
Abstract
Aerobic methane production from biological methylphosphonate degradation contributes to the observed supersaturation of oceanic methane in oxygenated surface waters. Atmospheric deposition is an important nutrient source and this process can affect nutrient ratios and biogeochemical cycle in the upper ocean. Here we present evidence that atmospheric nitrogen deposition modulates methane production by impacting microbial phosphorus acquisition. We found that methane production from methylphosphonate decomposition was enhanced (0.1–10.0 pmol L–1 d–1) by excessive nitrogen deposition from the atmosphere which altered nitrogen to phosphorus ratios (increase by ~27% in mixed layer) and exacerbated phosphorus limitation. Enhanced methane production could increase methane emissions from the open oceans, partially offsetting the decreased radiative forcing of carbon sequestration caused by atmospheric deposition. Our findings reveal an important linkage between atmospheric deposition and surface ocean methane cycling, suggesting a broader impact of atmospheric deposition on the feedback to the climate system that was not previously recognized.
Similar content being viewed by others
Introduction
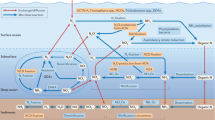
Methane (CH4) is a more potent greenhouse gas than carbon dioxide1, and the ocean is a source of atmospheric methane, with an annual emission of 6–12 Tg CH42. Methane supersaturation with respect to atmospheric concentrations in fully oxygenated surface waters has been termed the “methane paradox” since biological methane production was long considered a strictly anaerobic process conducted by archaeal methanogens3. The leading hypothesis to explain the oceanic methane paradox is that methane is produced through the carbon–phosphorus (C–P) lyase pathway from methylphosphonate (MPn) degradation4,5,6,7. Microbes under P limitation have evolved specific strategies to up-regulate production of inorganic phosphate (Pi) from the oceanic dissolved organic phosphorus (DOP) pool8, including the expression of the phn gene cluster that encodes a C–P lyase to degrade phosphonates or the increase of alkaline phosphatase (APA) activity to metabolize phosphate esters. Therefore, the decomposition of MPn with the phn gene would result in methane production as a byproduct5.
In the open ocean, nutrient limitation is regionally variable; while P limitation or co-limitation by trace metals such as iron has been observed in specific regions, nitrogen (N) is often the primary limiting nutrient in many oceanic regions9. The increasing atmospheric deposition of nitrogen to the ocean may alter local N to P ratios and lead to a potential shift from N limitation to P limitation10,11. In the northwestern Pacific Ocean, atmospheric depositional fluxes of inorganic nitrogen were at least two orders of magnitude higher than those of inorganic P12. This imbalance is largely driven by anthropogenic sources such as agricultural and fossil fuel emissions, which predominantly contribute reactive nitrogen to atmospheric aerosols10. The change of N/P ratios due to the imbalanced N and P deposition from mixed sources in the open ocean, may further affect microbial communities and biogeochemical processes10,13. The shift to P-limitation may accelerate DOP utilization, which could enhance microbial methane production during MPn degradation14. However, the impact of atmospheric deposition on aerobic methane production has not been recognized, nor has it been documented, and the significance of this process remains unknown on a global scale.
We conducted a variety of biogeochemical analyses in the Northwest Pacific Ocean (NWPO) and demonstrated the impact of atmospheric deposition on surface methane cycling. We also generated global predictions of the abundance of C–P cleavage pathway gene phnJ using a random forest regression (RFR) method and further assessed the effect of nitrogen deposition on phnJ abundance by incorporating atmospheric nitrogen deposition flux into the predictors in the well-trained model. Based on the conversion of MPn to methane under different N/P ratios, we tentatively estimated the increase in methane production and emission due to atmospheric deposition globally in the ocean. We further extrapolated the increase of methane production and emissions in global oceans over the period from 2025 to 2050 by incorporating the future deposition flux of atmospheric N. These results suggest that atmospheric deposition can enhance global marine methane production by impacting microbial P acquisition, revealing a feedback between atmospheric deposition and greenhouse gas cycling that was not previously recognized.
Results and discussion
Atmospheric deposition enhances aerobic methane production in the NWPO
In the NWPO, methane was oversaturated in surface waters relative to the concentration predicted from atmospheric equilibrium (Supplementary Fig. 1). Previous studies reported potential mechanisms for aerobic methane production in oxygenated seawater3, including archaeal methanogenesis in anoxic microhabitats15, the decomposition of DMSP or MPn4,5,6,7,8, or photochemical degradation of chromophoric dissolved organic matter (CDOM)16. Among these pathways, DMSP-driven methane production was not consistently detected in field studies3,8; the functional gene of anaerobic methanogens for archaeal methanogenesis (i.e., mcrA) was barely detected in oxic waters8; CDOM photodegradation contributes only partially to the observed methane supersaturation in the open ocean3,16. Instead, methane production from MPn degradation has been widely observed in global marine waters4,5,6,7,8. Therefore, we added different methylated substrates, including MPn, to surface waters to examine methane production pathways. No significant methane production was observed after the addition of these substrates during short-term incubation experiments (Supplementary Fig. 2a). Aerobic methane production from MPn usually occurred under phosphate-stressed conditions8, and the low N/P ratio might limit the utilization of exogenous MPn5,17. After imposing P limitation by adding glucose and nitrate5,7, the addition of MPn to seawater resulted in substantial methane production (Supplementary Fig. 2b). Furthermore, both genes involved in phosphonate biosynthesis and degradation, such as phosphoenolpyruvate mutase gene (PepM)18, methylphosphonate synthase (MPnS)19, and phosphonate degrading (phn) gene clusters (e.g., phnC-E, phnI-M)20,21, were present in our samples (Supplementary Table 1). Together, these results suggested that methane could be produced from MPn cleavage in surface waters of NWPO.
Given the increasing deposition of nutrients from the atmosphere to the open ocean10 and the influence of the N/P ratio on methane production, we hypothesized that atmospheric deposition could affect aerobic methane production through C–P lyase pathway. To test this hypothesis, we collected aerosol samples (Supplementary Table 2) and conducted incubations where we added aerosol and/or MPn to surface water that was driven to Pi limitation by amendment with 10.6 µM glucose and 1.6 µM nitrate. While no significant methane production was observed in aerosol or MPn+Pi amended samples, methane was produced in the MPn treatment, and the addition of aerosol with MPn further promoted methane production by 1.5-fold (Fig. 1a, d). Apparently, the addition of aerosol significantly increased the dissolved inorganic nitrogen (DIN) concentrations at the beginning of incubation (ANOVA, p < 0.01; Supplementary Fig. 3), and it is likely that the resultant change of N/P ratio induced the MPn decomposition to methane. Furthermore, we measured the enzymatic activity of APA during incubations to examine the Pi production, as APA has been widely used as an indicator for DOP utilization under Pi stress6,22. In the treatment with aerosol only, APA was much higher than in the control throughout the incubation. In contrast, APA was significantly elevated after 1-day incubation with the addition of aerosols+MPn (ANOVA, p < 0.01), and then rapidly decreased to a level lower than the control (Fig. 1c, f). These results suggested that aerosol addition promoted microbial utilization of DOP to cope with Pi deficiency, and this stress was relieved after Pi was produced from MPn degradation8. Furthermore, bacterial production (BP) rates increased following aerosol and/or MPn addition (Fig. 1b, e), suggesting the enhanced bacterial response to the amendment of aerosol and MPn. Molecular analysis demonstrated that Rhizobiales and Rhodobacterales, both containing phosphonate cleavage genes23, dominated microbial communities in the aerosol-amended samples after the amendment incubations (Supplementary Fig. 4). The enrichment of these bacteria with the capability to utilize phosphonate, also indicated that microorganisms tend to respond to the change of nutrient structure due to the deposition of aerosols into the ocean.

Changes of methane concentrations (a, d, CH4), heterotrophic bacterial production (b, e, BP) and APA activity (c, f, APA) during incubations with additions of 1.5 mg L−1 aerosol (aerosol), 5 µM MPn, 5 µM MPn + 1.5 mg L−1 aerosol (MPn+Aerosol), and 2.5 µM MPn + 2.5 µM phosphate (MPn + Pi) in surface seawater (amended with 10.6 µM glucose and 1.6 µM nitrate) at sites D5 (a–c; aerosol collected at site A1) and P2 (d–f; aerosol collected at site A2), respectively. Data are shown as mean ± standard deviation of triplicate incubations per treatment.
DIN was much more enriched than phosphate in the aerosol samples (Supplementary Table 2), and excess N could cause P limitation in the amended samples and enhance bacterial degradation of MPn with methane production as a byproduct. This notion was confirmed by another set of experiments with the addition of different nutrients to the MPn-amended samples. Compared to other nutrients, 1.0 µM ammonium + 1.0 µM nitrate (N) addition significantly increased APA and heterotrophic production rates in all N treatments (Supplementary Fig. 5; ANOVA, p < 0.01); this indicated that N addition drove more extreme Pi limitation and stimulated heterotrophic activity. The imposed extreme Pi limitation by N input would accelerate the degradation of MPn, and thus more methane was produced (increased by ~1.8-fold) during the utilization of MPn (Supplementary Fig. 5a). Consistently, a recent study demonstrated that nitrate addition significantly enhanced methane formation rates from 13C-MPn in the oligotrophic North Atlantic (i.e., the median rate of methane formation increased by ~2.4–6.3-fold), whereas co-addition of Pi with MPn inhibited methane production24. These results, together, confirmed the potential influence of N addition on microbial MPn degradation and resultant methane production. Furthermore, Fe addition stimulated BP rates, but not APA or methane production, compared to controls, suggesting that Fe might not be a primary limiting factor for MPn degradation and resultant methane production under our experimental conditions. However, Fe is an essential nutrient for microbial growth25, and it is a key cofactor required for the catalysis of the C–P lyase protein phnJ and MPn synthesis enzyme MpnS20,26. Therefore, the availability of Fe can modulate DOP cycling to some extent27 and potentially impacts the methane production in the upper ocean. Collectively, these results on microbial responses and methane production with N additions are consistent with those observed in the aerosol addition experiments, confirming that the input of exogenous N is the primary driver for observed increases in methane production with aerosol addition and suggesting that N deposition from the atmosphere could accelerate phosphonate decomposition under imposed P limitation, and thereby enhance aerobic methane production in the upper ocean.
Atmospheric deposition enhances the phn gene abundance and potential phosphonate utilization in the global ocean
As Pi concentrations were chronically low and the DOP pool can be orders of magnitude larger in surface oceans, microbes could acquire additional Pi from DOP to cope with P-limiting conditions by upregulating the expression of the phn gene cluster or APA28. Thus, the efficiency of marine DOP utilization can affect marine biological productivity at various temporal and spatial scales29. To explore the utilization of DOP containing C–P bonds in the surface ocean on a global scale, we collected the data of C–P cleavage pathway gene phnJ in global oceans from previous publications (Supplementary Fig. 6). Based on the pattern similarities between phnJ abundance and environmental factors, we developed an RFR model to predict global phnJ distributions (see “Methods” for details). Although the RFR modeling is challenging for identifying drivers as various factors might co-vary, the introduced N* value (defined as N* = [DIN] − 16 × [PO43−] in Eq. 1, which was used to characterize the relative N or P limitation and also to better incorporate atmospheric N deposition into the prediction model), emerged as a significant factor among the predictor variables (Supplementary Fig. 7). As the random forest models identify variables that best explain variance within a dataset rather than the mechanistic drivers, the relationship between N* and phnJ in our model results should be interpreted as a statistical correlation rather than a direct causal link. Furthermore, since the model captures primarily covariation patterns in heterogeneous global datasets, N* demonstrated better robustness to covariation and noise, thereby outperforming Pi as a predictor, even though Pi is also mechanistically relevant. The partial dependence plots reveal a nonlinear relationship between N* and phnJ abundance: phnJ abundance increases significantly only when N* approaches 0, whereas little variation is observed when N* is negative (Supplementary Fig. 8). With this approach, phnJ abundance was predicted by capturing complex nonlinear interactions between environmental variables.
Using RFR machine learning models, we first predicted the global distributions of phnJ abundance based on the collected database with other environmental factors. Generally, high phnJ abundance occurred in marine regions with low Pi concentrations, such as the North and South Atlantic (Fig. 2a and Supplementary Fig. 9). Similar to previous studies23, the abundance of phnJ correlated negatively with the concentrations of Pi (R2 = 0.30, p < 10–12). These results suggest that the degradation of DOP containing C–P bonds may be an important mode of bacterial P acquisition in the oceans with low Pi concentrations. Although the frequency of the phnJ gene occurrence cannot be directly interpreted as MPn utilization, as the C–P lyase pathway is not substrate-specific and can cleave multiple phosphonates, and metagenomic read abundance reflects genetic potential rather than in-situ activity, the predicted phnJ abundance still provides insight into the phosphonate-degrading potential of microbial communities on a global scale. To further estimate the influence of atmospheric deposition on phosphonate utilization through C–P cleavage, we first calculated the spatial correlation between our predicted phnJ abundance and global datasets of atmospheric inorganic N deposition. The significant positive correlation (Pearson r = 0.36, p < 0.0001) suggested the spatial consistency between phnJ distribution and observed deposition patterns. We then incorporated atmospheric nitrogen deposition based on global total inorganic N deposition fluxes during the period from 1984 to 2016 into the well-trained phnJ prediction model (Supplementary Fig. 10). We used mixed-layer depth (MLD) as the dilution depth to calculate the input concentration of atmospheric N fluxes to the surface ocean and the consequent influence on phnJ abundance. We noted that it is difficult to define a reference state with no atmospheric input, as atmospheric deposition is inherently a component of the standing DIN inventory, and the calculation of N** inevitably double-counts the atmospheric N component. Therefore, simulation results after incorporating N** should be regarded as a sensitivity diagnostic of the potential influence from atmospheric N deposition. After running the model, we found that phnJ abundance increased by 12.5–18.6% in the group with N deposition (Dep-MLD) compared to the control without considering additional N deposition (Fig. 2b and Supplementary Table 3). Elevated phnJ abundance was usually observed in offshore oceanic waters (water depths > 200 m), reflecting a more important role of atmospheric N deposition in phosphonate cycling in the open ocean. We used a similar approach to examine the impact of atmospheric deposition on mpnS gene abundance (see “Methods” for details). Although the genome frequency of mpnS is lower than other phosphonate biosynthesis pathways26, our analysis showed that the mpnS gene is widespread in the surface ocean. However, no significant differences between mpnS gene predictions were observed in the global surface ocean under scenarios with and without N deposition (Supplementary Fig. 11). Presumably, atmospheric deposition significantly changed the N/P ratios due to excessive N inputs, rather than Pi concentrations, and this significantly impacted the degradation of MPn (i.e., phnJ abundance), while the synthesis of MPn (i.e., mpnS abundance) was less influenced.

a Global map of the C-P cleavage pathway gene phnJ abundance (%) in marine surface water generated by the RFR method. b Probability distributions of predicted phnJ abundance in the groups without atmospheric nitrogen deposition added (phnJ) and with atmospheric nitrogen deposition added (Dep-MLD; influencing depth of atmospheric nitrogen inputs: mixed layer depth (MLD), see “Methods” section; n = 43,336 grid cells). The black dashed lines represent the 50th percentiles, and the red diamonds are the mean values. The boxes show the interquartile range, with whiskers spanning the maximum and minimum values excluding outliers (one-sided Wilcoxon rank-sum test, p = 3.1 × 10–64).
Furthermore, given the significant role of N inputs in driving the predicted increase in phnJ frequency, when exogenous N is available, phnJ expression may be up-regulated significantly, making phosphonate degradation an important Pi acquisition strategy in Pi-limited ecosystems. This implies that other biogeochemical processes that introduce N into Pi-limited regions may also affect phnJ abundance and the microbial utilization of phosphonates. Taken together, these results demonstrate that atmospheric aerosol deposition can impose Pi limitation and enhance DOP utilization in the upper ocean.
Global significance of atmospheric deposition on oceanic methane cycling
In certain oceanic regions where N limitation predominates, microbes develop a co-limitation by P9. In the (sub)-tropical North Atlantic, a shift to proximal P limitation could be driven by DIN supply through diazotrophy30,31,32, which may enhance MPn decomposition and methane production5. Moreover, our findings indicate that abundant N deposition elevated the heterotrophic activity and led to a stronger demand for bioavailable Pi in marine systems. Such a shortage of Pi tends to force microbial communities to up-regulate the expression of the C–P lyase and/or APA to degrade DOP, thereby supplying alternative bioavailable Pi for cell growth and releasing methane as a by-product in the case of MPn metabolism (Fig. 1 and Supplementary Fig. 3a). In addition to generating Pi limitation, atmospheric deposition drives a shift in the microbial communities towards an ecosystem dominated by phosphonate-utilizing prokaryotes (Supplementary Fig. 4). This shift may accelerate the utilization of DOP containing C–P bonds and potentially contribute to methane production.
Phosphonates are an important component of marine DOP that could account for ~10% of the oceanic DOP pool33. About ~15% of the bacterioplankton in the global surface ocean possess genes for phosphonate synthesis, thereby sustaining the surface ocean phosphonate pool34. MPn is a significant marine phosphonate, and its synthesis genes are widely distributed among bacteria and archaea (Supplementary Table 1)8,26,35. While genetic evidence shows that MPn synthesis gene frequencies in surface waters are much lower (<1.5%) compared to those of pepM (~15%) and the substrate-specific degradation of 2-aminoethylphosphonate (2-AEP) by phnZ is the primary strategy for phosphonate remineralization26, the presence of C–P lyase genes (phnIJM) supports the consistent availability of other organic phosphonates like MPn. Similar to the heterogeneous distribution of mpnS or phnJ, the abundance of MPn and potential methane production from MPn could also vary across marine environments. By combining laboratory results and model extrapolations, we sought to tentatively explore the impact of atmospheric input on oceanic aerobic methane production and emissions. As MPn concentration is rarely reported in the ocean, we approximated MPn concentration based on the proportion of MPn acid and phosphonate in oceanic DOP7. We note that the proportion of phosphonates in marine DOP could be variable33, and we used an average of 10% to estimate the phosphonate proportion in marine DOP and applied a range of 2–21% as upper and lower bounds (i.e., the reported minimum and maximum values; Supplementary Fig. 12)7,33,36. The results showed that MPn concentrations ranged from 0.4 to 26.6 nM (Supplementary Fig. 13a), and these estimates were generally consistent with the limited data reported in marine waters (~14 nM; ref. 37). In our incubation experiments, the conversion of MPn to methane was related to the N/P ratios (Fig. 3 and Supplementary Fig. 14). Irrespective of MPn concentration, the conversion of MPn to methane increased with N/P ratio < 16 and then leveled off under P-limited conditions. These results suggested that methane production from the decomposition of marine MPn may be predominantly governed by local N/P ratios. Based on these observations, we developed a sigmoid function to correlate the conversion of MPn with N/P ratios, using which methane production from the degradation of MPn can be extrapolated in global oceans (Fig. 3). Due to the inherent complexity and heterogeneity of natural ecosystems, where microbial community composition and nutrient availability vary across space and time, it is difficult and challenging to incorporate all relevant factors into a model to provide a global prediction. Therefore, we established a fundamental relationship between the N:P ratio and MPn degradation rates under controlled laboratory conditions to provide a simplified and preliminary extrapolation for the impact of atmospheric nitrogen deposition on marine methane cycling on a global scale. We first simulated the increase of nitrogen in surface waters within three depths of 1 m38, 10 m39, and MLD influenced by annual deposition flux of atmospheric nitrogen during the period from 1984 to 2016 (Supplementary Fig. 10). Then we incorporated the resultant change of N:P ratios (increasing by ~1108% at 1 m, ~111% at 10 m, and ~27% in the MLD) due to N deposition into the fitted sigmoid function, and extrapolated the increase of MPn decomposition and enhanced methane production in global oceans (Fig. 4). Again, it should be noted that this extrapolation is based on the results from laboratory incubation experiments, which are not identical to the natural conditions given the complexity and variability of natural water bodies. Though we incorporated experimental results into a probability function for global extrapolation, uncertainties could arise as the variability in environmental factors, microbial communities, and other ecological parameters may influence the N:P ratios and MPn degradation in situ. Therefore, our extrapolated results should be interpreted as a preliminary approximation of MPn degradation and methane production in response to atmospheric N inputs.

MPn conversion (%) in surface seawater after 6-day incubation with the addition of 0.1 µM (a), 0.5 µM (b), 1 µM (c), and 5 µM (d) MPn. Data are shown as mean ± standard deviation of triplicate incubations per treatment. e Sigmoid function fitting the MPn conversion rates at different nitrogen to phosphorus (N:P) ratios (adjusted with glucose and nitrate). The colors represent the different N:P ratios (i.e., N:P = 1, 5, 10, 16, 20, 25, and 32) set in the incubation experiments.

Increases in surface N:P ratios (a, after log normalization) and methane emissions (b; µmol m−2 yr−1) at the depth of 10 m influenced by annual deposition of atmospheric nitrogen.
Based on these calculations, methane production from MPn decomposition increased by 27.6%, 8.3%, and 2.5% respectively, with deposition depths at 1 m, 10 m, and MLD (Table 1). Approximately, a global increase of 0.05 Tg yr−1 in the mixed layer can be achieved for methane production from MPn decomposition due to the impact of atmospheric deposition (Table 1). As a result, methane production rates could increase by 0.1–10.0 pmol L−1 d−1 in global surface waters. Furthermore, assuming an upper-limit phosphonate fraction of 21% in marine DOP, we estimated that methane production could reach up to 40 pmol L−1 d−1 in the global surface ocean. This would correspond to ~0.1 Tg yr−1 of methane production in the mixed layer, implying the potential for methane production influenced by atmospheric deposition (Supplementary Fig. 12). Due to the low methanogenic activity17, in situ methane production rates are difficult to measure directly. At station ALOHA, gross methane production rates were estimated to be 0.01–0.05 nmol L−1 d−1 based on sea-air gas exchange and eddy diffusion17. Previous studies reported a wide range of C–P lyase activity in ALOHA surface waters from 1 to 223 pmol P L−1 d−1 (refs. 5,7,17,40). Furthermore, phosphate reduction to low-molecular-weight P(III) compounds (e.g., MPn) has been observed at rates of 18.0–42.2 pmol P L−1 h−1 in the tropical North Atlantic surface waters41, suggesting that both phosphonate production and degradation are highly dynamic in ocean environments. Assuming all observed C–P lyase activity is dedicated to MPn degradation, the potential methane production rate in ALOHA surface waters could range from 0.001 to 0.2 nmol L−1 d−1; this range encompasses previous estimates at station ALOHA (i.e., 0.01–0.05 nmol L−1 d−1)17. However, this estimate may represent an upper limit for methane production, as other phosphonates, such as 2-AEP, likely constitute a large fraction of the organic phosphonate pool26. Additionally, C–P lyase activity exhibited depth-dependent variability and was influenced by N:P flux ratios at the deep chlorophyll maximum42. This supports the rationale of linking MPn degradation to the N:P ratio in the present model extrapolation. Combining our model with field data (e.g., N:P ratio, MLD, MPn abundance) at station ALOHA, we estimate that atmospheric deposition leads to an increase of 0.2 pmol L−1 d−1 for methane production, accounting for ~0.4–2.6% of gross methane production17. This number is in good agreement with our estimated increase of global MPn decomposition within the MLD influenced by atmospheric deposition (i.e., 2.5%), confirming the validity of our prediction and the enhanced MPn decomposition and methane production by atmospheric deposition in the global ocean.
An increase in methane production due to N deposition would amplify the uncertainty of sea-air fluxes and have important implications for global methane emissions. Due to the seasonal variability and inherent uncertainty of MLD in the ocean43, we extrapolated the surface-enhanced methane production at deposition depths of 1 m38 and 10 m39 into a standard gas transfer model and approximated the influence of this process on global methane emissions. Globally, the sea-air methane flux could increase by 0.01 ± 0.0001 Tg yr−1 and 0.03 ± 0.001 Tg yr−1 due to excess atmospheric N deposition, respectively (Table 1). Elevated methane emissions occur primarily in low-phosphate regions (Supplementary Fig. 9), such as the Mediterranean Sea, the North Atlantic Ocean, and the NWPO (Fig. 4), where N2-fixation is also highly active44. Although excess N introduced by atmospheric deposition might influence biological N2 fixation, the resultant nutrient imbalances and changes of N/P ratios45 would ultimately lead to increased methane production and emissions. Specifically, the Atlantic Ocean, characterized by P limitation and substantial atmospheric deposition, exhibits an annual methane emission increase that is approximately 23% higher than that observed in the Pacific Ocean. Given the vast area of the open ocean (>2000 m), atmospheric N deposition could lead to an increase of 0.01–0.02 Tg yr−1 in methane emission flux. A recent study reported a global diffusive methane flux of 2–6 Tg CH4 yr−1 from the ocean to the atmosphere, with an emission of 0.6–1.4 Tg yr−1 for the open ocean (>2000 m; ref. 2). Based on our calculation, increased methane flux caused by atmospheric N deposition to the open ocean, would account for ~2% of the annual methane emission flux. Furthermore, although the extrapolation of our experimental data to a global scale indicates the regional differences in the impact of atmospheric N deposition, it should be noted that our data were derived from the open ocean, which might differ from coastal waters. The different biogeochemistry between coastal waters and open oceans could introduce uncertainties for global extrapolation, which should be considered for tentative interpretation and deserves further investigation.
In addition to regional variation, strong synoptic deposition episodes could deliver substantial nutrients to the ocean that are several orders of magnitude greater than general deposition and impact oceanic biological production and element cycling46,47. By adopting the N deposition rate of 2.8 mg N m−2 d−1 from a study used for the simulation of episodic dust deposition46, we estimated that methane production in surface waters could increase by ~0.01 nmol L−1 d−1 within the deposition depth of 10 m due to episodic deposition pulses. This would result in an increase of 2.2−5.1% in daily methane emissions from the open ocean, similar in magnitude to those observed in our incubation experiments, suggesting that the impact of aerosol deposition on methane cycling could be significantly amplified on a short timescale with strong episodic pulses.
Furthermore, as atmospheric deposition occurred persistently, we assessed the long-term impact of atmospheric N inputs on future oceanic methane cycling. We simulated the increase of N in surface waters within the depth of 10 m influenced by annual deposition flux of atmospheric N between 2025 and 2050 based on future N deposition data under the Representative Concentration Pathway 6.0 (RCP6.0) scenario from the Inter-Sectoral Impact Model Intercomparison Project (ISIMIP). By incorporating the changed N:P ratio into the fitted sigmoid function, we extrapolated the cumulative methane emissions in global oceans from 2025 to 2050. Approximately, an increase of 0.22 Tg in surface methane emissions caused by atmospheric N deposition to the global ocean would be achieved over this period (Supplementary Table 4). Likewise, a 0.20 Tg methane increase would be emitted from the open ocean due to the vast area, and this value accounted for 14.3–33.3% of the annual amount of methane released from the open ocean (0.6–1.4 Tg; ref. 2), suggesting that atmospheric deposition could increase methane emissions by the end of half century. Note that these estimates only account for N gains from atmospheric deposition and do not consider oceanic N-loss pathways (e.g., denitrification, anammox), thus representing an upper-bound, N-gain-only scenario for future prediction. While uncertainty remains as to whether the global ocean will experience a net gain of N in the future, the increased methane production and emissions should occur in certain regions with N input from atmospheric deposition.
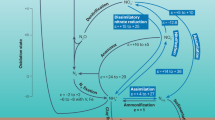
Biological N2 fixation and atmospheric deposition are two important N sources in the open ocean, and now the quantity of atmospheric N deposition is approaching N2 fixation due to the remarkable increase in anthropogenic emissions of reactive N45. Atmospheric N deposition may not only increase primary production, carbon dioxide (CO2) uptake, and carbon export to the deep ocean, but also enhance nitrous oxide (N2O) emissions and therefore offset the decreased radiative forcing from CO2 sequestration45. In addition to N2 fixation, the excess N over P from atmospheric deposition would further decouple N and P cycles and promote phosphate limitation. Karl et al. proposed a methane production pathway from MPn utilization under P-stressed conditions where Trichodesmium, a nitrogen-fixing cyanobacterium, produced methane from MPn, solving the oceanic methane paradox5. Here, we demonstrate a missing link between atmospheric deposition and surface methane production that was not previously known. Our results suggested that the impacts of atmospheric deposition on the open ocean and the feedbacks to the climate system could be expanded. The increase of methane production and emission due to atmospheric deposition, to some extent, would also offset the N input-induced decrease of radiative forcing (Fig. 5). Increasing atmospheric deposition and continued global warming would lead to enhanced ocean stratification and phosphate limitation48,49, which will further reinforce greenhouse gas emissions and increase radiative forcing from N2O5 and methane. This finding advances our understanding of oceanic response to atmospheric deposition and reveals an intricate interplay between atmospheric deposition, nutrient dynamics, greenhouse gas production, and elemental cycling in the open ocean.

C–O–P carbon-oxygen-phosphorus, C–P carbon–phosphorus, PON particulate organic nitrogen, NH4+ ammonium, N2O nitrous oxide, CO2 carbon dioxide, CH4 methane.
Methods
Sample collection and analysis
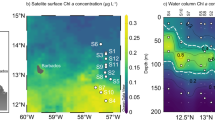
Samples were collected from the NWPO onboard the R/V “Dongfanghong 3” during the expedition in October 2019–January 2020 (Supplementary Fig. 15). Seawater for methane and nutrient analysis and incubation experiments was sampled using 12 L Niskin bottles deployed on a Seabird 911 conductivity-temperature-depth sensor rosette. Methane concentrations in surface water ([CH4]surface) were determined using a cryogenic purge-and-trap setup connected to an Agilent GC-8890 gas chromatograph according to the previous methods8. The saturation of dissolved methane (R, %) was calculated using the equation of R = [CH4]surface/[CH4]equilibrium × 100%, where [CH4]equilibrium is the calculated methane concentration equilibrated with atmospheric methane and corrected for in situ salinity and temperature50. Atmospheric methane data were obtained from the NOAA/ESRL Global Observations Project (http://www.esrl.noaa.gov/gmd). Nutrients, i.e., nitrate, nitrite, ammonium, silicate, and phosphate, were analyzed using standard spectrophotometric methods with an autoanalyzer (SEAL Analytical Gmbh, QuAAtro, Germany)51. The detection limits for these nutrients were 0.02 µM, 0.003 µM, 0.04 µM, 0.03 µM and 0.02 µM, respectively. The precision for these measurements was less than 0.3% and the correlation of the calibration curves was greater than 0.999.
Atmospheric aerosol samples were collected using a high-volume total suspended particulate sampler (Model KB-1000, Jinshida Electronic Technology Co., Ltd., China), with Whatman 41 filters (20.3 × 25.4 cm2, Cellulose) at an airflow rate of ~1.0 m3 min−1 for 24 h. The samples were stored at −80 °C and analyzed by Ion Chromatography (IC-940, Aptar, Switzerland)52.
Incubation experiment
Methane production experiments
Methane production experiments were conducted in surface seawater (sampling depth: 5 m) at three different sites (i.e., site P1, P2, D5; Supplementary Fig. 15 and Supplementary Tables 5 and 6). Seawater was filled directly into 60 mL acid-washed and sterilized serum vials with no headspace. The vials were sealed immediately, and a 20 mL headspace was created by replacing the same volume of high-purity helium (99.999%). Subsequently, different substrates including 10 µM dimethylsulfoniopropionate (DMSP), 10 µM MPn, 10 µM trimethylamine (TMA), and 10 µM methanethiol (MeSH) were added to the vials separately (Site P1; n = 3 each; Supplementary Table 5). To generate P-limiting conditions, we added 1060 µM glucose and 160 µM nitrate to seawater and subsequently added 10 µM MPn to monitor the methane production (Site P1; Supplementary Table 5). Glucose was added as a carbon source to adjust the C:N:P ratio following methods used in previous studies4,5,6,7,8. The addition of glucose ensured sufficient carbon supply and the high C(N): P ratio during the incubation, which allowed us to examine the effects of N:P ratios on methane production, with detectable methane produced from MPn degradation. Although the concentration of glucose in natural waters could be lower than that used in our study, our experiments are environmentally relevant as monosaccharides such as glucose are significant components of the DOM pool and they are widely produced by phytoplankton exudation and the decomposition of organic matter53. After substrate addition, the vials were incubated in the dark at the in situ temperature. Previous studies demonstrated that MPn degradation to methane production is predominantly mediated by heterotrophic bacteria, and this process was not significantly affected by light exposure4,5,7,8. Therefore, the incubation was conducted in the dark, and this is not likely to impact the methane production from MPn degradation. Methane concentrations in the headspace were measured at selected time points. Gas samples of 0.5 mL were taken from the headspace of vials and analyzed on the Agilent GC-8890 gas chromatography with flame ionization detection (HP-PLOT Q capillary column, 30 m × 0.32 mm × 20 μm, Agilent). Similarly, MPn conversion experiments were conducted in surface water at site P2 (Supplementary Fig. 15) by adding 0.1 µM, 0.5 µM, 1 µM, and 5 µM MPn to seawater at different N/P ratios (adjusted with glucose and nitrate, n = 3 each; Supplementary Table 7). The conversion rates of MPn were calculated by quantifying the methane production rates from MPn decomposition (Supplementary Fig. 14). We did not measure the MPn concentrations in these incubation experiments due to the analytical challenge in the available method, which required specialized instrumentation, complicated pretreatment, and a large sample volume (i.e., ≥3 L)54. Future studies should aim to quantify the MPn concentrations with sensitive methods to better constrain the linkage between MPn cycling and methane production in the ocean.
Aerosol addition experiments
The collected aerosol membranes were cut using a ceramic scissor and then ultrasonicated in centrifuge tubes containing Milli-Q water for 40 min to extract the supernatant and finally transferred to a volumetric flask. To examine the addition of aerosol on methane production, incubation experiments with aerosol addition (final concentration: 1.5 mg L−1)52 were conducted in 310 mL Schott bottles containing 250 mL seawater (n = 3; Supplementary Table 5). Different substrates, including 1.5 mg L−1 aerosol, 5 µM MPn, 2.5 µM Pi + 2.5 µM MPn, and 5 µM MPn + 1.5 mg L−1 aerosol were added to the seawater cultures (Supplementary Table 5). The seawater was amended with 10.6 µM glucose and 1.6 µM nitrate7, which was important to facilitate the degradation of added MPn and minimize interference from residual Pi. However, it may also enhance microbial activity and nutrient utilization, potentially influencing the efficiency of methane production from MPn decomposition. During incubation, headspace gas and seawater samples were taken through two sampling valves, a water outlet valve and a gas extraction valve, at different time points (0.25, 1, 3, 6, and 10 days). Seawater (~10 mL) was sampled for BP rate and APA activity measurements. APA was determined using the p-NPP colorimetric method55, and BP rates were determined by quantifying the bacterial protein synthesis using the 3H-leucine incorporation method56,57. Headspace gas for methane was analyzed with a GC as described above. In addition, we determined the response of microbial communities to atmospheric inputs. Natural surface seawater (sampling depth: 5 m; site D5) was amended with/without the 1.5 mg L–1 aerosol solution and incubated in 1-L sterile containers at in situ temperature (Supplementary Table 5). Due to limited sample volume and aerosol solution, the incubation experiment was conducted in replicates. In each case, seawater from the replicate incubation bottles was filtered onto a single membrane filter (0.2 µm; GSWP, Millipore) for downstream DNA extraction and 16S rRNA gene sequencing. The results were visualized using stacked bar plots to compare the relative abundances of bacterial taxa between treatments.
Nutrient addition experiments
We conducted incubation experiments with nutrients based on the composition of atmospheric aerosols (Supplementary Table 2). For methane production, 2.5 µM ammonium + 2.5 µM nitrate (N) and/or 50 nM ferric chloride (Fe) were added to 10.6 µM glucose+1.6 µM nitrate + 10 µM MPn amended surface seawater in 310 mL Schott bottles with 60 mL headspace (Supplementary Table 5). For APA and BP incubations, 1.0 µM ammonium + 1.0 µM nitrate (N) and/or 50 nM Fe were amended in 60 mL sterilized serum vials filled with seawater (Supplementary Table 5). The samples were then incubated in the dark at in situ temperature for 10 h. Determination procedures were the same as described above.
Quantification of global ocean phnJ and mpnS genes
To quantify the relative abundance of the C–P cleavage pathway gene phnJ in the global ocean, a total of 193 metagenomic sequence read archive (SRA) data were downloaded and collected from the GEOTRACES program (Section GA10, GP13, GA02, and GA03)58, ALOHA program continuous stations (HOT)58, and TARA ocean dataset59 (Supplementary Data 1). We analyzed the sampling data from surface seawater at these stations, and for stations without surface data, data within the mixed layer were used. Meanwhile, metagenome-assembled genomes (MAGs) of corresponding SRA data were downloaded from the iMicrobe database (https://www.imicrobe.us/). Prodigal v.2.6.360 was used to predict the protein-coding genes (CDS) of the MAGs. To identify the phnJ genes of each metagenomic dataset, the multiple sequence alignment of phnJ genes was downloaded as a homologous protein database23. HMMER v.3.3.261, based on the Hidden Markov model, was employed to identify the homologous sequences of the phnJ gene by searching against the homologous protein database above with a setting of E-value < 0.001 according to the ref. 62. FetchMG v.1.263 was used to find 40 single-copy marker genes from each MAG. For the raw sequencing data, Trimmomatic v.0.3964 was employed for quality control with the parameters as follows: ILLUMINACLIP: TruSeq3-PE-2.fa:2:30:10, LEADING:20, TRAILING:20, SLIDINGWINDOW:4:15, MINLEN:100. Minimap2 v.2.2465 was used for mapping the clean reads to metagenomic assemblies with default parameters. Bedtools v.2.30.066 was used to calculate the coverage of phnJ genes and 40 single-copy marker genes of each MAG, and normalized the relative abundance of phnJ genes as previously described in ref. 62. Specifically, the normalization was performed by calculating the median coverage of 40 universal single-copy marker genes identified by FetchMG. The coverage of phnJ was then divided by this median value to estimate the relative abundance of phnJ in genome equivalents, thereby accounting for variation in sequencing depth and genome completeness. The resulting normalized phnJ abundance reflects the relative abundance of microbial genomes possessing this gene, expressed as a percentage of total genome equivalents within each metagenomic sample. In total, we obtained 193 relative abundance of phnJ across the global surface ocean (Supplementary Fig. 6). Similarly, we quantified the abundance of the mpnS gene using the same analytical approach. Metagenomic datasets were obtained from the GEOTRACES program58, the ALOHA program continuous stations58, and the TARA Oceans dataset59 (Supplementary Data 1). Homologous mpnS sequences were retrieved from Lockwood et al.26, and gene identification, normalization, and abundance estimation followed the identical pipeline used for phnJ.
Machine-learning prediction of global phnJ and mpnS abundances
We used the compiled phnJ dataset together with environmental variables to train the prediction model and map the global distribution of phnJ. We used local environmental variables in the phnJ model, and for data points that lacked predictors or did not provide the environmental predictors required by the model, we paired them with publicly available environmental datasets. Predictors used in our model included sea surface temperature (SST, 0.25° resolution), salinity (SSS, 0.25° resolution) and phosphate ([PO43−], 1° resolution), taken from the World Ocean Atlas 2018 (WOA18) climatology, surface concentration of DOP (2° resolution) taken from the previous publication27, surface chlorophyll-a (Chla, 0.083° resolution) and particulate organic carbon (POC, 0.083° resolution) derived from NASA Aqua MODIS satellite climatology, and the surface N* values. The introduction of the N* value was to characterize the relative N or P limitation in surface seawater and also to better incorporate N deposited from the atmosphere into the prediction model, which is calculated using Eq. (1). We note that the phosphate data from WOA18 may not adequately capture extreme P-limited regions in the surface ocean67. As global phosphate data measured using high-sensitivity methods is lacking67, the WOA data provide a general overview for the regional variations of global phosphate distribution and serve as a viable alternative for global-scale modeling27,44.
In Eq. (1), [DIN] is the sum of surface seawater concentration of nitrate ([NO3−]) and total ammonia ([NHx]). [NO3−] is taken from WOA18 climatology, and [NHx] is taken from a previous publication68 at 1° resolution. All predictors were interpolated to the same 1° × 1° latitude/longitude grid.
We used the RFR method to predict the global phnJ distribution. Our RFR model was structured with 125 decision trees according to refs. 2,8, which was trained using the standard Classification and Regression Trees (CART) algorithm. We tested different levels of complexity of the model and chose the optimal parameters for the model. Specifically, we performed 10-fold cross-validation to optimize the mtry parameter, and evaluated model performance on different numbers of trees using the optimal mtry value (Supplementary Fig. 7). To ensure robustness, the dataset was split into 80% for training and 20% for testing. The final model performance was evaluated using different metrics, including root mean squared error (RMSE), R2, and mean absolute error (MAE). To enhance robustness, we also applied 10-fold cross-validation, where phnJ was standardized within each iteration using only the mean and standard deviation of the training subset (9 folds). This yielded a R2 value of 0.727 ± 0.145 (n = 10), indicating stable performance across folds. We also assessed the importance of predictor variables and conducted partial dependence analysis to examine the marginal effects of each predictor (Supplementary Figs. 7 and 8). Since phnJ data spans four orders of magnitude, we standardized the data using Z-score normalization to avoid a few data points with high phnJ abundance dominating the training process2. We also developed other models, including simple linear regression (LR, R2 < 0.15), multiple linear regression (MLR, R2 < 0.15), and support vector machine (SVM, R2 < 0.9) models for comparison against the RFR model. RFR model showed good predictive performance, and all members could be reproduced with R2 = 0.92 for training datasets (Supplementary Fig. 7). After training, the RFR model was used to generate a global map of surface ocean phnJ abundance at 1° × 1° resolution.
To assess the impact of atmospheric nitrogen deposition on global phnJ abundance, we interpolated the global total inorganic nitrogen deposition fluxes (2° × 2.5° resolution, in kg N km−2 yr−1) taken from the ref. 69 to a 1° × 1° grid. As the N deposition flux was estimated based on deposition data during the period from 1984 to 2016, the results simulated represent an annual average from 1984 to 2016. We calculated the input concentration of atmospheric nitrogen ([TIND], in μmol N L−1) by assuming that the deposition flux (kg N yr−1) was diluted into the volume represented by the surface area × MLD, and then calculated the changed N* values (N**) using Eq. (2)
MLD is derived from WOA18 climatology (0.25° resolution) and interpolated to the same 1° × 1° grid as the nitrogen deposition fluxes. By incorporating calculated N** values into our trained RFR model, we generate the corresponding global surface ocean phnJ abundance maps after including atmospheric nitrogen deposition.
Similarly, we used the RFR model to predict the global abundance of the mpnS gene. The predictor variables for the mpnS modeling were similar to those described above for the phnJ model, including SST, SSS, [PO43−], DOP, Chla, POC, N*, and silicate (1° resolution) taken from the World Ocean Atlas 2018 (WOA18). The mpnS abundance was standardized using Z-score normalization to account for differences in data scale and distribution. We performed 10-fold cross-validation to optimize the mtry parameter (mtry = 3), and evaluated model performance across different numbers of decision trees, with 350 trees selected for model construction. The dataset was randomly split into 80% training and 20% testing subsets to ensure robustness, and model performance was evaluated using RMSE, R2, and MAE. After training, the RFR model showed good predictive performance and was used to predict the global distribution of surface mpnS gene abundance. Similarly, by incorporating N** values into the well-trained RFR model, we further assessed the potential influence of atmospheric N deposition on global mpnS gene abundance. We quantified the aggregate relative change between the simulations with N** and N* using a ratio-of-sums approach. Specifically, the domain was partitioned into 10° × 10° latitude–longitude blocks, and a spatial block bootstrap was applied (B = 3000, blocks resampled with replacement). For each replicate, values from the resample blocks were pooled to form totals for each simulation, and the global percent changes of the N** total relative to the N* total were recalculated. Uncertainty was quantified by a 95% confidence interval, defined as the 2.5th–97.5th percentiles of the bootstrap distribution.
Global quantification of methane production and emissions due to atmospheric deposition
To estimate enhanced methane production rates from MPn decomposition due to atmospheric nitrogen inputs, we conducted experiments to examine methane production rates from the conversion of MPn under different N:P ratios by adding nitrate and MPn5. The conversion rates of MPn were calculated by quantifying the methane production over 6-day incubations. Based on the observations, we developed a probability function (f(N:P)) of methane production rates (Ratepro) from MPn decomposition (Fig. 3) and further computed the conversion of MPn by combining the global MPn concentrations and N:P ratios. We calculated the annual input of atmospheric nitrogen concentration (μmol N L−1) with influencing depths of 1 m38, 10 m39, and MLD and simulated the increase of nitrogen in surface waters. With the resultant change of N:P ratios, we quantitatively estimated the increased MPn decomposition and methane production rates due to atmospheric nitrogen inputs.
In Eq. (3), FraC–P is the proportion of MPn in the surface DOP concentration. We used an average of 10% to estimate the phosphonate proportion in marine DOP and applied a range of 2–21% as upper and lower bounds (reported minimum and maximum values)7,33,36. Based on the proportion of MPn acid and phosphonate7, we estimated the MPn concentrations.
We applied a sigmoid function to estimate the rate of MPn conversion based on the N:P ratio (DIN/[PO43−]) using Eq. (4). To estimate the optimal values for the parameters (i.e., a, b, c, and d) of the function, we developed a fitting procedure using nonlinear least squares (NLS) via the nlsLM function in R. The observed MPn decomposition ratios (%) used for fitting were 4.31% at N:P = 1, 12.89% at N:P = 5, 28.17% at N:P = 10, 52.35% at N:P = 16, 53.35% at N:P = 20, 58.92% at N:P = 25, and 63.29% at N:P = 32 (Fig. 3). The fitting procedure was designed to minimize the sum of squared errors between the predicted and observed values by adjusting the parameters. By leveraging the nlsLM function, we ensured a robust fitting while allowing for nonlinear relationships. This approach ensures that the optimization algorithm starts within a reasonable parameter space and converges to an optimal solution. After fitting, we calculated the R2 to evaluate the fit of the model, yielding an R2 of 0.99, and extracted the optimal parameter set (a = –2.04, b = 63.82, c = 0.24, and d = 10.12) for the sigmoid function.
Diffusive methane fluxes (F) across the sea-air interface were calculated using Eq. (5), where εice is the blocking efficiency of gas exchange through the ice cover, fice is the proportion of sea ice cover of each grid cell, k is the methane gas transfer velocity (in cm h−1), and ∆CH4 is increased annual methane production (in nmol L–1) due to atmospheric nitrogen input. Here, k was calculated using the W2014 empirical algorithms and εice was randomly matched from the range 0.9–12 using the Monte Carlo iterations method. Sea ice data were obtained from the GO8p7 Global Ocean-Sea Ice Model output (0.25° resolution) in the CEDA archive (https://data.ceda.ac.uk/bodc/SOC220065/GO8p7_JRA55_eORCA25). Monthly wind climatologies were derived from merged wind speed measurements including SSM/I, SSMIS, and WindSat Instrument in Remote Sensing Systems (https://www.remss.com/measurements/wind/). The fice and wind speed were interpolated into the same 1° × 1° grid as ∆CH4 and annual average data were calculated to apply Eq. (5).
To assess the long-term impact of atmospheric N inputs on future oceanic methane cycling, we estimated the increased methane production and emissions for the period from 2025 to 2050 based on future N deposition data. Inter-Sectoral Impact Model Intercomparison Project (ISIMIP) is a highly regarded international initiative that provides standardized climate impact projections across multiple sectors70. Specifically, we downloaded the nitrogen deposition datasets simulated for the period 2006–2099 under the RCP6.0 scenario combined with socioeconomic conditions (0.5° × 0.5° resolution, in g N m–2 yr–1) from the ISIMIP. We then interpolated these nitrogen deposition fluxes from 2025 to 2050 to the same 1° × 1° latitude/longitude grid and simulated the annual increase of nitrogen concentrations in surface waters within the depth of 10 m influenced by atmospheric deposition. By incorporating the resultant change of N:P ratios into the fitted sigmoid function, we quantitatively estimated the increased methane production due to atmospheric N inputs from 2025 to 2050. We then extrapolated the surface-enhanced methane production into the W2014 empirical gas transfer model and estimated the increase in global methane emissions over the period from 2025 to 2050. Similarly, to assess the impact of strong synoptic deposition episodes on methane cycling, we calculated the change of N:P ratios in surface waters within the depth of 10 m influenced by episodic deposition pulses by assuming a N deposition rate of 2.8 mg N m−2 d−1 (ref. 46). By incorporating the changed N:P ratios into the fitted function, we estimated the increased methane production and extrapolated resultant increase in daily methane emissions during episodic deposition pulses using the empirical gas transfer model.
Additionally, due to the high variability of our predictions, we used the Monte Carlo method to derive the more representative estimates. Specifically, we performed 100,000 Monte Carlo simulations for each model outcome. In each simulation, we randomly sampled 80% of the data and calculated the mean of the subset. This simulation generated 100,000 independent estimates for each model outcome, from which we derived the overall mean and standard deviation.
16S rRNA gene analysis
The DNA was extracted by E.Z.N.A.® Soil/Water DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) following the manufacturer’s instructions. The primers 515 F/806 R (5’-GTGYCAGCMGCCGCGGTAA-3’/5’-GGACTACNVGGGTWTCTAAT-3’)71 were used for bacterial 16S rRNA gene amplification. The cycling conditions were as follows: 95 °C pre-denaturation for 3 min, 27 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 45 s, and single extension at 72 °C for 10 min, and finally end at 4 °C by an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). Each sample was amplified three times and the PCR products were purified by the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA), and quantified by a Quantus™ Fluorometer (Promega, USA)72. Purified amplicons were pooled in equimolar and paired-end sequenced on an Illumina MiSeq (Illumina, San Diego, USA) platform according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Raw sequencing reads were processed using a standard bioinformatics pipeline. Quality control was performed using fastp v.0.19.673 to remove adapter sequences, trim low-quality bases, and filter short reads. Specifically, bases with a quality score < 20 at the read tails were trimmed using a 50 bp sliding window, and reads shorter than 50 bp after trimming or containing N bases were discarded. Paired-end reads were merged using FLASH v.1.2.1174 with a minimum overlap length of 10 bp and a maximum mismatch ratio of 0.2 in the overlap region. Sequences that did not meet these criteria were removed. The high-quality merged sequences were clustered into operational taxonomic units (OTUs) at a 97% similarity threshold using UPARSE v.7.175, and chimeric sequences were identified and removed using UCHIME76. Taxonomic classification of OTUs was performed using the Ribosomal Database Project (RDP) classifier v.2.1177 against the SILVA database v.138.178 with a confidence threshold of 70%. Sequences classified as chloroplasts or mitochondria were removed to eliminate potential contamination. Microbial community composition was analyzed by comparing the relative abundances of bacterial taxa between different samples. The relative abundance of bacterial taxa was calculated at the order level (>1%).
Bioinformatic analyses of genes involved in MPn and methane metabolism
Total genomic DNA was extracted from the environmental samples collected at different depths from the Mariana Trench (Supplementary Fig. 15) using the E.Z.N.A.® soil/water DNA kit (Omega Bio-tek, Norcross, GA, U.S.) following the manufacturer’s instructions79. DNA libraries were constructed and sequenced at Novogene Bioinformatics Technology Co., Ltd (Beijing, China). Raw 150 bp paired-end metagenomic reads were generated using the Illumina NovaSeq platform. Trimmomatic v.0.3964 was employed for quality control with the parameters as follows: ILLUMINACLIP: TruSeq3-PE-2.fa:2:30:10, LEADING:20, TRAILING:20, SLIDINGWINDOW:4:15, MINLEN:100. The resulting high-quality reads were then assembled into contigs using MEGAHIT v.1.2.980. Prodigal v.2.6.360 was performed to predict the CDSs of assemblies. To identify functional genes associated with MPn synthesis and degradation, we retrieved a set of functionally validated protein sequences from the National Center for Biotechnology Information (NCBI) database8. These included McrA, MmoX, MmoY, MpnS, PepM, PhnA, PhnB, PhnC, PhnD, PhnE, PhnF, PhnG, PhnH, PhnI, PhnJ, PhnK, PhnL, PhnM, PhnN, PhnO. The quantitative method for functional genes is consistent with the global phnJ quantification method described above. Specifically, the coverage of related genes and 40 single-copy marker genes of each MAG was quantified using Bedtools v.2.30.066. The relative abundances of these genes were normalized by dividing the coverage of each target gene by the median coverage of 40 universal single-copy marker genes identified using FetchMG.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All experimental data, model prediction outputs and datasets used for the model building presented in this study are available in the Source Data file and on Figshare (https://doi.org/10.6084/m9.figshare.30933482). Environmental predictor datasets, including temperature, salinity, nutrient and mixed layer depth climatologies were sourced from the World Ocean Atlas 2018. The surface total ammonia dataset was derived from the ESGF CMIP6 archive (https://aims2.llnl.gov/search/cmip6/). The DOP dataset was sourced from https://doi.org/10.1038/s41561-022-00988-1. Chlorophyll a and particulate organic carbon datasets were obtained from the NASA OceanColor website (https://oceandata.sci.gsfc.nasa.gov/). Global nitrogen deposition dataset was obtained from https://conservancy.umn.edu/handle/11299/197613. Future nitrogen deposition datasets were sourced from the ISIMIP database (https://data.isimip.org/; ndep_nhx_rcp60soc_annual + ndep_noy_rcp60soc_annual). The 16S rRNA gene sequencing data (PRJNA1255851) and metagenomic sequence data (PRJNA541485) are available through the NCBI Sequence Read Archive. Source data are provided with this paper.
Code availability
Code used to develop the model and reproduce the figures is archived at https://doi.org/10.6084/m9.figshare.30933482.
References
Shindell, D. T. et al. Improved attribution of climate forcing to emissions. Science 326, 716–718 (2009).
Weber, T., Wiseman, N. A. & Kock, A. Global ocean methane emissions dominated by shallow coastal waters. Nat. Commun. 10, 4584 (2019).
Mao, S., Wang, J., Joye, S. & Zhuang, G. Marine methane paradox: enigmatic production of methane in oxygenated waters. Innov. Geosci. 2, 100071 (2024).
Carini, P., White, A. E., Campbell, E. O. & Giovannoni, S. J. Methane production by phosphate-starved SAR11 chemoheterotrophic marine bacteria. Nat. Commun. 5, 4346 (2014).
Karl, D. et al. Aerobic production of methane in the sea. Nat. Geosci. 1, 473–478 (2008).
Teikari, J. E. et al. Strains of the toxic and bloom-forming Nodularia spumigena (cyanobacteria) can degrade methylphosphonate and release methane. ISME J. 12, 1619–1630 (2018).
Repeta, D. J. et al. Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nat. Geosci. 9, 884–887 (2016).
Mao, S. et al. Aerobic oxidation of methane significantly reduces global diffusive methane emissions from shallow marine waters. Nat. Commun. 13, 7309 (2022).
Browning, T. J. et al. Iron limitation of microbial phosphorus acquisition in the tropical North Atlantic. Nat. Commun. 8, 15465 (2017).
Kim, I. et al. Increasing anthropogenic nitrogen in the North Pacific Ocean. Science 346, 1102–1106 (2014).
Kim, T., Lee, K., Najjar, R. G., Jeong, H. & Jeong, H. J. Increasing N abundance in the Northwestern Pacific Ocean due to atmospheric nitrogen deposition. Science 334, 505–509 (2011).
Seok, M. et al. Atmospheric deposition of inorganic nutrients to the western North Pacific Ocean. Sci. Total Environ. 793, 148401 (2021).
Karl, D. M., Bidigare, R. R. & Letelier, R. M. Long-term changes in plankton community structure and productivity in the North Pacific Subtropical Gyre: the domain shift hypothesis. Deep-Sea Res. II 48, 1449–1470 (2001).
Duhamel, S. et al. Phosphorus as an integral component of global marine biogeochemistry. Nat. Geosci. 14, 359–368 (2021).
Karl, D. & Tilbrook, B. Production and transport of methane in oceanic particulate organic matter. Nature 368, 732–734 (1994).
Li, Y., Fichot, C. G., Geng, L., Scarratt, M. G. & Xie, H. The contribution of methane photoproduction to the oceanic methane paradox. Geophys. Res. Lett. 47, e2020GL088362 (2020).
Del Valle, D. & DM, K. Aerobic production of methane from dissolved water-column methylphosphonate and sinking particles in the North Pacific Subtropical Gyre. Aquat. Microb. Ecol. 73, 93–105 (2014).
Seidel, H. M., Freeman, S., Seto, H. & Knowles, J. R. Phosphonate biosynthesis: isolation of the enzyme responsible for the formation of a carbon–phosphorus bond. Nature 335, 457–458 (1988).
Born, D. A. et al. Structural basis for methylphosphonate biosynthesis. Science 358, 1336 (2017).
Kamat, S. S., Williams, H. J. & Raushel, F. M. Intermediates in the transformation of phosphonates to phosphate by bacteria. Nature 480, 570–573 (2011).
Metcalf, W. W. & Wanner, B. L. Mutational analysis of an Escherichia coli fourteen-gene operon for phosphonate degradation, using TnphoA’ elements. J. Bacteriol. 175, 3430–3442 (1993).
Li, H., Veldhuis, M. & Post, A. Alkaline phosphatase activities among planktonic communities in the northern Red Sea. Mar. Ecol. Progr. Ser. 173, 107–115 (1998).
Sosa, O. A., Repeta, D. J., DeLong, E. F., Ashkezari, M. D. & Karl, D. M. Phosphate-limited ocean regions select for bacterial populations enriched in the carbon–phosphorus lyase pathway for phosphonate degradation. Environ. Microbiol. 21, 2402–2414 (2019).
von Arx, J. N. et al. Methylphosphonate-driven methane formation and its link to primary production in the oligotrophic North Atlantic. Nat. Commun. 14, 6529 (2023).
Mahowald, N. M. et al. Aerosol trace metal leaching and impacts on marine microorganisms. Nat. Commun. 9, 2614 (2018).
Lockwood, S., Greening, C., Baltar, F. & Morales, S. E. Global and seasonal variation of marine phosphonate metabolism. ISME J. 16, 2198 (2022).
Liang, Z., Letscher, R. T. & Knapp, A. N. Dissolved organic phosphorus concentrations in the surface ocean controlled by both phosphate and iron stress. Nat. Geosci. 15, 651–657 (2022).
White, A. & Metcalf, W. Microbial metabolism of reduced phosphorus compounds. Annu. Rev. Microbiol. 61, 379–400 (2007).
Letscher, R. T., Wang, W., Liang, Z. & Knapp, A. N. Regionally variable contribution of dissolved organic phosphorus to marine annual net community production. Glob. Biogeochem. Cycles 36, e2022G–e7354G (2022).
Moore, C. M. et al. Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 6, 701–710 (2013).
Wu, J., Sunda, W., Boyle, E. A. & Karl, D. M. Phosphate depletion in the western North Atlantic Ocean. Science 289, 759–762 (2000).
Mark Moore, C. et al. Large-scale distribution of Atlantic nitrogen fixation controlled by iron availability. Nat. Geosci. 2, 867–871 (2009).
Young, C. L. & Ingall, E. D. Marine dissolved organic phosphorus composition: insights from samples recovered using combined electrodialysis/reverse osmosis. Aquat. Geochem. 16, 563–574 (2010).
Acker, M. et al. Phosphonate production by marine microbes: exploring new sources and potential function. Proc. Natl. Acad. Sci. USA 119, e2113386119 (2022).
Metcalf, W. et al. Synthesis of methylphosphonic acid by marine microbes: a source for methane in the aerobic ocean. Science 337, 1104 (2012).
Bell, D. W., Pellechia, P. J., Ingall, E. D. & Benitez-Nelson, C. R. Resolving marine dissolved organic phosphorus (DOP) composition in a coastal estuary. Limnol. Oceanogr. 65, 2787–2799 (2020).
Kanwischer, M., Klintzsch, T. & Schmale, O. Stable isotope approach to assess the production and consumption of methylphosphonate and its contribution to oxic methane formation in surface waters. Environ. Sci. Technol. 57, 15904 (2023).
Mills, M. M., Ridame, C., Davey, M., La Roche, J. & Geider, R. J. Iron and phosphorus co-limit nitrogen fixation in the eastern tropical North Atlantic. Nature 429, 292–294 (2004).
Chien, C. et al. Effects of African dust deposition on phytoplankton in the western tropical Atlantic Ocean off Barbados. Glob. Biogeochem. Cycles 30, 716–734 (2016).
Granzow, B. N. et al. A sensitive fluorescent assay for measuring carbon–phosphorus lyase activity in aquatic systems. Limnol. Oceanogr. Methods. 19, 235–244 (2021).
Van Mooy, B. A. S. et al. Major role of planktonic phosphate reduction in the marine phosphorus redox cycle. Science 348, 783–785 (2015).
Granzow, B. N. Chemical controls on the cycling and reactivity of marine dissolved organic matter. PhD thesis (Massachusetts Institute of Technology and Woods Hole Oceanographic Institution, 2023).
Sallée, J. et al. Summertime increases in upper-ocean stratification and mixed-layer depth. Nature 591, 592–598 (2021).
Wang, W. L., Moore, J. K., Martiny, A. C. & Primeau, F. W. Convergent estimates of marine nitrogen fixation. Nature 566, 205 (2019).
Duce, R. et al. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 320, 893–897 (2008).
Guieu, C. et al. The significance of the episodic nature of atmospheric deposition to Low Nutrient Low Chlorophyll regions. Glob. Biogeochem. Cycles 28, 1179 (2014).
Owens, N. et al. Episodic atmospheric nitrogen deposition to oligotrophic oceans. Nature 357, 397–399 (1992).
Karl, D. M. Microbial oceanography: paradigms, processes and promise. Nat. Rev. Microbiol. 5, 759–769 (2007).
Hutchins, D. A. et al. CO2 control of Trichodesmium N2 fixation, photosynthesis, growth rates, and elemental ratios: implications for past, present, and future ocean biogeochemistry. Limnol. Oceanogr. 52, 1293–1304 (2007).
Wiesenburg, D. A. & Guinasso, N. L. Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and sea water. J. Chem. Eng. Data 24, 356–360 (1979).
Parsons, Y., Parsons, T. R., Maita, Y. & Lalli, C. M. A Manual of Chemical and Biological Methods for Seawater Analysis (Elsevier Science & Technology Books, 1984).
Mao, S. et al. Microbial metabolism and environmental controls of acetate cycling in the Northwest Pacific Ocean. Geophys. Res. Lett. 51, e2024G–e109692G (2024).
Elifantz, H., Malmstrom, R. R., Cottrell, M. T. & Kirchman, D. L. Assimilation of polysaccharides and glucose by major bacterial groups in the Delaware estuary. Appl. Environ. Microb. 71, 7799–7805 (2005).
Lohrer, C., Cwierz, P. P., Wirth, M. A., Schulz-Bull, D. E. & Kanwischer, M. Methodological aspects of methylphosphonic acid analysis: determination in river and coastal water samples. Talanta 211, 120724 (2020).
Ray, J., Bhaya, D., Block, M. & Grossman, A. Isolation, transcription, and inactivation of the gene for an atypical alkaline phosphatase of Synechococcus sp. strain PCC 7942. J. Bacteriol 173, 4297–4309 (1991).
Kirchman, D. Measuring bacterial biomass production and growth rates from leucine incorporation in natural aquatic environments. Methods Microbiol. 30, 227–237 (2001).
Smith, D. & Azam, F. A simple, economical method for measuring bacterial protein synthesis rates in seawater using 3H-Leucine. Mar. Microb. Food Webs 6, 107–114 (1992).
Biller, S. J. et al. Marine microbial metagenomes sampled across space and time. Sci. Data 5, 180176 (2018).
Salazar, G. et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 179, 1068–1083 (2019).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Sosa, O. A. et al. Phosphonate cycling supports methane and ethylene supersaturation in the phosphate-depleted western North Atlantic Ocean. Limnol. Oceanogr. 65, 2443–2459 (2020).
Sunagawa, S. et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 10, 1196–1199 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Martiny, A. C. et al. Biogeochemical controls of surface ocean phosphate. Sci. Adv 5, eaax0341 (2019).
Paulot, F., Stock, C., John, J. G., Zadeh, N. & Horowitz, L. W. Ocean ammonia outgassing: modulation by CO2 and anthropogenic nitrogen deposition. J. Adv. Model. Earth Sy. 12, e2019M–e2026M (2020).
Ackerman, D., Millet, D. B. & Chen, X. Global estimates of inorganic nitrogen deposition across four decades. Glob. Biogeochem. Cycles 33, 100–107 (2019).
Warszawski, L. et al. The inter-sectoral impact model intercomparison project (ISIMIP): project framework. Proc. Natl. Acad. Sci. USA 111, 3228–3232 (2014).
Walters, W. et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, 10–1128 (2015).
Ye, W. W., Wang, X. L., Zhang, X. H. & Zhang, G. L. Methane production in oxic seawater of the western North Pacific and its marginal seas. Limnol. Oceanogr. 65, 2352–2365 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 73, 5261–5267 (2007).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196 (2007).
Zheng, Y. et al. Bacteria are important dimethylsulfoniopropionate producers in marine aphotic and high-pressure environments. Nat. Commun. 11, 4658 (2020).
Li, D., Liu, C., Luo, R., Sadakane, K. & Lam, T. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2014).
Acknowledgements
We acknowledge the captain, crew and scientists of the R/V ‘Dongfanghong 3’ for their kind assistance and cooperation during sampling. We gratefully acknowledge Fabien Paulot (National Oceanic and Atmospheric Administration, Princeton, NJ, USA) for providing modeled global surface seawater concentrations of total ammonium. We thank the 2019 Pacific Northwest Foundation Commission Shared Voyage for providing supporting data. This work was financially supported by the National Natural Science Foundation of China (Grant 42550168 to G.-C.Z.), the Scientific Research Innovation Capability Support Project for Young Faculty Grants (Grant SRICSPYF-ZY2025145 to G.-C.Z.), the Fundamental Research Funds for the Central Universities (Grants 202441002, 202172002 to G.-C.Z.), the National Key Research and Development Program of China (Grant 2022YFE0136300 to G.-C.Z.), the Taishan Scholars Program of Shandong Province (Grant tstp20250524 to G.-C.Z.).
Author information
Authors and Affiliations
Contributions
S.-H.M. and G.-C.Z. designed the experiments. S.-H.M., H.-H.Z., X.-X.G., Y.-Z.N., and Z.Z. collected seawater samples and performed the analyses. S.-H.M. conducted the incubation experiments. S.-H.M., W.-L.W., and G.-C.Z. developed machine learning prediction models. M.L. and J.S. conducted the metagenomic analyses. W.-L.W., K.-Y.L., X.-T.L., X.-H.Z., S.B.J., M.W.B., and G.P.Y. contributed to data reduction and analyses. S.-H.M. and G.-C.Z. wrote the paper and all authors provided critical feedback and editorial comments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Volker Brüchert, Fanghua Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhuang, GC., Mao, SH., Zhang, HH. et al. Atmospheric deposition enhances marine methane production and emissions from global oceans. Nat Commun 17, 1811 (2026). https://doi.org/10.1038/s41467-026-68527-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68527-9
