
Abstract
Neuronal intranuclear inclusion disease (NIID) is an adult-onset neurodegenerative disease caused by expanded GGC repeats in the 5’ untranslated region of the human-specific NOTCH2NLC gene. The high sequence similarity between NOTCH2NLC and its paralogs poses a significant challenge for precise gene editing. Here, we develop a CRISPR/spCas9-based gene-editing strategy that precisely excises the expanded GGC repeats in NOTCH2NLC without detectable off-target effects on the highly homologous NOTCH2/NOTCH2NL family genes (<2% sequence divergence at this locus). The efficacy, specificity and safety of this approach are rigorously validated across multiple experimental models, including human cell lines, NIID iPSCs, and our previously established transgenic NIID mouse model. Our results demonstrate that precise excision of the expanded GGC repeats effectively alleviates NIID-related neuropathological, molecular and behavioral abnormalities. This study establishes the proof of concept for genome editing as a therapeutic strategy for NIID and other related repeat expansion disorders.
Similar content being viewed by others

Introduction
Repeat expansion disorders (REDs) represent a class of genetic diseases characterized by the pathological expansion of tandem repeat sequences beyond their normal range1,2. To date, more than sixty REDs have been identified in humans, with the majority manifesting as neurodevelopmental or neurodegenerative diseases, such as fragile X syndrome, Huntington’s disease, spinocerebellar ataxia and C9ORF72-related amyotrophic lateral sclerosis and frontotemporal dementia1,3,4,5,6,7,8. Notably, recent advancements in long-read sequencing and whole-genome sequencing (WGS) technologies have significantly accelerated the discovery of new REDs9,10,11,12. Among these, GGC repeat expansions located within the “non-coding” regions of the human genome have garnered particular attention13. One such disorder, neuronal intranuclear inclusion disease (NIID) has attracted increasing attention in recent years.
NIID is a progressive neurodegenerative disorder characterized by widespread eosinophilic intranuclear inclusions in the nervous system and visceral organs14,15. It exhibits substantial clinical heterogeneity, with primarily neurological manifestations including dementia, muscle weakness, tremor, bradykinesia, cerebellar ataxia, periodic encephalitic episodes, and autonomic dysfunction9,14,15,16,17,18, as well as non-neurological involvement19,20,21. Severe cases can lead to loss of ambulation and premature death14,17,18. In 2019, the GGC repeat expansion (>60 repeats) in the 5′ untranslated region (5′UTR) of the NOTCH2NLC gene was reported as the genetic cause of NIID by several groups9,22,23,24. Furthermore, this mutation has also been implicated in a series of neurodegenerative and neuromuscular diseases22,25,26,27,28,29,30,31,32. Although comprehensive epidemiological data are limited, over 600 cases have been documented worldwide by the end of 202233. Despite these genetic insights, no effective treatments currently exist, and conventional pharmacological therapies provide only limited symptomatic relief without halting disease progression.
Previous studies have confirmed that the GGC repeat expansions in the NOTCH2NLC gene are located within an independent upstream open reading frame (uORF) positioned before the canonical ATG, and these expanded GGC repeats can be translated into multiple toxic polypeptides, with polyglycine (polyG) being the predominant one34. These toxic peptides accumulate as intranuclear inclusions in the central nervous system and visceral organs, driving disease pathology through mechanisms such as dysregulated alternative splicing, impaired nuclear-cytoplasmic transport, mitochondrial dysfunction, and abnormal ribosome biogenesis35,36,37,38,39,40,41. Consequently, targeting the expanded GGC repeats through gene editing represents a rational and potential therapeutic strategy for NIID.
The human-specific NOTCH2NL (NOTCH2 N-terminal like) gene family, which includes NOTCH2NLA, NOTCH2NLB, NOTCH2NLC and NOTCH2NLR, shares high sequence homology and plays critical roles in human brain development and homeostasis42,43,44. Specifically, NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC are expressed in cortical progenitors, where they enhance self-renewal and delay premature differentiation by potentiating canonical Notch signaling, thereby contributing to the evolutionary expansion of the human cerebral cortex. Abnormal dosage of NOTCH2NL genes has been linked to neurodevelopmental disorders, including microcephaly or cortical malformations upon loss and macrocephaly or autism spectrum disorders upon duplication. Therefore, it is essential to specifically target the GGC repeat expansion within NOTCH2NLC while preserving the normal expression and function of the NOTCH2NL family. However, the near-identical sequence flanking the GGC repeats (<2% divergence) among these homologous genes, combined with the large size and high GC content of the expanded repeats, pose significant challenges for precise and efficient gene editing.
In this study, we develop a CRISPR/spCas9-based gene-editing strategy to specifically target the GGC repeat expansions within the NOTCH2NLC gene. To ensure editing precision, we design sgRNAs with minimal sequence homology to NOTCH2NL paralogs and NOTCH2 and employed a dual-sgRNA approach. We systematically assess its efficacy, specificity and safety across multiple models, including HEK293 cells with normal GGC repeats or expanded GGC repeats in NOTCH2NLC, NIID patient-derived induced pluripotent stem cells (iPSCs) and neural progenitor cells (NPCs), as well as a previously established transgenic NIID mouse model. Importantly, targeted excision of the expanded GGC repeats markedly reduces polyG protein levels and significantly ameliorates neuropathological, molecular and behavioral phenotypes associated with NIID. Collectively, our work establishes a strong preclinical foundation for the development of genome-editing-based therapies for NIID.
Results
CRISPR/Cas9-based gene-editing specifically targeting GGC repeats within NOTCH2NLC in human cell lines with normal GGC repeats
Given the near-identical sequence flanking the GGC repeats among NOTCH2NLC, NOTCH2 and other members of the NOTCH2NL family (NOTCH2NLA, NOTCH2NLB, and NOTCH2NLR, NOTCH2NLA/B/R for short), we conducted comparative analysis and identified two mismatch regions flanking the GGC repeats (Supplementary Fig. 1A). To enable precise excision of the expanded GGC repeats of NOTCH2NLC, we designed multiple single guide RNAs (sgRNAs) targeting the upstream and downstream mismatch regions. Specifically, sgRNA 1-4 were designed to target sequences upstream of the GGC repeats, while sgRNA 5-8 targeted sequences downstream (Fig. 1A, Supplementary Fig. 1B). The details of sgRNAs are provide in Supplementary Data 1.

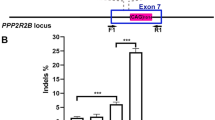
A Schematic diagram of a CRISPR/Cas9-based gene-editing strategy targeting the GGC repeats within the upstream open reading frame (uORF) of NOTCH2NLC. Two mismatch-flanking regions adjacent to the GGC repeats were selected for the design of specific sgRNAs. The coding sequences and untranslated regions of NOTCH2NLC are indicated in blue and pink, respectively. B–E HEK293T cells were co-transfected with Cas9 and different sgRNAs, and the T7EI assay was performed to evaluate the targeting efficiency of sgRNAs designed for the upstream (B&D) and downstream (C&E) mis-match regions. PCR amplification using NOTCH2NLC specific primers yielded a ~900 bp product. The T7EI cutting efficiency (%) = [1 − (gray value of cut band)/(gray value of uncut band)] × 100%. The cut and uncut bands are indicated by green arrow and blue arrow respectively. F, G Agarose gel electrophoresis evaluated the GGC deletion efficiency of selected sgRNA combinations. The GGC deletion efficiency (%) = [(gray value of edited band)/(gray value of edited band + unedited band)] × 100%. The edited and unedited bands were indicated by green arrow and blue arrow respectively. A reduction of ~150 bp in the PCR product size was observed due to the GGC deletion. H, I TA cloning and Sanger sequencing of PCR products from the homologous regions of NOTCH2 and NOTCH2NL family genes confirmed the specific deletion of GGC repeats in NOTCH2NLC following gene-editing. J Agarose gel electrophoresis evaluated the specificity of GGC deletion in NOTCH2NLC. The truncated PCR products (indicated by green arrow) were specifically detected in NOTCH2NLC, but were absent in NOTCH2 and NOTCH2NLA/B/R. One-way ANOVA test with multiple comparisons. In D, ****P < 0.0001, ns no significance, **P = 0.0012 (sgRNA3 vs mock), **P = 0.0026 (sgRNA4 vs mock); In E, **P = 0.0062 (sgRNA5 vs mock), **P = 0.0049 (sgRNA6 vs mock); In G, ****P < 0.0001, ***P = 0.0001. N = 3 independent experiments. Data are represented as mean ± SEM. Source data are provided as a Source Data file.
First, the gene-editing efficiency of each sgRNA was evaluated in human cell lines with normal GGC repeats (HEK293 cells). Forty-eight hours after co-transfection with Cas9 and sgRNA plasmids (Supplementary Fig. 1C) at a Cas9-to-single sgRNA ratios of 4:1, which was selected for preliminary screen based on our prior experience in genome editing of other genes45. We performed a T7 endonuclease I (T7EI) assay on the PCR products amplified using NOTCH2NLC-specific primers (Supplementary Fig. 1A and Supplementary Data 2). The results indicated that the upstream sgRNA1, as well as the downstream sgRNA5 and sgRNA6, exhibited optimal cutting efficiency (Fig. 1B–E). Intriguingly, unexpected T7EI cleavage fragments (400–500 bp) were observed in the control samples without CRISPR/Cas9 targeting (Fig. 1B, C). Since the extracted gDNA pooled multiple HEK293 cells, the unexpected bands may reflect intrinsic variations in the GGC repeat length of NOTCH2NLC among cells within the same population (Supplementary Fig. 2A).
To validate the efficacy of these sgRNAs mediating GGC repeats deletion, various sgRNA combinations were tested. T7EI assay performed on the PCR products amplified using NOTCH2NLC-specific primers demonstrated that both sgRNA1 + 5 and sgRNA1 + 6 combinations efficiently excised the GGC repeats (33–40%), as evidenced by truncated PCR products due to the GGC deletion (Fig. 1F–G) and mismatch cleavage products (Supplementary Fig. 2B). For each combination, we tested different Cas9-to-sgRNA combinations ratios (1:1, 2:1, and 4:1) to optimize the targeting efficiency. The 1:1 ratio was found to be the most effective (Supplementary Fig. 2C) and was subsequently used in all following experiments.
Then, we evaluated whether the Cas9-mediated deletion of GGC repeats was specific to NOTCH2NLC in HEK293 cells. Genomic DNA region encompassing GGC repeats was amplified using consensus primers that amplify NOTCH2NLA/B/C/R and NOTCH2 simultaneously (Supplementary Fig. 1A). The PCR products were subjected to TA cloning, and fifty randomly selected colonies per sgRNA combination were subjected to Sanger sequencing. The results confirmed that the deletion of GGC repeats occurred exclusively in the NOTCH2NLC gene, with no detectable off-target editing in homologous NOTCH2NLA/B/R and NOTCH2 (Fig. 1H, I). The specificity of gene-editing was further validated by PCR using primers that specifically amplify GGC repeats region of NOTCH2NLA/B/R and NOTCH2. The agarose gel electrophoresis confirmed the absence of truncated PCR products or deletions in these homologous genes (Fig. 1J and Supplementary Fig. 2D). In addition, quantitative PCR (qPCR) analysis using NOTCH2NLC-specific primers, or consensus primers, which amplify NOTCH2NLA/B/C/R and NOTCH2, demonstrated that the deletion of GGC repeats did not alter the expression of NOTCH2NLC or its homologs (Supplementary Fig. 3A), providing additional evidence for the specificity of the gene-editing strategy.
Gene-editing of expanded GGC repeats significantly reduced polyG levels in stable HEK293 cell line expressing NOTCH2NLC-98GGC
Accumulating evidence supports that the polyG polypeptides, translated from the expanded GGC repeats, play a central role in the pathogenesis of NIID34,35,36,37,38,39,40,41. To evaluate whether our gene-editing strategy could effectively target the expanded GGC repeats and reduce polyG levels, we established a HEK293 cell line stably expressing NOTCH2NLC-98GGC-GFP (Fig. 2A). The GFP tag (start codon removed) was fused to the polyG for detecting its expression. PCR analysis confirmed the expansion of GGC repeats in this cell line, and western blotting analysis showed the translation of the expanded GGC repeats into polyG-GFP (Fig. 2B, C). We then co-transfected the stable cell line with Cas9 and sgRNA combinations (1 + 6, 1 + 5) or control sgRNA to test the effect of our gene-editing strategy.

A Schematic diagram of the NOTCH2NLC-98GGC-GFP construct and establishment of stable HEK293 cells expressing NOTCH2NLC-98GGC-GFP (98polyG cell line for short). GFP tag was infused with polyG proteins. B PCR confirmed the expansion of GGC repeats in the 98polyG cell line. The experiment was repeated at least three times with consistent results. A representative image is shown. C Western blotting analysis of polyG protein expression in 98polyG cell lines using anti-GFP antibody. Positive control: HEK293 cells transiently transfected with NOTCH2NLC−98GGC-GFP construct. Vinculin served as the loading control. The experiment was repeated at least three times with consistent results. A representative image is shown. D, E Western blotting analysis demonstrated that co-transfection of Cas9 with selected sgRNA combinations significantly reduced both aggregated and soluble polyG proteins levels in the 98polyG cell line. Quantification of the results is shown in (E). N = 3 independent experiments. Data are represented as mean ± SEM. One-way ANOVA test with multiple comparisons. For aggregated protein, ***P = 0.0005 (sgRNA1 + 5 vs sgRNA_ctl), ***P = 0.0004 (sgRNA1 + 6 vs sgRNA_ctl); for soluble polyG protein, ***P = 0.0003 (sgRNA1 + 5 vs sgRNA_ctl), ***P = 0.0001 (sgRNA1 + 6 vs sgRNA_ctl). F, G Immunostaining demonstrated that co-transfection of Cas9 with selected sgRNA combinations significantly reduced the number and the size of polyG aggregates in the 98polyG cell line. Quantification of the results is shown in (G). Scale bar is 40 μm. One-way ANOVA test with multiple comparisons. For number of aggregates per image, ***P = 0.0001, ****P < 0.0001; for size of aggregate, **P = 0.004 (sgRNA1 + 6 vs sgRNA_ctl), **P = 0.0047 (sgRNA1 + 5 vs sgRNA_ctl). N = 3 independent experiments. Data are represented as mean ± SEM. The cell selection process in (A) were created in BioRender. xie, n. (2025) https://BioRender.com/dysupql. Source data are provided as a Source Data file.
Western blot analysis demonstrated a significant reduction in the levels of both aggregated and soluble forms of polyG (by 49.0–51.7% and 67.6–78.4%, respectively) following treatment with Cas9 and NOTCH2NLC sgRNA combinations (Fig. 2D, E). Consistent with these findings, immunofluorescence staining results showed that both NOTCH2NLC sgRNAs combinations significantly reduced the number (by 63.1–69.7%) of polyG-GFP aggregates compared to the control group (Fig. 2F, G). Further PCR and sequencing analyses confirmed that the decrease in polyG-GFP signal was consistent with the successful deletion of GGC repeats (Supplementary Fig. 4A–E). Collectively, all these results indicate that our gene-editing strategy targeting expanded GGC repeats effectively and significantly reduced polyG expression in vitro.
Gene-editing of expanded GGC repeats reversed transcriptomic changes in patient-derived cell model
Human iPSCs provide a more physiologically relevant system for studying the expression and function of human-specific disease genes that are expressed at the endogenous level. We therefore established iPSCs from peripheral blood mononuclear cells (PBMCs) of a NIID patient and a sex- and age-matched health control (Supplementary Fig. 5A). Repeat-primed polymerase chain reaction (RP-PCR) and GC-rich PCR (GC-PCR) assay, performed as previously described22, determined the GGC repeat lengths to be 113/17 repeats (patient) and 19/18 repeats (control) in the respective iPSC lines (Supplementary Fig. 5A, B). All iPSC lines maintained normal karyotypes and exhibited comparable expression of pluripotency markers (Supplementary Fig. 6A, B).
Recent studies have demonstrated a promising gene-therapy strategy involving the correction of expanded CAG repeats to normal repeats via homology directed repair (HDR), which has been successfully validated in a HD pig model46. However, whether this gene replacement strategy can be used in NIID models remains to be explored. To address this, Cas9 with sgRNA1 + 6 and a HDR donor template with normal GGC repeats were nucleofected into the NIID iPSC line (Supplementary Fig. 7A, B). Using fluorescence-activated cell sorting (FACS), serial dilution, and PCR-based genotyping, we successfully isolated four monoclonal lines in which the expanded GGC repeats were either deleted or repaired to normal-length repeats. The deleted or repaired GGC repeats were further confirmed by RP-PCR, GC-PCR and Sanger sequencing after TA cloning (Supplementary Fig. 5C–F and Supplementary Fig. 7C–F). Furthermore, all GGC-deletion and GGC-repair iPSC lines, retained normal pluripotency and karyotypic stability (Supplementary Fig. 6A, B).
To evaluate the specificity of our gene-editing system and investigate potential off-target effects, we selected two representative edited iPSC lines, one with GGC-deletion (line-22) and one with GGC-repair (line-37), using the unedited NIID iPSC line as a control. First, we employed deep-sequencing (deep-seq) to analysis top twenty potential off-target sites (ten for sgRNA1 and ten for sgRNA6), including the highly homologous NOTCH2 and NOTCH2NL gene family (Fig. 3A, B). These candidate sites were amplified by PCR using candidates-specific primers (Supplementary data 2) and subjected to deep-seq. Bioinformatic analysis confirmed precise gene-editing in NOTCH2NLC, with minimal off-target activity (Fig. 3A, B and Supplementary Fig. 8A, B). Second, to broadly assess genomic integrity and potential off-target activity, we performed WGS on the same iPSC lines. Our results revealed comparable and negligible extent of single-nucleotide polymorphism (SNP), insertion/deletion (InDel), structure variants and copy number variants (CNV) between edited iPSCs and unedited iPSC (Supplementary Fig. 8C). In addition, read-depth analysis of the additional 30 predicted off-target sites also showed no significant differences between groups (Supplementary Fig. 8D, E), further supporting the specificity of our editing strategy.

Deep-seq analysis revealed minimal off-target activity for sgRNA1 (A) and sgRNA6 (B) at the top 20 predicted off-target sites in both GGC-deleted and GGC-repaired iPSC colonies. Multiplex PCR-amplified potential off-target sites and target loci were deep sequenced, with off-target activity calculated as (mutated reads/total reads) *100% at each site. The heatmap analysis (C) and principal component analysis (PCA) (D) of RNA-seq data showing transcriptomic similarity among indicated groups. Transcriptomic profiles of GGC-deletion and GGC-repair NPCs more closely resembled those of WT NPCs than untreated NIID NPCs. E–G Volcano plots showing significantly up-regulated (depicted in red) and down-regulated (depicted in blue) DEGs among different groups (N = 3 biological replicates per group). The complete lists of DEGs are available in Supplementary Data 3. H, I Upset plot illustrating the overlap and unique DEGs among the different comparison groups shown in (E–G). The dark blue and dark red rectangles indicated genes down-regulated and up-regulated in the unedited NIID NPC, respectively. The light blue and pink rectangles indicated genes down-regulated and up-regulated in the edited NIID NPC, respectively. Among 1244 downregulated genes in NIID NPCs (relative to WT NPCs), 633 and 903 genes were upregulated after GGC-deletion (H) and GGC-repair (I); while among 1983 upregulated genes in NIID NPCs, 1482 genes and 1394 genes were downregulated after GGC-deletion (H) and GGC-repair (I). J Gene Ontology (GO) analysis on the subset genes exhibiting reversal trends revealed significant enrichment in neuronal function after GGC-deletion and GGC-repair. For all panels, differential expression analysis was performed using DESeq2 (two-sided Wald test), with DEGs defined as |log2(fold change)| ≥ 2 and FDR < 0.05 (Benjamini-Hochberg method). GO enrichment was assessed using a one-sided hypergeometric test with FDR adjustment. Source data are provided as a Source Data file.
Subsequently, we differentiated these edited iPSCs (both GGC-deletion and GGC-repair) along with NIID and WT iPSCs into NPCs. Immunofluorescence analysis showed similar expression of PAX6 and Nestin in GGC-deletion and GGC-repair NPCs relative to WT controls, suggesting preserved neural differentiation potential (Supplementary Fig. 9). Furthermore, no significant alterations were observed in the expression levels of NOTCH2NLA/B/C/R and NOTCH2 following gene editing (Supplementary Fig. 3B).
NIID develops neuronal pathology in an age-dependent manner. Although our previous studies revealed that NPCs derived from NIID iPSCs retained differentiation capacity, they displayed significant transcriptomic alterations that may be associated with the disease39. To investigate whether GGC-deletion or GGC-repair could reverse the transcriptomic alterations, we performed bulk RNA sequencing. Heatmap analysis and principal component analysis (PCA) revealed that the transcriptomic profiles of GGC-deletion and GGC-repair NPCs more closely resembled those of WT NPCs than NIID NPCs (Fig. 3C, D). Compared to WT NPCs, 1983 upregulated genes and 1244 downregulated genes were identified in NIID NPCs (Fig. 3E). However, after GGC repeats deletion, 50.88% (633/1244) of downregulated genes increased and 74.74% (1482/1983) of upregulated genes decreased; whereas following GGC repeats repair, 72.59% (903/1244) of downregulated genes increased and 70.30% (1394/1983) of upregulated genes decreased (Fig. 3F–I). Gene Ontology (GO) analysis on the subset genes that exhibit reversal trends revealed significant enrichment in neuronal function after GGC-deletion and GGC-repair (Fig. 3J). These findings suggest that removing or correcting the expanded GGC repeats not only preserves the pluripotency and differentiation capacity of iPSCs but also reverses the GGC expansion-mediated transcriptomic changes in iPSCs derived cells.
Gene-editing of expanded GGC repeats alleviated disease-related pathological and clinical phenotypes in transgenic NIID mouse model
To evaluate the in vivo efficacy of our gene-editing strategy, we leveraged a transgenic NIID mouse model, previously established in our laboratory, that ubiquitously expresses the NOTCH2NLC-98GGC39. This model faithfully recapitulates pathological hallmarks and clinical features of NIID, including widespread intranuclear inclusions, progressive neurodegeneration, motor deficits, as observed in patients with NIID39,40. To deliver the CRISPR/Cas9 system effectively across the blood-brain barrier, we packaged Cas9 and sgRNAs into AAV-PHP.eB, a capsid known for its high transduction efficiency in the central nervous system47.
First, we conducted the treatment in a specific brain region of NIID mice to evaluate the in vivo effectiveness of our gene-editing strategy. To minimize inter-individual variability, therapeutic AAV (Cas9 and sgRNA1 + 6) and control AAV (Cas9 and control sgRNA) were injected into the left and right striatum of the same NIID mouse, respectively. Injections were performed at postnatal day 8–10 (P8-10) with a dose of 1 × 1010 genomic copies (GC) per side. Four weeks post-injection, immunostaining revealed a significant reduction in the number of polyG aggregates on the left striatum injected with Cas9 and sgRNA1 + 6 compared to the right side injected with Cas9 and control sgRNA (Fig. 4A). Additionally, the striatum injected with Cas9 and control sgRNA showed significant decreased levels of GFAP and IBA1, markers of astrocyte and microglial activation, respectively, and a significant increased NeuN expression (Fig. 4B–D). Consistent results were observed when compared with age-matched non-disease controls, which further confirmed the therapeutic efficacy of our editing strategy (Supplementary Fig. 10).

A Immunofluorescence analysis using antibody against Flag revealed a significant reduction of polyG aggregates in the striatum injected with AAV-Cas9 and AAV-sgRNA1 + 6 (treated), compared to the contralateral side injected with AAV-Cas9 and AAV-control sgRNA (untreated). Immunofluorescence analysis using antibodies against GFAP, IBA1, and NeuN demonstrated reduced GFAP (B) and IBA1 (C) expression alongside increased NeuN (D) levels in the treated striatum, compared to the contralateral untreated side. ZsGreen fluorescence (reporter encoded in the AAV-sgRNA vector) confirmed efficient AAV transduction in targeted regions. Scale bar is 200 μm and 50 μm for low- and high-magnification images, respectively. ****P < 0.0001 (polyG), ***P = 0.0001 (GFAP & IBA1), *P = 0.0245 (NeuN), two-tailed t-test. N = 3 mice per group. Data are represented as mean ± SEM. Source data are provided as a Source Data file.
Subsequently, we conducted a systematic treatment via retro-orbital vein injection (ROV), a more clinically relevant injection method, in NIID mice. Previous studies have shown that NIID mice progressively develop phenotypic abnormalities starting around 40 days of age, with a median survival of ~2 months39. To assess the therapeutic potential of early intervention, we administered AAV (1 × 1011 GC per mouse) at P2 and performed a comprehensive evaluation on treated NIID mice (injected with Cas9 and sgRNA1 + 6), untreated NIID mice (injected with Cas9 and control sgRNA) and control mice (injected with Cas9 and sgRNAs) at P35-50 (Fig. 5A). Immunofluorescence analysis confirmed successful transduction of AAV throughout the whole brain (Supplementary Fig. 11A). PCR performed with primers that amplify exon1 of NOTCH2NLC confirmed removal of expanded GGC repeats in the genomic DNA of mice post ROV injection (Supplementary Fig. 11B).

A Schematic diagram of retro-orbital vein injection (ROV) in neonatal mice and the timeline for behavioral tests, pathological exams and RNA-seq. Three groups of mice were used. Treated group: NIID mice injected with AAV-Cas9 and AAV-sgRNA1 + 6; Untreated group: NIID mice injected with AAV-Cas9 and AAV-control sgRNA; Control group: littermate control mice injected with AAV-Cas9 and AAV-sgRNA. Motor functions of mice in different groups were evaluated using open field test (B–E), wire hang (F), and rotarod tests (G, H). Treated NIID, N = 18; untreated NIID, N = 13; control, N = 26. I Kaplan-Meier survival curve of mice with or without gene therapy (treated NIID, N = 13; untreated NIID, N = 8; control, N = 20). J, K Western blotting of cortical tissue revealed that treated NIID mice exhibited reduced polyG and GFAP expression alongside increased NeuN levels, compared to untreated NIID mice. The GFAP and NeuN levels of treated NIID mice were restored to levels comparable to those in the control group. Successful AAV transduction was confirmed by zsGreen expression in all experimental animals. Vinculin served as the loading control. N = 3 mice per group. One-way ANOVA test with multiple comparisons. In C, ***P = 0.0002, ****P < 0.0001. In D, *P = 0.0475. In E, ***P = 0.0001, ****P < 0.0001. In F, *P = 0.0263, ****P < 0.0001. In G, ****P < 0.0001. In H, *P = 0.0189, ****P < 0.0001. In K, for polyG aggregates levels, **P = 0.0013, ***P = 0.0003; for GFAP levels, *P = 0.0255, **P = 0.0059; for NeuN levels, **P = 0.0027 (treated vs untreated), **P = 0.0068 (untreated vs control). Data are represented as mean ± SEM. The neonatal mouse in Figure 5A was created in BioRender. xie, n. (2025) https://BioRender.com/dysupql. Source data are provided as a Source Data file.
We analyzed behavioral performance in mice at P35–P40 and found that, compared to the control mice, the untreated NIID mice exhibited significantly reduced moving distance and duration in the open-field test (Fig. 5B–E), as well as markedly poorer performance in motor coordination tests such as the wire hang tests and rotarod (Fig. 5F–H). In contrast, the treated NIID mice showed significant improvement in these behavioral tests, with their performance reaching levels comparable to those of the control group (Fig. 5B–H). In addition, Kaplan-Meier survival analysis showed that the treated NIID mice exhibited significantly longer lifespans compared to the untreated NIID mice (Fig. 5I). Furthermore, western blot results demonstrated that, compared to the untreated NIID mice, the treated NIID mice exhibited significantly reduced protein levels of polyG and GFAP, alongside increased NeuN levels in the cortex, reaching levels comparable to those of non-disease control mice (Fig. 5J, K).
Gene-editing of expanded GGC repeats alleviated disease-related transcriptomic alterations in transgenic NIID mouse model
Transcriptional dysregulation is a well-documented molecular hallmark of NIID, which is observed in patient tissues, patient-derived cells and animal models9,37,38,39. To investigate whether our CRISPR/Cas9-based strategy targeting expanded GGC repeats could reverse transcriptomic alterations associated with NIID, we performed bulk RNA-seq analysis on cortex tissues from the treated NIID mice (injected with Cas9 and sgRNA1 + 6), untreated NIID mice (injected with Cas9 and control sgRNA) and control mice (injected with Cas9 and sgRNAs). Global transcriptomic visualization via heatmap view and PCA analysis revealed the notably altered gene expression in untreated NIID mice as compared with the control mice, and these alterations could be reversed by NIID treatment with CRISPR/Cas9 (Fig. 6A, B). For example, 1789 upregulated and 334 downregulated genes were identified in the untreated NIID mice compared to the control mice (Fig. 6C). In contrast, treated NIID mice showed markedly fewer differential expressed genes (DEGs, 171 upregulated and 134 downregulated) (Fig. 6D). Among the downregulated genes, 52.10% (174/334) were increased, while among the upregulated genes, 58.69% (1050/1789) were decreased in the treated NIID mice (Fig. 6E, F). Further analysis revealed that the reversed DEGs were enriched in disease-associated pathways, including neural function and inflammatory response, suggesting a partial restoration following GGC repeat-deletion treatment (Fig. 6G–I).

The heatmap (A) and PCA (B) analysis of RNA-seq data on cortex showing transcriptomic similarity among indicated groups. Transcriptomic profiles of treated NIID mice more closely resembled those of control mice than untreated NIID mice (N = 3 biological replicates per group). C–E Volcano plots showing up-regulated (depicted in red) and down-regulated (depicted in blue) DEGs among different groups. The complete lists of DEGs are available in Supplementary Data 4. F Upset plot showing the overlap and unique DEGs among the different comparison groups shown in (C–E). Among 334 downregulated genes in untreated NIID mice (relative to control mice), 174 genes were upregulated after treatment; while among 1789 upregulated genes in untreated NIID mice, 1050 genes were downregulated after treatment. G GO enrichment analysis revealed that the reversed DEGs were enriched in neural-related pathways. H, I GO enrichment analysis comparing untreated NIID-vs-control and treated NIID-vs-control revealed that neural function and inflammatory response related pathway was partially ameliorated in treated NIID mice. For all panels, differential expression analysis was performed using DESeq2 (two-sided Wald test), with DEGs defined as |log2(foldchange)| ≥ 0.585 and P < 0.05. GO enrichment was assessed using a one-sided hypergeometric test with FDR adjustment (Benjamini-Hochberg method). Source data are provided as a Source Data file.
In addition, we assessed the impact of our gene editing strategy on peripheral tissue. Previous studies indicated that polyG aggregates are also present in cardiomyocytes, leading to cardiac abnormalities40. Western blot results showed a significant reduction (~79%) in cardiac polyG protein levels in the treated NIID mice compared to the untreated NIID mice (Supplementary Fig. 12A). Consistent with our observations in the central nervous system, transcriptomic profiling demonstrated that the treated NIID mice exhibited gene expression patterns more similar to the control mice, as evidenced by both heatmap visualization and PCA analysis (Supplementary Fig. 12B, C). Further analysis showed that the treated NIID mice had markedly fewer DEGs than the untreated NIID mice (571 vs 5372) when compared to the control mice (Supplementary Fig. 12D, E). Notably, 56.53% (277/490) - 64.03% (3126/4882) of originally dysregulated genes displayed reversal trends following treatment (Supplementary Fig. 12F, G), with these genes being significantly enriched in mitochondrial function and cardiac contraction pathways (Supplementary Fig. 12H). Taken together, analysis of NIID mice suggests that deletion of expanded GGC repeats effectively rescues transcriptomic dysregulation across both central nervous system and peripheral tissue.
Discussion
The GGC repeat expansion in the NOTCH2NLC gene is not only the genetic cause of NIID, but has also been implicated in a broad spectrum of neurological disorders, such as essential tremor48,49,50,51, amyotrophic lateral sclerosis26,52,53, cerebral small vascular disease54,55, multiple system atrophy30, leukoencephalopathy29,56, Alzheimer’s disease31,57, and Parkinson’s disease27,58, inherited peripheral neuropathy59,60 and oculopharyngodistal myopathy type 332,61. Collectively, these conditions are referred to as “NOTCH2NLC-related repeat expansion disorders (NREDs)”16,20,22,48,62,63. Mechanistically, expanded GGC repeats are mainly translated into polyG peptides, which constitute a major driver of intranuclear inclusion formation and cellular dysfunction35,36,37,38,39,40,41. Although administration of compounds or expression of downstream effector gene have been shown to partially ameliorate disease phenotypes in animal models37,41,64, no therapy with broad and durable efficacy has yet been established.
In this study, we developed a CRISPR/Cas9-based gene-editing strategy that precisely targets and excises the expanded GGC repeats in the NOTCH2NLC gene. This approach markedly reduced polyG aggregates in the 98G stable cell line, patient-derived iPSCs and NPCs, as well as in the transgenic NIID mouse model. Notably, in the mouse model, editing of the GGC expansion significantly ameliorated neurodegenerative phenotypes, including the restoration of neuronal proteins, attenuation of gliosis, behavioral improvement, and extended survival. At the molecular level, removal of the expanded repeats corrected transcriptional dysregulation in both NPCs and mouse brain tissues, particularly in pathways related to neuronal function and inflammatory response. In addition, owing to the ROV administration of the PHP.eB capsid AAV, our gene-editing strategy partially alleviated molecular abnormalities in peripheral tissue of the mouse model. Considering the multisystem involvement of NIID19,20, further optimization of delivery strategies may enhance the therapeutic efficacy of this approach. Together, these findings highlight the central pathogenic role of GGC repeats in NIID and support permanent correction at DNA-level as a promising therapeutic avenue.
Given the high sequence similarity among NOTCH2NLC, its NOTCH2NL paralogs, and NOTCH2, we carefully designed sgRNAs targeting regions with minimal sequence homology and implemented a dual-sgRNA strategy to ensure editing precision. This approach achieved highly specific targeting of the NOTCH2NLC locus with minimal off-target activity across HEK293 cell line, 98G stable cell line, patient-derived iPSCs and NPCs. The expression of homologous genes remained unaltered (Supplementary Fig. 3), and edited iPSCs preserved normal karyotype and differentiation potential (Supplementary Figs. 6 and 9). Moreover, by providing a repair template with normal repeat length, we achieved HDR-mediated correction of the expanded GGC repeats to normal repeat length in dividing cells like iPSCs. Although HDR-based repair remains limited in postmitotic neurons, it is more feasible in proliferative cells of both the nervous system and peripheral tissues. Refining repeat-targeted repair strategies may further improve the therapeutic outcomes of this approach.
In addition to NREDs, expansions of GGC or CCG repeats in the 5′ UTR of various genes have been implicated in an expanding spectrum of neurological and muscular disorders, such as FXS, FXTAS65, FRAXE intellectual disability66,67, movement disorders68,69, oculopharyngeal-related myopathies23,70,71. Although previous studies in FXS have explored CRISPR/Cas9-mediated editing of the FMR1 CGG repeats in cell line and iPSC72,73, these studies were restricted to in vitro settings and did not rigorously assess potential off-target effects. In contrast, our work demonstrates the feasibility and efficacy of this strategy in both cellular and animal models of NIID, while also addressing the additional challenge posed by high sequence homology. Importantly, our study provides the preclinical proof demonstrating the efficacy of gene editing for NOTCH2NLC GGC repeat expansion in NIID. Furthermore, it offers a conceptual framework for applying CRISPR/Cas9-mediated repeat excision to a broader class of GGC-driven REDs.
Despite the robust efficacy and safety of our gene-editing strategy demonstrated in both in vitro and in vivo NIID models, several limitations remain. First, the transgenic NIID mouse model used in this study reproduces key pathological hallmarks of the human disorder, providing a valuable platform for therapeutic assessment. However, its rapid disease progression and short lifespan contrast sharply with the late-onset, slowly progressive nature of NIID in patients16,74, likely reflecting species-specific differences and the absence of native regulatory mechanisms for the human-specific NOTCH2NLC gene. In addition, the model may not recapitulate the typical MRI signatures observed in NIID patients16,18, such as corticomedullary hyperintensity, which may be attributed to the relatively simple white matter architecture and lower glial density in mice75. Another limitation is the lack of long-term safety evaluation. Although we combined targeted deep-seq with WGS, and RNA-seq to assess safety in several human cell models and mouse model, these analyses were limited in duration and sensitivity. Incorporating unbiased, genome-wide approaches such as GUIDE-seq76 in future work will be essential for comprehensive off-target assessment prior to clinical translation. The development of large-animal NIID models, such as pigs or non-human primates, whose brain architecture and immune responses more closely resemble humans, will be indispensable for assessing the long-term safety, delivery efficiency, and immune compatibility of this CRISPR/Cas9-based therapy in a more clinically relevant context.
In conclusion, our study provides compelling evidence that precise removal of expanded GGC repeats in the NOTCH2NLC gene using CRISPR/Cas9 represents a safe and effective therapeutic strategy for NIID, laying the groundwork for future clinical translation.
Methods
Human participants
One NIID patient and one health control from mainland China were recruited in this study. Each participant underwent a neurological examination performed by at least two experienced neurologists. The patient was a 57-year-old male with a history of bradykinesia and hand tremors for over 10 years underwent physical examination, which revealed increased muscle tone in all four extremities and action tremors in the upper extremities. Cognitive assessment yielded an MMSE score of 28 and a MoCA score of 27. The health control was a 50 year-old male. Peripheral blood samples were collected from all participants after obtaining written informed consent. These samples were subsequently reprogrammed into iPSCs for research purposes.
GGC repeat size determination
The GGC repeat size within NOTCH2NLC was verified and determined using RP-PCR and GC-PCR, as previously described22. RP-PCR was performed with a FAM-labeled gene specific primer (CCTCAGCCCGATACTCACCAT) and a repeat-containing primer [TACCAATACGCATCCCGCGATTTGTCTTA(CGG)5]; GC-PCR was performed with a FAM-labeled forward primer (AGCGCCAGGGCCTGAGCCTTTGAAGCAG) and a reverse primer (TCGCCCCAGGTGGCAGCCCCGGGCGCCGCGGAC). Amplified products were analyzed by capillary electrophoresis (3500xL Genetic Analyzer, Applied Biosystems) with allele sizing based on the GeneScan 1000 ROX Size Standard.
Animals
The transgenic NIID mouse model (C57BL/6J background) that ubiquitously expresses NOTCH2NLC-98GGC was generated previously, and no significant differences in pathological or behavioral phenotypes between males and females39. Heterozygous NIID mice and the littermate controls were used in this study. Both sexes were used for stereotaxic injection, ROV, and behavioral tests. All mice were bred and maintained on a 12:12 h light/dark cycle (lights off at 7 p.m.) in the animal facility under specific pathogen-free conditions in the animal facility of Xiangya Hospital, Central South University. The temperature was maintained at 22 ± 1 °C with relative humidity (30–70%).
Derivation of iPSC and differentiation to NPCs
PBMCs from NIID patient and healthy control were reprogrammed into iPSCs using CytoTune-iPS 2.0 Sendai virus reprogramming kit according to the manufacturer’s protocol. The iPSCs were cultured on vitronectin-coated plates in StemPro-34 SFM (Gibco) medium. After 3–4 weeks, the medium was replaced by Essential 8 (Gibco) when iPSC colonies were established. The iPSC lines were validated by karyotyping to confirm chromosomal constitution, and the specific GGC repeat expansion within the patient-derived line was verified by RP-PCR and GC-PCR. IPSCs were differentiated toward the neural progenitor lineage as previously reported39. In brief, NPCs were derived from iPSCs using the monolayer culture protocol with STEMdiffTM Neural Induction Medium (Stemcell technologies). The generated NPCs were maintained in neural progenitor medium (STEMCELL Technologies) on Matrigel-coated and used for subsequent experiments. All cell lines were negative for mycoplasma.
Plasmids and viruses
The NOTCH2NLC-98GGC, Cas9, and sgRNAs plasmids were constructed in previous studies39,45,46. The sequences of sgRNA are listed in the Supplementary Data 1. The non-targeting control sgRNA was verified previously45. Cas9 and sgRNA vectors were sent to PackGene Biotech (Guangzhou, China) for viral packaging and production. All the viral vectors were packaged into the PHP.eB serotype. The titers of the viruses used for stereotactic injection and retro-orbital injection were 1 × 1013 GC/mL and 5 × 1013 GC/mL, respectively.
Antibodies
Primary antibodies used in this study include the following: Flag (Sigma-Aldrich, F1804, 1:1000), vinculin (Sigma-Aldrich, V9131, 1:1000), NeuN (Cell Signaling Technology, 24307s, 1:1000), GFAP (Cell Signaling Technology, 3670S, 1:1000), IBA1 (Wako, 019-19741, 1:1000), GFP (Invitrogen, A-11122, 1:2000), zsGreen (Sangon, D199984, 1:2000), OCT4 (Proteintech, 60242-1-Ig, 1:200), SOX2 (Proteintech, 11064-1-AP, 1:1000), PAX6 (Abcam, AB195045, 1:350), Nestin (CST, 33475S, 1:2000). Fluorescent secondary antibodies (1:200) used were donkey anti-rabbit and donkey anti-mouse antibodies conjugated with Alexa Fluor 488 or 594 from Jackson ImmunoResearch. HRP-conjugated secondary antibodies (1:10000) were donkey anti-rabbit and donkey anti-mouse from Jackson ImmunoResearch.
Cell culture and transfection
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 u/ml) at 37 °C with 5% CO2. For the transfection procedure, the cells were seeded onto 12-well plates at a density of ~200,000 cells per well. After 24 h, the cells were transfected with plasmids using Lipofectamine 3000 according to the manufacturer’s instructions. 48–72 h after the transfection, the cells were harvested for subsequent experiments. HEK293 cells (Procell, CL-0001) were authenticated by STR profiling and were tested negative for mycoplasma contamination.
Stable cell line establishment
200,000 HEK293 cells were co-transfected with a total of 1 μg NOTCH2NLC-98GGC-GFP plasmid. DMEM was supplemented with G418 (600 μg/mL) for selection. 48 h after transfection, the cells were treated with trypsin and then reseeded in 96-well plates containing selective medium. A serial dilution approach was adopted during this process. The selective medium was refreshed every 3–4 days to eliminate non-transfected cells while allowing resistant colonies to proliferate. Once visible G418-resistant foci emerged, they were isolated and expanded sequentially into 24-well plates and later into 6-well plates for validation of stable integration.
Nucleofection and monoclonal cell isolation
One day before the passage day, iPSCs were treated with 10 μM Y-27632 overnight. On the passage day, 1 × 106 iPSCs resuspended in human stem cell nucleofector solution I (Lonza, Basel, Switzerland) were nucleofected with 2 μg of plasmids (Cas9: sgRNA = 1:1) using program B16 in the nucleofector IIb device (Lonza). After nucleofection, the cells were transferred to a freshly Matrigel-coated plate and cultured in mTeSR1 medium containing 10 μM Y-27632 for 1 day, followed by regular mTeSR1 medium. 24–48 h after nucleofection, the iPSCs sorted on a Becton Dickinson FACS Aria II cell sorter were seeded onto a 24-well plate layered with mouse embryonic fibroblasts at a density of 100 cells per well. iPSC colonies emerged between 10-and 12-days post-sorting. These colonies were transferred to freshly Matrigel-coated plates for genotyping. A total of ninety clones were analyzed and four monoclonal lines, in which the expanded GGC repeats were either deleted or repaired to normal-length repeats, were selected for subsequent experiments.
T7EI assay
T7 Endonuclease I assay was performed as described previously77. Genomic DNA was extracted from HEK293 cells using a commercial DNA isolation kit (Magen). To amplify the target regions flanking the CRISPR-Cas9 editing site, PCR was performed with LA Taq (Takara) using primers specific to amplify NOTCH2NLC or NOTCH2/NOTCH2NLA/B/R. The T7EI primers are listed in the Supplementary Data 2. The PCR products were purified and subjected to heteroduplex formation by denaturation and gradual reannealing to allow mismatched base pairing between the wild-type and mutant alleles. The heteroduplex DNA was digested with T7 Endonuclease I (New England Biolabs) at 37 °C for 5 min, followed by enzyme inactivation. The digested fragments were separated on a 1.5% agarose gel, and the band intensities were quantified using the ImageJ software to calculate the cutting frequency.
TA cloning
Genomic DNA region encompassing GGC repeats was amplified using primers that amplify NOTCH2NLA/B/C/R and NOTCH2 simultaneously. The TA clone primers are listed in the Supplementary Data 2. PCR products were purified using a commercial gel extraction kit (Magen). TA cloning was performed using a commercial SuperTOPO TA clone kit (Vazyme). The ligation mixture was transformed into chemically competent E. coli DH5α cells, followed by plating onto LB agar containing ampicillin at 37 °C overnight for colony selection. The next day, colonies were picked and cultured in LB broth. Plasmid DNA was isolated and sequenced with M13 forward primer. Sanger sequencing results were aligned and analyzed using the SnapGene software.
Reverse transcriptase PCR and quantitative real-time PCR
Total RNA was extracted from cell pellets using a TRIzol reagent (Invitrogen) following the manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA using the RevertAid First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific) and oligo dT primers. Quantitative real-time PCR was performed and analyzed by applying the comparative cycle threshold method. The primers used for the quantitative real-time PCR are listed in the Supplementary Data 2.
Western blot
Mouse tissues or cell pellets were lysed in ice-cold RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA pH 8.0, 1 mM EGTA pH 8.0, 0.1% SDS, 0.5% DOC, and 1% Triton X-100) containing protease inhibitor. Protein concentration was determined with the BCA Protein Assay Kit (Thermo Scientific). Protein samples were separated by SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore), which was further blocked with 5% milk/PBS for 1 h at room temperature. The blot was incubated with primary antibodies overnight at 4 °C. After three washes in 0.05% PBST, the blot was incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. After three washes in 0.05% PBST, protein bands were developed using an ECL Prime Chemiluminescence kit (GE Healthcare) on a Bio-Rad developing and imaging platform. Protein band intensities were quantified using ImageJ to calculate expression levels relative to internal controls.
Immunofluorescent staining
Mice were anesthetized and perfused intracardially with 0.9% saline solution, followed by 4% paraformaldehyde in 0.1 M phosphate buffer at pH 7.2. The isolated mouse brains were dehydrated in 30% sucrose at 4 °C and sectioned at a thickness of 30 μm for the subsequent immunofluorescence study. Brain sections were blocked with a solution of 3% bovine serum albumin (BSA) in 0.3% Triton X-100/PBS for 60 min, followed by incubation with primary antibodies overnight at 4 °C. After incubation with fluorescent secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI), the brain sections were mounted onto coated glass slides and examined using a Zeiss microscope (Apotome 3).
For immunostaining of iPSCs and NPCs, cells grown on coverslips were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 15 min at room temperature. Throughout the entire staining process, a solution consisting of 2% normal goat serum and 3% BSA in PBS was used for blocking. Incubation with primary antibodies was carried out overnight at 4 °C and followed by fluorescent secondary antibody incubation for 45 min at room temperature. Images were captured using a Zeiss microscope (Apotome 3). The number of polyG aggregates and immunofluorescent intensities of GFAP, IBA1, and NeuN were quantified using ImageJ software.
Stereotaxic injection and retro-orbital vein injection
During the striatal stereotaxic injection, P8-10 mice were anesthetized via inhalation of 1.5% isoflurane and then firmly positioned in a stereotaxic instrument. AAV-sgRNA and AAV-Cas9 were mixed at a ratio of 1:1. 1 μl of the mixed viruses (1 × 1010 GC) was injected into one side of the mouse striatum according to the following coordinates adjusted to the flat skull position: 0.39 mm rostral to the bregma, 1.4 mm lateral to the midline, and 2.5 mm ventral from the dural surface over a period of 5 min. An RWD stereotaxic instrument (69100) and a Hamilton microsyringe (1700 series, 10 μl) were used to deliver the virus at a speed of 100 nl per min. After the surgery, the mice were placed on a heated blanket to recover. All stereotaxic surgeries were completed successfully, with incisions healing well and no adverse events observed.
ROV was conducted following a previously established protocol78. Briefly, P1-P2 neonatal mice were cryo-anesthetized on ice for 3 min. The viruses expressing sgRNA and Cas9 were mixed at a ratio of 1:1 and 2 μl of the mixed viruses (1 × 1011 GC) were injected into each pup. To perform the injection, place the pup in left lateral recumbency with its head facing to the right. Gently hold the pup’s head with the thumb and forefinger of the non-dominant hand. Identify the retro-orbital sinus region and insert ~1/3 of the length of a 10 μL needle, with the bevel facing down, at a 45° angle into the inner canthus. After the injection, the pups were placed on a heated blanket to recover. Among sixty-four injected pups, fewer than ten died due to poor rewarming after hypothermic anesthesia or cannibalization by the dam, while the remaining mice survived and underwent subsequent experiments. Injections were performed in a blinded manner, as the genotypes were unknown at P1-2. For each litter, either therapeutic or control AAV was administered uniformly. After genotyping, transgenic mice were assigned to either treated (with therapeutic AAV) or untreated (with control AAV) NIID groups, and all control littermates were assigned to the control group.
Mouse behavioral analysis
Behavioral tests were performed as previously described39. At least 13 mice per genotype were included. The open field test was performed in a Perspex box (40 cm in length, 40 cm in width, and 31 cm in height) with the floor divided into 16 equal-sized squares. During the 10-min test period, the movement of the mice was recorded on video and analyzed by an automated behavior analysis system. The total moving duration and distance were then compared between different groups. In the wire-hang test, mice were placed on the wire lid of a mouse cage, which was then inverted. During the 3-min test period, the latency to fall was recorded. Each mouse underwent the test twice, and the average hang time was compared across different groups. For the rotarod test, mice were trained for 3 min on the rotarod, with three trials per day for three consecutive days. On the testing day, each mouse underwent an accelerating-speed test (from 5 to 20 rpm) for 3 min, and this test was repeated twice. The intertrial interval was 10 min, and the latency to fall was recorded for each trial. The average time the mice spent on the rotarod was compared among different groups. The survival time of each mouse was monitored and recorded daily.
Deep-sequencing analysis
Potential off-target sites were predicted using the online tool CRISPOR. We generated a PCR library for next-generation sequencing (NGS). We first used specific primers to amplify genomic DNA containing the targeting region with LA Taq (Takara). The second PCR was then performed with the same primers containing Illumina forward and reverse adapters to generate the PCR library for NGS. The final 50 μl PCR products were purified using a commercial gel extraction kit (Magen) and qualified with an Agilent Bioanalyzer 2100. The PCR products were then sent to IGE Biotechnology (Guangzhou) for sequencing. The NGS data were analyzed by CRISPResso279. The top 20 potential off-target sites and primer sequences are listed in the Supplementary Data 2.
RNA-seq and analysis
Mice cortex and heart, and human NPCs were used for RNA extraction. An amount of 1 μg of total RNA per sample was processed for library preparation using VAHTS Universal V10 RNA-seq Library Prep Kit following the manufacturer’s recommendations. Libraries were sequenced on an Illumina NovaSeq X Plus (mice tissues) or Illumina Novaseq 6000 (human NPCs) platform. Raw reads were processed by fastp to get clean reads. Paired-end clean reads were mapped to GRCm38 or GRCh38 by HISAT2 and assembled by StringTie. Quantification of gene expression levels was estimated by TPM (mice tissues) or FPKM (human NPCs). Heatmap and PCA analysis were performed using R80. Differential expression analysis was performed using DESeq2. GO enrichment analysis of DEGs was performed using R81.
Whole-genome sequencing
WGS was performed by Novogene, Co., LTD. A total amount of 0.2 μg DNA per sample was used as input material for the DNA library preparations. Sequencing library was generated using NEB Next® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations and index codes were added to each sample. Next, the DNA libraries were sequenced on Illumina platform and 150 bp paired-end reads were generated. The effective sequencing data was aligned with the reference sequence through BWA82 software. Samtools83 and bcftools were used to extract all potential polymorphic SNPs and InDel sites from the entire genome with the mapped reads. BreakDancer84 software was used to detect insertion, deletion, inversion, intra-chromosomal translocation and inter-chromosomal translocation mutations, based on the reference genome mapping results and the detected insert size. CNVnator85 was used to detect CNVs of potential deletions and duplications. Cas-OFFinder86 was used to predict off-targets across the entire genome. Additional 30 off-target loci in the experimental group were compared with the control group. The relative depths were calculated by taking the number of mapped reads on these sites into the whole genome of the pointer reads.
Statistical analysis and reproducibility
All statistical analyses were performed with either two-tailed Student’s t test or the one-way ANOVA test with multiple comparisons using GraphPad Prism 9. All quantitative data were presented as mean ± SEM. A P value of less than 0.05 was considered statistically significant. Sample sizes were chosen based on established standards in the field to ensure robustness. No statistical method was used for predetermination, and no data were excluded. For in vivo studies (e.g., behavior, survival; N ≥ 8 per group), littermates were randomly allocated and investigators were blinded during procedures and assessment. For in vitro assays (N ≥ 3 independent replicates), while group allocation was defined by experimental conditions, sample processing and analysis order were randomized. Quantitative analysis for in vitro data was performed with blinding to group identity.
Ethics statement
All experimental protocols involving human participants and animals were conducted in compliance with all relevant ethical regulations. The study involving human participants was reviewed and approved by the Ethics Committee of Xiangya Hospital, Central South University (Approval No. 2024040420). Written informed consent was obtained from all human participants. The animal study was reviewed and approved by the Animal Ethics Committee of Xiangya Hospital, Central South University (Approval No. 202103138).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw RNA-seq data from mice have been deposited in the Gene Expression Omnibus under accession code GSE295763. The raw RNA-seq data from human NPCs and the raw WGS data from human iPSCs have been deposited in the Genome Sequence Archive for Human under accession code HRA011636 and HRA014016, respectively. The deposition and sharing of the raw data have been approved by the Human Genetics Resource Office in China (registration number: 2025BAT00839). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary data files. Source data are provided with this paper.
References
Depienne, C. & Mandel, J. L. 30 years of repeat expansion disorders: what have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785 (2021).
Malik, I., Kelley, C. P., Wang, E. T. & Todd, P. K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607 (2021).
Verkerk, A. J. M. H. et al. Identification of a gene containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991).
Pieretti, M. et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822 (1991).
Duyao, M. et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 4, 387–392 (1993).
Banfi, S. et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 7, 513–520 (1994).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Renton, A. E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
Sone, J. et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 51, 1215–1221 (2019).
Cortese, A. et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658 (2019).
Pellerin, D. et al. Deep intronic FGF14 GAA repeat expansion in late-onset cerebellar ataxia. N. Engl. J. Med. 388, 128–141 (2023).
Figueroa, K. P. et al. A GGC-repeat expansion in ZFHX3 encoding polyglycine causes spinocerebellar ataxia type 4 and impairs autophagy. Nat. Genet. 56, 1080–1089 (2024).
Zhou, Z.-D., Jankovic, J., Ashizawa, T. & Tan, E.-K. Neurodegenerative diseases associated with non-coding CGG tandem repeat expansions. Nat. Rev. Neurol. 18, 145–157 (2022).
Takahashi-Fujigasaki, J. Neuronal intranuclear hyaline inclusion disease. Neuropathology 23, 351–359 (2003).
Sone, J. et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 139, 3170–3186 (2016).
Tian, Y. et al. Clinical features of NOTCH2NLC-related neuronal intranuclear inclusion disease. J. Neurol. Neurosurg. Psychiatry 93, 1289–1298 (2022).
Liu, Y. H. et al. Neuronal intranuclear inclusion disease in patients with adult-onset non-vascular leukoencephalopathy. Brain 145, 3010–3021 (2022).
Tai, H. et al. Clinical features and classification of neuronal intranuclear inclusion disease. Neurol. Genet 9, e200057 (2023).
Chen, H. et al. Re-defining the clinicopathological spectrum of neuronal intranuclear inclusion disease. Ann. Clin. Transl. Neurol. 7, 1930–1941 (2020).
Huang, X. R., Tang, B. S., Jin, P. & Guo, J. F. The phenotypes and mechanisms of NOTCH2NLC-related GGC repeat expansion disorders: a comprehensive review. Mol. Neurobiol. 59, 523–534 (2022).
Liu, Y. et al. Clinical and mechanism advances of neuronal intranuclear inclusion disease. Front. Aging Neurosci. 14, 934725 (2022).
Tian, Y. et al. Expansion of human-specific GGC repeat in neuronal intranuclear inclusion disease-related disorders. Am. J. Hum. Genet. 105, 166–176 (2019).
Ishiura, H. et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 51, 1222–1232 (2019).
Deng, J. et al. Long-read sequencing identified repeat expansions in the 5’UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J. Med. Genet. 56, 758–764 (2019).
Sun, Q. Y. et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain 143, 222–233 (2019).
Yuan, Y. et al. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology 95, e3394–e3405 (2020).
Shi, C. H. et al. NOTCH2NLC intermediate-length repeat expansions are associated with Parkinson disease. Ann. Neurol. 89, 182–187 (2021).
Yan, Y. et al. Assessing the NOTCH2NLC GGC expansion in essential tremor patients from eastern China. Brain 144, e1–e1 (2021).
Okubo, M. et al. GGC repeat expansion of NOTCH2NLC in adult patients with leukoencephalopathy. Ann. Neurol. 86, 962–968 (2019).
Fang, P. et al. Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann. Clin. Transl. Neurol. 7, 517–526 (2020).
Jiao, B. et al. Identification of expanded repeats in NOTCH2NLC in neurodegenerative dementias. Neurobiol. Aging 89, 142.e1–142.e7 (2020).
Yu, J. et al. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain 144, 1819–1832 (2021).
Zeng, T. et al. Neuronal intranuclear inclusion disease with NOTCH2NLC GGC repeat expansion: a systematic review and challenges of phenotypic characterization. Aging Dis. 16, 578–597 (2024).
Ehrlich, M. E. & Ellerby, L. M. Neuronal intranuclear inclusion disease: polyglycine protein is the culprit. Neuron 109, 1757–1760 (2021).
Boivin, M. et al. Translation of GGC repeat expansions into a toxic polyglycine protein in NIID defines a novel class of human genetic disorders: the polyG diseases. Neuron 109, 1825–1835.e5 (2021).
Zhong, S. et al. Upstream open reading frame with NOTCH2NLC GGC expansion generates polyglycine aggregates and disrupts nucleocytoplasmic transport: implications for polyglycine diseases. Acta Neuropathol. 142, 1003–1023 (2021).
Yu, J. et al. CGG repeat expansion in NOTCH2NLC causes mitochondrial dysfunction and progressive neurodegeneration in Drosophila model. Proc. Natl. Acad. Sci. USA 119, e2208649119 (2022).
Fan, Y. et al. GGC repeat expansion in NOTCH2NLC induces dysfunction in ribosome biogenesis and translation. Brain 146, 3373–3391 (2023).
Liu, Q. et al. Expression of expanded GGC repeats within NOTCH2NLC causes behavioral deficits and neurodegeneration in a mouse model of neuronal intranuclear inclusion disease. Sci. Adv. 8, eadd6391 (2022).
Pan, Y. et al. Expression of expanded GGC repeats within NOTCH2NLC causes cardiac dysfunction in mouse models. Cell Biosci. 13, 157 (2023).
Zhong, S. et al. Microglia contribute to polyG-dependent neurodegeneration in neuronal intranuclear inclusion disease. Acta Neuropathol. 148, 21 (2024).
Fiddes, I. T. et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173, 1356 (2018).
Suzuki, I. K. et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through Delta/Notch regulation. Cell 173, 1370 (2018).
Florio, M. et al. Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex. Elife 7, e32332 (2018).
Yang, S. et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 127, 2719–2724 (2017).
Yan, S. et al. Cas9-mediated replacement of expanded CAG repeats in a pig model of Huntington’s disease. Nat. Biomed. Eng. 7, 629–646 (2023).
Chan, K. Y. et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179 (2017).
Sun, Q. Y. et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain 143, 222–233 (2020).
Ng, A. S. L. et al. NOTCH2NLC GGC repeat expansions are associated with sporadic essential tremor: variable disease expressivity on long-term follow-up. Ann. Neurol. 88, 614–618 (2020).
Yan, Y. et al. Assessing the NOTCH2NLC GGC expansion in essential tremor patients from eastern China. Brain 144, e1 (2021).
Zhou, X. et al. Clinical features and reclassification of essential tremor with NOTCH2NLC GGC repeat expansions based on a long-term follow-up. Eur. J. Neurol. 29, 3600–3610 (2022).
Zhang, W. et al. GGC repeat expansions in NOTCH2NLC causing a phenotype of lower motor neuron syndrome. J. Neurol. 269, 4469–4477 (2022).
Wan, M. et al. Intermediate-length GGC repeat expansion in NOTCH2NLC was identified in chinese patients with amyotrophic lateral sclerosis. Brain Sci. 13, 85 (2023).
Wang, Y. C. et al. NOTCH2NLC expanded GGC repeats in patients with cerebral small vessel disease. Stroke Vasc. Neurol. 8, 161–168 (2022).
Liao, Y. C. et al. NOTCH2NLC GGC repeat expansion in patients with vascular leukoencephalopathy. Stroke 54, 1236–1245 (2023).
Wu, C. et al. The genetic and phenotypic spectra of adult genetic leukoencephalopathies in a cohort of 309 patients. Brain 146, 2364–2376 (2022).
Wu, W. et al. Intermediate-length CGG repeat expansion in NOTCH2NLC is associated with pathologically confirmed Alzheimer’s disease. Neurobiol. Aging 120, 189–195 (2022).
Ma, D. et al. Association of NOTCH2NLC repeat expansions with Parkinson disease. JAMA Neurol. 77, 1559–1563 (2020).
Wang, H. et al. GGC repeat expansion in the NOTCH2NLC gene is associated with a phenotype of predominant motor-sensory and autonomic neuropathy. Front. Genet. 12, 694790 (2021).
Liao, Y. C. et al. GGC repeat expansion of NOTCH2NLC in Taiwanese patients with inherited neuropathies. Neurology 98, e199–e206 (2022).
Ogasawara, M. et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol. Commun. 8, 204 (2020).
Cao, L., Yan, Y. & Zhao, G. NOTCH2NLC-related repeat expansion disorders: an expanding group of neurodegenerative disorders. Neurol. Sci. 42, 4055–4062 (2021).
Fan, Y., Xu, Y. & Shi, C. NOTCH2NLC-related disorders: the widening spectrum and genotype-phenotype correlation. J. Med. Genet. 59, 1–9 (2022).
Pan, Y. et al. HnRNP M expression rescues neurodegeneration in neuronal intranuclear inclusion disease mouse model by restoring dysregulated RNA splicing and transcription. Cell Biosci. 15, 134 (2025).
Hagerman, R. J. et al. Intention tremor, Parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130 (2001).
Gu, Y., Shen, Y., Gibbs, R. A. & Nelson, D. L. Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat. Genet. 13, 109–113 (1996).
Gecz, J., Gedeon, A. K., Sutherland, G. R. & Mulley, J. C. Identification of the gene FMR2, associated with FRAXE mental retardation. Nat. Genet. 13, 105–108 (1996).
Fan, Y. et al. GIPC1 CGG repeat expansion is associated with movement disorders. Ann. Neurol. 91, 704–715 (2022).
Pan, Y. et al. Assessment of GGC repeat expansion in GIPC1 in patients with Parkinson’s disease. Mov. Disord. 37, 1557–1559 (2022).
Brais, B. et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 18, 164–167 (1998).
Ishiura, H. Recent progress in oculopharyngodistal myopathy research from clinical and genetic viewpoints. J. Neuromuscul. Dis. 12, 303–311 (2025).
Park, C. Y. et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 13, 234–241 (2015).
Xie, N. et al. Reactivation of FMR1 by CRISPR/Cas9-mediated deletion of the expanded CGG-repeat of the fragile X chromosome. PLoS ONE 11, e0165499 (2016).
Wang, X. H. et al. Modeling neuronal intranuclear inclusion disease: a review of animal and human-derived cellular models and mechanistic insights. Zool. Res. 46, 1565–1574 (2025).
Yin, P., Li, S., Li, X. J. & Yang, W. New pathogenic insights from large animal models of neurodegenerative diseases. Protein Cell 13, 707–720 (2022).
Malinin, N. L. et al. Defining genome-wide CRISPR-Cas genome-editing nuclease activity with GUIDE-seq. Nat. Protoc. 16, 5592–5615 (2021).
Liu, Q. et al. Cerebellum-enriched protein INPP5A contributes to selective neuropathology in mouse model of spinocerebellar ataxias type 17. Nat. Commun. 11, 1101 (2020).
Yardeni, T., Eckhaus, M., Morris, H. D., Huizing, M. & Hoogstraten-Miller, S. Retro-orbital injections in mice. Lab Anim. (NY) 40, 155–160 (2011).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Xu, K. et al. Detecting anomalous anatomic regions in spatial transcriptomics with STANDS. Nat. Commun. 15, 8223 (2024).
Deng, T. et al. LEGEND: Identifying co-expressed genes in multimodal transcriptomic sequencing data. Genom. Proteom. Bioinform. 23, qzaf056 (2025).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Acknowledgements
The authors would like to thank Peng Jin and Yujing Li from School of Medicine, Emory University; Hao Wu from School of Computer Science and Control Engineering, Shenzhen University of Advanced Technology; Su Yang from Guangdong-Hongkong-Macau Institute of CNS Regeneration, Jinan University; Ying Cheng from Institute of biomedical research, Yunnan University for providing valuable advice to the study. This study was supported by the National Natural Science Foundation of China (82394421 and 82394420 to B.T., 82394422 and 82371874 to X.-J.L., 82271902 and U24A6013 to S.L., 32071037 to Q.L., 82171843 to Y.P., 82101946 to N.X., 82171256 to Q.S.), the National Key R&D Program of China (2021YFA0805200 to H.J.), and the Natural Science Foundation of Hunan Province (2023JJ10097 to Q.L., 2025JJ20079 to Y.P., 2022JJ40832 to N.X.).
Author information
Authors and Affiliations
Contributions
Q.L., Y.P., S.L., and X.-J.L. designed the study, supervised the study, and revised the manuscript; N.X., Y.P., and Q.L. performed experiments, analyzed data, and wrote the manuscript; H.T., Y.L., Y.J., Z.W., J.W., W.Z., X.W., X.S., S.Y., P.Y., Q.S., C.Q., and Y.T. provided important technical assistance to the study; L.S., H.J., D.L., and B.T. provided insightful advice to the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hye Young Lee, who co-reviewed with Seo-Jun Kang; the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, N., Pan, Y., Tong, H. et al. Precise excision of expanded GGC repeats in NOTCH2NLC via CRISPR/Cas9 for treating neuronal intranuclear inclusion disease. Nat Commun 17, 1683 (2026). https://doi.org/10.1038/s41467-026-68385-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68385-5
