
Abstract
Co(IV) = O-mediated Fenton-like processes show great potential for water remediation but are fundamentally limited by the “oxo-wall” effect, which imposes prohibitive activation energies for Co(IV) = O bond formation and stabilization. Herein, by unifying thermodynamic analysis with the “oxo-wall” constraint mechanism, we establish the comprehensive theoretical framework for Co(IV) = O-dominated non-radical Fenton-like oxidation pathways. We design a Ce-Co tetra-(4-carboxyphenyl) porphyrin framework (Ce-Co TCPP), where Ce(IV)-based oxide linkers induce long-range electronic modulation, enhancing electronic delocalization at Co–N4 sites. This significantly reduces electron occupancy in Co–O antibonding orbitals, thereby effectively circumventing “oxo-wall” constraints. Combined experimental and computational analyses confirm that Co(IV) = O species dominate in the Ce-Co TCPP/peroxymonosulfate (PMS) system, where synergistic electron transfer and proton transfer processes significantly lower activation barriers. Practically, the lamellar Ce-Co TCPP membrane/PMS system achieves desirable water permeability (126.97 L·m−2·h−1·bar−1 (LMHB)), high pollutant degradation efficiency (0.0717 ms−1), robust anti-interference capability, and long-term operational stability (95 h), which can be attributed to the shortened mass transport pathways and the approximately 1000-fold enrichment of Co(IV) = O complexes within membrane nanoconfined channels. This work offers an innovative strategy for sustainable Co(IV) = O-mediated advanced oxidation processes in water treatment.
Similar content being viewed by others


Introduction
Organic micropollutants in wastewater present significant ecological and public health risks even at trace concentrations, necessitating urgent development of efficient removal technologies1,2. Recent advances have highlighted non-radical Fenton-like oxidation processes as particularly promising solutions, offering anti-interference capacity and selective degradation performance for addressing complex water contamination challenges3,4,5. Among these technologies, high-valent metal-oxo species offer distinct advantages for water treatment, including extended operational lifetimes (~7 s), elevated steady-state concentrations (~10−8 M), versatile reaction pathways (involving hydrogen atom transfer, oxygen atom transfer, electron transfer, and electrophilic addition), and strong resistance to matrix interference6,7,8. Notably, high-valent cobalt-oxo (Co(IV) = O) complexes exhibit redox potentials than the iron (Fe(IV) = O) and manganese (Mn(IV) = O) counterparts, making them particularly effective for degrading refractory organic micropollutants in peroxymonosulfate (PMS)-based Fenton-like processes9,10. However, the transformation from Co(II) to Co(IV) = O involves a substantial energy barrier with regard to the required O–O bond cleavage in PMS coupled with a two-electron transfer process11. Meanwhile, the high bond dissociation energy of the O–H bond in PMS favors direct O–O cleavage, preferentially generating •OH/SO4•− radicals over the desired Co(IV) = O species12,13. Moreover, the well-established yet frequently overlooked “oxo-wall” rule for late-transition metals (e.g., Co, Ni, Cu) establishes that high-valent metal-oxo complexes become inherently unstable when the d-orbital electron occupancy exceeds four electrons (five electrons for Co(IV)) in a tetragonal (C4v) ligand field14,15,16. The development of a unified theoretical framework for Co(IV) = O-dominated Fenton-like oxidation requires systematic integration of two critical aspects: (1) catalytic processes (e.g., adsorption energetics, reaction pathways, and electron transfer) and (2) fundamental formation principles governed by the “oxo-wall” rule. However, current studies often treat these aspects in isolation, either neglecting the “oxo-wall” constraint or failing to correlate it with practical catalytic parameters13,14.
Single-atom catalysts (SACs) have emerged as a transformative platform in catalysis, offering maximal atomic utilization efficiency and precisely tunable coordination environments that enable optimized reaction selectivity17,18. Extensive research has focused on engineering the first coordination shell of SACs—including configuration, coordination number, and ligand identity—enabling a strategic shift from radical-dominated to non-radical advanced oxidation processes (AOPs)10,12,14. Despite considerable advances in Co(IV) = O-mediated non-radical Fenton-like systems, a critical knowledge gap persists in systematically bridging catalytic processes with the structural constraints imposed by the “oxo-wall” rule. Moreover, alternative strategies emphasizing long-range electronic tuning (beyond first-shell coordination) to facilitate the formation of high-valent metal-oxo species warrant further exploration6,7. In particular, non-covalent modulations originating from high coordination shells—such as functional groups, organic ligands, intrinsic carbon defects, and heteroatom doping sites—hold considerable promise for elucidating structure-function relationships and underlying catalytic mechanisms19,20,21.
Porphyrin-based framework structures, assembled from porphyrin-derived ligands, have emerged as highly promising catalytic platforms due to their unique capacity to stabilize isolated metal atoms within tetrapyrrole coordination sites, thereby effectively preventing active site aggregation22,23,24. Meanwhile, the π-electrons of both phenyl and pyrrole moieties in the porphyrin framework exhibit extensive conjugation, creating an extended delocalized electronic network. This conjugated system enables long-range electronic modulation of the central metal site—despite being separated by multiple bonds—through efficient transmission of microenvironmental perturbations23. Therefore, the strategic incorporation of functionally tunable remote linkers into porphyrin frameworks enables precise electronic modulation of active sites through long-range interactions, bypassing direct metal coordination. By leveraging microenvironment-mediated support-metal interactions and extended π-conjugation pathways within the framework architecture, this approach subtly tailors the electronic properties of metal centers while preserving their primary coordination environment, offering a sophisticated method for catalyst design25,26,27. For instance, Zhang et al. demonstrated that electron-withdrawing substituents on cobalt porphyrins enhance electronic delocalization at the Co center, thereby modulating *OOH adsorption and improving H2O2 electrosynthesis28. Similarly, Huang et al. reported that tailored linkers in iron phthalocyanine-based polymers regulate the spin state of Fe 3d orbitals via long-range modulations, optimizing *OH binding and boosting oxygen reduction performance29. Inspired by these findings, the rational design of cobalt porphyrin-based frameworks may represent a promising strategy for Co(IV) = O-dominated non-radical Fenton-like oxidation. In particular, the long-range modulation between active cobalt centers and remote functional linkers may induce electronic delocalization at the Co sites, which could serve as an effective strategy to enhance Co–oxo bond strength and promote the catalytic process through energetically favorable pathways.
Herein, as a proof of concept, we design the metal tetra-(4-carboxyphenyl) porphyrin (H2TCPP) framework—Ce-Co TCPP—by coordinating Co centers within the tetrapyrrole structure of H2TCPP for efficient PMS activation (Fig. 1)30. Initially, density functional theory (DFT) calculations evaluate the thermodynamic feasibility of Co(IV) = O formation within the designed frameworks. The simulations reveal that Ce-based oxide linkers induced long-range electronic modulation, promoting electron delocalization at Co active sites. This effect significantly reduces electron occupancy in Co-O antibonding π* orbitals while increasing the Co-oxo bond order, thereby effectively circumventing the “oxo-wall” limitation. Subsequently, experimental validation demonstrates that the Ce-Co TCPP/PMS system effectively degraded organic pollutants through a Co(IV) = O species-dominated non-radical pathway. Further catalytic process analysis shows that the enhanced electronic delocalization at the Co center facilitated the formation of low-energy intermediates and promoted two-electron transfer processes, both of which are critical for the efficient generation of Co(IV) = O complexes. Based on these theoretical insights and experimental evidence, we develop a lamellar Ce-Co TCPP membrane incorporating two-dimensional (2D) nanoconfined channels, which was rationally engineered to achieve the approximately 1000-fold enrichment of Co(IV) = O complexes compared to conventional heterogeneous systems. Consequently, benefiting from the synergistic effect of nanoconfinement and the Co(IV) = O complexes, the membrane/PMS system exhibits water permeability (126.97 LMHB), rapid pollutant degradation kinetics (k = 0.0717 ms−¹), strong resistance to environmental interferences, and long-term operational stability over 95 h. Furthermore, molecular dynamics (MD) simulations indicate that the enrichment of reactants and shortened mass-transfer distances induced by nanoconfinement effects were the primary mechanisms underlying the efficient degradation of organic pollutants in the Ce-Co TCPP membrane/PMS system (Fig. 1). This work lays the groundwork for efficient and sustainable Co(IV) = O-mediated Fenton-like oxidation for water remediation.

Schematic of the 2D lamellar Ce-Co TCPP membrane integrating Co(IV) = O-dominated non-radical pathways and nanoconfinement effects for efficient removal of organic pollutants.
Results
Theoretical predications for the formation of Co(IV) = O species
To validate the influence of long-range modulations on the electronic structure of the Co catalytic sites, Ce-Co TCPP and Al-Co TCPP were synthesized as model compounds30 (Figs. S1 and S2). Binding energy comparisons identified a periodic Ce-Co TCPP structural model—featuring trans, corner-sharing Ce(IV) clusters that are μ-bridged via benzoate oxygen atoms and positioned beyond the first coordination shell of the Co center—as the thermodynamically most favorable configuration (Fig. S1b)30. The electron-withdrawing capability of Ce(IV) oxide clusters was found to be substantially stronger than that of Ce(III) clusters, which exhibited an electronic effect comparable to Al(III) (Fig. S3). These results establish Ce(IV) as the dominant contributor to long-range electronic modulation31. The electrostatic potential maps (Fig. S4) further reveal that this long-range tuning effect from Ce(IV) clusters induces pronounced electron depletion at the Co center relative to Al-Co TCPP. This observation is quantitatively supported by the Bader charge analysis (−1.142 e for Ce-Co TCPP vs. −1.097 e for Al-Co TCPP; Fig. S5), confirming the enhanced electronic delocalization within the Ce-Co TCPP framework28,32. Density of states (DOS) calculations (Fig. 2a) further show that Ce-Co TCPP features a greater number of unoccupied Co 3d orbitals, suggesting lower π* orbital occupancy and a more favorable electronic configuration for Co(IV) = O formation. Crystal orbital Hamilton population (COHP) analysis (Figs. 2b and S6) confirms this bond-strengthening effect, with Ce-Co TCPP exhibiting a significantly more negative ICOHP (–1.28 vs. –0.65 for Al-Co) in the spin-up channel, indicating stronger Co–O bonding32. Moreover, orbital-resolved DOS analysis (Fig. 2c, d) reveals that upon coordination with the oxo dianion (O2−) (Fig. S7), Ce-Co TCPP exhibits a higher density of unoccupied Co–O antibonding orbitals above the Fermi level, thereby further increasing the Co–oxo bond order. Collectively, these theoretical results demonstrate that long-range electronic modulation induced by Ce(IV)-based oxide linkers enabled the generation of additional unoccupied Co 3d states and reduced the electronic occupancy of Co–O antibonding π* orbitals during Co(IV) = O formation, thereby effectively circumventing the “oxo-wall” constraint16 (Fig. 2e).

a Density of states (DOS) of Co 3d orbitals for Al-Co tetra(4-carboxyphenyl) porphyrin (TCPP) and Ce-Co TCPP. b Crystal orbital Hamilton population (COHP) analysis of the Co–O orbital bonding. c, d DOS of Co 3d orbitals and O 2p orbitals for the Co–O bonding in Al-Co TCPP and Ce-Co TCPP. e Schematic bonding formation of the Co–O bonding for Al-Co TCPP and Ce-Co TCPP. Source data are provided as a Source Data file.
Synthesis and characterization of Ce-Co TCPP
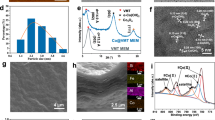
Guided by DFT simulations, we synthesized Ce-Co TCPP nanosheets and a corresponding two-dimensional (2D) lamellar membrane as an effective strategy to achieve Co(IV) = O species-dominated Fenton-like oxidation (Fig. 3a)33,34. The 2D Ce-Co TCPP catalyst was fabricated via a solvothermal approach, utilizing Ce oxide clusters as metal linkers that coordinate with H2TCPP organic ligands to form the Ce TCPP framework with four pyrrolic nitrogen coordination sites33. Subsequently, Co atoms were atomically integrated into the N4 coordination sites within the porphyrin ring, resulting in a precisely defined Co–N4 structure24. Upon exfoliation, scanning electron microscopy (SEM) indicated that the as-prepared Ce TCPP possessed distinct 2D structural characteristics (Fig. 3b), which were retained after cobalt modification (Fig. 3c). Atomic force microscopy (AFM, Fig. 3d) further validated the 2D nature of Ce-Co TCPP nanosheets, with lateral dimensions around 500 nm and a thickness of approximately 4.69 nm, highlighting a high aspect ratio. Advanced nanoscale characterization techniques confirmed the atomic dispersion of Co within the Ce-Co TCPP framework. High-resolution transmission electron microscopy (HR-TEM, Fig. 3e) demonstrated a homogeneous crystalline structure with no observable metal clusters or agglomeration, consistent with X-ray diffraction (XRD) results (Fig. S8). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Fig. 3f) unequivocally verified atomically dispersed Co species. Complementary elemental mapping confirmed uniform spatial distribution of C, N, O, Ce, and Co throughout the nanosheets (Fig. 3g). Nitrogen adsorption-desorption isotherms of Ce-Co TCPP nanosheets measured at 77 K (Fig. S9a), analyzed via Brunauer–Emmett–Teller (BET) and non-local density functional theory methods, revealed a pore volume of 0.093 cm3/g and pore size distribution ranging between 0.5 and 1.3 nm for Ce-Co TCPP nanosheets (Fig. S9b and Table S1). Additionally, the resulting Ce-Co TCPP nanosheets exhibited colloidal dispersibility, remaining stable in ethanol suspension for more than one month (Fig. S10). Control samples (Al-Co TCPP and Al TCPP) were synthesized analogously by replacing Ce with Al as the central metal. The Al-Co TCPP material was characterized under identical conditions (Fig. S11)34.

a Schematic diagram for the preparation of 2D Ce-Co TCPP nanosheets and lamellar membrane. b SEM image of Ce TCPP nanosheets (inset: photograph of the Ce TCPP nanosheet suspension). c SEM image of Ce-Co TCPP nanosheets (inset: photograph of the Ce-Co TCPP nanosheet suspension). d AFM image, e HR-TEM image, f HADDF-STEM image, and g EDX elemental mapping images of Ce-Co TCPP nanosheets. h XANES (inset: magnification of local areas) and i Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra of Co in R-space for Ce-Co TCPP, Al-Co TCPP, and references. j Wavelet-transformed (WT)-EXAFS plots of Co K-edge for different samples. k Corresponding FT-EXAFS fitting of Co K-edge in R-space of Ce-Co TCPP (inset: theoretical model structure). Blue, brown, red, white, gray, and yellow-green spheres represent Co, C, O, H, N, and Ce atoms, respectively. Source data are provided as a Source Data file.
Chemical state and atomic structure of Ce-Co TCPP
The chemical composition and coordination environment within Ce-Co TCPP were systematically investigated by spectroscopic methods. UV–Vis absorption spectra (Fig. S12) of Ce TCPP and Ce-Co TCPP displayed a characteristic Soret band at approximately 420 nm, confirming the preservation of the tetrapyrrole porphyrin macrocycle. Importantly, the incorporation of Co atoms into the porphyrin center reduced the number of Q-band peaks from four (Ce TCPP) to one (Ce-Co TCPP) due to an enhanced molecular symmetry upon metal coordination35. Fourier transform infrared spectroscopy (FT-IR, Fig. S13) provided further structural confirmation, evidenced by the diminished C = O stretching vibration at 1700 cm−1, indicative of successful ligand-metal coordination forming the Ce-Co TCPP framework36. Additionally, the combined information, including the disappearance of the N–H stretching vibration at 3315 cm−1, attenuation of the in-plane N–H peak at 964 cm−1, and emergence of a new absorption band at approximately 1000 cm−1 strongly supported the formation of the Co–N4 coordination structure36. X-ray photoelectron spectroscopy (XPS, Fig. S14a) elucidated the chemical states and coordination environments of key elements. Ce 3d spectra (Fig. S14b) revealed the coexistence of Ce3+ (45.33%) and Ce(IV) (54.67%), supporting the DFT simulation results that indicate the presence of a high oxidation state of Ce(IV) in the Ce-Co TCPP37. While Co 2p spectra (Fig. S14c) demonstrated characteristic peaks at 781.8 eV and 796.8 eV (Co2+, 71.18%) and 780.6 eV and 795.7 eV (Co3+, 28.80%)24. Additionally, C 1 s and O 1 s spectra confirmed the presence of C–C, C–O, and C = O functional groups (Fig. S14d, e), while the fitted N 1 s peaks at 400.1 eV and 398.5 eV (Fig. S14f) confirmed Co–N structure in the porphyrin macrocycle24,29. In particular, the clear 0.3 eV shift of the Co 2p peak to higher binding energies indicates that the construction of Ce oxide clusters within the Ce-Co TCPP framework (Fig. S15) promotes electronic delocalization at Co sites, markedly lowering their local electron density32. X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses provided further insights into the chemical states and coordination geometries. Co K-edge XANES analysis revealed that both Ce-Co TCPP and Al-Co TCPP exhibited absorption energies higher than that of CoO (Fig. 3h), suggesting Co oxidation states above +224,28. Notably, Ce-Co TCPP displayed even higher absorption energies than Al-Co TCPP, indicative of more pronounced electronic delocalization. FT-EXAFS analyses revealed a prominent coordination peak around 1.44 Å corresponding to Co–N scattering paths, confirming the Co–N first-shell coordination in both Ce-Co TCPP and Al-Co TCPP structures (Fig. 3i)24,28. The absence of detectable peaks at ~2.18 Å (Co–Co scattering) or ~1.64 Å (Co–O scattering) excluded the presence of metallic cobalt or oxide aggregates. Wavelet-transform (WT) contour plots further supported the atomic-level dispersion of Co in a Co–N coordination environment, matching closely with the cobalt phthalocyanine (CoPc) reference (Fig. 3j). Finally, FT-EXAFS fitting in R-space aligned accurately with the theoretical Co–N4 scattering model, confirming that cobalt atoms occupy the N4 coordination sites in both Ce-Co TCPP and Al-Co TCPP structures (Figs. 3k and S16 and Table S2)28.
PMS activation and reactive species analysis
To systematically evaluate the PMS activation performance of Ce-Co TCPP and Al-Co TCPP catalysts in heterogeneous systems, the degradation kinetics of ranitidine (RNTD) as the model pollutant were thoroughly investigated. Initial adsorption tests revealed a negligible (<5%) contribution to RNTD removal for both catalysts (Fig. 4a). Additionally, pristine Ce TCPP showed negligible catalytic activity for PMS activation, with the performance level similar to the background degradation due to PMS self-decomposition (Fig. 4a). Remarkably, the Ce-Co TCPP/PMS system achieved near-complete degradation of RNTD within only 4 min, exhibiting observed rate constant (kobs) of 0.0265 s−1—approximately 4.1-fold greater than that obtained in the Al-Co TCPP/PMS system (Figs. 4a and S17). Meanwhile, the comparable atomic ratios of Co in both Al-Co TCPP and Ce-Co TCPP catalysts (Table S3) indicate that Co content is unlikely to be the primary factor influencing the observed differences in pollutant degradation rates. Electron paramagnetic resonance (EPR) spectroscopy was employed to identify potential reactive oxygen species (ROS) formed during PMS activation (Fig. 4b). To ensure experimental reliability and avoid misleading conclusions, periodic in-situ EPR measurements were conducted38. As expected, no evident ROS signals were detected for Ce TCPP/PMS system using spin-trapping agents such as 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), 2,2,6,6-tetramethylpiperidine (TEMP), and DMPO dissolved in methanol, aligning with its limited catalytic activity (Fig. S18). Conversely, multiple ROS signals, including DMPO-•OH adducts, DMPO-SO4•– adducts, DMPO-O2•– adducts, and TEMP-1O2 adducts, were detected prominently in the Al-Co TCPP/PMS system (Figs. 4b and S19). Intriguingly, only the characteristic 1O2 triplet peak appeared in the Ce-Co TCPP/PMS system. (Figs. 4b and S20). These results suggested that the distinct catalytic selectivity of Ce-Co TCPP and Al-Co TCPP toward PMS accounts for the differences in both the composition and concentration of reactive species within their respective systems. The introduction of quenching agents into the pollutant degradation system induced competitive reactions between the quenchers and ROS, inhibiting the degradation of the target pollutant (Table S4)3,38,39. Therefore, subsequent quenching experiments were carried out to qualitatively evaluate the roles of specific ROS in RNTD degradation (Fig. S21). Different quenching agents at varied concentrations were tested to enhance interpretative accuracy and avoid misleading conclusions due to quencher selectivity overlaps (Table S4). Specifically, methanol (MeOH) exhibited less than 15% inhibition of RNTD removal in the Ce-Co TCPP/PMS system, while tert-butanol (TBA) displayed negligible suppression. These findings suggest that •OH and SO4•–—dominant in the Al-Co TCPP/PMS system (as evidenced by 87.2% and 79.1% inhibition of RNTD removal by MeOH and TBA, respectively)—are not the primary reactive species in the Ce-Co TCPP/PMS system (Fig. 4c). Likewise, the negligible inhibition observed upon chloroform (CHCl3) addition excluded O2•– as a significant contributor for the Ce-Co TCPP/PMS system, while it contributed substantially (~60% suppression) in the Al-Co TCPP/PMS system. Additionally, the removal of RNTD was suppressed to varying extents by the introduction of L-histidine (L-his) and ferrous sulfate (FFA) into both systems (Fig. 4c), suggesting potential involvement of singlet oxygen (1O2). Moreover, the degradation kinetics of RNTD remained consistent in both H2O and D2O solvents (Fig. S22), despite the significantly longer lifetime of 1O2 in D2O (20–32 µs) compared to H2O (2 µs), indicating the limited role of 1O2 in the Ce-Co TCPP/PMS system10. To probe for electron transfer pathways, PMS and RNTD were introduced into separate chambers. The near-constant current (Fig. S23a) and minimal RNTD depletion (Fig. S23b) demonstrate that this pathway is negligible. Collectively, these results establish a fundamental dichotomy: while conventional reactive species (•OH, SO4•–, 1O2, and O2•–) dominate oxidation processes in the Al-Co TCPP/PMS system, they contribute negligibly in the Ce-Co TCPP/PMS system. This mechanistic divergence motivated our focus on high-valent cobalt-oxo complexes (Co(IV) = O) as the primary reactive species.

a Ranitidine (RNTD) removal performance in various systems. b Qualitative analysis of reactive species based on electron paramagnetic resonance (EPR) spectra in Ce-Co TCPP/PMS and Al-Co TCPP/PMS systems. Dot symbol sizes represent the concentration of reactive species. c Comparison of RNTD removal efficiencies under different quenching conditions in Ce-Co TCPP/PMS and Al-Co TCPP/PMS systems. d In-situ Raman spectra of Ce-Co TCPP/PMS and Al-Co TCPP/PMS systems. e Degradation profiles of different probe organic compounds in the Ce-Co TCPP/PMS system. f Degradation of methyl phenyl sulfoxide (PMSO) and formation of PMSO2 in the Ce-Co TCPP/PMS system. g Steady-state concentrations of various reactive species in the Ce-Co TCPP/PMS system (inset: oxidation contributions of different reactive species to RNTD degradation). Reaction condition: [Ce-Co TCPP] = 20 mg/L; [RNTD] or [probe organic compounds] = 5 mg/L; [PMSO] = 0.15 mM; [peroxymonosulfate (PMS)] = 0.2 mM; Temperature = 298.15 K; pH = 4.3. Data are presented as mean values, and error bars represent the standard deviation calculated from three independent measurements (n = 3). Source data are provided as a Source Data file.
Identification of Co(IV) = O species in Ce-Co TCPP
Methyl phenyl sulfoxide (PMSO), which exhibits a very high second-order rate constant (2.0 × 106 M−¹s−¹) for reacting with Co(IV) = O species and a much lower rate with PMS (0.1 M−¹s−¹), was employed as an alternative scavenger to demonstrate the contribution of Co(IV) = O species in the catalytic system (Fig. S24)40. Although PMSO effectively inhibited RNTD removal in the Al-Co TCPP/PMS system (Fig. 4c), this observation alone can not definitively attribute the activity to Co(IV) = O in the Al-Co TCPP/PMS system. As shown in Table S4, PMSO is a known scavenger for both •OH and SO4•–, which were previously identified as the dominant reactive species in the Al-Co TCPP/PMS system (Fig. 4b, c). Furthermore, in-situ Raman spectroscopy failed to detect any characteristic signals of Co(IV) = O species in the Al-Co TCPP/PMS system (Fig. 4d)12. In marked contrast, in the Ce-Co TCPP/PMS system—characterized by negligible ROS formation—introduction of PMSO significantly suppressed approximately 90% of RNTD degradation (Figs. 4c and S21), strongly implying the prominent role of Co(IV) = O species14. Notably, in-situ Raman spectroscopy of the Ce-Co TCPP/PMS system identified a distinctive peak near 550 cm−1, explicitly indicative of Co(IV) = O species formation (Fig. 4d)12. To further validate and quantify Co(IV) = O production, several probe organic molecules including nitrobenzene (NB), benzoic acid (BA), para-chlorobenzoic acid (p-CBA), fluorescein isothiocyanate (FFA), and PMSO were employed to experimentally determine the steady-state concentrations of various reactive species ([•OH]ss, [SO4•–]ss, [1O2]ss, [O2•–]ss and [Co(IV) = O]ss) in the Ce-Co TCPP/PMS system (detailed in Text S1.11, Tables S5 and S6)10,41. Degradation profiles of NB and BA indicated negligible [•OH]ss, [SO4•–]ss (Figs. 4e and S25). Moderately enhanced degradation rates for p-CBA and FFA suggested a minor contribution from 1O2 (Fig. S25). Importantly, PMSO exhibited the highest observed reaction rate constant (kobs = 3.6 × 10−3 s−1) with nearly 100% selective oxidation to PMSO2 in the Ce-Co TCPP/PMS system (Figs. 4f and S26), confirming the presence of Co(IV) = O species. As summarized in Fig. 4g, the [Co(IV) = O]ss was determined to be 1.98 × 10−9 M, surpassing [•OH]ss, [SO4•–]ss, [1O2]ss, [O2•–]ss by 3–5 orders of magnitude. Additionally, quantitative contributions of Co(IV) = O species to the overall degradation of RNTD reached 97.84% (Fig. 4g). In conclusion, comprehensive evidence derived from EPR tests, quenching experiments, in-situ Raman spectroscopy, and steady-state ROS quantification robustly supports the dominant role of Co(IV) = O species in the Ce-Co TCPP/PMS system, accounting for the RNTD degradation efficiency via non-radical pathways.
Theoretical investigation of the catalytic mechanism
Previous theoretical predictions have demonstrated that enhanced electronic delocalization reinforces the Co–O bond order, laying a fundamental basis for the formation of Co(IV) = O species. To further elucidate this effect, we quantified the influences of electronic delocalization to the catalytic process governing Co(IV) = O generation. As illustrated in Fig. 5a, PMS spontaneously adsorbs onto Co sites of both Al-Co TCPP (left) and Ce-Co TCPP (right). Following adsorption, the O–O bond length of PMS elongates from its original value of 1.356 Å to 1.497 Å on Al-Co TCPP and 1.482 Å on Ce-Co TCPP, indicative of peroxide bond dissociation42. Interestingly, the more negative adsorption energy (Eads) of PMS on Al-Co TCPP (−2.69 eV) compared to Ce-Co TCPP (−2.53 eV) suggests a stronger interaction between PMS and Co active sites in the Al-Co TCPP structure. This discrepancy arises from intensified electronic delocalization at the Co center within Ce-Co TCPP, inducing a downward shift of the d-band center (−1.16 eV for Ce-Co TCPP vs. −0.59 eV for Al-Co TCPP), thereby weakening its affinity toward PMS during catalysis (Fig. S27)43. Nonetheless, electronic structure modulation and symmetry adjustments play critical roles in catalyst activity and selectivity44. As shown in Fig. S28, Ce-Co TCPP exhibits a higher magnetic moment (1.789 μB), reflecting a greater abundance of unpaired electrons compared to Al-Co TCPP (1.397 μB). EPR spectra further experimentally confirmed this theoretical insight, displaying intensified signal peaks for Ce-Co TCPP (Fig. S29). Collectively, these results reveal that Ce(IV)-mediated long-range modulations in Ce-Co TCPP promote greater electronic delocalization and more significant Co electronic structure/spin-state modifications relative to Al-Co TCPP14,29. Consequently, when PMS adsorbs onto the catalytic sites of Ce-Co TCPP, a greater extent of electron transfer occurs toward PMS (0.9149 e for Ce-Co TCPP vs. 0.8726 e for Al-Co TCPP) (Fig. 5a). This simulation result aligns well with the more pronounced current jump observed in the i-t curve of the Ce-Co TCPP/PMS system compared to the Al-Co TCPP counterpart (Fig. S30), suggesting a more favorable two-electron transfer pathway for the formation of Co(IV) = O species13. Furthermore, upon PMS addition, the open-circuit potential (Fig. S31) exhibited a markedly greater increase in the Ce-Co TCPP/PMS system than in the Al-Co TCPP/PMS system, underscoring significantly accelerated electron transfer kinetics and the potential generation of potent reactive complexes (i.e., Co(IV) = O) on the catalyst12.

a Differential charge density for PMS adsorption on Al-Co TCPP (left) and Ce-Co TCPP (right). Yellow and cyan regions indicate electron accumulation and depletion, respectively. b, c Schematic illustration of the reaction pathways for Co(IV) = O generation on Ce-Co TCPP and Al-Co TCPP via PMS activation, respectively. The (*) denotes the adsorption site. d Energy profiles of the reaction pathways. Blue, brown, red, white, gray, light blue, and yellow-green spheres represent Co, C, O, H, N, Al, and Ce atoms. Source data are provided as a Source Data file.
Next, to gain deeper mechanistic insights into PMS activation on Al-Co TCPP and Ce-Co TCPP, computational simulations of free energies associated with potential intermediates and reaction pathways were performed (Fig. 5b–d). Initially, PMS (Fig. 5b, c, state I) spontaneously adsorbs onto Co sites in both Al-Co TCPP and Ce-Co TCPP via one terminal peroxyl oxygen, forming the adsorbed intermediate PMS* (state II) (Fig. 5b, c). Subsequently, the adsorbed PMS* in the Ce-Co TCPP/PMS system follows a distinct activation pathway compared to the Al-Co TCPP/PMS system (Fig. 5b, c, state III). Notably, the proton in the H–O* bond of PMS* (HO*–O–SO3–) adsorbed on Ce-Co TCPP undergoes facile intramolecular transfer, forming O*–O–(H)SO3– (Fig. 5b, state III)12. In stark contrast, the proton in the H–O* bond of PMS* (HO*–O–SO3–) activation on Al-Co TCPP requires energetically demanding deprotonation (∆EII to III = 1.37 eV, Fig. 5d) for the formation of O*–O–SO3•– (Fig. 5c, state III). Optimization via the climbing-image nudged elastic band method (Fig. S32) revealed that despite encountering a transition state with an elevated energy barrier, the overall intramolecular proton transfer reaction follows a thermodynamically favorable path toward a lower-energy state in the Ce-Co TCPP/PMS system (∆EII to III = −1.38 eV, Fig. 5d), demonstrating a spontaneous and energetically favorable process45. This reaction is likely facilitated by a local electrostatic potential gradient generated by the coexistence of Ce(III) and Ce(IV), which enhances O–H bond polarization, promoting spontaneous intramolecular proton transfer12. Ultimately, the O–O bond in O*–O–(H)SO3− cleaves (Fig. 5b, state IV), driven by substantial electron donation from Co sites on Ce-Co TCPP, yielding Co(IV) = O species (∆Etotal = −1.45 eV, Fig. 5d). The energy required for this final bond cleavage is likely offset by the energy released in the preceding proton transfer step. In contrast, the corresponding reaction pathway in the Al-Co TCPP/PMS system is energetically unfavorable for forming Co(IV) = O species (∆Etotal = 0.73 eV; Fig. 5d) but favors radical formation (∆E = −1.26 eV; Fig. S33)46. Overall, varying degrees of long-range electronic modulation govern the extent of electronic delocalization at Co catalytic centers, thereby steering PMS-based Fenton-like processes along distinct mechanistic trajectories. A comprehensive understanding of these phenomena offers valuable insights into the effect of higher coordination shells in modulating catalytic selectivity and mediating the preferential generation of specific reactive species.
Catalytic performance of 2D lamellar Ce-Co TCPP membrane
From a practical application perspective, a Ce-Co TCPP membrane with a smooth, defect-free surface (Fig. 6a) was fabricated via vacuum filtration to evaluate its catalytic performance toward pollutants in aqueous environments. The 2D Ce-Co TCPP nanosheets were assembled into a well-defined lamellar architecture (Fig. 6b), forming interconnected nanochannels that significantly enhance mass transport42. The relationship between nanosheet concentration and absorbance was used to determine the loading of membranes prepared at different suspension concentrations (Fig. S34), while cross-sectional (Fig. S35) and surface (Fig. S36) SEM images were employed to establish the correlation between membrane loading and thickness (Fig. S37). The Ce-Co TCPP membrane/PMS system with different catalyst loadings consistently demonstrated efficient degradation for RNTD, accompanied by different water permeability (Figs. 6c and S38). Among them, the membrane with an optimal catalyst loading of 2.4 μg mm−2 (denoted as Ce-Co TCPP MEM) was selected for further investigation due to the nearly complete RNTD removal and appropriate water permeance (126.97 LMHB). As depicted in Fig. 6d, the Ce-Co TCPP MEM alone (in the absence of PMS) exhibited only 20.24% RNTD removal efficiency. Meanwhile, lowering the pH to 4.3 (matching 0.2 mM PMS conditions) did not significantly enhance ranitidine removal by the Ce-Co TCPP membrane, indicating minimal impact from the Donnan effect or surface charge interactions (Fig. S39). Impressively, the introduction of PMS into the membrane system resulted in nearly complete RNTD degradation within 1 min, achieving a kobs of 0.0717 ms−1 (calculation details are provided in Text S1.12 and Fig. S40)47. This value is approximately 2700-fold higher than that observed in the corresponding heterogeneous Ce-Co TCPP/PMS suspension system (Figs. 6e and S17), and significantly outperforms other state-of-the-art.

a SEM image of the surface of the Ce-Co TCPP membrane (MEM). (inset: photograph of the Ce-Co TCPP MEM). b SEM image of the cross-section of the Ce-Co TCPP MEM. (inset: local magnification). c Water permeance and RNTD removal efficiency of the Ce-Co TCPP membrane/PMS system at different catalyst loadings. d RNTD removal efficiency by various treatment systems. e Correlation between RNTD removal efficiency and retention time (inset: pseudo-first-order kinetic model fitting of RNTD degradation in the Ce-Co TCPP MEM/PMS system). f Comparison of pseudo-first-order rate constants for RNTD removal by different Fenton-like systems (Details are shown in Table S7, and reference numbers herein represent the references listed in Table S7). g RNTD removal performance of the Ce-Co membrane/PMS system under different treatment conditions. h Selective removal of various pollutants by the Ce-Co TCPP MEM/PMS system. i Long-term stability of water permeance and removal efficiency under continuous operation. Conditions for all membrane tests: [Ranitidine] or [Other pollutants] = 5 mg/L; [PMS] = 0.2 mM; pH = 4.3; [Anions] = 10 mM; Pressure = 1 bar; Temperature = 298.15 K. Data are presented as mean values, and error bars represent the standard deviation calculated from three independent measurements (n = 3). Source data are provided as a Source Data file.
Environmental applicability of 2D lamellar Ce-Co TCPP membrane
A series of immersion experiments were conducted to investigate the structural stability and robustness of the Ce-Co TCPP MEM, an essential factor for the practical long-term application. After immersing the catalyst in 0.2 mM PMS solution for 10 days, the Ce-Co TCPP nanosheets consistently maintained their porous structure without noticeable degradation (Fig. S41). Moreover, minimal leaching of Ce and Co ions further underscored the stability of the metal-organic coordination frameworks (Table S8). Both the negligible changes of the macroscopic lamellar structure and microscopic lattice integrity of the Ce-Co TCPP MEM after 10 days of immersion were confirmed by SEM (Fig. S42) and XRD results (Fig. S43), respectively. Furthermore, degradation experiments under diverse operational conditions and real-world environmental water matrices were performed to evaluate the practical applicability of the Ce-Co TCPP MEM/PMS system. The effective degradation of RNTD over a wide range of water quality conditions highlighted the broad adaptability of the system across pH 4 to 9 (Fig. S44), substantial resistance to coexisting anions (Fig. 6g), and sustained high degradation of RNTD in both lake and tap water matrices (Fig. S45 and Table S9). These findings suggest that the targeted generation of Co(IV) = O species significantly contribute to the improved stability and practical utility of the Ce-Co TCPP MEM. Additionally, consistent PMS consumption (~80.76%) and high mineralization rates (61.29% in lake water and 63.69% in tap water) further demonstrate the robustness of the system (Fig. S45). Importantly, the Ce-Co TCPP MEM/PMS system exhibited selective oxidation towards pollutants containing electron-donating functional groups, such as hydroxyl and amine groups, efficiently degrading contaminants including oxytetracycline, tetracycline, moxifloxacin, carbamazepine, and ofloxacin (Figs. 6h and S46), aligning well with previously reported Co(IV) = O catalytic systems8,12,13. However, the removal efficiency for electrophilic pollutants such as BA and NB remained below 10% (Fig. 6h), consistent with their intrinsic electrophilic nature and the nucleophile-selective oxidation mechanisms of the dominant reactive species of Co(IV) = O in the Ce-Co TCPP/PMS system12,13. The toxicological assessment encompassed fish, daphnia, and algae47. As illustrated in Fig. S47, eight degradation intermediates of ranitidine (P1–P8) were identified (Table S10), with proposed transformation pathways in the Ce-Co TCPP MEM/PMS system including oxygen addition, dehydrogenation, denitrification, and oxidative ring-opening reactions (Fig. S48). Toxicity predictions indicated that these intermediates exhibited significantly reduced toxicity and were considered non-harmful (Fig. S49). Moreover, actual toxicity analysis using luminescent bacteria (Vibrio fischeri) showed that water samples treated for 30 min in the Ce-Co TCPP MEM/PMS system displayed inhibition rates below 3%, in stark contrast to the higher inhibition rates observed in untreated samples (0 min), confirming effective pollutant degradation and substantial reduction of toxicity48 (Fig. S50).
The Ce-Co TCPP MEM/PMS system maintained stable water permeability (128.3 LMHB) and sustained efficient removal of RNTD over an extended operation of 95 h under continuous cross-flow filtration conditions (Fig. 6i). Notably, Co ion concentrations in the permeate (89.4 μg L−1) (Table S11) remained significantly below the regulatory discharge standard in China (1.0 mg L−1, GB 25467-2010)49. This long-term operational stability originates from the synergistic effect between Co(IV) = O-dominated Fenton-like oxidation and the 2D lamellar structure of Ce-Co TCPP MEM, which mitigates membrane fouling and concentration polarization through direct oxidative degradation, ensuring stable flux performance42. Simultaneously, the single-atom Co active sites within the nanoconfined channels effectively prevent catalyst aggregation and maximize the exposure of active sites, thereby supporting prolonged efficient pollutant removal24. The Co 2p XPS spectra of the deactivated Ce-Co TCPP membrane were fitted and compared with those of the fresh sample. A marked decrease in the proportion of Co2+ species was observed after long-term operation, which correlates with the diminished RNTD removal efficiency after 95 h (Fig. S51 and Table S12). During prolonged operation, the sluggish reduction of Co3+ species led to a significant decline in the Co2+ fraction, thereby suppressing the formation of active Co(IV) = O species via the two-electron transfer pathway and ultimately resulting in catalyst deactivation. In contrast, the oxidation state of Ce remained essentially unchanged after extended use (Fig. S52 and Table S13), indicating that Ce did not directly participate in the catalytic redox process. Meanwhile, the membrane maintained stable water permeance, confirming that its structural integrity was preserved. As such, the performance loss was primarily originated from the deactivation of catalytic sites rather than membrane deterioration47. Importantly, mild chemical reduction (Fig. S53) effectively restored the catalytic activity, further confirming that the depletion of Co2+ species is the primary cause of deactivation for the Ce-Co TCPP membrane and demonstrating the feasibility of this regeneration strategy for long-term reusable operation. Moreover, the feasibility of large-scale production of the Ce-Co TCPP membrane was also demonstrated (Fig. S54a). The scaled-up Ce-Co TCPP membrane achieved nearly 100% removal efficiency for RNTD with a water permeability of 138 LMHB (Fig. S54), confirming that the catalytic activity was retained after scale-up.
Theoretical insights of nanoconfinement effects
Compared to an unrestricted batch system, the encapsulation of reactants within the nanostructured channels of the Ce-Co TCPP MEM enhances their local concentration by reducing the reaction volume, while maintaining the overall quantity of reactants. Furthermore, the confined spatial environment significantly reduces the diffusion distance and increases the collision frequency between reactants and active catalytic sites42,50. Specifically, during permeation through the Ce-Co TCPP MEM, both PMS and target pollutants are constrained to navigate the abundant 2D nanochannels formed between adjacent Ce-Co TCPP nanosheets, which are densely populated with atomically dispersed Co–N4 catalytic sites. Such configuration notably improves accessibility to catalytic centers and facilitates degradation reactions47,50. To verify the proposed mechanisms, MD simulations were conducted to elucidate the diffusion and interaction behaviors of PMS, ranitidine, and water within the Ce-Co TCPP MEM. As illustrated in Fig. 7a, PMS molecules (0.315 × 0.305 × 0.350 nm), ranitidine molecules (0.570 × 0.460 × 1.68 nm), and water molecules successfully penetrate the nanochannels (1.25 nm) within the Ce-Co TCPP MEM (Fig. S55). The nanochannel confinement effect significantly enhances PMS concentration, with local mass density reaching 0.05 g/cm3 in 1.25 nm channels—approximately 1.3–2.5 times higher than in larger spacings (0.038 g/cm3 for 5 nm and 0.02 g/cm3 for 10 nm channels). Furthermore, the Mean square displacement (MSD) analysis (Fig. 7b) reveals that the diffusion distance of PMS molecules to catalytic sites is significantly shortened within the 1.25 nm nanochannels compared to larger channels, substantially increasing the collision frequency between PMS molecules and catalytic sites47. Such elevated PMS concentration and intensified collision frequency lead to an approximately four-order-of-magnitude higher steady-state concentration of Co(IV) = O species in the Ce-Co TCPP MEM/PMS system compared to the conventional heterogeneous system (Figs. 7c and S56). Ultimately, pollutants such as RNTD are effectively degraded with significantly enhanced reaction kinetics during their permeation through the 2D lamellar Ce-Co TCPP MEM (Fig. 7d)47,50.

a Molecular dynamics (MD) simulation of the diffusion behavior of PMS, RNTD, and H2O molecules, along with the mass density distribution of PMS within 2D lamellar Ce-Co TCPP channels at three different interlayer free spacings (1.25, 5, and 10 nm). Blue, red, white, yellow, black, light blue, and yellow-green represent N, O, H, S, C, Co, and Ce, respectively. b Mean square displacement (MSD) curves of PMS molecules within the Ce-Co TCPP MEM. c Steady-state concentrations of various reactive species in different Fenton-like systems. d Schematic illustration of mass transport behavior in the Ce-Co TCPP MEM/PMS system during the degradation of RNTD. Source data are provided as a Source Data file.
Discussion
This work addressed the challenging formation of Co(IV) = O species by exploiting electronic modulation in Ce-Co TCPP. Computational predictions underscored the essential role of Ce(IV)-induced electronic delocalization around the Co–N4 structure through long-range interactions, effectively reducing electron occupancy in Co–O antibonding orbitals, thus overcoming the intrinsic constraints imposed by the “oxo-wall” rule. Complementary experimental evidence, including EPR spectroscopy, quenching tests, in-situ Raman characterization, and steady-state ROS quantification, consistently validated the dominant formation of Co(IV) = O species in the Ce-Co TCPP/PMS system. DFT simulations further clarified that enhanced electronic delocalization in Ce-Co TCPP promotes electron transfer from Co sites to PMS, accompanied by a thermodynamically favorable intramolecular proton transfer mechanism within the adsorbed PMS intermediate, significantly lowering the overall reaction barrier and promoting the generation of Co(IV) = O species. Practically, the engineered Ce-Co TCPP MEM/PMS system exhibited considerable water permeability (126.97 LMHB), efficient degradation of RNTD (0.0717 ms−1), and long-term operational stability (95 h), leveraging nanoconfinement effects within its 2D nanochannels and the intrinsic activity of the Co(IV) = O complexes. MD simulations confirmed that this nanoconfinement significantly shortens diffusion pathways and increases local concentration of Co(IV) = O complexes, markedly accelerating RNTD degradation kinetics. This study presents a promising strategy for designing efficient, sustainable Co(IV) = O-dominated Fenton-like water treatment processes.
Methods
Materials and reagents
Reagents and materials are provided in Supplementary Materials.
Synthesis of Ce TCPP nanosheets
The Ce TCPP nanosheets were synthesized using a combined method that incorporates thermal solvent processes, ultrasound treatment, and centrifugation. Initially, 0.3 mM (93.23 mg) of Ce(NO3)3·6H2O was dissolved in a mixed solvent of 25 mL ethanol and 75 mL N,N-dimethylformamide (DMF), followed by sonication for 10 min at room temperature. Subsequently, 400 µL of a 60 wt% acetic acid aqueous solution was added, and the mixture was sonicated for an additional 5 min to ensure complete dissolution of the Ce precursor. Next, the ligand solution was prepared by dispersing 50 mg of H2TCPP in 50 mL DMF, with 30 min of sonication to achieve a uniform dispersion. The Ce precursor solution (8 mL) and the ligand solution (4 mL) were then combined in a 20 mL capped vial, which was sealed and heated to 80 °C for 16 h in an oil bath, without stirring. After the reaction, a purple Ce TCPP bulk solution was obtained by centrifugation at 8000 rpm for 10 min, followed by washing with DMF and ethanol. Monolayer Ce TCPP nanosheets were then isolated by sonicating the Ce TCPP bulk material in DMF for 1 h, followed by centrifugation at 2690 × g for 30 min. The resulting suspension containing Ce TCPP nanosheets was used for the subsequent synthesis of Ce-Co TCPP. The synthesis of Al TCPP followed a similar procedure, with the substitution of Ce(NO3)3·6H2O by Al(NO3)3·9H2O, and the replacement of 400 µL of 60 wt% acetic acid with 7 mg of pyrazine.
Preparation of Ce-Co TCPP nanosheets
Ce-Co TCPP nanosheets were synthesized via a post-synthetic modification of Ce TCPP in DMF, whereby cobalt atoms were incorporated into the porphyrin rings of Ce TCPP, resulting in the formation of the Co–N4 coordination moieties. Specifically, 0.2 mM (93.23 mg) of Co(NO3)2·6H2O was introduced into 50 mL of a 1 mg/mL Ce TCPP solution. The mixture was subjected to sonication for 20 min to ensure homogeneity, then transferred to a 100 mL Teflon-lined autoclave, where it was heated at 120 °C for 24 h. Upon completion of the reaction, the resultant bulk product was isolated by centrifugation and thoroughly washed with DMF and ethanol to remove any unreacted metal ions and residual by-products. Monolayer Ce-Co TCPP nanosheets were then obtained by sonicating the Ce-Co bulk material in ethanol for 1 h, followed by centrifugation at 2690 × g for 30 min to separate the nanosheets from the bulk phase. The synthesis of Al-Co TCPP was carried out using a similar methodology, with the Ce TCPP solution being substituted by an Al TCPP solution.
Fabrication of 2D lamellar membranes
The 2D lamellar Ce-Co TCPP membranes were synthesized through vacuum filtration. A diluted suspension of Ce-Co TCPP nanosheets (0.1 mg/mL) in ethanol was filtered onto a hydrophilic PVDF membrane with a pore size of 0.22 μm and a diameter of 47 mm. The high aspect ratio 2D Ce-Co TCPP nanosheets spontaneously self-assembled into a compact, lamellar catalytic membrane driven by van der Waals interactions. The catalyst loading (mg·cm−2) could be precisely controlled by varying the volume of the suspension of Ce-Co TCPP nanosheets during filtration. Importantly, the concentration of the suspension was determined using the absorbance-concentration relationship (Lambert-Beer law) rather than by drying and re-dispersing the powdered sample, ensuring optimal dispersion of the suspension. The resulting catalytic membrane was subsequently dried overnight at 60 °C in a vacuum oven prior to characterization and testing.
Experimental test of catalytic performance
Further details are provided in Supplementary Materials.
Analytical methods
Detailed information on the characterization techniques (SEM, TEM, AFM, HAADF-STEM, UV, XRD, BET, XPS, FTIR, XANES, and XAFS), and analytical methods (steady-state ROS concentration measurement, the PMS concentration, in-situ Raman spectroscopy, the analysis of the reaction intermediates and toxicity evaluation, the open-circuit potential, i-t analysis, two-chamber galvanic oxidation process, DFT, and MD simulations) are provided in Supplementary Materials.
Statistics and reproducibility
All micrographs were acquired by randomly selecting regions from independently prepared samples. Imaging was repeated across different sample areas and consistently yielded similar microstructural features. The most representative images were chosen for display. All experimental data are presented as mean values, and error bars represent the standard deviation calculated from three independent measurements (n = 3).
Data availability
The data generated in this study are provided in the Supplementary Materials/source data file. All the raw data relevant to the study are available from the corresponding author upon request. Source data are provided with this paper. The optimized DFT computational models and molecular dynamics trajectories generated in this study are publicly available on GitHub: https://github.com/JomerMatirays/Breakingtheoxo-wallforCo-IV--oxospeciesandtheirnanoconfinedcatalyticperformancewithinCe-Comembrane/. These data are also permanently archived in the Zenodo repository (https://doi.org/10.5281/zenodo.17895247). Source data are provided with this paper.
References
Duan, P. J. et al. Polymeric products deactivate carbon-based catalysts in catalytic oxidation reactions. Nat. Water. 3, 178–190 (2025).
Li, S., Zhu, Y., Zhong, G., Huang, Y. & Jones, K. C. Comprehensive assessment of environmental missions, fate, and risks of veterinary antibiotics in China: an environmental fate modeling approach. Environ. Sci. Technol. 58, 5534–5547 (2024).
Zhao, X. & Zhang, Z. Heterogeneous peroxymonosulfate-based advanced oxidation mechanisms: new wine in old bottles? Environ. Sci. Technol. 59, 5913–5924 (2025).
Gu, C. H. et al. Tuning electronic structure of metal-free dual-site catalyst enables exclusive singlet oxygen production and in-situ utilization. Nat. Commun. 15, 5771 (2024).
Chen, Z. et al. Organic carbon transfer process in advanced oxidation systems for water clean-up. Nat. Water. 3, 334–344 (2025).
Chen, T. et al. Robust Fe-N4-C6O2 single atom sites for efficient PMS activation and enhanced FeIV=O reactivity. Nat. Commun. 16, 2402 (2025).
Wu, Z. et al. Long-range interactions driving neighboring Fe-N4 sites in Fenton-like reactions for sustainable water decontamination. Nat. Commun. 15, 7775 (2024).
Wang, Z. et al. Aqueous iron(IV)-oxo complex: an emerging powerful reactive oxidant formed by iron(II)-based advanced oxidation processes for oxidative water treatment. Environ. Sci. Technol. 56, 1492–1509 (2022).
Huang, M. et al. Facilely tuning the intrinsic catalytic sites of the spinel oxide for peroxymonosulfate activation: from fundamental investigation to pilot-scale demonstration. Proc. Natl. Acad. Sci. USA 119, e2202682119 (2022).
Wu, Q. Y., Yang, Z. W., Wang, Z. W. & Wang, W. L. Oxygen doping of cobalt-single-atom coordination enhances peroxymonosulfate activation and high-valent cobalt-oxo species formation. Proc. Natl. Acad. Sci. USA 120, e2219923120 (2023).
Jiang, J. et al. Nitrogen vacancy-modulated peroxymonosulfate nonradical activation for organic contaminant removal via high-valent cobalt-oxo species. Environ. Sci. Technol. 56, 5611–5619 (2022).
Song, J. et al. Asymmetrically coordinated CoB1N3 moieties for selective generation of high-valence Co-oxo species via coupled electron-proton transfer in Fenton-like reactions. Adv. Mater. 35, 2209552 (2023).
Shi, Z. et al. High-entropy effect breaking the oxo wall for selective high-valent metal−oxo species generation. ACS Catal 14, 14796–14806 (2024).
Li, X. et al. CoN1O2 Single-atom catalyst for efficient peroxymonosulfate activation and selective cobalt(IV)=O generation. Angew. Chem. Int. Ed. 62, e202303267 (2023).
Andris, E. et al. M−O bonding beyond the oxo wall: spectroscopy and reactivity of cobalt(III)-oxyl and cobalt(III)-oxo complexes. Angew. Chem. Int. Ed. 58, 9619–9624 (2019).
Larson, V. A., Battistella, B., Ray, K., Lehnert, N. & Nam, W. Iron and manganese oxo complexes, oxo wall and beyond. Nat. Rev. Chem 4, 404–419 (2020).
Guo, J. et al. Fenton-like activity and pathway modulation via single-atom sites and pollutants comediates the electron transfer process. Proc. Natl. Acad. Sci. USA 121, e2313387121 (2023).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 50, 5281–5322 (2021).
Wei, S. et al. Self-carbon-thermal-reduction strategy for boosting the Fenton-like activity of single Fe-N4 sites by carbon-defect engineering. Nat. Commun. 14, 7549 (2023).
Zhang, Y. et al. The role of long-range interactions between high-entropy single-atoms in catalyzing sulfur conversion reactions. Adv. Mater. 37, 2413653 (2025).
Qi, Z., Zhou, Y., Guan, R., Fu, Y. & Baek, J. B. Tuning the coordination environment of carbon-based single-atom catalysts via doping with multiple heteroatoms and their applications in electrocatalysis. Adv. Mater. 35, 2210575 (2023).
Lu, Y. et al. Recent advances in bonding regulation of metalloporphyrin-modified carbon-based catalysts for accelerating energy electrocatalytic applications. Small 20, 2406180 (2024).
Lv, N. et al. Electrocatalytic porphyrin/phthalocyanine-based organic frameworks: building blocks, coordination microenvironments, structure-performance relationships. Adv. Sci. 10, 2206239 (2023).
Yang, C. et al. Incorporation of atomically dispersed cobalt in the 2D metal-organic framework of a lamellar membrane for highly efficient peroxymonosulfate activation. Appl. Catal. B: Environ. 325, 122344 (2023).
Jin, Z. et al. Ultrafast photogenerated charge dynamics driven by synergistic dual-metal atomic sites in porphyrinic metal-organic frameworks. Appl. Catal. B: Environ. 379, 125670 (2025).
Zhang, Y., Guan, X., Meng, Z. & Jiang, H. L. Supramolecular built local electric field microenvironment around cobalt phthalocyanine in covalent organic frameworks for enhanced photocatalysis. J. Am. Chem. Soc. 147, 3776–3785 (2025).
Wang, Q. et al. Modulating active center microenvironment in phthalocyanine-based covalent organic frameworks for enhanced electrocatalytic CO2 to CH3OH. Adv. Mater. 37, 2502644 (2025).
Zhang, J. et al. Suppressing the hydrogen bonding interaction with *OOH toward efficient H2O2 electrosynthesis via remote electronic tuning of Co-N4. Appl. Catal. B: Environ. 358, 124448 (2024).
Huang, M. et al. Linkage microenvironment and oxygen electroreduction reaction performance correlationship of iron phthalocyanine-based polymers. Angew. Chem. Int. Ed. 64, e202501506 (2025).
Jacobsen, J., Ienco, A., D’Amato, R., Costantino, F. & Stock, N. The chemistry of Ce-based metal-organic frameworks. Dalton Trans 49, 16551–16586 (2020).
Ma, Y. et al. Design of hierarchical CeO2@Co-Ni3S2 catalyst for rapid high-valent Ni3+ generation and optimized organic adsorption toward enhanced biomass conversion. Adv. Funct. Mater. 35, e12292 (2025).
Zhang, Y. L. et al. Electronic delocalization regulates the occupancy and energy level of Co 3dz2 orbitals to enhance bifunctional oxygen catalytic activity. Adv. Funct. Mater. 32, 2209499 (2022).
Jiang, Z. W. et al. Controllable synthesis of porphyrin-based 2D lanthanide metal-organic frameworks with thickness and metal-node-dependent photocatalytic performance. Angew. Chem. Int. Ed. 59, 3300–3306 (2020).
Jian, M. et al. Ultrathin water-stable metal-organic framework membranes for ion separation. Sci. Adv. 6, eaay3998 (2020).
Wang, J. et al. Light-responsive and ultrapermeable two-dimensional metal-organic framework membrane for efficient ionic energy harvesting. Nat. Commun. 15, 2125 (2024).
Zhang, Y. et al. Bidirectional light-driven ion transport through porphyrin metal-organic framework-based van der Waals heterostructures via pH-induced band alignment inversion. CCS Chem 4, 3329–3341 (2022).
Li, M. et al. Ce-induced differentiated regulation of Co sites via gradient orbital coupling for bifunctional water-splitting reactions. Adv. Energy Mater. 13, 2301162 (2023).
Wu, J. H. & Yu, H. Q. Confronting the mysteries of oxidative reactive species in advanced oxidation processes: an elephant in the room. Environ. Sci. Technol. 58, 18496–18507 (2024).
Lei, Y. et al. Assessing the use of probes and quenchers for understanding the reactive species in advanced oxidation processes. Environ. Sci. Technol. 57, 5433–5444 (2023).
Zong, Y. et al. High-valent cobalt-oxo species triggers hydroxyl radical for collaborative environmental decontamination. Appl. Catal. B: Environ. 300, 120722 (2022).
Pei, J. et al. Manipulating high-valent cobalt-oxo generation on Co/N cooped carbon beads via PMS activation for micropollutants degradation. ACS EST Eng 3, 1997–2007 (2023).
Tian, M. et al. Overcoming the permeability-selectivity challenge in water purification using two-dimensional cobalt-functionalized vermiculite membrane. Nat. Commun. 15, 391 (2024).
Zou, Y. et al. Unveiling the long-range interaction of sulfur in the second shell of Fe-N4 single-atom sites for highly selective generation of high-valent iron-oxo species in peroxymonosulfate activation. Chem. Eng. J. 505, 159684 (2025).
Cao, P. et al. Breaking symmetry for better catalysis: insights into single-atom catalyst design. Chem. Soc. Rev. 54, 3848–3905 (2025).
Fu, S. et al. Iron single-atom catalyst actuates PMS/O3 activation process: nonradical generation path for synergistic multi-peroxides. ACS Appl. Eng. Mater. 3, 202–213 (2025).
Lin, Y. et al. Coordination engineering of heterogeneous high-valent Fe(IV)-oxo for safe removal of pollutants via powerful Fenton-like reactions. Nat. Commun. 15, 10032 (2024).
Zhang, J., Liu, Y. & Zhang, Z. Nanoconfined catalytic water purification within CoFeCu LDH-assembled membrane nanochannels. Appl. Catal. B: Environ. 357, 124290 (2024).
Li, Y. et al. Peracetic acid-induced nanoengineering of Fe-based metallic glass ribbon in application of efficient drinking water treatment. Appl. Catal. B: Environ. 355, 124161 (2024).
Jiang, J. et al. Angstrom confinement-triggered adaptive spin state transition of CoMn dual single atoms for efficient singlet oxygen generation. Adv. Mater. 37, 2417834 (2025).
Meng, C. et al. Angstrom-confined catalytic water purification within Co-TiOx laminar membrane nanochannels. Nat. Commun. 13, 4010 (2022).
Acknowledgements
The research was supported by the National Natural Science Foundation of China (52170041, Z.Z.), Tsinghua SIGS Cross-disciplinary Research and Innovation Fund (JC2022006, Z.Z.), the Committee of Science and Technology Innovation of Shenzhen (JCYJ20230807111705011, Z.Z.), and the Guangdong Natural Science Fund for Distinguished Young Scholars (2024B1515020085, Z.Z.).
Author information
Authors and Affiliations
Contributions
Z.Z. conceived the project. M.T. synthesized and characterized the nanosheets and membranes and performed the catalytic activity experiments. L.L. and Z.C. helped with experiments and result analysis. C.Y. performed the XAFS analysis. H.Z. and Y.L. performed the theoretical calculations. M.T. and Z.Z. analyzed the results and wrote the manuscript. Z.Z. revised the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tian, M., Zhang, H., Liu, Y. et al. Breaking the oxo-wall for Co(IV)-oxo species and their nanoconfined catalytic performance within Ce-Co lamellar membrane. Nat Commun 17, 1767 (2026). https://doi.org/10.1038/s41467-026-68471-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68471-8
