
Abstract
The simultaneous utilization of methane and carbon dioxide via dry reforming holds promise for sustainable syngas production, yet conventional thermocatalytic processes suffer from energy-intensive operation and catalyst deactivation. Here, we report a light-driven methane dry reforming strategy utilizing sinter-resistant nano-island alloys catalyst, which are dynamically evolved from partially oxidized NiIr nanoclusters anchored on TiO2 under photoexcitation. In situ characterization reveals interfacial charge oscillations on the catalyst induce a support-Ni-Ir electron transfer pathway, stabilizing oxidized Ni linkages while electronically modulating surface Ir sites. This dual functionality promotes CHxO* intermediate formation, suppressing coking during 100-h operation under intermittent illumination. By decoupling photoelectric and photothermal contributions, we demonstrate that localized photogenerated electrons dominate balanced syngas production, whereas photothermal effects enhance molecular vibrations. The optimized catalyst achieves a syngas rate of 10841 mmol gcat-1 h-1 with 25.0% light-to-fuel efficiency, establishing a design paradigm for solar-driven alloy catalysts in greenhouse gas valorization.
Similar content being viewed by others

Introduction
Mitigating greenhouse gas emissions while securing sustainable routes for chemical energy conversion remains a central challenge in the transition toward a low-carbon future. Dry reforming of methane (DRM), which simultaneously converts methane (CH4) and carbon dioxide (CO2) into syngas (CO and H2) as valuable chemical feedstocks, offers a promising pathway for greenhouse gas utilization1,2. Yet, the highly endothermic nature of DRM (CH4 (g) + CO2 (g) → 2CO (g) + 2H2 (g), ΔHo298K = +247.3 kJ mol-1) necessitates harsh operating conditions (typically 700–1000 °C) in conventional thermocatalysis. Such high temperatures consume substantial fossil fuels, undermining environmental benefits of CO2 utilization3,4,5. In addition, the reverse water-gas shift (RWGS) reaction (CO2 (g) + H2 (g) → CO (g) + H2O (g), ΔHo298K = +41.2 kJ mol-1), being thermodynamically favored under the high-temperature conditions, compromises syngas selectivity by lowering the H2/CO ratio6. Solar light-driven catalysis has emerged as an energy-efficient alternative by synergistically combining photoelectric effects and photothermal heating to promote the dissociation of CH4 and CO2 without external heating7,8,9.
In materials terms, noble metals as active sites display high intrinsic activity toward CH4 and CO2 activation but suffer from low atom efficiency and over-strong intermediate binding, often leading to side reactions10. Nickel-based catalysts are more economically viable and exhibit notable CH4 activation capacity, which are prone to deactivation via carbon deposition11. Alloying noble metals with Ni modulates the electronic structure to optimize the balance between reactant activation and intermediate adsorption, thereby suppressing side reactions and improving noble metal utilization efficiency. Moreover, ultrafine nanoclusters (<2 nm) could further enhance catalytic efficiency through maximized surface exposure and metal utilization12,13. However, stabilizing such clusters against aggregation under reaction conditions remains challenging. Recent studies14,15,16 have demonstrated that integrating oxide nano-islands at metal-support interfaces can effectively suppress sintering through strong charge-mediated interactions. For instance, anchoring Pt14 or Pd16 on CeOx nano-islands has been shown to spatially confine metal mobility. However, extending this nano-island strategy to ultrafine alloy architectures requiring both interfacial stabilization and electronic tuning has yet to be reported.
In addition, catalyst structural stability at the atomic scale under intense light irradiation and elevated temperatures, which are conditions inherent to photothermal catalysis, remains challenging for elucidating catalytic mechanisms. Whether such conditions preserve the active phase or drive transformations from metastable intermediates to thermodynamically favored structures is still poorly understood. Beyond thermal vibrations that commonly induce surface reconstruction at high temperatures, photoexcitation can trigger substantial charge redistribution between the support and active site17,18,19,20. This photoinduced charge transfer may alter the electronic structure of surface chemical bonds, reducing bond energies and promoting dynamic surface reconstruction21,22. Despite growing interest, elucidating the nature and formation of the active phase under photothermal conditions as well as disentangling the respective roles of photoelectric and photothermal effects in light-driven DRM, remains a critical yet unresolved challenge, limiting progress toward rational catalyst design.
In this work, we demonstrate that photoinduced interfacial charge oscillations in NiIr/TiO2 trigger dynamic structural reconstruction into TiO2-supported NiIr nano-island alloys stabilized by oxidized Ni linkages (NiIr/TiO2-LNi). This structural evolution arises from accelerated electron transfer across the support-Ni-Ir interface under light excitation. The spatially and electronically synergistic configuration promotes CHxO* intermediate formation, effectively mitigates carbon deposition, and maintains stable performance for 100 h under intermittent illumination. Mechanistic investigations reveal that photoelectric effects dominate the reaction, contributing 57.5% of the syngas production rate and 91.7% of the H2/CO ratio by facilitating C-H/C = O scission and H-H coupling, while photothermal effects enhance molecular vibrations and reactant diffusion. Under full feed gas flow, NiIr/TiO2-LNi achieves a syngas production rate of 10841 mmol gcat-1 h-1, with a light-to-fuel efficiency (LTFE) of 25.0% and a turnover frequency (TOF) of 23 s-1.
Results
Synthesis and atomic-scale structural analysis of partially oxidized NiIr nanoclusters on TiO2
To construct bimetallic NiIr nanoclusters on TiO2 with precise atomic pairing, we adopted a directional adsorption strategy23 (Fig. 1a; see Methods for synthesis details). This approach leverages sequential electrostatic adsorption of cationic Ni complexes and anionic Ir precursors onto alkalized TiO2 nanosheets, ensuring preferential Ir anchoring onto pre-adsorbed Ni sites. For the Ir precursor, we used a species complexed with quaternary ammonium cations to enable its phase transfer into an aprotic solvent. This controlled process prevents Ir ripening and leaching. Consequently, by conducting adsorption in this manner, we minimized competitive adsorption of Ni and Ir, effectively overcoming a key limitation of conventional co-impregnation methods. Subsequent H2 calcination eliminated organic ligands while strengthening Ni-Ir metallic interactions, yielding ultrafine bimetallic clusters with uniform dispersion on the TiO2 support (Fig. 1b). Inductively coupled plasma-atomic emission spectrometry (ICP-AES) confirms controlled metal loadings across NiIrx/TiO2 samples (Supplementary Fig. 1 and Table S1), where x denotes the Ir/Ni theoretical atomic ratio. The sublinear increase in Ir/Ni ratios with x suggests saturation of Ir adsorption onto pre-adsorbed Ni sites, excluding independent Ir deposition. For comparison, monometallic Ni/TiO2 and Ir/TiO2 were prepared with a theoretical loading of 1 wt% for each metal. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis revealed that Ni species or Ir species on TiO2 exhibited sub-1.2 nm clusters (Supplementary Figs. 2–3), whereas the NiIrx nanoclusters on TiO2 showed an increase in size, progressively reaching up to 1.61 nm with higher Ir content (Supplementary Figs. 4). This trend supports the co-stabilization of Ni and Ir atoms within the bimetallic clusters. This size evolution directly supports the formation of bimetallic NiIr nanoclusters, as the incorporation of Ir stabilizes the clusters and promotes their growth. Atomic-scale characterization of NiIr0.5/TiO2 provided further insights into the spatial distribution of Ni and Ir atoms. Filtered and color-coded HAADF-STEM images and the corresponding intensity profiles (Fig. 1c, d, Supplementary Fig. 5b, c) revealed intertwined Ni-Ir distributions within individual clusters. The 3D surface plot of a selected region (Fig. 1f) highlighted Ir atoms (whitish-yellow peaks within blue dashed marks) surrounding Ni atoms (darker yellow bumps), demonstrating cohesive bimetallic nanocluster formation. Conversely, monometallic samples exhibited only metal-TiO2 contrast (Supplementary Figs. 2g-h, 3e-f). Moreover, EDS line scans (Fig.1g) and elemental mapping (Fig. 1h and Supplementary Fig. 5d-i) confirmed the intimate contact between Ni and Ir atoms. Owing to the higher atomic ratio of Ni to Ir, trace amounts of isolated Ni species inevitably persist. Crucially, all Ir atoms preferentially occupy Ni-associated sites, with no detectable isolated Ir single atoms observed on the TiO2 support (Fig. 1b).

a Schematic of the directional adsorption strategy for synthesizing NiIr nanoclusters supported on TiO2. b Aberration-corrected HAADF-STEM image of NiIr0.5/TiO2 and cluster size distribution (inset). c Color-coded atomic map of region 1 (marked by the white dashed box in b after image filtering. d Intensity profiles along the white line in c. e Aberration-corrected HAADF-STEM image of NiIr nanoclusters on TiO2. f 3D surface plot with atomic overlapping region 3 in e. Blue dashed circles indicate the Ir atoms in NiIr nanocluster. Inset: Filtered image of region 3. g EDS line-scan profiles of Ni and Ir across a cluster along the yellow arrow in e. h HAADF-STEM image and corresponding EDS elemental mapping of NiIr0.5/TiO2. i k2-weighted Fourier transform of Ir L3-edge EXAFS spectra (without phase correction) for Ir foil, IrO2, NiIrx/TiO2, and Ir/TiO2. j In situ CO-probe DRIFTS spectra of NiIrx/TiO2, Ni/TiO2, and Ir/TiO2 at the 15th min of Ar-flow during desorption (25 °C); schematic models of surface structures are included in j.
Next, we elucidate the chemical structure and coordination environment of the NiIr nanoclusters. X-ray diffraction (XRD) patterns confirm exclusive anatase-phase TiO2 (Supplementary Fig. 6), with no detectable reflections from metallic Ni or Ir phases, consistent with HAADF-STEM evidence of dispersed sub-2 nm clusters. Normalized Ir L3-edge and Ni K-edge X-ray absorption near-edge spectroscopy (XANES) analysis reveal partially oxidized states of Ni and Ir (Supplementary Fig. 7a, b). Linear combination fitting (Supplementary Fig. 7c-d) shows higher Ir oxidation in Ir/TiO2 (+3.6) than NiIr0.5/TiO2 (+2.7), and more reduced Ni in Ni/TiO2 (+1.6) versus NiIr0.5/TiO2 (+1.8), indicating electron transfer from Ni to Ir, later corroborated by in situ analyses. Extended X-ray absorption fine structure (EXAFS) at the Ir L3-edge provides atomic-scale coordination details (Fig. 1i). All samples exhibit a prominent peak at ~1.60 Å and a relatively weaker peak at ~2.56 Å, corresponding to Ir-O scattering and Ir-Ir scattering, respectively. Notably, NiIrx/TiO2 samples exhibit an additional peak at 2.18 Å, which can be attributed to Ni-Ir scattering. These results demonstrate that NiIr nanoclusters exhibit a partially oxidized state, with electronic modulation between Ni and Ir atoms. The partial oxidation probably arises from the interface reconstruction between nanocluster and the surface lattice O atoms of TiO2 during the calcination process and the exposure to air during storage. This electronic structure, coupled with their ultrafine size, positions these partially oxidized NiIr nanoclusters (Ox-NiIr) as promising candidates for the adsorption of CH4 and CO2 due to their inherent thermal instability19.
In situ CO probe molecule diffuse reflectance infrared fourier transform spectroscopy (DRIFTS) analyses were conducted to unravel the surface structure and adsorption behavior of NiIr nanoclusters (Supplementary Fig. 8 and Fig. 1j). Ni/TiO2 exhibited negligible *CO chemisorption signals (<2100 cm-1), which vanished entirely upon heating to 100 °C. This confirms the inadequacy of individual Ni sites for CO adsorption. In contrast, Ir-containing samples (NiIrx/TiO2, Ir/TiO2) display intense *CO chemisorption bands (2100–1990 cm-1), with adsorption strength scaling with Ir content. Heating-up desorption revealed a volcanic trend: *CO intensity peaked at 200 °C and declined continuously to 400 °C. It demonstrates reversible CO binding, which is a critical feature for sustaining DRM activity. Detailed comparison of the desorption spectra at 25 °C (Fig. 1j) highlights distinct Ir coordination environments. For Ir/TiO2, three CO species were resolved: (i) multi-carbonyl Ir-(CO)2 (2068 cm-1, low-coordination sites)24, (ii) linear CO on metallic Ir (Ir-CO, 2033 cm-1)25, and (iii) bridged CO at Ir-TiO2 interfaces (Irif-CO, 1994 cm-1)26. For Ni/TiO2, only weak peaks of Ni-CO and Niif-CO can be observed. In NiIrx/TiO2, the dominant Ir-(CO)2 peak progressively shifts downward (2063 → 2044 cm-1) with increasing Ir content, signaling enhanced π-backdonation from Ir to CO. The observed redshift arises from Ni-to-Ir electron transfer, which elevates Ir’s d-orbital occupancy, strengthens Ir-C bonds, and weakens C-O bonds. These results confirm that in partially oxidized NiIr nanoclusters, Ir serves as the main center for CO adsorption, while Ni modulates Ir’s electronic properties to optimize the balance between adsorption strength and desorption kinetics.
Photoelectric-photothermal synergy in light-driven dry reforming of methane over NiIr/TiO2
Light-driven DRM over TiO2-supported NiIr nanoclusters was investigated under full-spectrum irradiation (Supplementary Fig. 9). Ni and Ir modification significantly enhanced DRM activity compared to pure TiO2 (Fig. 2a, Supplementary Fig. 10). NiIr0.5/TiO2 achieved optimal performance (Fig. 2a, Supplementary Fig. 11 and Note 1) with syngas generation rates of 2512 mmol gcat-1 h-1 and feed gas conversions near 60% at the condition of CH4: CO2: Ar =1: 1: 8. Increasing the Ir/Ni ratio (x) from 0.05 to 1.0 elevated H2/CO from 0.68 to 1.03, demonstrating Ir-Ni synergy in balancing CH4-CO2 activation (Fig. 2a). NiIr0.15/TiO2 (equivalent Ir loading to 0.5% Ir/TiO2) exhibited 2.13-fold higher activity (CH4 conversion: 17% → 42%), underscoring the critical role of bimetallic interactions. Highly diluted conditions (CH4: CO2: Ar =1: 1: 98) achieved 90% conversion (Fig. 2b). Strikingly, at undiluted feed gas, the system achieved syngas generation rates of 10841 mmol gcat-1 h-1, with a LTFE of 25.0% and a TOF of 23 s-1. However, for gas conservation and safety considerations, subsequent tests were conducted using a feed gas mixture of CH4: CO2: Ar =1: 1: 8. Light irradiation dramatically reduced the apparent activation energies for CO and H2 production to 35.33 and 44.65 kJ mol-1, respectively, which is less than half those of thermal catalysis (68.76 and 99.19 kJ mol-1), effectively breaking the limitation of thermal catalysis (Fig. 2c, Supplementary Fig. 12).

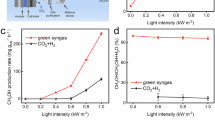
a Syngas generation rates and H2/CO ratios of TiO2, Ni/TiO2, Ir/TiO2, and NiIrx/TiO2 under 3.89 W cm-2 light irradiation (feed gas: CH4: CO2: Ar =1: 1: 8). Note: the same * or # marks indicate that the actual Ir loading amounts of the two samples are similar. b Feed-gas composition dependence of syngas generation rates, H2/CO ratios, and conversion rates for NiIr0.5/TiO2 (light intensity: 3.89 W cm-2). c Syngas generation rates of NiIr0.5/TiO2 under light irradiation or external heating. (CH4:CO2:Ar =1:1:8, the heating temperature is near to the detected light irradiation temperature at catalyst center.) d DRM activity per-unit light intensity, detected temperatures, and conversion rates of NiIr0.5/TiO2 under varied light intensities (CH4:CO2:Ar =1:1:8). Note that the TAVG and TCenter indicate the average and the center temperature of catalyst surface measured by a thermocouple, which agree with the results from the IR camera (Supplementary Fig. 13). e Syngas generation rates and H2/CO ratio of NiIr0.5/TiO2 with (blue lines) or without (yellow lines) condensation equipment. (CH4: CO2: Ar =1: 1: 8, condensate temperatures noted in blue font) Note that the PT and PE represent photothermal contribution and photoelectric contribution, respectively. f UV-Vis-NIR spectrum and wavelength-dependent DRM activity (per W cm-2) of NiIr0.5/TiO2 under monochromatic light (with 500 °C external heating, CH4: CO2: Ar =1: 1: 8). g 100-hour stability of NiIr0.5/TiO2 under light on/off cycles (Light on: 3.89 W cm-2, CH4: CO2: Ar =1: 1: 8; Light off: exposed to air). Reaction conditions for a-g: 5 mg catalyst, flow rate = 20 ml min-1; The error bars indicate that there were at least three independent tests.
Activity surged above a threshold light intensity of 2 W cm-2, marking the onset of photoelectric-photothermal synergy (Fig. 2d and Supplementary Fig. 13). Specifically, at lower intensities (<1.95 W cm-2, TCenter < 336 °C), the observed activity originated predominantly from photoelectric effects. This assignment is confirmed by the sustained syngas production under illumination, which stands in clear contrast to the negligible activity observed in dark thermal catalysis experiments conducted below the 350 °C threshold (as shown in Fig. 2c and Supplementary Fig. 12a). The interplay between photothermal and photoelectric contributions peaked at 3.89 W cm-2 (TCenter = 506 °C; TAVG = 403 °C). Beyond 4.27 W cm-2, the system approached a state of photon absorption saturation. At this point, the catalyst could no longer utilize the additional photons for charge carrier generation, and consequently, the excess energy was redirected into thermal pathways. This mechanistic shift is evidenced by a distinct decline in the per-unit-light efficiency (syngas generation rates/light intensity), which occurred despite the continued rise in the catalyst temperature (TCenter exceeding 600 °C). This nonlinear response highlights the delicate balance between photocarriers generation and thermal dissipation in light-driven systems.
Controlled condensation experiments (Fig. 2e) decoupled photothermal and photoelectric effects by selectively suppressing thermal contributions. The blue trend line maintained constant light intensity (3.89 W cm-2) to preserve the photoelectric effect and used condensation cooling to suppress photothermal contributions, while the yellow trend line varied light intensity (from 3.89 to 1.30 cm-2) without condensation and diminished both effects simultaneously. By comparing the syngas production rates and H2/CO ratios from these two paths at matched catalyst surface temperatures, it was determined that the photoelectric-dominated system (with condensation) contributed 57.5% to syngas rates and 91.7% to H2/CO balance, compared to the photothermal-assisted system (without condensation). This contrast confirms that photogenerated carriers preferentially drive H-H coupling and carbon intermediate oxidation, while thermal energy primarily enhances vibration and mobility of reactants. Comparative tests on NiIr0.5/TiO2 and NiIr0.5/SiO2 (Supplementary Fig. 14 and Note 2) further confirmed that TiO2-supported NiIr nanoclusters uniquely harness coupled photothermal-photoelectric effects to achieve efficient and stable DRM catalysis.
The UV-Vis-NIR spectra (Supplementary Fig. 15) revealed that the introduction of Ni and Ir significantly enhanced broadband light absorption across the entire spectral range compared to TiO2. Crucially, the absorption profile across the full spectral range of NiIr0.5/TiO2 exhibited a strong correlation with the wavelength-dependent DRM activity (Fig. 2f), confirming that the reaction is primarily driven by photoexcitation rather than thermal effects and evidencing its light-driven nature. Long-term stability tests (Supplementary Fig. 16 and Note 3) demonstrated considerable durability as NiIr0.5/TiO2 retained >97% activity over 720 min under light, with near-unity carbon balance. In contrast, thermal catalysis at 500 °C suffered a 20% activity drop and significant carbon deposition, emphasizing light’s role in suppressing deactivation. Moreover, NiIr/TiO2 sustained 100-hour stability under 20% reactant flow during five light/dark cycles (Fig. 2g), retaining >80% syngas generation rates with H2/CO > 0.9 throughout the test. Monometallic controls (Supplementary Fig. 17) further validated Ni-Ir synergy that Ni/TiO2 rapidly deactivated due to CHx accumulation, while Ir/TiO2 maintained 70% stability over 720 min but exhibited suppressed H2/CO ratios, probably due to RWGS side reaction.
These results reveal that UV-Vis- induced hot electrons at the NiIr-TiO2 interface, consistent with ultrafast (<10 fs) UV photoinduced electrons in TiO227, collaboratively cleave C-H/C = O bonds and mediate H-H coupling. Meanwhile, infrared absorption enhances local temperatures. This photoelectric-photothermal synergy suppresses carbon deposition and stabilizes active sites, achieving DRM efficiency in photothermal and even certain thermal systems (Supplementary Table 3)7,8,9,28,29,30,31,32,33,34,35,36,37,38,39,40.
Light-induced dynamic restructuring to form NiIr nano-island alloy on TiO2
Alloy structural evolution under operando light conditions is critical to unraveling true catalytic mechanisms, which was therefore systematically investigated. In situ X-ray absorption spectroscopy (XAS) and near-ambient pressure XPS (NAP-XPS) analyses revealed the light-induced structural reorganization of NiIr nanoclusters. Under light irradiation, Ir L3-edge EXAFS spectra of NiIr0.5/TiO2 (Fig. 3a) show a dominant Ir-Ni scattering peak at 2.18 Å, replacing the initial Ir-O coordination observed in the dark. Concurrently, Ni K-edge EXAFS of NiIr0.5/TiO2 (Fig. 3b) retains Ni-O (1.60 Å) and Ni-Ni (2.09 Å) scattering alongside emerging Ni-Ir (2.18 Å) coordination, indicating alloy formation without complete Ni reduction. In contrast, monometallic Ni/TiO2 under identical conditions transitions fully to metallic Ni-Ni bonds, while Ir/TiO2 retains residual Ir-O coordination (Supplementary Fig. 18). This indicates that during the dynamic alloy formation under light, electrons are transferred from Ni to Ir. Quantitative EXAFS fitting analysis (Fig. 3c, Supplementary Fig. 19–20, Supplementary Tables 4–5) quantifies this structural evolution. Under light irradiation, NiIr0.5/TiO2 exhibits nearly 100% Ir-Ni coordination at the Ir L3-edge and 32% Ir-Ni coordination at the Ni K-edge, alongside 37% Ni-O interfacial bonds. This contrasts sharply with monometallic Ni/TiO2. The nearly 100% Ni-Ni coordination in Ni/TiO2 likely drives its rapid deactivation (Supplementary Fig. 18), as non-oxidized Ni sites exhibit diminished ability to activate CH4 and oxidize carbonaceous intermediates, leading to CHx accumulation and irreversible poisoning. The absence of Ir-O coordination in NiIr0.5/TiO2 under light confirms that Ir in the alloy phase does not directly interact with TiO2, while Ni maintains interfacial Ni-O bonds, forming a nano-island architecture that isolates the NiIr alloy from the support (Supplementary Fig. 21). This structure formation is critical for efficient and stable DRM catalysis.

a, b In situ k2-weighted Fourier transform of Ir L3-edge a and Ni K-edge b EXAFS spectra for NiIr0.5/TiO2 during light-driven DRM conditions (CH4: CO2: Ar =1: 1: 8, 3.89 W cm-2; data are not phase-corrected). c Normalized coordination numbers (CN) of different samples from EXAFS fitting. d In situ NAP-XPS spectra of Ir 4 f for NiIr0.5/TiO2 under different irradiation conditions in a gas flow of CH4: CO2: Ar =1: 1: 8 (Unfocused light: 1.58 W cm-2; focused light: 3.89 W cm-2). e Ir0/Irδ+ ratio evolution of NiIr0.5/TiO2 during in situ NAP-XPS conditions and post-air exposure, derived from deconvolution of the XPS spectra. f Schematics of light induced oscillatory of NiIr/TiO2-LNi formation. g DFT-optimized structures of NiIr/TiO2-LNi and NiIr/TiO2-LIr. The key distances between Ni/Ir linkages and TiO2 are annotated in black font. Ir, Ni, Ti, and O atoms are shown in pink, yellow, grey, and blue colors.
In situ NAP-XPS analyses corroborate the XAS findings. To decouple photochemical and photothermal contributions, we first monitored structural evolution under unfocused light. The Ir 4 f spectra (Fig. 3d) reveal a pronounced shift toward lower binding energy, with the Ir0 fraction increasing from 36.96% (dark) to 60.65%. Upon switching to focused light, the Ir0 fraction further rises to 62.31%. Figure 3e quantifies this trend: the Ir0/Irδ⁺ ratio increases by 0.95 under unfocused light, with only an additional 0.11 gain under focused irradiation, demonstrating that low-intensity light suffices to initiate alloying. Monometallic Ir/TiO2 exhibits a smaller Ir0/Irδ⁺ increase of 0.61 under unfocused light (Supplementary Fig. 22), aligning with its higher oxidation state observed in in situ XAS. Moreover, air exposure restores the initial oxidized state (Fig. 3e, Supplementary Figs. 22–23), confirming the reversibility of the NiIr-LNi phase. Parallel in situ Ni 2p NAP-XPS (Supplementary Fig. 24a-c) shows analogous trends, with Ni0/Ni2+ ratios tracking Ir’s redox dynamics. While Ti 2p and O 1 s exhibit the positive shift in the binding energy of Ti and OLattice under light irradiation (Supplementary Fig. 24d-e), this demonstrates that electrons migrate from the TiO2 support to Ni-Ir under illumination. Post-reaction characterizations (Supplementary Figs. 25–26) reveal that prolonged light-driven DRM preserves Ni-Ir intimacy. Re-oxidation in post-EXAFS (Supplementary Fig. 27 and Note 4) and reversible NAP-XPS trends confirm that NiIr/TiO2-LNi exists transiently only under light irradiation. Thermal catalysis induces limited Ni or Ir reduction (Ir0/Irδ⁺=0.61 at 500 °C vs. 1.54 under unfocused light) and fails to form obvious Ni-Ir bonds (Supplementary Figs. 28–29), resulting in poor DRM performance. Conversely, light irradiation in pure Ar still forms dominant Ni-Ir scattering (Supplementary Figs. 30–31 and Note 5), proving that reactant gases are non-essential for NiIr alloying. Moreover, light cessation triggers rapid re-oxidation, with full structural recovery after air exposure.
Figure 3f illustrates the light-induced structural dynamics in NiIr/TiO2. Initial Ox-NiIr/TiO2 adsorbs CH4/CO2 but remains inefficient and prone to coking. Under light irradiation, interfacial oscillations drive electrons from Ni to Ir, and Ir atoms migrate away from the interface, forming NiIr island alloy anchored via oxidized Ni linkages on TiO2. The Ir-Ni binary phase diagram (Supplementary Fig. 32) and positive segregation energy (0.23 eV)41 confirm alloy feasibility under photoexcitation. We further constructed DFT structures (Supplementary Fig. 33) for a series of samples based on the above analysis and actual Ni/Ir atomic ratios. Figure 3g confirms NiIr/TiO2-LNi’s stability with a lower formation energy than the hypothetical Ir-linked model (NiIr/TiO2-LIr), where Ir-O bonds elongate (black arrow indication, >3.0 Å), destabilizing the interface. These results demonstrate that light excitation dynamically restructures Ox-NiIr/TiO2 into NiIr/TiO2-LNi. The reversibility of NiIr/TiO2-LNi under ambient conditions highlights its transient, light-governed property, serving as a suitable model for further investigation of photoelectric-dominated reaction mechanisms.
It is worth elucidating that the light-induced surface restructuring observed in this study is supported by prior findings regarding bimetallic systems. It is established that electron transfer in bimetallic clusters can drive surface segregation of one of the metals42,43. A pertinent mechanism involves atomic size, in which the introduction of a larger-radius atom (e.g., Sn in PtCu) can promote surface extraction and alloy reorganization21,44. In our system, Ir has a larger inherent atomic radius than Ni. Critically, the light-induced electron transfer from Ni to Ir reduces Ir’s oxidation state, further exacerbating this radius disparity (Ir0 > Irδ⁺ > Niδ⁺) and thereby strongly promoting the surface exposure of Ir. This process is further enabled by the photo-weakening of metal-support bonds, a phenomenon known to induce structural dynamics under illumination22,44,45. To confirm that the improved catalytic activity is induced by light-driven structural reorganization, DRM reaction experiment with external heating (Supplementary Fig. 34 and Note 6) were conducted. Under light irradiation with additional heating, a gradual decline in activity is observed over 30 min after the light is turned off. This is attributed to the immediate loss of the photoelectric–photothermal synergy, while the structural reversion of the alloy occurs more slowly, underscoring that light-induced structural reorganization together with the photoelectric-photothermal effect jointly contribute to the high DRM performance of the NiIr0.5/TiO2 catalyst.
Intermediates tracking and photoexcited charge transfer investigation
In situ synchrotron radiation-diffuse reflectance infrared Fourier transform spectroscopy (SR-DRIFTS) analysis during CO2-CH4 co-adsorption revealed distinct molecular activation patterns on NiIr0.5/TiO2 under dark conditions (Fig. 4a and Supplementary Fig. 35a). Characteristic vibrational modes emerged at 3014/1304 cm-1 (CH4 deformation), 2346 cm-1 (C = O stretching), 1354 cm-1 (OCO stretching vibration, ν(OCO)), and 1536 cm-1 (monodentate carbonate, m-CO32-), confirming robust adsorption of both reactants on partially oxidized NiIr nanoclusters. Temperature-programmed desorption (TPD) (Fig. 4b, Supplementary Fig. 35b) further demonstrated that NiIr0.5/TiO2 exhibited high CH4 chemisorption with a prominent desorption peak at 417 °C attributed to Ni-Ir dual sites, while maintaining stable CO2 adsorption through ensemble-dependent Ni or Ir adsorption at 546 °C.

a In-situ SR-DRIFTS of NiIr0.5/TiO2 during CO2-CH4 co-adsorption in the dark (25 °C, CH4: CO2: Ar =1: 1: 8). b CH4-TPD profiles of TiO2, Ni/TiO2, NiIr0.5/TiO2, and Ir/TiO2. c, d In-situ SR-DRIFTS (4000–1200 cm-1) of NiIr0.5/TiO2 under 3.89 W cm-2 focused light (c) vs. 500 °C dark heating (d) in a gas flow of CH4: CO2: Ar =1: 1: 8. e, f Time-resolved evolution of dominant sub-1200 cm-1 SR-DRIFTS peaks under focused light e and dark heating f. g 2D fs-TA spectroscopy mapping of NiIr0.5/TiO2. h Normalized fs-TA decay kinetics of NiIr0.5/TiO2 at 560 nm. i TD-DFT calculated electron-hole pairs distribution on NiIr/TiO2-LNi under S0 → S1/S2 excitations, and the corresponding electron/hole (e-/h+) ratio statistics of total Ir and Ni sites. The green and purple group represent the distribution of electrons and holes, respectively. The isosurface level is 0.005 e Å-3. Ir, Ni, Ti, and O atoms are shown in pink, yellow, grey, and blue colors, respectively.
Light irradiation or external heating induced surface intermediates over NiIr/TiO2-LNi during DRM reaction as monitored by time-resolved SR-DRIFTS (Fig. 4c-f). Under focused light, the complete conversion of m-CO32- to intensified ν(OCO) indicates light-triggered formation of NiIr/TiO2-LNi, optimizing CO2 adsorption configuration on NiIr alloy sites. Continuously detected gaseous CO (2024 cm-1), characteristic vibrations of HCO* (1786 cm-1) and *CH2 (1490 cm-1)8,46, confirmed efficient light-driven DRM activity (Fig. 4c). In contrast, dark thermal activation at 500 °C resulted in gradual emergence of attenuated CO signals after 12 min (Fig. 4d), aligning with its poor catalytic performance. Notably, transient intermediate signatures between 1120-920 cm-1 (Fig. 4e, Supplementary Fig. 36a) demonstrated ultrafast generation and decomposition of CHxO* species37,47 under illumination. Conversely, thermal catalysis produced prominent C-C stretching vibrations (974 cm-1)48 (Fig. 4f, Supplementary Fig. 36b), consistent with coke accumulation observed in Supplementary Fig. 16. Isotopic mass spectrometry (Supplementary Fig. 37) revealed that photoactivated *O species primarily originated from CO2 dissociation; these *O species combined with *CHx fragments to form metastable CHxO* intermediates, establishing a dehydrogenation-dominated pathway that suppressed carbon deposition. Comparative studies of monometallic systems (Supplementary Fig. 38) highlighted the necessity of Ni-Ir synergy. Ni/TiO2 exhibited negligible CO production with C-C coupling signals (873/893 cm-¹), while Ir/TiO2 promoted *COOH formation (1678 cm-1) via RWGS pathways. These findings underscore the necessity of Ni-Ir interaction in maintaining both C-H activation kinetics and intermediate stabilization.
To dissect photogenerated charge dynamics, femtosecond transient absorption (fs-TA) spectroscopy was employed (Fig. 4g, Supplementary Fig. 39a-c). Upon 325 nm excitation, all samples exhibited a ground-state bleach (GSB) at ca. 415 nm, reflecting the excited state relaxation49. Accompanied by excited state absorption (SA) between 460–570 nm, arising from electronic transitions from the singlet state (S1) to higher energy excited states (Sn)50. Notably, NiIr0.5/TiO2 displayed the pronounced TA intensity, confirming NiIr alloy-enhanced charge separation. Time-decay fitting of the SA signal at 560 nm (Fig. 4h and Supplementary Fig. 39d-f) revealed a 520.3 fs lifetime for NiIr0.5/TiO2, which was markedly longer than TiO2 (110.9 fs) or Ni/TiO2 (233.1 fs), directly evidencing retarded charge recombination via Ni-Ir interaction. In addition, compared to pure TiO2, NiIr nanoclusters show enhanced photocurrent density, lower electrochemical impedance, and quenched PL intensity; these respectively indicate stronger charge separation, faster charge transfer, and a greater number of effective photocarriers collected for reactions (Supplementary Fig. 39g-i and Note 7).
Ground-state and excited-state calculations resolved the interfacial charge transfer mechanism. Electron-density distributions (Supplementary Fig. 40a-d and Note 8) indicate Ni-mediated charge polarization enriches electrons at Ir sites. Simultaneously, density-of-states profiles (Supplementary Fig. 40e-h) reveal that NiIr alloy-induced d-band center modulation optimizes the trade-off between adsorption strength and intermediate desorption kinetics, suppressing both carbon deposition and parasitic reactions. Time-dependent DFT (TD-DFT) simulations (Fig. 4i) further identified excitation-dependent charge redistribution on NiIr/TiO2-LNi. Electrons in photoexcited states preferentially accumulate at Ir sites distal to the TiO2 interface, designating these Ir atoms as primary active centers for CO2 reduction or H-H coupling during DRM. Electron/hole (e-/h+) ratio statistics reveal preferential electron capture at both Ir and Ni sites under the S0 → S1 excited state, evidencing electron migration from the TiO2 support to NiIr nanoclusters. The S0 → S2 excited state exhibits a smaller Sr index (the geometric mean overlap function of hole and electron distributions), representing less overlap between the electron and hole, and further indicating superior charge separation. Mechanistically, S0 → S2 promotes hole accumulation at Ni sites alongside electron enrichment at Ir sites, which creates a spatial charge segregation that drives photoexcited electrons toward Ir centers in higher-energy states. Computational analysis of chemical bond strength changes between the ground and excited states (Supplementary Table 6) provides further mechanistic insight. The photoinduced charge transfer weakens the chemical bonds, which synergizes with thermal effects, facilitates atomic rearrangement into a more stable configuration. A detailed comparison reveals a more pronounced bond energy difference between the excited state and the ground state in Ir-O bond (-28.06 kJ mol-1) compared to that of the Ni-O bond (-9.36 kJ mol-1). This indicates a preferential destabilization and breaking of the Ir-O bonds, thereby facilitating the migration of Ir atoms. Furthermore, under the excited state, the Ni-Ir bond (92.35 kJ mol-1) is stronger than the Ir-O bond (69.64 kJ mol-1). These bond energy differences provide support for the formation of the Ni-Ir alloy under light irradiation.
Synergy between Ni-mediated charge transfer and Ir-tailored adsorption establishes a self-optimizing catalytic cycle under illumination. Transient intermediate tracking demonstrates light-driven suppression of coking pathways through metastable CHxO* formation, while fs-TA reveals hierarchical charge transfer from TiO2 → Ni→Ir. This dual functionality, achieved through NiIr nano-island architecture, enables simultaneous enhancement of photogenerated carrier utilization and surface reaction kinetics.
Theoretical calculation of reaction mechanism and natural sunlight validation
DFT calculations (Supplementary data 1) coupled with in situ spectroscopic evidence unraveled the light-induced dynamic restructuring mechanism governing reaction pathway selectivity. Site-resolved adsorption analysis on the NiIr/TiO2-LNi model (Supplementary Fig. 41) revealed that the Ir1-Ni3 asymmetric sites exhibited optimal CO2 adsorption (Eads = -1.22 eV) via ν(OCO) configuration. In contrast, the Ni1-Ni2-O domain exhibited negligible CO2 adsorption in the form of m-CO32- (Eads = −0.03 eV). Charge complementarity between Niδ+ and Irδ- drives complete CO2 adsorption mode transition from m-CO32- under dark to ν(OCO) upon illumination, as evidenced by time-resolved SR-DRIFTS. CH4 preferentially anchors at Ir2 sites through H···Ir interaction, where accumulated electrons from Ni-mediated interfacial transfer stabilize the C-H scission transition state. Comparative studies confirmed NiIr/TiO2-LNi’s superior adsorption (Supplementary Fig. 42), with Ni/TiO2 performing poorest, which is consistent with TPD trends.
For the initial bond cleavage of CO2 and CH4 molecules as shown in Fig. 5a-b and Supplementary Figs. 43–44, NiIr/TiO2-LNi demonstrates both thermodynamic and kinetic advantages. Compared to monometallic Ni/TiO2 and Ir/TiO2, NiIr/TiO2-LNi exhibits a significantly greater energy reduction after direct C = O scission of CO2 and the first C-H scission of CH4, indicating its superior thermodynamic favorability. Meanwhile, NiIr/TiO2-LNi exhibits activation energies (Ea) of the transition states at +0.26 eV for C = O scission and +0.30 eV for C-H scission. These values are lower than the transition state barriers for the first C-H scission of CH4 and direct CO2 scission on Ni/TiO2 and Ir/TiO2, indicating the kinetic advantage of the nanoisland NiIr alloy. Supplementary Fig. 45 further compares the pathway of direct C = O scission of CO2 with the indirect activation route via *COOH pathways. The results reveal Ir/TiO2’s propensity for *COOH formation, aligning with in situ SR-DRIFTS detected RWGS activity and a suboptimal H2/CO ratio. In contrast, NiIr/TiO2-LNi enables concerted C = O scission, where Ni site stabilizes dissociative oxygen and Ir site coordinates carbon, directly yielding CO and *O species.

a Reaction energy profiles (including transition states) for C = O bond scission of CO2 on NiIr/TiO2-LNi, Ni/TiO2, and Ir/TiO2 models. b Reaction energy profiles (including transition states) for C-H bond scission of CH4 on NiIr/TiO2-LNi, Ni/TiO2, and Ir/TiO2 models. c Reaction energy profiles for different CH4 activation pathways on NiIr/TiO2-LNi model. d Energy evolution of key steps (pink-shaded indicator in Supplementary Fig. 46a) during the first C-H scission of CH4 and the formation of *CH3O intermediate. Ir, Ni, Ti, and O atoms are shown in pink, yellow, grey, and blue colors, respectively. e DRM performance over NiIr0.5/TiO2 under concentrated natural sunlight, with measured reaction temperature and incident light intensity (CH4: CO2: Ar =1: 1: 8, 20 ml min-1). Inset: experimental setup. f DRM reaction demonstration under concentrated sunlight, with gas outlet directly interfaced to a PdCl2 indicator solution for CO detection. g Schematic illustration of light-driven DRM mechanism.
Regarding the subsequent activation process after the initial bond cleavage of CH4, *CH3 may undergo further direct C-H scission until a C atom remains. Alternatively, *CH3 can be assisted by activation through combination with O species from CO2 activation (OCO2) or lattice oxygen (OL). Figure 5c compares the thermodynamic energy changes before and after the reaction for these three possible activation pathways. The results show that the sequential direct C-H scission pathway (*CH3 → *CH2 → *CH → *C) on NiIr/TiO2-LNi requires prohibitively high energy, particularly 1.18 eV for cleaving the second C-H bond. For the *O-assisted C-H scission routes, OL-assisted pathways proved unfavorable due to the high *CH3 migration energy of 1.30 eV. Conversely, *O species from CO2 dissociation adsorbed on Ni sites in NiIr/TiO2-LNi enabled thermodynamically favorable *CH3O formation with ΔE = -0.61 eV, and subsequent dehydrogenation required only 0.29 eV, consistent with isotopic labeling results. Comparative studies between NiIr/TiO2-LNi and monometallic Ni/TiO2 and Ir/TiO2 in Supplementary Fig. 46 further highlight the optimal performance of NiIr/TiO2-LNi. Specifically, in *OCO2-assisted pathways, while Ni/TiO2 struggled with CH4 activation exhibiting a second C-H barrier of 0.75 eV and Ir/TiO2 suffered from poor *O stabilization, the NiIr alloy achieved balanced kinetics through asymmetric Ni-Ir sites. Figure 5d quantitatively contrasts key steps that Ir/TiO2 excels in initial C-H scission but falters in *CH3O stabilization, whereas NiIr/TiO2-LNi synergizes Ir’s C-H activation prowess with Ni’s oxygen affinity, enabling stepwise dehydrogenation without carbon buildup. Product desorption studies (Supplementary Fig. 47) revealed moderate H2/CO adsorption on NiIr/TiO2-LNi, ensuring sustained catalytic turnover. In addition, Ox-NiIr/TiO2 exhibited excessive CO2 adsorption but poor C = O scission capacity (Supplementary Fig. 48), while thermal conditions promoted direct CH4 cracking into carbon deposits (Supplementary Fig. 49). This starkly contrasts with photoinduced NiIr nano-island alloy, underscoring light’s critical role in structural optimization. We note that our DFT calculations are based on idealized slab models and static configurations and therefore provide qualitative insight into trends rather than a complete description of the real catalyst under operando conditions.
We also conducted an outdoor DRM experiment under actual sunlight using a Fresnel lens for light concentration (Fig. 5e-f). During 2-hour sunlight irradiation, light intensity varied from 3.22 to 4.08 W cm-2, catalyst temperature ranged 438–505 °C. NiIr0.5/TiO2 exhibits a maximum syngas generation rate of 2782 mmol gcat-1 h-1 (65.7% CO2 conversion) with 20% feed gas flow, indicating the feasibility of this system in the utilization of sunlight. Figure 5f and the Supplementary Movie 1 show the real-time colorimetric response of the PdCl2 indicator during the DRM reaction under focused natural sunlight. After illumination, the originally yellow PdCl2 solution gradually turns black, indicating that a large amount of CO generated in the system adsorbs on the Pd2+ sites and reduces them to Pd0, verifying the high efficiency of the NiIr/TiO2 system under natural sunlight.
Figure 5g illustrates the overall reaction mechanism. Under light excitation, Ox-NiIr/TiO2 undergoes interfacial charge oscillations, with electrons sequentially transferring from the TiO2 to Ni sites and subsequently to Ir atoms. This triggers the upward migration of Ir atoms, leading to the formation of NiIr nano-island alloys. A portion of Ni atoms act as linkages, spatially isolating the NiIr alloy from the support to form the NiIr/TiO2-LNi configuration. During light-driven DRM reaction, photoelectric effects dominate the activation of C-H and C = O bonds at NiIr alloy sites, coupled with efficient H-H coupling via localized photogenerated electrons. Concurrently, the oxygen affinity of the Ni linkages steers the reaction pathway toward the formation of key CHxO* intermediates, thereby achieving robust coking resistance. Meanwhile, photothermal effects facilitate molecular vibrations, intermediate migration, and product desorption. The synergistic interplay of photoelectric and photothermal effects on the NiIr nano-island alloy enables highly efficient and stable DRM performance.
In summary, this work reveals that light-induced NiIr nano-island alloy formation enables efficient DRM by synergizing photoelectric and photothermal effects. The interfacial charge oscillation mechanism not only stabilizes ultrafine alloy architectures against sintering but also creates dual-active sites. Specifically, Ni linkages with oxygen affinity favor coke suppression, while Ir sites modulated by Ni optimize reactant activation. Our experimental method for decoupling photoelectric/photothermal contributions reveals that localized electron transfer dominates bond cleavage selectivity, whereas thermal effects primarily govern mass transport. The achieved 25.0% LTFE and 100-h stability under practical feed conditions underscore the viability of solar-driven DRM. Importantly, the methodology combining in situ structural analysis with charge dynamics kinetics provides a universal approach to probe light–matter interactions in heterogeneous catalysis.
Methods
Catalyst preparation
Ox-NiIr/TiO2 catalysts were synthesized via an optimized directional adsorption strategy23. Typically, 200 mg of ground TiO2 nanosheets was dispersed in 0.01 M NaOH aqueous solution. After 3 h, the TiO2 was centrifuged and redispersed in deionized (DI) water through multiple cycles until the supernatant reached pH=9, ensuring a negatively charged TiO2 surface for cation adsorption. NiCl2 (Sigma-Aldrich) aqueous solution (containing Ni equivalent to 1 wt% of TiO2) was added to the TiO2 suspension and stirred for 12 h prior to centrifugation. Complete metal cation adsorption was confirmed by testing the first supernatant with NaBH4 (Thermo Fisher), which showed no color change. The cation-loaded TiO2 was washed with DI water and dried at 60 °C for 12 h. Co2+ (Co(NO3)2·6H2O, Macklin), Fe3+(FeCl2·6H2O, Aladdin), and Pt(NH3)42+ ([Pt(NH3)4]Cl2, Sigma-Aldrich) cations were similarly adsorbed using their respective chloride salts.
The dried cation-loaded TiO2 was subsequently used for complex metal anion adsorption IrCl63-(Na3IrCl6, Sigma-Aldrich), PtCl42- (K2PtCl4, Sigma-Aldrich), PdCl42- (Na2PdCl4, Sigma-Aldrich), AuCl4- (HAuCl4·3H2O, Sigma-Aldrich), and RuCl62-(NH4)2RuCl6, Sigma-Aldrich) with controlled theoretical anion/cation atomic ratios. Anion solutions were prepared via a phase-transfer method using tetrabutylammonium cations (TBA+) in dichloromethane (TBA⁺/anion molar ratio = 3) under vortex mixing. After centrifugation, the dichloromethane phase was retained, and mixed with cation-loaded TiO2 in dichloromethane. After 12 h of stirring, TiO2-supported double complex salts were centrifuged, dried at room temperature, and reduced under 5% H2 flow at 400 °C for 30 min. Monometallic TiO2-supported catalysts (Ni2+ and Ir3+) were prepared analogously without anion adsorption steps.
Catalytic reactivity measurements
Light-driven DRM experiments were performed in a custom quartz reactor (Supplementary Fig. 9) at 0.1 MPa without extra heating. The reactor features an outer layer for condensate circulation and an inner chamber containing a quartz sand bed for supporting catalyst and optimize gas distribution. A type-K thermocouple (0–1150 °C range, 0.5 mm diameter, ±0.1 °C accuracy) was embedded in the quartz sand bed, with a 90°-bent tip protruding 1.0 mm above the surface. The thermocouple was pre-calibrated against standard reference sources, and its performance was verified at multiple temperature setpoints to ensure consistency across the experimental range. TCenter (the center temperature of catalyst surface) was detected by the thermocouple probe placed in direct contact with the upper surface of the catalyst at its center, enabling capture of the highest local temperature under irradiation (as shown by the red indicator line representing the thermocouple in Supplementary Fig. 9). This arrangement minimizes the thermal resistance effect by avoiding indirect measurement through the gas phase or reactor materials. Infrared (IR) thermography was used to validate the accuracy of TCenter measurements from the thermocouple probe. TAVG (average surface temperature) was obtained by moving the thermocouple probe, measuring temperatures at five different points on the catalyst surface (center and edges) and using the formula below:
Catalyst (5 mg) was dispersed in DI water (1.0 mL), spread on a quartz fiber membrane, and vacuum-dried. The membrane was positioned on the quartz sand bed, and depressed by a tweezer align the thermocouple tip flush with the catalyst surface. Real-time temperature monitoring at catalyst center and edge positions was achieved by adjusting the thermocouple position via its stretchable connector. Note that unlike conventional temperature probes positioned below the catalyst, our small-diameter temperature probes are placed directly on the catalyst surface, enabling more accurate and sensitive temperature measurement. Prior to irradiation using a 300 W Xe lamp (PLS-SXE 300 + , Beijing PERFECTLIGHT; spectral range: 300–1000 nm), the reactor was introduced with CH4: CO2 (1:1) gas mixture at 20 mL min-1. A biconvex lens (diameter: 70 mm) was employed for light focusing. Light intensity was controlled by adjusting lamp current and measured using an optical power meter (range: 0–20 W, resolution: 1 mW).
Thermal catalytic tests were conducted in a dedicated reactor (PLR-PTSR II, Beijing PERFECTLIGHT) under dark conditions, using identical catalyst supporting and thermocouple configurations to those in the custom quartz reactor. Natural sunlight experiments utilized the custom quartz reactor with Fresnel lens focusing. All other experimental parameters (gas flow, catalyst weight, etc.) remained consistent across light-driven, thermal, and natural sunlight conditions.
The monochromatic light activity tests were carried out in a dedicated reactor (PLR-PTSR II, Beijing PERFECTLIGHT) with external heating at 500 °C. A 300 W Xenon lamp was used as the light source, coupled with optical filters to obtain monochromatic light of specific wavelengths. In each test, an optical power meter was used to accurately measure and calibrate the incident light intensity in front of the reactor window. Dividing the measured syngas generation rates by the light intensity under the corresponding monochromatic light yields the contributions of monochromatic light at different wavelength bands to the activity per unit light intensity. All other test conditions were consistent with those of the light-driven conditions.
The CO products were analyzed by GC with a flame-ionization detector (FID). H2 was analyzed by GC with a high-sensitivity thermal conductivity detector (TCD). The H2 and CO generation rates were defined as follows:
Where \({R}_{{{\mbox{H}}}_{2}}\) (mmol gcat-1 h-1) and \({R}_{{\mbox{CO}}}\) (mmol gcat-1 h-1) are the H2 and CO generation rates, respectively. The \({C}_{{{\mbox{H}}}_{2}}^{{\mbox{out}}}\) (dimensionless) and \({C}_{{\mbox{CO}}}^{{\mbox{out}}}\) (dimensionless) are the concentrations of H2 and CO detected by gas chromatography. \({F}_{{\mbox{out}}}\) (ml/min) is the space velocity of the gas exiting the reactor, \({V}_{{\mbox{m}}}\) (L/mol) is the gas molar volume under standard conditions, and \(M\) (g) is the mass of catalyst. 60 refers to the exponent needed to convert ml/min into the standard unit ml/h.
The CH4 and CO2 conversions were defined as follows:
Where \({C}_{{{\mbox{CH}}}_{4}}^{{\mbox{in}}}\) (dimensionless) and \({C}_{{{\mbox{CH}}}_{4}}^{{\mbox{out}}}\) (dimensionless) indicate the inlet and outlet concentration of products detected by gas chromatography, respectively.
Total carbon balance is considered using the following equation:
TOF under kinetics-controlled regime per second can be calculated with the following equation:
The LTFE (η) was determined with the following equation:
Where \({R}_{{{\mbox{H}}}_{2}}\) (mmol gcat-1 h-1), \({R}_{{\mbox{CO}}}\) (mmol gcat-1 h-1) and \({R}_{{\mbox{C}}{{\mbox{H}}}_{4}}\) (mmol gcat-1 h-1) are the H2 generation rates, CO generation rates, and CH4 conversion rates. \({\triangle }_{{\mbox{C}}}{{\mbox{H}}}_{{{\mbox{H}}}_{2}}^{{\mbox{o}}}\), \({\triangle }_{{\mbox{C}}}{{\mbox{H}}}_{{\mbox{CO}}}^{{\mbox{o}}}\), and \({\triangle }_{{\mbox{C}}}{{\mbox{H}}}_{{\mbox{C}}{{\mbox{H}}}_{4}}^{{\mbox{o}}}\) are the standard combustion heats (298.15 K) of H2, CO, and CH4 as fuels, respectively. Note that the \({\triangle }_{{\mbox{C}}}{{\mbox{H}}}_{{\mbox{C}}{{\mbox{O}}}_{2}}^{{\mbox{o}}}\) is 0. \(M\) (g) is the mass of catalyst. \({P}_{{\mbox{irradiation}}}\) (W) is the irradiation power.
Characterization
HAADF-STEM imaging was performed on an aberration-corrected FEI Themis Z instrument, with a convergence semi-angle set to 11 mrad and annular detection ranges of 59 mrad (inner) and 200 mrad (outer). Elemental mapping and EDS analysis were carried out utilizing the integrated four-quadrant Super-X detector system. Powder XRD measurements were obtained using a Rigaku D/MAX 2550 diffractometer equipped with Cu Kα radiation (λ = 1.5406 Å), operating at 40 kV and 40 mA, with 2θ scanned from 10° to 80°. Raman spectroscopy was performed on a Renishaw in Via system using a 532 nm laser excitation source. UV–Vis-NIR diffuse reflectance spectra were recorded using a Varian Cary 500 spectrophotometer equipped with an integrating sphere, using BaSO4 as a reference. For sample preparation, the powder sample was pressed into a flat, uniform pellet in a culture dish to ensure data reliability and reproducibility. The absorbance was represented by the Kubelka-Munk function:
which is derived from the measured reflectance data. Inductively coupled plasma atomic emission spectrometry (ICP-AES, Varian 730-ES, Agilent) quantified the metal loadings of catalysts. Thermogravimetric analysis (TGA) of spent catalysts was conducted on a Netzsch STA 449 F5 analyzer under air flow (50 mL min-1), heating from 100 °C to 800 °C at 10 °C∙min-1. Temperature-programmed desorption (TPD) experiments were performed on an Auto Chem II 2920 (Micromeritics, USA) in pure He, ramping to 770 °C at 10 °C∙min-1. H2-O2 titration was performed on a temperature programmed chemisorption analyzer (VDSorb-91i) to measure metal dispersion. Photoluminescence (PL) spectra were acquired at room temperature using a Cary Eclipse fluorescence spectrometer (Agilent). Femtosecond transient absorption (fs-TA) spectroscopy was performed on a Helios pump-probe system (Ultrafast Systems, USA) with an 800 nm Ti:sapphire laser (Coherent Legend, 85 fs pulse width, 1 kHz repetition rate). The pump beam (325 nm, 80 μW) was generated via frequency doubling in a β-barium borate (BBO) crystal, while the probe beam (near-infrared continuum) monitored photoexcited states. Isotope labeling experiments employed a high-precision gas isotope mass spectrometer (Nu Instruments Horizon, UK): a 1:1 mixture of CH4 and C18O2 was introduced into a batch reactor in the dark, followed by 15-min irradiation (300 W Xe lamp, 3.89 W cm-2). Reaction products were cryogenically separated and analyzed for C16O and C18O isotopes.
In situ XAS measurements conditions
In situ XAS measurements at Ir L3-edge and Ni K-edge were performed with Si (111) crystal monochromators at the BL14W Beam line at the Shanghai Synchrotron Radiation Facility (SSRF) (Shanghai, China). Typically, the powdered sample was pressed into a self-supporting wafer and placed into a reaction microdevice equipped with an extra window for light irradiation. Before the test, the sample surface was purged with Ar gas flow (50 ml min-1). Afterwards, the gas mixture (CH4: CO2: Ar=1: 1: 8) or pure Ar was continuously introduced into the reactor with a flow rate of 20 mL min-1. The microdevice was irradiated by the 300 W Xe lamp at the light intensity of 3.89 W cm-2. The XAFS spectra were recorded in situ in the dark or after 10-min light irradiation using a Bruker 5040 4-channel Silicon Drift Detector (SDD) in fluorescence mode. All spectra were collected in ambient conditions. The data reduction and analysis of the XAFS spectra were conducted using the Demeter software package (ATHENA and ARTEMIS, respectively)51. All fits were performed in the R space with a k-weight of 2.
In situ NAP-XPS conditions
In situ NAP-XPS experiments were conducted on a SPECS Proven-X NAP instrument (Germany) equipped with a monochromatic X-ray source µ-FOCUS 600 NAP. Prior to the test, the sample surface was cleaned using an Ar gas flow (50 ml min-1). Subsequently, a gas mixture (CH4:CO2:Ar=1:1:8) or pure Ar was continuously introduced into the reactor with a flow rate of 20 mL min-1. The NAP-XPS spectra were recorded in situ in the dark or after 10-min light irradiation. The irradiation started with unfocused light at an intensity of 1.58 W cm-2 and then switched to focused light at 3.89 W cm-2. Noted that under light conditions, no additional heating was applied, while under thermal conditions, the heating temperature was set at 500 °C. The NAP - XPS spectra were obtained with charge correction, using the C 1 s binding energy of 284.60 eV as the energy reference.
In situ SR-DRIFTS conditions
In situ synchrotron radiation DRIFTS were performed at the BL01B Beam line at the Shanghai Synchrotron Radiation Facility (SSRF) (Shanghai, China). The spectra were recorded by a Bruker infrared spectrometer (70 V) equipped with a liquid nitrogen-cooled mercury-cadmium-telluride (MCT) detector. Integrated with a high-brightness synchrotron radiation source, the setup achieved a spatial resolution near the theoretical diffraction limit, producing infrared spectra with high intensity and signal-to-noise ratio. The catalysts were first sealed in the chamber and purged with an Ar gas flow (50 ml min-1). Subsequently, a flowed mixture gas (CH4: CO2: Ar=1: 1: 8) or pure Ar was introduced into the chamber with a flow rate of 20 mL min-1. The chamber was then irradiated with the 300 W Xe lamp at a light intensity of 3.89 W cm-2.
In situ CO-probe DRIFTS conditions
In situ CO-probe DRIFTS was recorded by a Fourier-transform infrared spectrometer (Bruker VERTEX 80 v, Germany) equipped with a designed reaction cell. Catalysts were initially degassed at 100 °C for 1 h and then cooled to 25 °C under a flow of Ar (50 ml min-1). Subsequently, catalysts were exposed to a mixture of 20% CO in Ar (30 ml min-1) for 30 min at 25 °C, which proved sufficient to saturate the catalyst surface. After saturation, the CO flow was replaced with an Ar flow (30 ml min-1), and CO DRIFTS spectra were collected at 25 °C for 25 min. Then, the temperature was sequentially increased to 100 °C, 200 °C, 300 °C, and 400 °C, with spectra recorded at each temperature for 10 min.
Electrochemical measurements
Electrochemical measurements were conducted at room temperature using a Zahner Zennium potentiostat (Zahner-elektrik GmbH, Germany) with a three-electrode configuration: a fluorine-doped tin oxide (FTO) working electrode, a graphite counter electrode and a saturated calomel reference electrode (SCE). For working electrode preparation, 5 mg of catalyst was dispersed in 0.5 mL ethanol, and 20 μL of the slurry was drop-cast onto FTO (1 cm2 active area) and dried under ambient conditions. Transient photocurrent responses were measured in N2-saturated 0.5 M Na2SO4 under 300 W Xe lamp irradiation. The pH of the 0.5 M Na2SO4 electrolyte was measured to be 6.8 ± 0.1 (n = 3). Electrochemical impedance spectroscopy (EIS) was performed in the frequency range from 100 kHz to 0.1 Hz using an N2-saturated potassium ferricyanide mixed electrolyte (25 mM K3Fe(CN)6, 25 mM K4Fe(CN)6, and 0.1 M KCl), whose pH was 7.0 ± 0.1 (n = 3). All electrochemical measurements were performed without iR correction. All potentials were converted to the reversible hydrogen electrode (RHE) scale according to the formula below.
where 0.241 V is the standard potential of the saturated calomel electrode versus RHE at 25 °C.
Theoretical calculations
All density functional theory (DFT) calculations were performed using the Materials Studio software and the Vienna ab initio simulation package (VASP)52,53,54. The exchange-correlation interactions were treated within the generalized gradient approximation (GGA), employing the Perdew–Burke–Ernzerhof (PBE) functional55,56. The projector augmented-wave (PAW) method was used to describe core-valence electron interactions, with a plane-wave basis set and a kinetic energy cutoff set to 400 eV. The Gaussian smearing scheme with a width of 0.05 eV was applied for partial orbital occupancy. Convergence criteria were set to 10-5 eV for electronic self-consistent iterations and 0.05 eV/Å for ionic relaxation during geometry optimization. A vacuum layer of 15 Å was added perpendicular to the surface model to avoid periodic interactions. For surface calculations, the Brillouin zone was sampled using a 2 × 2 × 1 Monkhorst–Pack k-point mesh. The adsorption energy was calculated according to the following formula:
where EA-S is the energy of the slab together with the adsorbate, EA is the total energy of the free adsorbate and ES is the total energy of the bare slab.
The d band center (εd) was calculated as follow formula:
The transition states corresponding to elementary reaction steps were located using a sequential approach combining the nudged elastic band (NEB) and dimer methods57. First, the reaction pathway connecting the reactant and product states was sampled using the NEB method, which represents the path as a discrete set of structural images. From this set, the image possessing the highest energy, indicative of the most probable transition state configuration, was selected. This structure subsequently served as the initial guess for precise transition-state optimization using the dimer method.
Excitation-dependent charge redistribution calculations are performed with ORCA software58. The functional employed was PBE0, and the basis set used was def2-SVP. (This basis set is an all-electron basis set for the first four periods and a pseudopotential basis set starting from the fifth period, so no mixed basis set is required.) The electron-hole structures were calculated using Multiwfn59, and the visualization data were obtained using the visual molecular dynamics VMD software60. The geometric mean overlap function of hole and electron distributions (Sr) was calculated as follows:
ρhole and ρele represent the electron density of the hole and the excited electron at the spatial position r, respectively.
Data availability
All data generated in this study are provided in the main text, supplementary information, source data and supplementary data 1 file. Source data are provided with this paper.
References
Tavasoli, A. & Ozin, G. Green syngas by solar dry reforming. Joule 2, 571–575 (2018).
Song, Y. et al. Dry reforming of methane by stable Ni-Mo nanocatalysts on single-crystalline MgO. Science 367, 777–781 (2020).
Meng, X. et al. Direct methane conversion under mild condition by thermo-, electro-, or photocatalysis. Chem 5, 2296–2325 (2019).
Aresta, M., Dibenedetto, A. & Angelini, A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. technological use of CO2. Chem. Rev. 114, 1709–1742 (2014).
Palmer, C. et al. Dry reforming of methane catalysed by molten metal alloys. Nat. Catal. 3, 83–89 (2020).
Zhu, Q. et al. Enhanced CO2 utilization in dry reforming of methane achieved through nickel-mediated hydrogen spillover in zeolite crystals. Nat. Catal. 5, 1030–1037 (2022).
Zhou, L. et al. Light-driven methane dry reforming with single atomic site antenna-reactor plasmonic photocatalysts. Nat. Energy 5, 61–70 (2020).
Xiong, H. et al. Highly efficient and selective light-driven dry reforming of methane by a carbon exchange mechanism. J. Am. Chem. Soc. 146, 9465–9475 (2024).
Li, Q. et al. Suppressive strong metal-support interactions on ruthenium/TiO2 promote light-driven photothermal CO2 reduction with methane. Angew. Chem. Int. Ed. 62, e202300129 (2023).
Pakhare, D. & Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 43, 7813–7837 (2014).
Mattos, L. V., Jacobs, G., Davis, B. H. & Noronha, F. B. Production of hydrogen from ethanol: review of reaction mechanism and catalyst deactivation. Chem. Rev. 112, 4094–4123 (2012).
Li, X. et al. Advances in heterogeneous single-cluster catalysis. Nat. Rev. Chem. 7, 754–767 (2023).
He, C. et al. Regulating atomically-precise Pt sites for boosting light-driven dry reforming of methane. Angew. Chem. Int. Ed. 63, e202412308 (2024).
Li, X. et al. Functional CeOx nanoglues for robust atomically dispersed catalysts. Nature 611, 284–288 (2022).
Zhou, T. et al. Ultrafine metal nanoparticles isolated on oxide nano-islands as exceptional sintering-resistant catalysts. Nat. Mater. (2025).
Gashnikova, D. et al. Highly active oxidation catalysts through confining Pd clusters on CeO2 nano-islands. Angew. Chem. Int. Ed. 63, e202408511 (2024).
Marimuthu, A., Zhang, J. & Linic, S. Tuning selectivity in propylene epoxidation by plasmon mediated photo-switching of Cu oxidation state. Science 339, 1590–1593 (2013).
Lee, B.-H. et al. Reversible and cooperative photoactivation of single-atom Cu/TiO2 photocatalysts. Nat. Mater. 18, 620–626 (2019).
Luo, S. et al. Light-induced dynamic restructuring of Cu active sites on TiO2 for low-temperature H2 production from methanol and water. J. Am. Chem. Soc. 145, 20530–20538 (2023).
Ren, C., Li, Q., Ling, C. & Wang, J. Mechanism-guided design of photocatalysts for CO2 reduction toward multicarbon products. J. Am. Chem. Soc. 145, 28276–28283 (2023).
Jiang, H. et al. Light-driven CO2 methanation over Au-grafted Ce0.95Ru0.05O2 solid-solution catalysts with activities approaching the thermodynamic limit. Nat. Catal. 6, 519–530 (2023).
Li, J. et al. Activating lattice oxygen in perovskite ferrite for efficient and stable photothermal dry reforming of methane. J. Am. Chem. Soc. 147, 14705–14714 (2025).
Ding, K. et al. A general synthesis approach for supported bimetallic nanoparticles via surface inorganometallic chemistry. Science 362, 560–564 (2018).
Zou, S. et al. Manufacturing single-atom alloy catalysts for selective CO2 hydrogenation via refinement of isolated-alloy-islands. Angew. Chem. Int. Ed. 64, e202412835 (2025).
Zhou, J. et al. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 13, 327 (2022).
Li, X. et al. Controlling CO2 hydrogenation selectivity by metal-supported electron transfer. Angew. Chem. Int. Ed. 59, 19983–19989 (2020).
Tan, S. et al. Plasmonic coupling at a metal/semiconductor interface. Nat. Photonics 11, 806–812 (2017).
Han, J. W., Park, J. S., Choi, M. S. & Lee, H. Uncoupling the size and support effects of Ni catalysts for dry reforming of methane. Appl. Catal. B Environ. 203, 625–632 (2017).
Li, X. et al. Dry reforming of methane over Ni/La2O3 nanorod catalysts with stabilized Ni nanoparticles. Appl. Catal. B Environ. 202, 683–694 (2017).
Pan, F. et al. A novel photo-thermochemical approach for enhanced carbon dioxide reforming of methane. ChemCatChem 10, 940–945 (2018).
Zhang, Q. et al. Novel photoactivation promoted light-driven CO2 reduction by CH4 on Ni/CeO2 nanocomposite with high light-to-fuel efficiency and enhanced stability. Appl. Catal. B Environ. 239, 555–564 (2018).
Akri, M. et al. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat. Commun. 10, 5181 (2019).
Gao, Y. et al. Light-driven efficient dry reforming of methane over Pt/La2O3 with long-term durability. J. Mater. Chem. A 10, 16016–16028 (2022).
Kushida, M., Yamaguchi, A. & Miyauchi, M. Photocatalytic dry reforming of methane by rhodium supported monoclinic TiO2-B nanobelts. J. Energy Chem. 71, 562–571 (2022).
Wu, J. et al. Engineering the oxygen vacancies enables Ni single-atom catalyst for stable and efficient C-H activation. Appl. Catal. B Environ. 314, 121516 (2022).
Yang, Y. et al. Light-induced redox looping of a rhodium/CeWO3 photocatalyst for highly active and robust dry reforming of methane. Angew. Chem. Int. Ed. 61, e202200567 (2022).
Rao, Z. et al. Light-reinforced key intermediate for anticoking to boost highly durable methane dry reforming over single atom Ni active sites on CeO2. J. Am. Chem. Soc. 145, 24625–24635 (2023).
Tang, Y. et al. Enhanced dry reforming of CO2 and CH4 on photothermal catalyst Ru/SrTiO3. Appl. Catal. B Environ. 338, 123054 (2023).
Yao, Y. et al. Highly efficient solar-driven dry reforming of methane on a Rh/LaNiO3 Catalyst through a light-induced metal-to-metal charge transfer process. Adv. Mater. 35, 2303654 (2023).
He, Z. et al. Engineering an Ni-TiOx interface for highly active and durable solar-driven dry reforming of methane. Chem Catal 4, 101040 (2024).
Wang, B. et al. Single atom iridium decorated nickel alloys supported on segregated MoO2 for alkaline water electrolysis. Adv. Mater. 36, 2305437 (2024).
Zhao, G. F. & Zeng, Z. Geometrical and electronic structures of AumAgn (2 ≤ M + N ≤ 8). J. Chem. Phys. 125, 014303 (2006).
Liu, L. & Corma, A. Bimetallic sites for catalysis: from binuclear metal sites to bimetallic nanoclusters and nanoparticles. Chem. Rev. 123, 4855–4933 (2023).
Sun, G. et al. Full utilization of noble metals by atom abstraction for propane dehydrogenation. Science, eadw3053 (2025). https://doi.org/10.1126/science.adw3053.
Guo, C. et al. Light-driven metal exsolution-redissolution of high-entropy oxide enabling high-performance dry reforming of methane. Adv. Mater. 37, 2500928 (2025).
Zhang, W. et al. High-performance photocatalytic nonoxidative conversion of methane to ethane and hydrogen by heteroatoms-engineered TiO2. Nat. Commun. 13, 2806 (2022).
Li, X. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Jiang, W. et al. Pd-modified ZnO–Au enabling alkoxy intermediates formation and dehydrogenation for photocatalytic conversion of methane to ethylene. J. Am. Chem. Soc. 143, 269–278 (2021).
Wolff, C. M. et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 3, 862–869 (2018).
Wang, A. et al. Enhanced and synergistic catalytic activation by photoexcitation driven S−scheme heterojunction hydrogel interface electric field. Nat. Commun. 14, 6733 (2023).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Han, L. et al. Two-in-one template-assisted construction of hollow phosphide nanotubes for electrochemical energy storage. Inorg. Chem. Front. 9, 3398–3411 (2022).
Selcuk, S. & Selloni, A. Facet-dependent trapping and dynamics of excess electrons at anatase TiO2 surfaces and aqueous interfaces. Nat. Mater. 15, 1107–1112 (2016).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Si, X. et al. Catalytic production of low-carbon footprint sustainable natural gas. Nat. Commun. 13, 258 (2022).
Neese, F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 8, e1327 (2018).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
This work was supported by National Key Research and Development Program of China (2022YFE0107900, 2022YFB3803600 to J.Z.), the National Natural Science Foundation of China (22461142136 to J.Z., 22572051 to S.W.), the Innovation Program of Shanghai Municipal Education Commission (2021-01-07-00-02-E00106 to J.Z.), the Science and Technology Commission of Shanghai Municipality (22230780200, 20DZ2250400, 2018SHZDZX03 to J.Z.), the Chenguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (24CGA30 to S.W.), and Fundamental Research Funds for the Central Universities (222201717003 to J.Z.). Thanks to the Shanghai Synchrotron Radiation Facility of BL14W1 (https://cstr.cn/31124.02.SSRF. BL14W1) for providing beam time. We thank the staff members of the BL01B beamline (https://cstr.cn/31129.02.NFPS.BL01B) at the National Facility for Protein Science in Shanghai (https://cstr.cn/31129.02.NFPS), for providing technical support and assistance in data collection and analysis.
Author information
Authors and Affiliations
Contributions
J.Z., S.W., and C.H. conceived and designed the project. C.H. performed the experiments and analyzed the data. J.Z., S.W. gave suggestions on the research and supervised the project. C.H. performed the theoretical calculations and wrote the manuscript. Z.Y. provided guidance in theoretical calculations. R.Y., C.Z., Y.D., W.C., L.C., and Z.W. conducted part of light-driven experiments. S.W. and J.Z. reviewed the manuscript, and gave suggestions. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, C., Yang, R., Zhong, C. et al. Light-driven restructuring generates nanoisland NiIr alloy for efficient methane dry reforming. Nat Commun 17, 1730 (2026). https://doi.org/10.1038/s41467-026-68429-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68429-w
