange membrane electrolysis. Nat. Commun. 15, 7861 (2024).
Zhao, Y. et al. Correlating alkaline hydrogen electrocatalysis and hydroxide binding energies on Mo-modified Ru catalysts. ACS Sustain. Chem. Eng. 10, 1616–1623 (2022).
Xu, J. et al. Size-dependent core-shell fine structures and oxygen evolution activity of electrochemical IrOx nanoparticles revealed by cryogenic electron microscopy. ACS Nano 18, 29140–29151 (2024).
Chen, Z. et al. Isolated copper atoms boost *NO3 adsorption and active hydrogen retention over zinc oxide for ammonia electrosynthesis at ampere-level current densities. J. Am. Chem. Soc. 147, 18737–18746 (2025).
Gao, J. et al. Transition-metal substitution doping in synthetic atomically thin semiconductors. Adv. Mater. 28, 9735–9743 (2016).
Zhang, F. et al. Dynamic reconstruction of Ce-doped Fe2P/NiCoP hybrid for ampere-level oxygen evolution in anion exchange membrane water electrolysis. Adv. Funct. Mater. 35, 2500861 (2025).
Yan, Q. et al. Chromium-induced high covalent Co-O bonds for efficient anodic catalysts in PEM electrolyzer. Adv. Sci. 11, 202402356 (2024).
Wang, Z. et al. Synergistic Zn and MoS2 tailored Co-N/C environments enabling bifunctional ORR/OER electrocatalysis for advanced Li-O2 batteries. Angew. Chem. Int. Ed. 64, e202425502 (2025).
Wang, Y. et al. Gd-induced oxygen vacancy creation activates lattice oxygen oxidation for water electrolysis. Adv. Funct. Mater. 35, 2500118 (2025).
Liu, S. Q. et al. Electronic delocalization of bismuth oxide induced by sulfur doping for efficient CO2 electroreduction to formate. ACS Catal. 11, 7604–7612 (2021).
Yuan, P. et al. Design of single-atom Co-N5 catalytic site: a robust electrocatalyst for CO2 reduction with nearly 100% CO selectivity and remarkable stability. J. Am. Chem. Soc. 140, 4218–4221 (2018).
Zheng, X. B. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Gao, X. et al. Boosting urea electrooxidation on oxyanion-engineered nickel sites via inhibited water oxidation. Nat. Commun. 14, 5842 (2023).
Zhang, H. J. et al. Electron delocalized Ni active sites in spinel catalysts enable efficient urea oxidation. Angew. Chem. Int. Ed. 64, e202421027 (2024).
Wu, Z. P. et al. Operando unveiling the activity origin via preferential structural evolution in Ni-Fe (oxy)phosphides for efficient oxygen evolution. Sci. Adv. 11, 5370 (2025).
Gautam, J. et al. Cutting-edge optimization strategies and in situ characterization techniques for urea oxidation reaction catalysts: a comprehensive review. Adv. Energy Mater. 15, 2406047 (2025).
Guo, X. et al. Accelerating the generation of NiOOH by in-situ surface phosphating nickel sulfide for promoting the proton-coupled electron transfer kinetics of urea electrolysis. Chem. Eng. J. 483, 149264 (2024).
Zheng, J. X. et al. In-situ Raman spectroscopic studies on electrochemical oxidation behavior of chromium in alkaline solution. J. Electroanal. Chem. 921, 116622 (2022).
Lee, S. et al. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 58, 10295–10299 (2019).
Zhou, P. et al. Cost-effective fabrication of fluorite tailings-based calcium silicate hydrate for excellent adsorption performance of Cr (III) from aqueous solution. Environ. Res. 285, 122318 (2025).
Khawaji, M. et al. Catalytic oxidation of glucose over highly stable AuxPdy NPs immobilised on ceria nanorods. Catal. Today 365, 257–264 (2021).
Chen, L. et al. Artificial heterointerfaces with regulated charge distribution of Ni active sites for urea oxidation reaction. Small Methods 8, 2400108 (2024).
Han, W. K. et al. Activating lattice oxygen in layered lithium oxides through cation vacancies for enhanced urea electrolysis. Angew. Chem. Int. Ed. 61, e202206050 (2022).
Yan, Z. T. et al. Unlocking efficient alkaline hydrogen evolution through Ru-Sn dual metal sites and a novel hydroxyl spillover effect. Adv. Mater. 36, 2411942 (2024).
Fan, Y. et al. Nickel-based hollow spheres with optimized interfacial electronic structures by highly dispersed MoN for efficient urea electrolysis. Adv. Funct. Mater. 35, 2421222 (2024).
Zhang, L. et al. A lattice-oxygen-involved reaction pathway to boost urea oxidation. Angew. Chem. Int. Ed. 58, 16820–16825 (2019).
Zhang, R. et al. Electrochemical nitrate reduction in acid enables high-efficiency ammonia synthesis and high-voltage pollutes-based fuel cells. Nat. Commun. 14, 8036 (2023).
Fan, C. et al. Rapid B-Ni charge transfer pathway induced Ni3+/Ni2+ sites reversible conversions enabling efficient urea oxidation assisted hydrogen production. J. Colloid Interface Sci. 690, 137355 (2025).
Wang, X. et al. In situ electrochemical oxyanion steering of water oxidation electrocatalysts for optimized activity and stability. Adv. Energy Mater. 13, 2300765 (2023).
Niu, S. et al. Constructing a built-in electric field to accelerate water dissociation for efficient alkaline hydrogen evolution. ACS Appl. Mater. Interfaces 16, 31480–31488 (2024).
Kresse, G. et al. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Acknowledgements
This work is supported by the financial support from NSFC (Grant 22162020), the 2024 Youth Science and Technology Talent Development Project (Young Science and Technology Talents) (NJYT24007), and the Inner Mongolia autonomous region natural science fund project (2024MS02013), and the Program for Science & Technology Innovation Talents in Universities of Henan Province (25HASTIT006), and the Young Backbone Teacher Foundation of Henan Educational Committee (2023GGJS091).
Author information
Authors and Affiliations
Contributions
C.F. and M.Z. contributed equally to this work. C.F. performed the experiments. M.Z. conducted the Density Functional Theory (DFT) calculations and in situ Roman spectroscopy tests. C.F., and M.Z. wrote the manuscript. Y.Li provided advice to the improvement of the manuscript. Y.Z. discussed the techno-economic analysis (TEA). Y.Q.W. and F.G. supervised the project, conceived the idea, and analyzed the data. J.L. supervised the project. All authors participate in the analysis with constructive discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guanjie He, Qinghua Liu, and Pengtang Wang for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, C., Zhang, M., Li, Y. et al. Lewis acid-triggered hydroxyl spillover enables selective urea electrooxidation to nitrite with concurrent energy-saving hydrogen production. Nat Commun 17, 1585 (2026). https://doi.org/10.1038/s41467-026-68302-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68302-w
