
Abstract
Conventional closed batteries are constrained by the electrical energy efficiency of 100%, inevitably leading to the reduction of electricity storage. In contrast, open decoupled batteries offer the possibility to break this limitation, but remain unexplored. Here, we develop a highly efficient and sustainable open decoupled battery through a three-electrodynamic-potential (3E) design, simultaneously realizing waste-to-energy conversion, power generation and energy storage. For decoupled electrodes, we engineer high discharge voltage (ED) incorporating zinc oxidation and oxygen reduction reactions, and low charge voltage (EC) involving zinc-ion reduction and hydrazine (waste) oxidation reactions. Furthermore, we introduce reverse electrodialysis potential (ERED) by decoupling electrolytes. Consequently, the assembled battery demonstrates stability for 1000 cycles at the fast-charging current density of 300 mA cm−2. Moreover, a scaled 20-Ah-capacity battery was performed achieving a high electrical energy efficiency of 375% at 10 mA cm−2. Techno-economic analyses reveal that storing one megawatt-hour of electricity using the open decoupled battery can reduce the cost and carbon emissions of power generation by over 80% compared to conventional batteries. This work establishes a foundation for designing electricity-amplified batteries with economic and environmental benefits.
Similar content being viewed by others

Introduction
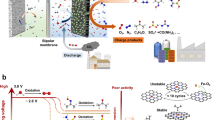
Energy storage systems (ESSs) are critical in developing smart-/micro-grids and the universal application of intermittent renewable energy1,2,3,4. To mitigate energy losses (e.g., activation loss and mass transport loss) during storage, researchers have proposed various strategies, including electrodes’ material designs5,6,7,8, electrolyte modification9,10,11 and battery structure exploration12,13, to improve ESSs’ electrical energy efficiency (ηEEE). Since conventional ESSs operate as closed systems that permit energy transfer with the external environment while preventing matter exchange, their upper limit of ηEEE is 100% (Fig. 1a)14. By comparison, open decoupled batteries (ODBs) allow the input of free or low-cost external matter during operation, potentially realizing ηEEE greater than 100% (ηEEE>100%) (Fig. 1b). A cell with ηEEE>100% can output more power than its inputs, thereby enabling electricity generation and amplification, which hopefully optimizes the energy mix and accomplishes a more economically and environmentally efficient energy supply. However, designing ODBs with ηEEE>100% has not been explored.

a The exchange of matter and energy in conventional closed batteries with the external environment, and the corresponding cell reaction. b The exchange of matter and energy in ODBs with the external environment, and the corresponding cell reactions. For designing ODBs with ηEEE>100%, first of all, we decouple electrodes into three parts including the redox electrode, CPE and DPE, and select appropriate redox pairs as electrodes to achieve low EC, high ED and thus high ηEEE. All the potentials are relative to SHE in alkaline environments. After determining electrodes (Zn-ORR for the discharging reaction and Zn-HzOR for the charging reaction), we further improve ηEEE by introducing ERED between different concentrations of electrolytes. Additionally, we find that nucleophilic sites of N2H4 specifically adsorb on Zn surface to diminish its corrosion, thus enhancing available Zn capacity. Overall, our “3E” design (ED, EC and ERED) renders ODBs with ηEEE exceeding 300%. Win and Wout refer to the input electrical energy and output electrical energy.
Converting waste to energy can conserve significant resources, reduce waste disposal costs and contribute to a sustainable economy15. Generally, wastewater contains abundant low-potential reducing substances (e.g., hydrazine, methanol, glucose, etc.), making it suitable as the charging positive electrode (CPE) of ODBs to achieve low charge voltage (EC). For the redox electrode, zinc (Zn) metal is a promising candidate, owing to its high theoretical capacity (820 mAh g−1 or 5854 mAh cm−3)16, a low working potential of −1.25 V (zincate ion (Zn(OH)42−)/Zn5,17, versus the standard hydrogen electrode (SHE)), abundant reserves and environmental benignity18. Additionally, discharging positive electrodes (DPE) should possess high potential to attain high discharge voltage (ED), whose active materials need to be accessible and economical. Although DPE in acidic conditions typically presents a higher potential than in alkaline conditions, acid-base crossover often leads to battery instability and complexity. Hence, this study primarily focuses on redox couples in alkaline environments. After decoupling electrodes, reverse electrodialysis potential (ERED) can be introduced by decoupling electrolytes, further increasing total discharge voltage (Udischarge = ED + ERED) or decreasing total charge voltage (Ucharge = EC − ERED) to maximize ηEEE.
Here, we develop an ODB achieving a high ηEEE of 445% at 1 mA cm−2, based on the three-electrodynamic-potential (3E: ED, EC and ERED) design. The battery utilizes hydrazine oxidation reaction (HzOR) electrode as CPE and oxygen reduction reaction (ORR) electrode as DPE, respectively. Two electrolytes with distinct concentrations are used separately for Zn electrode and DPE to create an ERED and further increase Udischarge (Udischarge = EZn-ORR + ERED), separated by a cation-exchange membrane (CEM). We also reveal that nucleophilic sites of hydrazine (N2H4) characteristically adsorb on Zn and then alter local charge distribution around it to inhibit its corrosion, improving Zn utilization and total capacity. As a result, the assembled ODB exhibits long-term durability, which works stably for over 1000 h at the fast-charging current density of 100 mA cm−2 with high Zn utilization of 56.7% (typically <10%)19, and for 1000 cycles at the fast-charging current density of 300 mA cm−2. Furthermore, we also demonstrate a large-scale ODB with 20 Ah capacity. Techno-economic analyses (TEAs) illustrate that benefitting from electricity amplification of ODBs, the cost and carbon emissions of the input electricity required to store one megawatt-hour (1 MWh) in ODBs are lower than 20% of those of conventional closed batteries. Notably, using ODB for waste-to-energy conversion also reduces more than 40% of oxidant expense to degrade hydrazine pollutants.
Results
Determination of decoupled components of ODB
The first step in developing a decoupled system is to identify whether the reactions of decoupled electrodes interfere with each other when placed in the same chamber. Therefore, a chronoamperometric curve of commercial platinum/carbon (Pt/C) catalyst was performed and exhibits the conflict between ORR and HzOR by introducing 1 mL N2H4 to 100 mL oxygen-saturated 1 mol L−1 (M) potassium hydroxide (KOH) at 300 s (Fig. 2b). Next, we evaluate the use of decoupled electrolytes to fully exploit the voltage advantage of the two electrodes. For the ORR electrode, its Pourbaix diagram suggests that a lower pH value of the electrolyte leads to a higher EZn-ORR (Fig. 2a). However, when pH values of electrolytes are less than 14 (corresponding to 0.01 M KOH and 0.1 M KOH), EZn-ORR of primary Zn-ORR batteries rapidly drops to approximately 0 V at 1 mA cm−2 (insets of Fig. 2a), probably resulting from sluggish ion transport of limited hydroxide ion (OH−). Hence, 1 M KOH is a suitable electrolyte for the ORR electrode. For the HzOR electrode using cobalt catalyst loaded on nickel foam (Co/NF, Supplementary Fig. 1), linear sweep voltammetry (LSV) curves in Fig. 2c show a higher concentration of OH− results in a lower reaction potential at the same current density, and thereby a smaller EZn-HzOR. In terms of Zn electrode, it is prone to passivation in 1 M KOH (Supplementary Fig. 2), resulting in a low depth of discharge (DoD), while the discharge capacity of Zn in 6 M KOH is four times that in 1 M KOH (Fig. 2d). Consequently, Zn and HzOR electrodes should be placed in one chamber, with the ORR electrode in the other. In typical alkaline Zn-based batteries (TAZBs), saturated zinc oxide is usually added to electrolytes to decrease the ionic concentration polarization, weaken the activity of water and hinder hydrogen evolution reaction (HER) that occurs during charging. Consequently, 6 M KOH, saturated zinc oxide and 0.5 M N2H4 (6 M KOH+sat. ZnO+0.5 M N2H4) are utilized as an electrolyte both for Zn and HzOR electrodes. As shown in Fig. 2e, Ucharge (equivalent to EZn-HzOR as no ERED for charging process) of Zn-HzOR are 0.33 V at 1 mA cm−2 and 0.88 V at 300 mA cm−2, far lower than that of TAZBs (~2 V)13,20,21. Due to decoupling electrolytes, an ion exchange membrane is needed to separate them (Fig. 2f). The direction of the produced ERED using CEM is consistent with that of EZn-ORR, thus boosting Udischarge; while using an anion-exchange membrane (AEM) makes the direction of ERED opposite to that of EZn-ORR, cutting down Udischarge. Zn-CEM-ORR with favorable ERED demonstrates a high voltage of 1.49 V at 1 mA cm−2 in Fig. 2g, better than that in single 1 M KOH (1.34 V) or 6 M KOH (1.44 V) in Supplementary Fig. 3. Notably, all ηEEE of ODB at different current densities ranging from 1 to 20 mA cm−2 exceed 300% and the maximum ηEEE is 445%.

a The Pourbaix diagram of ORR under alkaline conditions. Insets: Zn-ORR discharge voltage profiles of different pH values (pH = 12, 13 and 14) at a current density of 1 mA cm−2. b Chronoamperometric responses of commercial Pt/C by adding 1 mL N2H4 (at 300 s) at 0.74 V (versus RHE) in 100 mL oxygen-saturated 1 M KOH. c LSV curves of HzOR with a scan rate of 5 mV s−1 in 0.5 M N2H4 electrolytes containing different concentrations of KOH. d Long discharge curves of Zn-ORR with 1 M and 6 M KOH at a current density of 20 mA cm−2. The inserted schematic illustrates that the Zn electrode loses a large amount of capacity in 1 M KOH electrolyte because of easy passivation. e Ucharge of Zn-HzOR at different current densities from 1 to 500 mA cm−2 in 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes. Values are means and error bars were calculated by taking the standard errors from one sample for 10 measurements. f Schematics and discharge profiles of decoupled Zn-ORR using CEM and AEM at a current density of 10 mA cm−2 in 1 M KOH electrolytes for ORR electrode and 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes for Zn electrode. g Udischarge of Zn-CEM-ORR at different current densities from 1 to 50 mA cm−2 in 1 M KOH electrolytes for ORR electrode and 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes for Zn electrode. All of the above electrochemical tests were conducted at 25 ± 5 oC. Source data are provided as a Source Data file.
Revelation of corrosion inhibition mechanism
Figure 3a and Supplementary Fig. 4 exhibit that the specific capacity and DoD of Zn-CEM-ORR with N2H4 electrolytes are both greater than those without N2H4 electrolytes, on account of suppressed Zn corrosion by N2H4 molecules. Next, corrosion potential (Ecorr) and corrosion current (Icorr) were examined by Tafel plots (Fig. 3c). The Ecorr of Zn electrodes in N2H4 electrolytes is −1.39 V (versus the mercury/mercury oxide electrode (Hg/HgO)), which is more positive than that of blank electrolytes (−1.41 V versus Hg/HgO). Moreover, the Icorr in N2H4 electrolytes (37.1 μA cm−2) is much lower than that in electrolytes without N2H4 (165.4 μA cm−2), demonstrating the inhibition efficiency of 77.5% and confirming the prominent corrosion resistance of Zn electrode using N2H4 electrolytes (Supplementary Fig. 5). We further investigated the inhibition mechanism by density functional theory (DFT) calculations. Corrosion processes occur at the interface between Zn electrode and electrolyte, summarized as a “trilogy”, involving water dissociation, HER and Zn dissolution (Fig. 3b). According to calculations (Supplementary Fig. 6), adsorption energies of water (−0.29 eV) and N2H4 (−0.42 eV) molecules are negative, which means that both of them spontaneously adsorb on the surface of Zn (Supplementary Data 1,2). The differential charge density analysis shows that the adsorption of nucleophilic sites of N2H4 significantly alters the local charge distribution on Zn surface, increasing negative charge density around the adsorbed molecules (inset of Fig. 3d and Supplementary Fig. 6c). This rearranged local charge distribution resulting from N2H4 adsorption leads to three effects. First, it increases the activation energy of water dissociation (Supplementary Fig. 6d and Supplementary Data 3–5). The projected density of states (PDOS) further displays less overlap of Zn 3d and O 2p orbitals, demonstrating the diminished interaction between Zn surface and water molecule (Fig. 3d). Additionally, the calculated Milliken charges of O and Zn are both decreased, again confirming the weakened Zn-O bond to render water dissociation more challenging (Fig. 3e). Second, the increasing negative charge density strengthens the interaction between H and Zn, resulting in a higher energy barrier for HER compared to pure Zn surface (Fig. 3f and Supplementary Data 6–8). PDOS analysis also reveals that the presence of adsorbed N2H4 molecules makes Zn 3d orbital center closer to the Fermi level (Fig. 3e) and enhances the binding interaction between Zn and H*, causing H dissociation during HER process more difficult22. Third, the Zn dissolution process involves the detachment of Zn atoms from the crystal lattice and the solvation of the detached Zn atoms23,24. For the first process, we calculated the detachment energies of a Zn atom under the adsorption of H2O or N2H4 molecules (Fig. 3g and Supplementary Data 9, 10). For the second process, the negative solvation energies indicate that the solvation process is spontaneous (Supplementary Fig. 6e and Supplementary Data 11–13)25,26. Hence, the whole dissolution process is determined by the first process. The stable adsorption of N2H4 induces electronic rearrangement and spatial hindrance, raising the passivation effect on the Zn surface. As a result, detachment of Zn atoms requires overcoming a higher energy barrier, thus impeding Zn dissolution.

a Specific capacity of Zn-CEM-ORR with 6 M KOH+sat. ZnO and 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes at a current density of 20 mA cm−2. Values are means and error bars were calculated by taking the standard errors from three samples for measurements. b Schematic of corrosion process including water dissociation, HER and Zn dissolution. c Typical Tafel plots demonstrating corrosion of Zn electrodes in 6 M KOH+sat. ZnO and 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes and calculated corrosion inhibition efficiency of N2H4-containing electrolyte. All of the above electrochemical tests were conducted at 25 ± 5 oC. d The PDOS of Zn 3d orbitals and O 2p orbitals of water with and without N2H4 adsorption. Inset: the differential charge density of N2H4 molecule on Zn surface. e The calculated Milliken charges of O and Zn with and without N2H4 adsorption. f Free energy diagram of HER process on Zn surface with and without N2H4 adsorption. The insets are the corresponding optimized structural models. g Detachment energies of Zn atom with H2O and N2H4 adsorption. h In situ Raman spectra of interfacial water in 6 M KOH+sat. ZnO and 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes measured under different current densities. The insets are the schematics of interfacial molecule (H2O and N2H4) arrangements in different electrolytes. Source data are provided as a Source Data file.
To experimentally verify the effect of N2H4 specific adsorption on the water distribution of Zn surface, we conducted in situ Raman spectroscopy under different cathodic current densities (Fig. 3h and Supplementary Fig. 7). For 6 M KOH+sat. ZnO electrolyte, the peak of ~470 cm−1 attributed to the libration mode of H2O is evident regardless of whether a current is applied, which implies that interfacial water maintains a specially ordered structure throughout27,28. In contrast, the peak of interfacial water is almost absent at the beginning in 6 M KOH+sat. ZnO+0.5 M N2H4 electrolyte, which might be ascribed to the interfacial adsorption of N2H4 interfering with the formation of organized structures of water. When the current increases to 50 mA cm−2, a relatively strong peak emerges in the spectrum. We speculate that the elevated current intensifies HER, allowing more water to accumulate at the interface and thereby forming an ordered structure again. Benefiting from the suppressed corrosion, Zn electrode immersing in 6 M KOH+sat. ZnO+0.5 M N2H4 electrolyte for 2, 5, and 10 days presents a flatter and smoother surface as shown in confocal laser scanning microscope (CLSM) images, while severe corrosion with abundant pits and protrusions is observed on Zn surface without N2H4 protection (Supplementary Fig. 8). The above evidence again identifies that N2H4 effectively mitigates Zn corrosion, improving the capacity of ODB.
ODB system performance
Unlike traditional systems where Ucharge is always higher than Udischarge, the Udischarge of our system is higher than Ucharge (Fig. 4a). As illustrated in Fig. 2e, Ucharge of ODB is so low that fast charging is easily achieved. Specifically, ODB operates stably for 1000 cycles (1100 h) at the fast-charging current density of 100 mA cm−2 (10 mA cm−2 for discharging) with the capacity of 10 mAh cm−2 and enables recoverable ηEEE of 261% after replacing depleted electrolytes (Supplementary Fig. 9). During the cycle, ηEEE gradually decreases because of the continuous consumption of active substances in the simulated N2H4 wastewater and changes of the concentration of OH− in both electrolytes. After replacing electrolytes on both sides, the ODB nearly returns to its initial state of high Udischarge and low Ucharge, indicating the long-term stability and recoverability of the system. Furthermore, even when the charging current is increased to 300 mA cm−2 (20 mA cm−2 for discharging), ODB still demonstrates durability for 1000 cycles with a high initial ηEEE of 138% (Fig. 4c). In addition, the fast charging performance of ODB with high Zn utilization of 56.7% (typically <10%)19 is shown in Fig. 4b, steadily working for 1000 h. Considering the operating requirements of batteries in winter, we also tested the rate performance of ODB at 0 oC. As illustrated in Fig. 4d, ηEEE>100% is still achieved at the fast-charging current density of 100 mA cm−2 (10 mA cm−2 for discharging), demonstrating the feasibility of ODB at low temperatures. Notably, the ηEEE and lifespan performance of ODB exhibit advantages compared to previously reported flow battery systems (Fig. 4e, see Supplementary Table 1 for details)20,21,29,30,31,32,33,34,35. To further validate the practical application of our system, a scaled-up ODB system is shown in Fig. 4f and Supplementary Fig. 10. Since ODB with ηEEE>100% endows Wout greater than Win, after satisfying charging energy requirements for treating wastes, the excess electricity (Wout − Win) can be used for other stationary electrical equipment, such as lighting devices and charging stations. The assembled 11 × 11 cm2 ODB shows a large capacity of 20 Ah per cycle (over 18 days) (Fig. 4g).

a Schematic of ηEEE higher than 100% in our system and ηEEE lower than 100% in traditional batteries. b Fast-charging cycling performance of ODB at current densities of 10 mA cm−2 for discharging and 100 mA cm−2 for charging with the same capacity of 100 mAh cm−2. c Fast-charging cycling performance of ODB at current densities of 20 mA cm−2 for discharging and 300 mA cm−2 for charging with the same capacity of 10 mAh cm−2. d Rate performance of ODB at 0 oC under different current densities. e Comparison of flow battery performance in our open system and typical closed systems. f Digital photograph of the large-scale ODB system with ηEEE>100%, which means in addition to wastewater treatment for the charging process, the system has excess electricity for the plant’s stationary electrical equipment (such as lamps and charging stations). g Galvanostatic voltage profiles of the 20 Ah ODB at a current density of 10 mA cm−2. All of the above electrochemical tests (except where otherwise specified) were conducted at 25 ± 5 oC. Source data are provided as a Source Data file.
Economic and environmental benefits of ODB
The two operational modes of the ODB are shown in Fig. 5a. On the one hand, owing to ODBs with ηEEE>100%, all-ODB systems are self-driven, integrating power generation, energy storage and wastewater treatment. Intermittent renewable energies, such as solar energy (SE) and wind energy (WE), require supporting ESSs to ensure a continuous energy supply. Moreover, regulation equipment, such as inverters, is needed between the two, thereby increasing the complexity and cost of the entire system. For the fossil energy power systems, although they are simpler, but emit a large amount of greenhouse gases, which are harmful to the environment. In contrast, all-ODB devices not only simplify infrastructure, but also generate green electricity while treating waste, providing substantial economic and environmental benefits. On the other hand, the ODB can synergize with other power generation technologies to amplify electricity output, further reducing costs and carbon emissions. In detail, TEAs show that the required electricity generated by power generation for storing 1 MWH of electricity in ODBs with ηEEE of 300% is 70%, 73%, 75%, and 73% less than lithium-ion (Li-ion), lead-acid, nickel-iron (Ni-Fe) and all vanadium batteries36,37, respectively, greatly cutting down the expense of power generation technologies (Fig. 5b, Supplementary Fig. 11, Supplementary Note 1 and Supplementary Tables 2–6). Furthermore, due to the significant savings in wastewater disposal, ODB can achieve considerable profits throughout its entire service life, which is totally different from the losses incurred by other ESSs, demonstrating huge potential for application. Additionally, compared to traditional methods for degrading N2H4 in wastewater through oxidants such as ozone (36.4 US$ m−3) and hydrogen peroxide (10.2 US$ m−3), the operation of all-ODB systems significantly shortens disposal expenses (5.3 US$ m−3), thus creating additional economic benefits (Fig. 5c and Supplementary Note 2). Regarding environmental impacts (Fig. 5d), the carbon emission of an ODB is 12.7 kg CO2 eq. MWh−1, better than SE (30 kg CO2 eq. MWh−1) and comparable to WE (10 kg CO2 eq. MWh−1). The combination of SE and ODB emits only 15.3% of the CO2 emissions of the combination of SE and Li-ion. It is worth mentioning that traditional natural gas energy combined with ODB reduces carbon emissions per megawatt-hour of electricity by 63.7% compared with the original all-fuel energy, which is comparable to the combination of SE and Li-ion38,39,40. Based on this, we believe that scaling up ODBs can further diminish costs and emissions, enhancing their dual economic and environmental benefits.

a Schematic of power generation and transportation of all-ODB system, intermittent renewable energy and continuous fossil energy (blue background). And schematic of electricity amplification after combining intermittent renewable energy or continuous fossil energy with ODB, and electricity loss after combining intermittent renewable energy and conventional ESSs (green background). b TEAs of generating 1 MWh of electricity combining different power generation technologies with ESSs. c Disposal costs of hydrazine wastewater with different methods. d CO2 emissions generated by combining different power generation technologies and ESSs to generate 1 MWh of electricity.
Discussion
In summary, the proposed alkaline ODB, based on the “3E” design, combines three electrodes including Zn redox electrode, ORR as DPE and HzOR as CPE, specifically designing ED and EC. Additionally, ERED is engineered between ORR and HzOR electrolytes using CEM. Furthermore, we experimentally and computationally uncover the corrosion inhibition mechanism of N2H4 molecules based on the “corrosion trilogy”, thus promoting the DoD of Zn and the cell’s capacity. Consequently, the assembled ODB shows durability for 1000 cycles at the fast-charging current density of 300 mA cm−2. To illustrate the universality of our “3E” design, we also assembled an acid-ODB using 0.5 M sulfuric acid (H2SO4) as the ORR electrolyte to realize a higher ηEEE, as ORR exhibits a more positive potential in an acid environment. As illustrated in Supplementary Fig. 12, acid-ODB shows high Udischarge of 2.19 V and ηEEE of 665% at 1 mA cm−2. Moreover, even at 0 oC, acid-ODB still maintains a ηEEE of 417% at 20 mA cm−2. Eventually, we performed a scaled-up ODB with 20 Ah capacity. TEAs suggest that the ODB, functioning as both power generation and electricity amplification, demonstrates significant application potential in both economic and environmental aspects. This work represents a revolutionary shift in ESSs from electricity reduction to electricity enlargement.
Methods
Materials
The following chemicals and materials are all commercially available and were used as received: zinc plate (Zn, 99.9%, ZhengYinKeYan), zinc oxide (ZnO, 99%, Aladdin), potassium hydroxide (KOH, 95%, Macklin), hydrazine hydrate (N2H4·H2O, ≥ 80%, ShangHai LingFeng Co., Ltd), commercial platinum/carbon electrode (Pt/C, 1 mg cm−2, thickness of 0.235 mm, SEN23K, SuZhou SINERO Tech. Co., Ltd), commercial manganese dioxide electrode (MnO2, 20 mg cm−2, thickness of 1 mm, ChangSha SiPuLin), gas diffusion layer (ChangSha SiPuLin), cation-exchange membrane (CEM, Nafion 117, thickness of 0.183 mm, Dupont), anion-exchange membrane (AEM, FAA-3-PK-130, Fumasep), nickel foam (NF, thickness of 0.3 mm, ShenZhen KeJing), nickel foil (Ni foil, thickness of 0.05 mm, 99.9%, ZhengYinKeYan), acetone (AR, ShangHai LingFeng Co., Ltd), hydrochloric acid (HCl, AR, ρ = 1.19 g mL−1, XiHua), ethanol (99.7%, Rhawn), deionized water (18.2 MΩ, Millipore system), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99%, Macklin), ammonium fluoride (NH4F, 99.99%, Macklin), urea (CO(NH2)2, 99%, Macklin), sulfuric acid (H2SO4, AR, GuangZhou Chemical Reagent Factory).
Preparation of Co/NF electrocatalyst for HzOR
The Co precursor grown on NF as the substrate was prepared by the hydrothermal method41. Specifically, at the beginning, the NF was thoroughly washed with acetone, 1 M HCl, ethanol and deionized water with ultrasonic treatment for 10 min to wipe off the greasy dirt and oxide layer on the surface. Subsequently, 3 mmol of Co(NO3)2·6H2O, 4 mmol of NH4F and 10 mmol of CO(NH2)2 were dissolved in 60 mL of deionized water under stirring. Then, the resulting solution and the NF (2 × 3.5 cm2) were transferred to a 100 mL Teflon-lined stainless-steel autoclave and maintained at 120 °C for 6 h. After naturally cooling to room temperature, the obtained Co precursor was washed with deionized water and ethanol three times, and dried in a vacuum. After that, the precursor was heated at 325 °C for 2 h with a heating speed of 5 °C min−1 with an Ar-5% H2 mixture atmosphere in a tube furnace. Finally, a black Co/NF was collected after cooling to room temperature. The mass loading of Co/NF was about 2 mg cm−2.
Electrochemical measurements
All electrochemical measurements were performed in a standard three-electrode system using CHI760E electrochemical workstation. A Hg/HgO electrode (1 M KOH) was used as the reference electrode and a graphite rod was used as the counter electrode, respectively. A chronoamperometry test was performed to verify whether ORR and HzOR interfere with each other at 0.74 V versus the reversible hydrogen electrode (RHE). Linear sweep voltammetry (LSV) measurements of HzOR were conducted at a scan speed of 5 mV s−1 without iR compensation. Corrosion property was tested by Tafel plot from −1.30 V to −1.50 V versus Hg/HgO with the scan speed of 5 mV s−1. The corrosion inhibition efficiency (\({\eta }_{{\mbox{CIE}}}\)) is calculated as follows:
where Icorr-without and Icorr-with represent the corrosion current of Zn electrode in 6 M KOH+sat. ZnO and 6 M KOH+sat. ZnO+0.5 M N2H4 electrolytes, respectively.
Battery performance
In a homemade ODB flow system, Zn plate (4.5 cm2, thickness of 0.3 mm) and HzOR electrode with Co/NF electrocatalyst (1 cm2) and a current collector of Ni foil were in the same chamber, using 6 M KOH+sat. ZnO+0.5 M N2H4 (300 mL) as the HzOR electrolyte. ORR electrode including commercial Pt/C electrocatalyst (1 cm2), a gas diffusion layer and a current collector of NF was in a separate chamber, using 1 M KOH (300 mL) as the ORR electrolyte. Zn electrode was placed between HzOR electrode and ORR electrode. CEM was used to separate HzOR electrolyte and ORR electrolyte. The flow rate applied was about 52 mL min−1. Long discharge curves of primary Zn-ORR batteries were used to determine the discharge capacity of Zn in 1 M and 6 M KOH electrolytes without CEM. Galvanostatic charge-discharge measurements of ODB system were evaluated using NEWARE battery testing systems (CT-4008T-5V1A-TWS, Shenzhen, China) or LAND (CT3001A) at different current densities. The above tests were performed at 25 ± 5 oC. The rate performance of ODB at a low temperature (0 oC) was measured using an electrochemical workstation (BioLogic SP-150e). The large-scale ODB device (11 × 11 cm2) was assembled using a 3 × 3 cm2 HzOR electrode, 6 × 6 cm2 Zn electrode (thickness of 1 mm) and 3 × 3 cm2 ORR electrode. The volume of the electrolyte on both sides was 300 mL and the flow rate was also set to about 52 mL min−1. In the acid-ODB flow system, except that the ORR electrolyte was changed from 1 M KOH to 0.5 M H2SO4, other conditions remained the same as in ODB system. All tests are performed in an open-air environment.
The electrical energy efficiency (ηEEE) is calculated as follows:
Since the capacity (It) for charging and discharging is set to the same, the above equation is converted to:
where W, U, I, and t represent the electrical energy, voltage, current and time of the battery during charging and discharging, respectively.
Characterizations
In situ Raman spectra were conducted to characterize the interfacial water of Zn electrodes in electrolytes in order to reveal the corrosion mechanism, recorded by Raman spectrometer (HORIBA LabRAM HR800) with a 532 nm laser. The in situ three-electrode test device is shown in Supplementary Fig. 7. The Zn plate (size: 1 × 1 cm2), a Pt wire and a Hg/HgO electrode (1 M KOH) were used as the working, counter and reference electrode, respectively. The working electrode was oxidized by applying different currents (each current maintained for 6 min) through an electrochemical workstation. A confocal laser scanning microscope (CLSM, VK-X1000, KEYENCE) was used to determine the calendar corrosion of electrodes by analyzing surface roughness. The X-ray diffraction was performed on Bruker D8 Advance with Cu Kα radiation at a scan rate of 10° min−1. All tests were performed in the atmosphere at 25 ± 5 oC.
Theoretical calculations
DFT calculations were carried out using the CP2K/Quickstep software package. For electronic structure calculations, the Perdew-Burke-Ernzerhof (PBE) functional was applied, with Van der Waals forces accounted for through the Grimme D3 dispersion correction. A plane wave energy cutoff of 800 Ry was chosen to ensure precise wavefunction expansion, employing the Triple-Zeta Valence with Polarization (TZVP) basis sets. To achieve high accuracy in electronic calculations, the self-consistent field (SCF) convergence criterion was set at a stringent 1 × 10−6 Hartree. The Brillouin zone sampling was performed using a 3 × 3 × 1 k-point mesh. For slab relaxations, the bottom two layers were kept fixed, while the top two layers and any adsorbates were allowed to relax. Solvation effects were modeled through a hybrid computational approach combining explicit molecular representation with continuum electrostatics. The first two solvation shells were explicitly simulated using OH− and water molecules, selected for their specific coordination geometry and binding energetics with ions and critical roles in hydration thermodynamics. Surrounding solvent effects were incorporated via the Polarizable Continuum Model (PCM), where bulk water was represented as a uniform dielectric medium characterized by its experimentally determined static permittivity of ε = 78.5. The adsorption free energies of hydrogen and water on Zn surface in this work are calculated using the expression:
where ΔE represents the adsorption energy of H or water. The terms ΔZPE and TΔS account for the corrections due to zero-point energy and entropy, respectively. The calculated values for and H₂O, H* and H2 are consistent with previous studies 42.
Data availability
The data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Yao, Y., Lei, J., Shi, Y., Ai, F. & Lu, Y.-C. Assessment methods and performance metrics for redox flow batteries. Nat. Energy 6, 582–588 (2021).
Zhou, G., Chen, H. & Cui, Y. Formulating energy density for designing practical lithium–sulfur batteries. Nat. Energy 7, 312–319 (2022).
Wang, J. et al. Toward direct regeneration of spent lithium-ion batteries: a next-generation recycling method. Chem. Rev. 124, 2839–2887 (2024).
Jia, Y. et al. Variable and intelligent catalyst design based on local chemical environments in sulfur redox reactions. Joule 9, 101878 (2025).
Zhang, Q. et al. Constructing bipolar dual-active sites through high-entropy-induced electric dipole transition for decoupling oxygen redox. Adv. Mater. 36, 2401018 (2024).
Li, Y. et al. In situ formation of liquid crystal interphase in electrolytes with soft templating effects for aqueous dual-electrode-free batteries. Nat. Energy 9, 1350–1359 (2024).
Wang, B. et al. Sulfion oxidation assisting self-powered hydrogen production system based on efficient catalysts from spent lithium-ion batteries. Proc. Natl. Acad. Sci. USA 120, e2317174120 (2023).
Liu, J.-N. et al. A data-driven bifunctional oxygen electrocatalyst with a record-breaking ΔE = 0.57 V for ampere-hour-scale zinc-air batteries. Joule 8, 1804–1819 (2024).
Choi, G. et al. Soft-hard zwitterionic additives for aqueous halide flow batteries. Nature 635, 89–95 (2024).
Yang, C. et al. All-temperature zinc batteries with high-entropy aqueous electrolyte. Nat. Sustain. 6, 325–335 (2023).
Song, Y.-W. et al. Phase equilibrium thermodynamics of lithium–sulfur batteries. Nat. Chem. Eng. 1, 588–596 (2024).
Zhong, C. et al. Decoupling electrolytes towards stable and high-energy rechargeable aqueous zinc–manganese dioxide batteries. Nat. Energy 5, 440–449 (2020).
Zhong, X. et al. Rechargeable zinc-air batteries with an ultra-large discharge capacity per cycle and an ultra-long cycle life. Adv. Mater. 35, e2301952 (2023).
Xi, D. et al. Mild pH-decoupling aqueous flow battery with practical pH recovery. Nat. Energy 9, 479–490 (2024).
Gong, A. et al. Energy recovery and saving in municipal wastewater treatment engineering practices. Nat. Sustain. 8, 112–119 (2024).
Liu, Z. et al. Unraveling paradoxical effects of large current density on Zn deposition. Adv. Mater. 36 (2024).
Xiao, X. et al. Rational design of flexible Zn-based batteries for wearable electronic devices. ACS Nano 17, 1764–1802 (2023).
Zheng, Z. et al. An extended substrate screening strategy enabling a low lattice mismatch for highly reversible zinc anodes. Nat. Commun. 15, 753 (2024).
Parker, J. F., Ko, J. S., Rolison, D. R. & Long, J. W. Translating materials-level performance into device-relevant metrics for zinc-based batteries. Joule 2, 2519–2527 (2018).
Huang, S. et al. A universal coulombic efficiency compensation strategy for zinc-based flow batteries. Adv. Mater. 36, e2406366 (2024).
Wang, Q. et al. Rescue of dead MnO2 for stable electrolytic Zn-Mn redox-flow battery: a metric of mediated and catalytic kinetics. Natl. Sci. Rev. 11, nwae230 (2024).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238–240 (1995).
Taylor, C. D. The transition from metal–metal bonding to metal–solvent interactions during a dissolution event as assessed from electronic structure. Chem. Phys. Lett. 469, 99–103 (2009).
Sharma, S. et al. Metal dissolution from first principles: potential-dependent kinetics and charge transfer. Electrochim. Acta 437, 141443 (2023).
Cao, L. et al. Solvation structure design for aqueous Zn metal batteries. J. Am. Chem. Soc. 142, 21404–21409 (2020).
Roy, K. et al. How solvation energetics dampen the hydrogen evolution reaction to maximize zinc anode stability. Adv. Energy Mater. 14, 2303998 (2024).
Toney, M. F. et al. Voltage-dependent ordering of water molecules at an electrode–electrolyte interface. Nature 368, 444–446 (1994).
Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Li, Y. et al. Main-group element-boosted oxygen electrocatalysis of Cu-N-C sites for zinc-air battery with cycling over 5000 h. Nat. Commun. 15, 8365 (2024).
Wei, Z. et al. Starch-mediated colloidal chemistry for highly reversible zinc-based polyiodide redox flow batteries. Nat. Commun. 15, 3841 (2024).
Wang, C. et al. High-voltage and dendrite-free zinc-iodine flow battery. Nat. Commun. 15, 6234 (2024).
Xie, C. et al. Reversible multielectron transfer I−/IO3− cathode enabled by a hetero-halogen electrolyte for high-energy-density aqueous batteries. Nat. Energy 9, 714–724 (2024).
Lv, X.-L. et al. Modular dimerization of organic radicals for stable and dense flow battery catholyte. Nat. Energy 8, 1109–1118 (2023).
Zhao, Z. et al. Air-stable naphthalene derivative-based electrolytes for sustainable aqueous flow batteries. Nat. Sustain. 7, 1273–1282 (2024).
Lei, J. et al. An active and durable molecular catalyst for aqueous polysulfide-based redox flow batteries. Nat. Energy 8, 1355–1364 (2023).
Schmidt, O., Hawkes, A., Gambhir, A. & Staffell, I. The future cost of electrical energy storage based on experience rates. Nat. Energy 2, 1–8 (2017).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Yokoi, R., Watari, T. & Motoshita, M. Future greenhouse gas emissions from metal production: gaps and opportunities towards climate goals. Energy Environ. Sci. 15, 146–157 (2022).
Turconi, R., Boldrin, A. & Astrup, T. Life cycle assessment (LCA) of electricity generation technologies: overview, comparability and limitations. Renew. Sustain. Energy Rev. 28, 555–565 (2013).
Wang, L. et al. A comparative life-cycle assessment of hydro-, nuclear and wind power: a China study. Appl. Energy 249, 37–45 (2019).
Zhu, L. et al. Active site recovery and N-N bond breakage during hydrazine oxidation boosting the electrochemical hydrogen production. Nat. Commun. 14, 1997 (2023).
Nørskov, J. K. et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152 (2005).
Acknowledgements
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2023B1515120099, G.Z.), Guangdong Innovative and Entrepreneurial Research Team Program (Grant No. 2021ZT09L197, G.Z.), Shenzhen Science and Technology Program (Grant No. KQTD20210811090112002, G.Z.), Guangxi Young Elite Scientist Sponsorship Program (Grant No. GXYESS2025063, B.W.), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2025A1515011765, X.Z.), Shenzhen Science and Technology Program (Grant No. JCYJ20240813094606009, X.Z.), and the National Natural Science Foundation of China (Grant No. 22309077 and No. 22579077, X.Z.). The first author would like to thank Jianyu Xie from Southern University of Science and Technology, Zhiyuan Zhang, Zhiyuan Han and Yeyang Jia from Tsinghua University for their useful discussion.
Author information
Authors and Affiliations
Contributions
G.Z., Z.Z., B.W. and X.Z. conceived the project. Z.Z. and B.W. synthesized the materials. Z.Z. and F.Z. carried out the materials characterization and analyzed the data. B.W. conducted the TEAs. B.H. conducted theoretical simulations. J.X., Z.X., Z.L. and J.L. provided experimental insights. All authors participated in manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Michael Purdy, and the other, anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, Z., Zheng, FY., Huang, B. et al. An open decoupled cell design achieving electricity generation and amplification through waste-to-energy conversion. Nat Commun 17, 1838 (2026). https://doi.org/10.1038/s41467-026-68550-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68550-w
