
Abstract
Hundreds of thousands of tons of epoxy-containing carbon fibre (CF) wastes are generated from the manufacture of CF reinforced epoxy composites to their end-of-use. Today, most recycling strategies of these CF wastes aim at recovering CFs to the ones as virgin as possible, while techniques for efficiently converting CF wastes to materials of higher value, i.e., upcycling of CF wastes, are underexplored. Here, we develop a solid-flames upcycling technique using Mg and CaCO3 powders as reactants to convert CF wastes into graphene-grafted CFs (GCFs) and graphene powders within a few seconds. In Mg/CaCO3 solid-flames, Mg accelerates disconnection of C-O bonds in epoxy resins, promoting interconnection of C-C bonds that lead to the graphene-CF grafting microstructures. This solid-flames upcycling technique, with its superior sustainability metrics and attractiveness in commercial applications including reinforced graphite composites and electromagnetic interference shielding, presents a viable strategy for long-term management of accumulating CF wastes.
Similar content being viewed by others


Introduction
The extensive manufacture of carbon fibre (CF) reinforced epoxy composites (CFRECs) results in vast epoxy-containing CF wastes, typically including trimmed offcuts, out-of-date prepregs and end-of-use composites1. High-performance CFRECs are durable in service, whereas in turn, the epoxy-containing CF wastes are hard to recycle2. Since the demand for CFRECs used in aircraft and wind power industry is rising, the amount of CF wastes is predicted to reach 478,000 tons by 20503.
In recent years, researchers have devoted substantial efforts to developing sustainable strategies for managing the accumulating CF wastes because traditional methodologies, such as incineration and landfilling, have been prohibited in some countries due to environmental unsustainability4,5. Mechanical recycling is sustainable, but mechanically pulverising CF wastes only leads to low-value fillers2. Currently, chemical and thermal approaches are the most adopted recycling techniques for sustainably managing CF wastes1. In chemical recycling techniques, specific chemical reagents (such as noble metal catalysts or oxidizing solvents) are selected to release the CFs by slowly destroying the crosslinked epoxy resin matrix2. Although chemical approaches can recover the CFs with near-virgin properties for reuse, their low efficiency and reagent toxicity make them unsuitable for recycling accumulating CF wastes at scale5,6. Moreover, the chemical products obtained from epoxy decomposition generally have lower value than their petroleum-derived homologues, reducing the commercial attractiveness of chemical recycling methods7. Thermal recycling techniques rely on pyrolyzing the epoxy resins into gaseous or liquid products under limited or no oxygen atmosphere at 400-1000 °C for several hours, followed by oxidation to remove the residual char from CF surface1,3. These lengthy processes result in high energy consumption as well as reduced quality of recycled CFs that jointly reduce the economic potential of thermal recycling techniques. Beyond these conventional recycling techniques, some recent approaches to converting CF wastes into products with other value-added forms have emerged. Flash Joule heating, for example, applies a large current for rapid heating (~104 K s-1) that enables the co-upcycling of CF wastes and glass fibre wastes to SiC6; however, the need for high-power generator increases energy consumption. Moreover, using CF wastes as carbon sources for synthesizing SiC by flash Joule heating compromises the value of CFs. Due to these shortcomings of existing methods, the urgency of addressing sustainability challenges for efficient management of CF wastes continues to intensify.
Here we report a solid-flames upcycling technique to rapidly convert epoxy-containing CF wastes, including CF offcuts, CF prepreg and CFRECs, into graphene-grafted CFs (GCFs) and graphene powders based on the self-propagating combustion of Mg and CaCO3 powders. We characterize the chemical states at the graphene-CF grafting interface and quantify the contributions of different bonding types to grafting stiffness. Using molecular dynamics (MD) simulations and density functional theory (DFT) calculations, we propose a Mg-mediated carbonization mechanism that drives the decomposition of epoxy resins to graphene formation in solid-flames. The sustainability metrics of this solid-flames upcycling technique, including environmental and economic impacts, are also evaluated. We demonstrate the commercial potential of solid-flames upcycling technique by reusing GCFs to reinforce graphite materials and employing graphene powders for electromagnetic interference shielding.
Results
Methodology description
Our methodology is based on the self-propagating high-temperature synthesis (SHS) of graphene powders from solid reactants (Mg and CaCO3 powders) as reported in our previous study8. The chemical reaction between Mg and CaCO3 is as follows:
Using the above Mg/CaCO3-based SHS method, we design a four-step protocol to upcycle CF wastes into GCFs and graphene powders: reactant mixing, electrically igniting, self-propagating combustion, and product purification (Fig. 1a). Firstly, a mixture of CF wastes and Mg/CaCO3 powders is prepared. The mixture is then transferred into a graphite crucible and electrically ignited inside a vacuum container. A total energy of only 0.009 kJ g-1 or less is required for ignition (see “Methods”), granting this main upcycling step higher energy efficiency compared to processing CF wastes by full electrical heating (1-22.5 kJ g-1)6. After self-propagating combustion, the products comprise GCFs, graphene powders, and metal oxides (MgO and CaO). The HCl solution is then used to remove the metal oxides. After acid dissolution, filtration, and drying, the purified graphene powders and GCFs are separated by sieving. Since the SHS method is also phenomenologically recognized as solid-flames (defined as the flames from solid reactants to solid products despite some gaseous or liquid intermediates during combustion9), we name this SHS-based protocol as solid-flames upcycling technique.

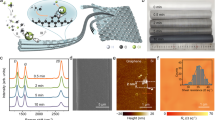
a Schematic diagram of solid-flames upcycling technique. b–d Experimental photos and SEM images of three typical CF wastes before and after upcycling: b CF offcuts and GCF1. c CF prepreg and GCF2. d Milled CFRECs and GCF3. Scale bars, 5 μm (b–d). e Raman spectra of VCF and three GCF samples (GCF1-3). f The ID/IG and I2D/IG values of Raman spectra in (e). g BET adsorption-desorption curves of GCF1 and VCF. CF, carbon fibre. GCF graphene-grafted carbon fibre. CFREC carbon fibre reinforced epoxy composites. VCF virgin carbon fibre. SSA, specific surface area.
Characterizations of upcycled products
Leveraging this solid-flames upcycling technique, we first upcycled the CF offcuts (T800S CF) containing ~2.7 wt.% epoxy sizing agents (Supplementary Fig. 1) into GCF1 and graphene powders. X-ray diffraction (XRD) pattern shows no signals of Mg or CaCO3 peaks in the products before acid dissolution (Supplementary Fig. 2), indicating thorough reactions in solid-flames. Scanning electron microscopy (SEM) results show that well-distributed flakes were synthesized on CF surface (Fig. 1b), making the average roughness of GCF1 surface (144 nm) surpass that of virgin CF (VCF, 4 nm) by 36 times (Supplementary Fig. 3). Raman spectroscopy reveals D, G and 2D peaks (ID/IG = 0.28 and I2D/IG = 0.99) in the spectra of the GCF1 sample as the graphene characteristics while no 2D peak is detected in the VCF sample (Fig. 1e, f), demonstrating the flakes on GCF1 surface are mainly composed of graphene10. The surface graphene flakes promote the specific surface area (88.75 m2 g-1) of GCF1 to 170 times greater than that of VCF (0.52 m2 g-1) (Fig. 1g). In addition, SEM and transmission electron microscopy (TEM) images show that graphene powders exhibit flower-like morphologies with few-layer nanostructures (Supplementary Fig. 4), which is consistent with previous study8.
To highlight the universality of our solid-flames upcycling technique, we then upcycled another two typical CF wastes, CF prepreg (T300 CF, containing ~38.6 wt.% partially cured epoxy resins) and CFRECs (T300 CF, containing ~35.2 wt.% cured epoxy resins), into GCFs (denoted as GCF2 and GCF3, respectively) and graphene powders by this solid-flames upcycling technique (Supplementary Fig. 1 and Fig. 1c, d). SEM results and Raman spectra show that the graphene flakes were likewise synthesized on GCF2 and GCF3 surface (Fig. 1c–f). These on-surface graphene flakes also promote the average surface roughness of GCFs (86 nm for GCF2 and 82 nm for GCF3) by over 20 times than that of VCF (Supplementary Fig. 3). However, the ID/IG values of GCF2 (0.83) and GCF3 (1.23) are higher than that of GCF1, implying more defects in the graphene flakes on GCF2 and GCF3 surface10. This could be explained by the higher contents of epoxy resins and increased curing degrees that jointly make the crystallization of graphene flakes harder under the ultrafast cooling stage after self-propagating combustion. We also processed the VCFs (without epoxy resins) in solid-flames (denoted as SCF), barely finding graphene attached to SCF surface (Supplementary Fig. 5). In the above three experiments, graphene powders were also synthesized. These results elucidate that a combination of recycling of CF wastes, CF surface modification by graphene flakes, and synthesis of graphene powders, i.e., upcycling of CF wastes, could be achieved in one step by this solid-flames upcycling technique. In the following discussions related to GCF, the experimental results and simulation models are taken from GCF1 unless otherwise stated.
Microstructural analysis of GCF surface
We first used focused ion beam (FIB) technology to prepare a longitudinal cross-sectional GCF sample to study the microstructures of graphene flakes on GCF surface. Figure 2a shows a rough covering with a thickness of 100-200 nm on CF surface, corresponding to the atomic force microscopy (AFM) roughness results (144 nm). High-resolution TEM (HRTEM) image further verifies the graphene lattice fringe (d = 0.35 nm) perpendicular to the CF graphitic surface within the covering (Fig. 2b).

a SEM image of GCF sideview morphology. The inset shows the GCF cross-section after FIB sampling. b HRTEM image of graphene-CF microstructure. c HRTEM image of the EELS acquisition map area of the graphene-CF interface (middle), estimated sp3 percentages (left) and bond lengths (right). The points shown in the HRTEM image are seven EELS acquisition positions (top three for graphene, one for interface, and bottom three for CF substrate). The distance between two adjacent points is 1 nm. d C K-edge XANES spectra of GCF sample, GCF fitting, SCF sample, SCF fitting and HOPG sample. SCF solid-flames-processed carbon fibre. HOPG highly-oriented pyrolytic graphite. e Nanoscratch testing lateral force versus displacement curve, SEM image and AFM morphological mapping. The yellow and blue dashed boxes shown in SEM and AFM image are the same acquisition area corresponding to the grey dash box in the nanoscratch load-displacement curve. f Root-mean-square displacement (RMSD) of sp2C and sp3C computed from MD grafting model with simulated stiffness to be 733 N m-1. g Calculated force constants for sp3C-sp3C, sp3C-sp2C, sp2C-sp2C bonding, and sp2 out-of-plane configuration. Blue atoms refer to sp3C and orange atoms refer to sp2C. Scale bars, 1 μm (a), 200 nm (a, inset), 5 nm (b), 1 nm (c), 2 μm (e).
To further identify the chemical states at graphene-CF interface, we employed electron energy loss spectroscopy (EELS) using an aberration-corrected electron microscope (Fig. 2c and Supplementary Fig. 6), targeting a representative region within the FIB-prepared sample. C K-edge EELS spectra acquired from seven positions along the graphene-interface-CF trajectory consistently display characteristic π* (~285.7 eV) and σ* (~292.9 eV) peaks, corresponding to π bonding and σ bonding in carbon materials, respectively. From the C K-edge electron energy loss near-edge structure (ELNES) spectra (280-310 eV), we observed a slight increase in the σ* peak intensity at the interface, indicating reduced sp2-hybridized carbon (sp2C) fraction and increased sp3-hybridized carbon (sp3C) fraction. By fitting the ELNES spectra, we estimated interfacial sp3% to be at a maximum of 19.14% among the seven positions (Fig. 2c and Supplementary Table 1). To estimate the bond length distribution at graphene-CF interface, we fitted the C K-edge extended electron energy loss fine structure (EXELFS) spectra at 310-500 eV. The interfacial bond length reached 1.457 Å, slightly greater than that at adjacent positions (1.453-1.456 Å) (Supplementary Table 2). In addition, O K-edge EELS spectra showed no obvious peaks, excluding significant C-O (~1.43 Å) and C=O bonding (~1.21 Å). Since C-C bonds typically span ~1.42 Å (in graphene) to ~1.54 Å (in diamond) in carbon materials, we confirmed the chemical states at this observed graphene-CF interface to be C-C bonding (namely grafting) with a higher interfacial sp3%. The grafting nature is much stronger than van der Waals interactions and thus effectively prevents graphene from peeling off the CF graphitic surface.
Next, C K-edge X-ray absorption near-edge structure (XANES) spectroscopy under synchrotron radiation was used to verify whether the grafting phenomenon universally occurs at graphene-CF interface. Highly-oriented pyrolytic graphite (HOPG, 100% sp2) was measured as the reference standard and graphene-free SCF served as the control sample. Considering five possible bonding components (sp2C-sp2C, sp3C-sp3C, C-O-C, C=O, and C-Mg), we performed linear combination fitting (LCF) of GCF and SCF XANES spectra using the HOPG reference XANES spectra and four other calculated XANES spectra (Fig. 2d, Supplementary Fig. 7 and Supplementary Table 3). Both GCF and SCF were dominated by sp2C-sp2C bonds (93.79% and 94.35%, respectively). Similar fractions of C-O-C, C=O, and C-Mg bonding in both samples imply these bonds arose from the reactions in solid-flames and subsequent processing rather than a characteristic result from graphene-CF grafting. By contrast, a higher proportion of sp3C-sp3C bonds in GCF sample (3.19%) than in SCF (2.48%) was detected, corresponding to C-C bonding with increased sp3% at grafting interface, thereby corroborating the universal grafting nature at graphene-CF interface.
We then measured the graphene-CF grafting stiffness by nanoscratch testing and analyzed how sp2C and sp3C influence grafting stiffness. The nanoscratch lateral force-displacement curve for monofilament GCF (Fig. 2e) shows multiple “T-shaped” characteristics caused by the indenter moving across graphene flakes, with a pronounced force jump at ~8.5 μm corresponding to a stiffness of 727.65 N m-1. AFM morphological mapping at the same location reveals nanoscale protrusions (tens of nanometres), suggesting that the grafting sites are responsible for the increased lateral force. We performed an MD shear simulation based on graphene-CF grafting model under shear stress. The grafting model displays a simulated grafting stiffness of 733 N m-1 (Supplementary Figs. 8-9), matching well with the nanoscratch testing result. We next computed the root-mean-square displacement (RMSD) of sp2C and sp3C from the MD results, discovering that sp3C exhibits better resistance to deformation than sp2C (Fig. 2f). To understand the atomic nature behind this contrast, we conducted DFT calculations to compare the force constants (k) of sp3C-sp3C, sp3C-sp2C, sp2C-sp2C bonding, and sp2 out-of-plane configuration (Fig. 2g). The highest stiffness occurs in sp2C-sp2C bonding (k = 831.48 N m-1), but this configuration also exhibits poor out-of-plane stiffness with only k = 5.95 N m-1 due to anisotropy. By contrast, the sp3C-related configurations present stable stiffness of k = 396.27-397.17 N m-1 from sp3C-sp3C and sp3C-sp2C bonding. which is ~66.7 times greater than sp2C out-of-plane stiffness.
To quantify the relationship between bonding types and grafting stiffness, a theoretical model based on ideal random mixing assumption and Voigt parallel averaging scheme11 was established. Supposing that sp2C and sp3C are randomly distributed within the grafting interface, we determined the bonding fractions to be 65.38% for sp2C-sp2C bonding, 30.96% for sp3C-sp2C bonding and 3.66% for sp3C-sp3C bonding based on ELNES estimation (sp3% = 19.14%). According to Voigt parallel averaging scheme, we multiplied the bonding force constants to their fractions and summed them up to a theoretical maximal grafting stiffness of kT = 681.08 N m-1. The stiffness surplus of 46.57 N m-1 from measured value (727.65 N m-1) is therefore assigned to the deformation resistibility from CF surface graphitic structures. This model further highlights that the applied load could be efficiently transferred to CF substrate by virtue of the C-C bonding at graphene-CF grafting interface.
Mechanism of GCF formation in solid-flames
To understand GCF formation mechanism from epoxy-containing CF wastes, we tracked the epoxy decomposition pathways to investigate C-C bonding formation in graphene growth and grafting. Theoretical calculations indicate that the adiabatic temperature (Tad) of Mg/CaCO3 solid-flames reaches 3098 K, and CF near-surface temperature maintains 2318 K for ~16 μs (Supplementary Note 1, Supplementary Fig. 10 and Supplementary Table 4). This duration is too short for direct experimental probing, so we modelled this process using MD simulations at 2500 K in combination with DFT calculations to study epoxy decomposition pathways in solid-flames.
To begin with, we targeted the pyrolysis of a single diglycidyl ether of bisphenol A (DGEBA) chain (main component of epoxy resin) through reactive force field (ReaxFF) MD simulation. Because of higher bond dissociation energy (BDE) of aryl-O bond (104.89 kcal mol-1) than alkyl-O bond (73.02 kcal mol-1), the ReaxFF-MD trajectory shows that DGEBA chain mainly breaks at alkyl-O bonds, yielding bisphenol A-like (BPA-like) intermediates (Fig. 3a).

a Decomposition of single diglycidyl ether of bisphenol A (DGEBA) epoxy single chain to BPA-like intermediates and subsequent reactions. b Molecular structure, aryl-O bond lengths and aryl-O bond dissociation energies (BDE) of model 1 and model 2. c Dynamic estimation of aryl-O bonds cleavage probabilities of model 1 and model 2. d The variation of C-C and C-O bonds over time. e The variation of monocyclic rings, polycyclic rings and carbon atoms in the largest cluster over time. f Investigation of grafting of epoxy carbon clusters to CF graphitic surface. The blue dashed circles show some C-C bonds form between graphite carbon atoms and epoxy carbon atoms. The snapshot of C-C bonding is taken from 155 ps.
The subsequent evolution of these BPA-like intermediates was also analyzed. The ReaxFF-MD trajectories indicate that ring-opening and methyl removal are two key elementary reactions driving further epoxy decomposition. Ring-opening generates flexible carbon chains with dangling bonds that promote interconnections among carbon clusters, while methyl removal induces sp3-to-sp2 conversion of quaternary carbons that align the aromatic rings towards planarity (Supplementary Fig. 11). However, we also noticed the persistence of aryl-O bonds in BPA-like intermediate over time because of increased BDE from 104.89 kcal mol-1 to 185.75 kcal mol-1, hindering further graphitization of epoxy-derived carbon clusters during pyrolysis12.
Given the inadequacy of thermal effects alone for graphitization of epoxy resins, our focus turned to the role of Mg, which might act as a reducing agent affecting the epoxy decomposition pathways. Energy-dispersive spectroscopy (EDS) mapping of the GCF and SCF samples prior to acid dissolution revealed a distinguishable co-enrichment of Mg and O signals on the GCF surface but minimal in the SCF sample (Supplementary Fig. 12), providing initial evidence for Mg-epoxy reactions resulting in MgO formation. Following acid dissolution, XRD analysis showing the emergence of broadened MgO (200) peak at ~43° indicated the persistence of some MgO nanocrystalline in GCF sample (Supplementary Fig. 13), with a residual content of 0.25 wt.% quantified by inductively coupled plasma optical emission spectrometry (ICP-OES). The near-absence of Mg 1 s signals in X-ray photoelectron spectroscopy (XPS) suggested the encapsulation of MgO nanocrystalline within the graphene flakes, an inference verified by HRTEM imaging where close spatial adjacency between MgO and graphene domains was observed (Supplementary Fig. 14). Scherrer analysis determined the average size of the MgO nanocrystalline to be 4.2 nm (Supplementary Table 5), matching the nanopore size distribution (centred around 3 nm) acquired in BET analysis of the GCF sample (Supplementary Fig. 15). The absence of similar nanopores in the SCF sample further suggests that MgO nanocrystalline formed from the Mg-epoxy reaction functioned as templates for epoxy-to-graphene growth.
To theoretically validate the Mg-epoxy reaction mechanism, we compared the BPA-like intermediate (model 1) with a Mg-integrated analogue (model 2) (Fig. 3b). DFT calculations reveal that aryl-O bond elongates from 124.8 pm (in model 1) to 133.0 pm (in model 2) and a reduction in BDE from 185.75 kcal mol-1 (in model 1) to 134.68 kcal mol-1 (in model 2). Through second-order perturbation theory analysis of the natural bond orbital, we found that the electron acceptance by Mg (\({E}_{{\rm{O}}\to {\rm{Mg}}}^{\mathrm{(2)}}\) = 19.68 kcal mol−1) reduces π-delocalization energy from oxygen to the aryl ring (\({\Delta {E}_{{\rm{O}}\to {\rm{aryl}}}^{(2)}|}_{{{\mathbf{model}}}\,{{\mathbf{1}}}}^{{{\mathbf{model}}}\,{{\mathbf{2}}}}\) = -40.93 kcal mol−1), confirming that Mg weakens aryl-O bond in model 2. For kinetic assessment of this Mg-mediated weakening of aryl-O bond, we computed bond cleavage probabilities across a temperature range (Supplementary Note 2 and Supplementary Tables 6-8). Figure 3c shows that Mg greatly enhances aryl-O bond disconnection efficiency at high temperatures (2000-2500 K), where 90.0% aryl-O bond cleavage probability for model 2 was estimated at 2318 K, significantly surpassing that for model 1 (24.2%). These experimental and computational results demonstrate that Mg promotes the cleavage of aryl-O bonds in BPA-like intermediates for subsequent graphitization.
Building on the insights from the single-chain analysis, we next scaled up our study using a larger ReaxFF-MD model to simulate the reactions at the CF near-surface in solid-flames (Supplementary Fig. 16). We first performed a model comprising 70 DGEBA chains at 2500 K and cross-verified the pyrolytic products with thermogravimetric mass spectral (TG-MS) data. Both ReaxFF-MD trajectory and TG-MS data show the presence of CH3· radicals (Supplementary Fig. 17), verifying the key elementary reactions of single DGEBA chain. Meanwhile, the gaseous products (H2, CH4 and CO) revealed from MD trajectory are also detected in TG-MS data. These released epoxy pyrolytic products are consistent with reported literature13, validating this ReaxFF-MD model for tracking epoxy decomposition pathways in solid-flames.
We next supplied 900 Mg atoms and a 5-layer graphene flake representing CF graphitic surface into this model for subsequent simulations (Supplementary Fig. 18). Tracking bond evolution over time reveals a gradual increase in C-C bonds and a continuous decrease in C-O bonds (Fig. 3d). The latter proceeds in two stages: rapid cleavage of ether alkyl-O bonds (54% cleavage within 25-30 ps), followed by slower cleavage of aryl-O bonds. Concurrently, the gradual increase in C-C bonds implies progressive interconnection of epoxy-derived carbon clusters. Analysis of monocyclic and polycyclic rings suggests that decrease in aromatic rings occurs through two processes: ring-opening, which promotes interconnection among growing clusters, and subsequent growth into larger polycyclic structures that serve as graphene nuclei (Fig. 3e). These pathways enable the largest carbon cluster to grow beyond 1000 atoms with various polycyclic rings, further demonstrating that this Mg-mediated carbonization mechanism promotes transformation from epoxy resins into graphene in solid-flames.
With the formation of large carbon clusters, we tracked their grafting onto the CF surface and found that it occurs preferentially at graphitic edge carbon atoms through C-C bonding (Fig. 3f). This is because graphitic edges have higher reaction activity14, making them ideal sites for the grafting process. Meanwhile, the excellent consistence between grafting incidents and the emergence of sp3C provided simulated dynamic evidence for the increased sp3% at grafting interface from ELNES estimation. Since CF surface defects are typically graphitic edges, we inferred that these defects account for the formation of sp3C and C-C bonds at graphene-CF interface.
Sustainability of solid-flames upcycling technique
We conducted a cradle-to-gate life cycle assessment (LCA) study to evaluate the environmental impacts of this solid-flames upcycling technique (Supplementary Note 3 and Supplementary Tables 9-21). Except for solid-flames upcycling technique, another two CF waste management strategies, thermal recycling and incineration, were also compared in this LCA study (Fig. 4a). The contents of epoxy resins in CF wastes were set to 25 wt.% as a compromise for CF offcut, CF prepreg and CFRECs. Compared to cumulative energy demand (CED) for VCF manufacture (183-286 MJ kg-1)15, the CED for upcycling CF wastes by this solid-flames upcycling technique (10.8 MJ kg-1) is far lower (Supplementary Table 14). In terms of the global warming potential (GWP), the results of the solid-flames upcycling technique (2.1 kg CO2eq kg-1) are relatively lower than another two scenarios (both are 3.4 kg CO2eq kg-1) (Fig. 4b). In addition, synthesizing graphene powders in solid-flames causes less environmental impacts (including CED and GWP) than conventional strategies of graphene synthesis, such as Hummer’s method or CVD (Fig. 4c, d)4,16. Meanwhile, the waste solvents (mainly MgCl2 and CaCl2) generated from acid dissolution are subsequently recyclable17,18.

a Materials flow and environmental impacts of three scenarios, including solid-flames upcycling, thermal recycling and incineration of CF wastes. b Global warming potential of three scenarios for CF waste management in LCA study. c-d Global warming potential and cumulative energy demand of graphene production by solid-flames, Hummer’s method and CVD. The data are cited from refs. 4,16.
The economic benefits of upcycling 1 kg CF wastes by this solid-flames upcycling technique were analyzed by a technoeconomic assessment (TEA) study (Supplementary Tables 17-19). According to the materials flow in the LCA study, 1 kg GCFs and 1 kg graphene powders were synthesized by this solid-flames upcycling technique. Compared to mere synthesis of graphene powders in solid-flames (cost: $17.11 kg-1), the incorporation of CF wastes significantly alleviates the costs of upcycled products into $8.68 kg-1 (Supplementary Table 18). Supposing the selling price of GCFs approaches the market price of recycled CFs ($15 kg-1), we found as low as $11.62 kg-1 can be a profitable selling price of graphene powders with an interest rate of 15% at the tax rate of 25%. Such selling price even makes our graphene powders economically competent with graphite powders (export price: $3-7 kg-1). Overall, the value-added GCFs and graphene powders make this solid-flames upcycling technique economically attractive.
To exemplify the reuse potential of GCFs, we prepared a series of GCF reinforced graphite (GCF/G) composites as a proof-of-concept demonstration for GCF reuse. For GCF1 samples (~1.80 g cm-3), we found the density of GCF/G composites increases to a maximum of 1.79 ± 0.01 g cm-3 at 10 wt.% GCFs compared to that of graphite matrix (~1.65 g cm-3) (Fig. 5a). Three-point bending tests reveal a maximal flexural strength of 106.9 ± 12.9 MPa at 10 wt.% GCFs that substantially surpasses that of the unreinforced graphite (UG) material (24.7 ± 2.9 MPa), demonstrating well reinforcing effects of GCFs. However, adding more than 10 wt.% GCFs led to simultaneous decreases in density and flexural strength. These declines are attributed to void formation among cross-bridged GCFs (Fig. 5a, SEM image) due to the poor deformability of GCFs and poor fluidity of carbon powders under sintering, while the reinforcing benefits of additional GCFs could not compensate for the structural deterioration. For that reason, 10 wt.% fraction was chosen for GCF2 and GCF3 samples, where reinforcing effects in GCF/G composites were also observed (Supplementary Table 22).

a Influences of GCF contents on the densities and flexural strengths of the GCF/G composites. The SEM images show the cross-section of GCF/G composites with 10 wt.% and 20 wt.% GCFs. The error bars in a represent the standard deviation (SD) from n = 4 independent experiments. Data are presented as the mean ± SD. b SEM image and finite element simulations of GCF-matrix and SCF-matrix interface. c Flexural strength-versus-density landscape of GCF/G composite and various graphite materials. CNT, carbon nanotube. ND, nanodiamond. CB, carbon black. d Apparent reinforcing factor (ARF) benchmarking of GCFs against VCF and other reinforcements in graphite materials. e ID/IG distribution acquired from Raman mapping results of graphene powders. f Electrical conductivities of graphene powders under different pressures. The fitting method is based on effective media theory37. g BET adsorption-desorption curve of graphene powders. h EMI shielding efficiency (SE) of the graphene platelet. SET for total SE. SEA, SE for absorption loss. SER, SE for reflection loss. Scale bars, 5 μm (a), 2 μm (b).
We further conducted contrast experiment versus graphene-free SCFs together with finite element (FE) simulations to study how on-surface graphene flakes influence the mechanical performance of GCF/G composites (Fig. 5b, and Supplementary Figs. 19–22 and Supplementary Table 23). Under a flexural stress of 30 MPa, we found interfacial debonding in the SCF/G sample, while the GCF-matrix interface remained intact. FE simulations indicated that graphene flakes effectively disperse the circumfibrous interfacial von Mises stress to 6.17 MPa. In contrast, SCF/G composites exhibited localized stress concentrations with a maximum value of 33.03 MPa, corresponding to the experimentally observed debonding regions which lead to poor flexural performances of SCF/G composites (35.5 ± 6.4 MPa). Therefore, the stress distribution modulated by GCF-matrix interfacial graphene flakes enhances load transfer from matrix to GCF, accounting for the higher mechanical performance of GCF/G composites.
We evaluated the reinforcing performance of GCFs against VCF and other reinforcements used in graphite materials. GCF/G composites exhibit superior flexural strength (106.9 ± 12.9 MPa) than VCF/G composites (89.0 ± 11.7 MPa) due to stronger interfacial GCF-matrix binding (Supplementary Figs. 23–25). With 10 wt.% GCFs, the GCF/G composite also outperforms most commercial graphite materials19 and a variety of reinforced graphite composites, including CF20,21,22,23,24,25,26-, carbon nanotube27,28,29,30-, nanodiamond31-, and carbon black22,32,33-reinforced graphite composites (Fig. 5c). The superior mechanical performance of our GCF/G composites highlights their commercial potential in applications such as metallurgical furnaces and photovoltaic thermal systems. To quantitatively assess the reinforcing performance of GCFs, we defined the apparent reinforcing factor (ARF) as the ratio of composite flexural strength to matrix strength. By benchmarking GCFs (5-20 wt.%) against VCF and other reinforcements20,21,22,23,24,25,26,27,28,29,30,31,32,33,34, we found a maximal ARF of 4.33 at 10 wt.% GCF, higher than VCF (3.60) and other reinforcements (1.04-3.91) (Fig. 5d). Despite variations in material systems and sintering conditions, these results still demonstrate that our GCFs are effective reinforcements for graphite composites.
Except for GCF reuse, we also measured the fundamental properties of graphene powders to assess their functional potential. Raman spectroscopy mapping (Fig. 5e and Supplementary Fig. 26) reveals the ID/IG values of graphene powders to be 0.21-0.43, approaching the benchmark for high-quality graphene (ID/IG < 0.2)35. These ID/IG values also imply low defect densities of graphene powders, resulting in a carrier mobility of 326 cm2 V-1 s-1 (not optimal compared to pristine graphene but still a high value of scalable graphene powders), and a powdery electrical conductivity of up to 3953 S m-1 (Fig. 5f) that exceeds that of activated carbon (10-100 S m-1) and activated graphene (500 S m-1)36. Coupled with a BET specific surface area of 130 m2 g-1 (Fig. 5g), we inferred that the combination of high conductivity and a porous microstructure would make these graphene powders promising candidates for electromagnetic interference (EMI) shielding by virtue of both effective charge conduction and interfacial polarization. In addition, the graphene powders showed great thermal stability with only slight weight loss (~3.26 wt.%) in N2 atmosphere under 800 °C due to the removal of residual oxygen functional groups (Supplementary Figs. 27–28).
In light of these fundamental properties, we sintered these graphene powders to a platelet towards EMI shielding applications. The graphene platelet exhibited an electrical conductivity of 20,635 S m-1 over the estimated powdery conductivity (14,139 S m-1 under 100 MPa according to effective medium theory37), demonstrating the formation of conductive graphene networks within the platelet. With a total shielding efficiency (SET) of 33.3 dB at Ku-band (12.4-18 GHz) (Fig. 5h), this graphene platelet could block more than 99.95% of electromagnetic radiation, comparable to some reported graphene films (SET = 20-48.6 dB38,39). Mechanistic analysis revealed absorption (SEA = 19.1 dB) as the dominant contributor over reflection (SER = 14.2 dB), consistent with the powder’s porous morphology and high specific surface area that promote multiple internal reflections and dipolar polarization. Deconvolution of the absorption mechanism through frequency-dependent complex permittivity quantified the loss contributions as 57.72% from conductive loss and 42.28% from polarization loss at 18 GHz (Supplementary Note 4 and Supplementary Fig. 29). These results corroborate that the high EMI absorption efficiency stems from a synergistic effect between network conductivity inherited from the powder’s graphitic domains and interfacial polarization arising from residual defects and functional groups. Furthermore, the low cost of our graphene powders (less than $17.11 kg-1, see TEA results) is orders of magnitude cheaper than $1000-500,000 kg-1 for commercial graphene or graphene oxide, making them ideal for mass-market applications such as EMI shielding components in electric vehicles and consumer electronics.
Discussion
In view of the deficiency in conventional recycling techniques for managing accumulating CF wastes, we develop an efficient and scalable solid-flames upcycling technique to upcycle epoxy-containing CF wastes into value-added GCFs and graphene powders using Mg and CaCO3 as reactants. By confirming the C-C bonding nature at graphene-CF grafting interface and estimating the bonding contributions to grafting stiffness, we demonstrate the effective load transfer capability of the grafting structures. The proposed Mg-mediated carbonization mechanism that Mg promotes C-O bond disconnection and C-C bond interconnections in epoxy decomposition to graphene pathways in solid-flames could be otherwise inspired for designing sophisticated carbon materials from polymer or biomass based on their interactions with metals at high temperatures. In terms of sustainability, the reagents used in solid-flames upcycling technique are all cheap base chemicals, generating only few greenhouse gas emissions and recyclable waste solvents in the upcycling process. Moreover, the giant released thermal energy from the solid-flames is expected to be harvested for heating supply and subsequent energy conversion. The as-upcycled GCFs, except for their excel reuse in graphite materials, are likewise promising to reinforce polymeric, metal and cementitious matrices. The as-synthesized low-cost graphene powders are also promising candidates for commercial applications. We foresee that our solid-flames upcycling technique could provide a sustainable supply chain from substantial CF wastes to industrial products in the coming future, thereby alleviating environmental burdens and facilitating circular economy.
Methods
Materials
The CF offcuts (T800S CF), CF prepreg (T300 CF) and CFRECs (T300 CF) were purchased from Guangwei Group Co., Ltd. (Weihai, China). The contents of epoxy resins were measured by thermogravimetric analysis (Rigaku TG-DTA8122) under air atmosphere with a heating rate of 10 K min-1. Before upcycling, the CF offcuts were chopped into 3-mm-length short CFs; the CF prepreg was trimmed into 70 × 30 × 0.8 mm3; and the CFRECs were milled into small pieces in a pulverizer. The chemical reagents: Mg (99.5%), CaCO3 (99.5%) and HCl (37%), were purchased from Aladdin (Shanghai, China). The HOPG standard sample was purchased from XFNANO (Nanjing, China).
Upcycling of CF wastes and purification
In the reactant mixing step as shown in Fig. 1a, we prepared three CF waste reactant mixtures for upcycling, respectively, whose compositions are listed in Supplementary Table 24. The mixture was put into a graphite crucible within the 5 L container (Ankoclave SHS5-16-180). The air in the container was pumped out until the pressure less than 1.0 Torr. The mixture was ignited by electrically heating the resistance with a voltage of U = 20 V and a current of I = 20 A for t = 2 seconds. Using Q = UIt, we could calculate the energy consumption of ignition is Q = 800 J. For a typical reactant patch of ~87 grams in this study (or more), the average energy use is normalized to ~0.009 kJ g-1 (or less). After self-propagating combustion, the mixed products were collected in a beaker. The products were immersed with 6 M HCl solution, followed by ultrasonication for 6 h and magnetic stirring for 12 h. Next, the products were filtrated and washed by deionized water, then dried at 110 °C in a vacuum oven for 12 h. Lastly, after separating the products in a 150-mesh sieve, GCFs and graphene powders were collected. During the entire upcycling process, the container was thoroughly protected by a water-cooling system. While Mg and CaCO3 powders were adopted as the reactants in this study, they might not be the only choice for solid-flames upcycling technique because some other substances, such as Mg/MgCO3 or Li/(C2F4)n as reported in refs. 9,40, could also undergo self-propagating combustion and release giant heat to decompose the epoxy resins in the CF wastes. We chose the Mg/CaCO3 system in this study because of the low reactant costs and high-quality graphene products.
Characterizations
SEM images of the fibre surface morphologies were taken by a Tescan Magna field scanning electron microscope. AFM (Bruker Dimension Icon) was used to characterize the surface morphology of GCF in tapping mode. The Raman spectra of the upcycled products were measured by a 532-nm laser with the power of 50 mW equipped in the Raman spectroscopy (Rainshaw inVia-Reflex). The specific surface area of the CFs was measured by Brunauer-Emmet-Teller (BET) adsorption-desorption method in the Micromeritics ASAP 2460 Surface Area and Porosity Analyzer. XRD measurement was carried out in a range of detection 10° <2θ < 90° with Cu Kα radiation (λ = 0.15418 nm, Panalytical Empyrean).
The cross-section of GCF sample was carved by FIB etching (FEI Strata 400S). By depositing organometallic Pt glue onto the FIB-etched sample, the sample was subsequently transferred to a TEM grid, then thinned to less than 100 nm with a progressive FIB etching process. The voltage and currents used in FIB etching were reduced from 20 kV/0.6-1 nA to 5 kV/20-30 pA. The final polish was performed using a voltage of 5 kV and a current of 10 pA. The microstructure of the GCF sample was characterized using a transmission electron microscope (Thermo Fisher Talos F200) with an accelerating voltage of 300 kV. The STEM-EELS characterization was conducted with a spherical aberration-corrected transmission electron microscope (Thermo Fisher Themis Z) at an acceleration voltage of 200 kV. The methods for estimation of sp3% from ELNES spectra and bond lengths from EXELFS spectra are provided in the Supplementary Information. The XANES characterizations were conducted at Shanghai Synchrotron Radiation Facility (SSRF). The four reference XANES spectra for sp3C-sp3C, C=O, C-O-C and C-Mg bonding were calculated using FDMNES codes41 (see the following sections).
The nanoscratch testing to measure graphene-CF grafting stiffness requires a sampling step to prevent GCF from slipping under nanoindentation. The GCFs were dispersed in ethanol and ultrasonicated for 10 min, then fixed onto a copper-coated Si substrate by UV-curing agents. An additional FIB Pt deposition for welding the GCF onto the substrate was employed to guarantee both adhesion and conduction. The parametres for nanoscratch testing over GCF were loading of 10 mN, scratch velocity of 1 μm s-1 and scratch displacement of 10 μm under Berkovich nano-indenter using Keysight G200 Nano Indenter.
Life cycle assessment and techno-economic assessment
The description of the model, calculations and considered parametres of the assessments are provided in Supplementary Information.
Fabrication of graphite materials and measurements
The carbon powders for fabricating graphite materials were purchased from Xinhe New Material Co., Ltd (Quanzhou, China). For GCF/G composites, the GCFs (varying from 5 wt.% to 20 wt.%) and carbon powders were mixed together and then loaded into a graphite die with 20 mm inner diametre. The UG material, SCF/G and VCF/G composite were composed of pure carbon powders, SCFs (10 wt.%)/carbon powders and VCFs (10 wt.%)/carbon powders with the same parametres, respectively. All the graphite materials were fabricated at 2573 K/100 MPa by spark plasma sintering (Shanghai Chenhua SPS-10t-10 furnace) with a heating rate of 100 K min-1 and annealing time of 10 min.
The densities of graphite materials were measured by Archimedes drainage method. The graphite materials were cut into samples of 2 × 2 × 15 mm3, then performed three-point bending test on an AGXplus electronic universal material testing machine with a constant displacement rate of 0.5 mm min-1 and support span of 10 mm. The final density and flexural strength of the graphite materials were determined by the average results of 4 samples.
The CF monofilament was chopped into 40 mm and fixed onto paper supports by UV-cured structural adhesives, then performed monofilament tensile test on Microforce Testing System (ED-Test) with a constant displacement rate of 1 mm min-1. The final CF monofilament tensile strength was determined by the average results of 10 samples.
Testing of graphene powders
The XPS measurements were conducted with a K-alpha (Thermo Fisher) system. The thermal stability of graphene powders was studied by thermogravimetric analysis (Rigaku TG-DTA8122) under N2 atmosphere with a heating rate of 10 K min-1. The powder electrical conductivity measurement was performed by firstly moulding a 1-cm-diametre platelet from graphene powders, then measuring the electrical conductivity of this platelet on a four-probe electrical conductivity instrument at room temperature. The parametre used for sintering graphene powders to a platelet (thickness: 0.6 mm) is also 2,573 K/100 MPa. The EMI SE testing was conducted by waveguide method using a Ceyear 3672B microwave vector network analyzer. The SET, SEA, SER were calculated from S parametres based on the following equations:
The conductive loss and polarization loss behind the absorption loss were calculated from the imaginary permittivity. The detailed analysis is provided in Supplementary Note 4.
Theoretical calculations and simulations
The reference XANES spectra for sp3C-sp3C (diamond), C=O (formaldehyde), C-O-C (diethyl ether) and C-Mg (Grignard reagent) bonds were otherwise calculated using FDMNES codes under three steps: a self-consistent field calculation to determine the ground-state electronic structure, followed by a finite difference method simulation to compute the absorption cross-section, and finally a convolution to account for core-hole lifetime and experimental broadening effects.
The MD simulations were implemented using the Large-scale Atomic-Molecular Massively Parallel Simulator (LAMMPS) software package42. The primary grafting model contains one flat graphene sheet and two curved graphene sheets with amorphous carbon embedded at interface, where amorphous carbon was built from quenching of as-relaxed randomly-located carbon under 5500 K for 10 ps using the reactive forcefield (ReaxFF) potential43. By putting carbon clusters surrounding this grafting model and supplying two rigid graphene sheets as cooling layers, we built an all-carbon shearing model. This shear model was then optimized to a local configuration with an energy and force tolerances of 1.0 × 10-4 kcal mol-1 and 1.0 × 10-4 kcal mol-1 Å−1, and relaxed under NPT ensemble for 100,000 timesteps at 300 K using AIREBO potential44. Next, we exerted a force of x-directional 10-4 eV Å−1 to the top rigid layer and ran the model under NVT ensemble to simulate the grafting sites under shearing stress. The simulated grafting stiffness was calculated from average atom stresses at interface.
The single-chain epoxy pyrolytic model contains a DGEBA polymer chain composed of 6 monomers. This single epoxy chain was optimized to a local configuration with an energy and force tolerances of 1.0 × 10-4 kcal mol-1 and 1.0 × 10-4 kcal mol-1 Å−1. ReaxFF potential was employed to investigate its pyrolytic behaviour under NVT ensemble at 2500 K for 5 ps. Similarly, the large pyrolytic model containing 70 DGEBA models were investigated by the same process.
The simulation of epoxy decomposition pathways in solid-flames was conducted under DGEBA/graphene/Mg nanocomposite subjected to condition of high-temperature from molecular level. The MD box with dimensions of around 50 × 50 × 160 Å3 were created, in which 5-layered graphene flake with 2D dimensions of 36 × 36 Å2 was placed at middle. The graphene flake was surrounded by 70 DGEBA polymer chains and 900 randomly distributed Mg atoms. ReaxFF potential was used to describe the atomic interactions in the DGEBA/graphite/Mg nanocomposite system. The as-generated nanocomposite system was optimized to a local configuration with energy and force tolerances of 1.0 × 10-4 kcal mol-1 and 1.0 × 10-4 kcal mol-1 Å−1. Then, MD simulations were performed to further relax the samples with 100,000 timesteps at temperature of 300 K and z-directional pressure of 1 bar under NPT ensemble. Finally, as-relaxed sample was equilibrated at 2500 K for 150 ps under NVT ensemble. The atom motions in the nanocomposite followed the classical Newton’s motion, in which the velocity-Verlet algorithm with timestep of 0.25 fs was applied to integrate the classic Newton’s equation. All the MD simulation results were visualized by open-source OVITO software45.
The DFT calculations were implemented by ORCA software46. The bond dissociation energy (BDE), defined as the standard energy change at 0 K for the homolytic cleavage of a chemical bond in a molecule (A-B) into two radicals (A· and B·), could be calculated by:
The force constants of the C-C bonding configurations are calculated from secondary derivative of energy (E) variations to displacement near the equilibrium position (x), where the formula is given by:
The E(2) (second-order perturbation energy) as a key quantitative metric derived from natural bond orbital (NBO) analysis could provide an energy estimate for the donor-acceptor hyper-conjugative interaction between a filled NBO (donor, i) and an empty NBO (acceptor, j). The E(2) energy for a specific donor (i) → acceptor (j) interaction is calculated using the following perturbative expression:
where qi is the occupancy (typically ~2 for a bonding or lone pair orbital) of the donor NBO i;F(i, j) is the off-diagonal element (the Fock or Kohn-Sham matrix element) between the donor NBO i and the acceptor NBO j, representing the quantum mechanical coupling or interaction between the two orbitals; εi and εj are the orbital energy of the donor NBO i and acceptor NBO j.
The Gibbs energy barrier (ΔG‡) for reaction A → B under temperature (T) was determined through high-precision single point energy (ESPE) accompanied by thermal corrections (GTC) to Gibbs free energy:
The primary geometric optimization and thermal corrections were calculated at UM06-2X/6-31g(d) level of theory, while UM06-2X/6-311++g(d,p) level of theory was adopted to calculate single point energy for computing bond dissociation energy2, force constants47 and second-order perturbation energy48.
The FE simulation was used to analyze the stress distribution modulated by graphene flakes at GCF-graphite interface. The model comprised 13 fibres (5 μm) embedded within a square matrix domain of 30 μm × 30 μm. The GCF-matrix interface and SCF-matrix interface were both set to 150 nm. Specific parametres are provided in Supplementary Table 23. The domain was discretized using a structured mesh of trilateral elements. A critical, refined mesh was implemented along the fibre-matrix interface to accurately capture the high stress gradients. A detailed discussion on the selection of parametres for FE simulation and related FE sensitivity analysis is provided in Supplementary Note 5. The FE simulations were performed by FEniCS49 codes and the circumfibrous von Mises stress azimuthal integral was computed by self-written python script50.
Data availability
The data generated in the main text and Supplementary Information, the DFT structures and MD model files are provided as a Source Data zip file. Source data are provided with this paper.
Code availability
The post-processing python script for integration of circumfibrous stresses from finite-element simulation results is available at https://doi.org/10.5281/zenodo.17959215 (ref. 50).
References
De, B., Bera, M., Bhattacharjee, D., Ray, B. C. & Mukherjee, S. A comprehensive review on fiber-reinforced polymer composites: Raw materials to applications, recycling, and waste management. Prog. Mater. Sci. 146, 101326 (2024).
Ahrens, A. et al. Catalytic disconnection of C–O bonds in epoxy resins and composites. Nature 617, 730–737 (2023).
Hecker, M. D., Longana, M. L., Thomsen, O. & Hamerton, I. Recycling of carbon fibre reinforced polymer composites with superheated steam – a review. J. Clean. Prod. 428, 139320 (2023).
Munuera, J., Britnell, L., Santoro, C., Cuéllar-Franca, R. & Casiraghi, C. A review on sustainable production of graphene and related life cycle assessment. 2D Mater. 9, 012002 (2021).
Li, H. et al. The challenge of recycling fast-growing fibre-reinforced polymer waste. Nat. Rev. Mater. https://doi.org/10.1038/s41578-024-00762-2 (2025).
Cheng, Y. et al. Flash upcycling of waste glass fibre-reinforced plastics to silicon carbide. Nat. Sustainability 7, 452–462 (2024).
Weckhuysen, B. M. Creating value from plastic waste. Science 370, 400–401 (2020).
Wang, L. et al. Large-scale synthesis of few-layer graphene from magnesium and different carbon sources and its application in dye-sensitized solar cells. Mater. Des. 92, 462–470 (2016).
Nersisyan, H. H. & Lee, J. H. Synthesis of carbon nanostructures in solid-flame: a review of opportunities and challenges. Carbon 226, 119238 (2024).
Ferrari, A. C. & Basko, D. M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 8, 235–246 (2013).
Bak, J. H. et al. High-frequency micromechanical resonators from aluminium–carbon nanotube nanolaminates. Nat. Mater. 7, 459–463 (2008).
Dou, X. et al. Hard carbons for sodium-ion batteries: Structure, analysis, sustainability, and electrochemistry. Mater. Today 23, 87–104 (2019).
Xu, M. et al. Insights into the pyrolysis mechanisms of epoxy resin polymers based on the combination of experiments and ReaxFF-MD simulation. Chem. Eng. J. 473, 145404 (2023).
Liu, L. et al. Heteroepitaxial growth of two-dimensional hexagonal boron nitride templated by graphene edges. Science 343, 163–167 (2014).
Oliveux, G., Dandy, L. O. & Leeke, G. A. Current status of recycling of fibre reinforced polymers: review of technologies, reuse and resulting properties. Prog. Mater. Sci. 72, 61–99 (2015).
Zhang, S. et al. Sustainable production of value-added carbon nanomaterials from biomass pyrolysis. Nat. Sustainability 3, 753–760 (2020).
Wang, Q. et al. Full recycling of MgCl2 wastewater generated in rare earth extraction and separation process by spray pyrolysis. Process Saf. Environ. Protect. 183, 544–554 (2024).
Lu, W. et al. Preparation of α-hemihydrate gypsum from phosphogypsum in recycling CaCl2 solution. Constr. Build. Mater. 214, 399–412 (2019).
Inagaki, M., Kang, F., Toyoda, M. & Konno, H. Chapter 17 - Isotropic High-density Graphite and Nuclear Applications. in Advanced Materials Science and Engineering of Carbon (eds Inagaki, M., Kang, F., Toyoda, M. & Konno, H.) 387–410 (Butterworth-Heinemann, Boston, 2014).
Zhao, Y. et al. Microstructure and thermal/mechanical properties of short carbon fiber-reinforced natural graphite flake composites with mesophase pitch as the binder. Carbon 53, 313–320 (2013).
Lin, X. et al. Investigation of recycled carbon fiber-reinforced ultrafine-grain carbon-matrix composites. Sustain. Mater. Technol. 41, e01033 (2024).
Zheng, J. et al. Study on carbon matrix composite bipolar plates with balance of conductivity and flexural strength. Chin. Chem. Lett. 34, 107616 (2023).
Roh, J. U., Ko, J. H., Hwang, Y.-M. & Roh, J.-S. Mechanical and electrical properties of milled carbon fiber reinforced artificial graphite scrap composites with phenol resin as the binder. Fibers Polym. 24, 741–747 (2023).
Chen, Q. et al. Scalable, low-cost, and environment-friendly preparation of high strength carbon-matrix composites with tree-root-like structured reinforcements. Compos. Commun. 32, 101149 (2022).
Li, J. et al. Improving carbon/carbon composites mechanical and thermal properties by the co-carbonization of pre-oxidized carbon fiber and pitch. J. Appl. Polym. Sci. 139, 51846 (2022).
Wei, H., Chang, G., Xu, S. & Liu, J. Muti-filler composites reinforced with multiwalled carbon nanotubes and chopped carbon fibers for the bipolar plate of fuel cells. Energies 17, 1603 (2024).
Zhao, Y. et al. A sandwich structure graphite block with excellent thermal and mechanical properties reinforced by in-situ grown carbon nanotubes. Carbon 51, 427–430 (2013).
Li, J. et al. Improving the performance of carbon/graphite composites through the synergistic effect of electrostatic self-assembled carbon nanotubes and nano carbon black. Ceram. Int. 48, 36029–36037 (2022).
Lin, X. et al. TiC-modified CNTs as reinforcing fillers for isotropic graphite produced from mesocarbon microbeads. N. Carbon Mater. 36, 961–969 (2021).
Sun, C. et al. Effect of the structure and properties for carbon graphite materials with oleic acid modified multi-walled carbon nanotubes. Appl. Phys. A 128, 901 (2022).
Lin, K. et al. Nanoburl Graphites. Adv. Mater. 33, 2007513 (2021).
Tan, J. et al. Enhanced self-sintering mechanical strength of carbon blocks via removal of light molecular-weight compound in green petroleum coke. Fuel 343, 127912 (2023).
Mathur, R. B., Dhakate, S. R., Gupta, D. K., Dhami, T. L. & Aggarwal, R. K. Effect of different carbon fillers on the properties of graphite composite bipolar plate. J. Mater. Process. Technol. 203, 184–192 (2008).
Chen, Y. et al. Microstructure and mechanical properties of carbon graphite composites reinforced by carbon nanofibers. Carbon Lett. 33, 561–571 (2023).
Wang, X.-Y., Narita, A. & Müllen, K. Precision synthesis versus bulk-scale fabrication of graphenes. Nat. Rev. Chem. 2, 0100 (2017).
Xu, Y. et al. Holey graphene frameworks for highly efficient capacitive energy storage. Nat. Commun. 5, 4554 (2014).
Sreekanth, K. V. et al. Extreme sensitivity biosensing platform based on hyperbolic metamaterials. Nat. Mater. 15, 621–627 (2016).
Shen, B., Zhai, W. & Zheng, W. Ultrathin flexible graphene film: an excellent thermal conducting material with efficient EMI shielding. Adv. Funct. Mater. 24, 4542–4548 (2014).
Wan, S. et al. High-strength scalable graphene sheets by freezing stretch-induced alignment. Nat. Mater. 20, 624–631 (2021).
Zhang, H. et al. Sub-millisecond lithiothermal synthesis of graphitic meso–microporous carbon. Nat. Commun. 15, 3491 (2024).
Bunău, O. & Joly, Y. Self-consistent aspects of x-ray absorption calculations. J. Phys.: Condens. Matter 21, 345501 (2009).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995).
van Duin, A. C. T., Dasgupta, S., Lorant, F. & Goddard, W. A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 105, 9396–9409 (2001).
Stuart, S. J., Tutein, A. B. & Harrison, J. A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 112, 6472–6486 (2000).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2009).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Shimajiri, T., Kawaguchi, S., Suzuki, T. & Ishigaki, Y. Direct evidence for a carbon–carbon one-electron σ-bond. Nature 634, 347–351 (2024).
Rojas, J. J. et al. Amino-oxetanes as amide isosteres by an alternative defluorosulfonylative coupling of sulfonyl fluorides. Nat. Chem. 14, 160–169 (2022).
Logg, A., Mardal, K.-A. & Wells, G. Automated Solution of Differential Equations by the Finite Element Method: The FEniCS Book. 84 (Springer Science & Business Media, 2012).
Ren, Q. Circumfibrous stress integration for finite- element vtu files. https://doi.org/10.5281/zenodo.17959215 (2025).
Acknowledgements
This work was supported by Stable Support Project Funding (grant KJD231170) awarded to J.S. Q. Ren thanks Ruilin Mao of Peking University for helpful discussions of the MD simulation results and Jiapeng Wang of Harbin Institute of Technology for helpful discussions of STEM-EELS characterizations. We thank Dr. Zhaoyuan Sun from the Centre of Analysis and Measurement from Harbin Institute of Technology for the help in nanoscratch testing.
Author information
Authors and Affiliations
Contributions
L.W. and Q. Ren conceived the idea and designed the experiments. W.F. supervised the project as an expert. Z.S. provided and processed the CF wastes. Q. Ren, B.L. and X.B. performed the upcycling of CF wastes. Q. Ren, J.L., B.L., T.Z. and Y.W. conducted the characterizations. W.F., L.W., Q. Ren and C.X. proposed the mechanisms. Q. Ren conducted the functional testing, theoretical/DFT calculations, MD/FE simulations, and LCA/TEA study. Q. Ren, J.S., W.C. and L.Z. conducted the CF monofilament tensile testing. Q. Ren, J.L. and B.L. fabricated the graphite materials and conducted the measurements. L.W. and Q. Ren wrote and revised the paper. All aspects of the research were overseen by W.F., L.W. and J.S. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
L.W., Q. Ren and W.F. are listed as the inventors for two Chinese patents (CN119639079A for invention of a method to upcycle and reuse of CF wastes, and CN119824678A for invention a method for GCF production) filed by Harbin Institute of Technology (patents status: pending). The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, Q., Sheng, J., Li, J. et al. Upcycling carbon fibre wastes in solid-flames. Nat Commun 17, 1443 (2026). https://doi.org/10.1038/s41467-026-68528-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68528-8
