
Abstract
Nanopore direct RNA sequencing (DRS) offers distinct advantages for transcriptome analysis over the traditional high-throughput RNA sequencing methods by preserving native RNA modifications, eliminating polymerase chain reaction bias, and simplifying the workflow. However, its high basecalling error rate remains a significant hurdle. Here we introduce Coral, a dual context-aware nanopore DRS basecaller that uses a Transformer-based encoder-decoder architecture to capture contextual dependencies at both the signal and sequence levels, substantially improving accuracy. Coral achieves up to a 6.17% improvement in accuracy on human RNA samples compared to Oxford Nanopore Technologies’ Dorado basecaller. This improved accuracy enables the detection of 26% more annotated transcript isoforms. Coral also enhances the downstream haplotype phasing, reducing switch errors by up to 78.8% and Hamming errors by 76%, while phasing 36% more single nucleotide polymorphisms.
Similar content being viewed by others

Introduction
Advancements in RNA sequencing technologies have significantly enhanced our understanding of transcriptomes, revealing intricate details about gene expression, splicing events, and RNA modifications1. Traditional high-throughput RNA sequencing (RNA-seq) typically involves the synthesis of complementary DNA (cDNA) followed by sequencing. However, the cDNA synthesis and amplification tend to introduce biases that potentially affect the accuracy of the transcript quantification2. Moreover, the critical information of native RNA modifications is often lost in the cDNA conversion process, hindering comprehensive epitranscriptomics analyzes3. Nanopore direct RNA Sequencing (DRS) developed by Oxford Nanopore Technologies (ONT), enables the direct sequencing of RNA molecules without the need for reverse transcription into cDNA4,5. It has emerged as a powerful alternative to traditional RNA-seq methods, yielding full-length and strand-specific RNA sequences and enabling the direct detection of nucleotide analogs in RNA6,7,8,9,10,11.
Recent studies have demonstrated the versatility and power of nanopore DRS in transcript structure identification, novel transcripts discovery, and allele-specific analysis. For example, Glinos et al.12 used nanopore DRS to elucidate the genetic effects of rare and common variants on the transcriptome. The analysis provided new insights into how genetic variants influence transcript structure and splicing, contributing to our understanding of gene regulation and disease risk. Berger et al.13 demonstrated that haplotype phasing could be significantly enhanced using the allele-specific information within nanopore DRS data, offering longer and more accurate haplotypes. In a systematic comparison of long-read RNA sequencing methods, Palacios et al.14 demonstrated that libraries with longer reads can improve transcript identification accuracy, further solidifying nanopore DRS as a preferred method for transcriptomic analysis over relying solely on increased sequencing depth. Wang et al.15 investigated a coupled nanopore DRS with the adaptive sampling function to enable the selective enrichment of target transcriptome and identification of unknown transcripts.
Despite significant advancements, a major limitation of nanopore DRS is its inherently high error rate16. The errors primarily arise from noisy ionic-current signals and complex sequence structures (e.g., long homopolymer stretches, hairpins) passing through the pore17. To improve the basecalling accuracy of nanopore sequencing, deep learning-based basecallers have been widely proposed to handle complex noise interference. For example, Chiron18 combines Convolutional Neural Network (CNN) and Recurrent Neural Network to achieve end-to-end basecalling without event segmentation. Causalcall19 employs a modified temporal CNN with a Connectionist Temporal Classification (CTC) decoder to model long-range sequence features. SACall20 incorporates self-attention mechanisms to capture long-term dependencies of electrical currents. Compared to nanopore DNA sequencing basecalling, nanopore DRS basecalling is under-explored5. The early ONT-developed basecaller Guppy employs an alternating gated recurrent unit (GRU) and reverse-GRU layers network architecture combined with a CTC decoder21 to predict variable-length nanopore current signals17. The Guppy DRS model was reported to achieve around 90% accuracy across various organisms in both native and in vitro transcribed samples16. Dorado22 is the latest commercially released basecaller developed by ONT, featuring code optimization across multiple platforms and support for the latest DRS kits (e.g., RNA004). RODAN23 represents one of the most successful third-party open source basecallers of nanopore DRS for demonstrating competitive performance with Guppy. It adopts a fully convolutional architecture and CTC decoder to ensure efficient parallel processing. GCRTcall24 is a recent third-party DRS basecaller that integrates a Transformer25 encoder with gated convolutional networks and relative positional encoding, achieving state-of-the-art performance.
Deep learning-based basecallers have achieved promising results, especially in nanopore DNA sequencing26, yet the accuracy of nanopore DRS is unsatisfactory. This discrepancy is primarily due to the structural differences between RNA and DNA molecules7. The inherent noise in raw electrical signals, compounded by RNA-specific features such as chemical modifications3 and secondary structures27, and slower nanopore translocation speed (70 bp per second for RNA002 and 130 bp per second for RNA004) than DNA (450 bp per second)28, necessitate more specific models for nanopore DRS. Despite numerous basecalling models, most are predominantly designed for nanopore DNA sequencing and rely on an encoder-CTC architecture that estimates symbol probability based on signal information alone. While the reliance on the conditional independence assumption between output symbols in CTC enables parallel decoding, this restricts the ability of the model to capture dependencies among output symbols during decoding.
To address the aforementioned issues, in this study, we propose a dual context-aware basecalling model namely Coral for nanopore DRS. Coral employs an autoregressive Transformer-based encoder-decoder architecture to learn the contextual dependencies at both the signal and sequence levels, and utilizes the dual attention mechanisms (self-attention and cross-attention) for richer sequence contextual learning and capturing the alignment between the raw signals and output RNA sequence. Incorporating dual contextual dependencies introduces an inductive bias that helps direct the model toward a more plausible decoding pathway. Beam search and parallel attention computation are incorporated in decoding to enhance computational efficiency. A fast mode, enabling the simultaneous prediction of multiple bases, is also provided to optimize throughput for large-scale analyzes. Coral was systematically validated against the latest versions of ONT’s basecallers Guppy and Dorado, as well as third-party basecallers RODAN and GCRTcall, across various species and sequencing kits, demonstrating a substantial improvement in basecalling accuracy. Coral achieved an advancement in basecalling accuracy on RNA001/002 reads with an average median read accuracy of ~97% across six test datasets and an accuracy of 99.52% on RNA004 reads. Particularly, Coral achieved up to a 6.17% improvement in accuracy on human RNA samples compared to Dorado. The improvement in basecalling substantially benefits the downstream transcriptome analyzes. Coral’s enhanced performance led to a 26% increase in the number of annotated transcripts discovered compared to Dorado on the human NA12878 sample. Coral also improved the phasing of single-nucleotide polymorphisms (SNPs) by up to 36%, and reduced up to 78.8% switch errors and 76% Hamming errors within haplotype blocks, compared to other basecallers.
Results
Overview of Coral
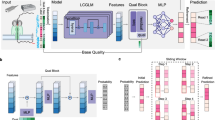
Coral employs an encoder-decoder framework for basecalling (Fig. 1a). The encoder leverages a CNN-Transformer network to process the raw electrical signal effectively, capturing complex temporal dependencies and generating high-level signal features (Fig. 1b). The decoder operates in an auto-regressive manner, predicting the current base conditioned on the previously decoded bases (Fig. 1c). Specifically, the decoder utilizes a Transformer network incorporating dual attention mechanisms, i.e., causal self-attention and cross-attention. Causal self-attention models intricate relationships within the output sequence, enabling richer contextual learning, while cross-attention implicitly learns the alignment between signal and sequence contextual representation. The base probabilities are derived from the final decoder features, and the optimal sequence is identified using a beam search strategy. During the training of Coral, an additional regularization loss is introduced to encourage the alignment produced by the final cross-attention layer to be monotonic, reflecting the nature of the sequencing process. Coral can work in two modes: the default mode predicts a single base per step to achieve maximum precision; while the fast mode, denoted as Coral-FAST, predicts k consecutive bases simultaneously to optimize throughput. More details on the implementation of Coral and Coral-FAST can be found in the “Methods” section and the Supplementary Materials. In the following sections, we demonstrate Coral’s performance on real nanopore DRS data of various species, showing substantial improvements in basecalling accuracy and downstream transcriptome analyzes, i.e., transcript discovery and quantification, allele-specific expression (ASE) analysis, and haplotype phasing.

a Basecalling workflow in Coral. Native RNA sequences are first sequenced using nanopore DRS, obtaining raw signal data. Then the signals are normalized and converted into temporal features by the CNN-Transformer encoder. Then, the decoder combines these temporal features and the contextual dependencies of decoded sequences to predict the next base. b Framework of encoder. The signal encoder extracts temporal features through multiple CNN and Transformer layers with multihead self-attention mechanisms. c Framework of decoder. Previously decoded bases are embedded into high-dimensional vectors via an embedding layer and processed by N stacked decoder blocks. Each block contains two Transformer layers with multihead causal self-attention, followed by a fusion layer that integrates signal context via multihead cross-attention. Finally, a linear layer with softmax produces a probability distribution over the output symbols (A, C, G, U, start-of-sequence symbol 〈sos〉, and end-of-sequence symbol 〈eos〉) for beam search decoding.
Basecalling accuracy
To validate the basecalling performance, Coral was compared with the latest version of Guppy (v6.5.7)29, RODAN (v1.0)23, GCRTcall (v1.0)24, and Dorado (v0.8.3)22 using real nanopore DRS datasets of both RNA001/002 and RNA004 kits. Guppy and Dorado are the official basecallers provided by ONT and have been trained with broadly representative data. RODAN and GCRTcall models were well trained in the work of refs. 23, 24, respectively. For RNA001/002 reads basecalling, Coral was trained on different sequencing samples of Human (from the NA12878 project, BHAM_Run1)7, Arabidopsis23, Epinano synthetic constructs30, Caenorhabditis elegans (C. elegans)31, Escherichia coli (E. coli)32, Mus musculus (Mouse)33, Populus trichocarpa (Poplar)34, Saccharomyces cerevisiae S288C (Yeast)35, 36, and Danio rerio (Zebrafish)37. RNA004 sequencing is featured by increased translocation speed (130bp/s) and improved throughput compared to RNA001/002. Also, the RNA004 kit chemistry operates at a 4 kHz sampling rate, compared to 3 kHz in RNA001/002, enhancing the resolution of the recorded electrical signal. These advancements necessitate the development of specialized basecalling models to interpret the distinct signal characteristics of RNA004 data accurately. To address this, a distinct Coral model was trained on a publicly available Universal Human Reference RNA004 sample38.
All basecallers were tested on RNA001/002 sequencing datasets from six representative species namely Human (from the NA12878 project, HOPKINS_RUN1)7, Arabidopsis39, Mouse40, Yeast36, Poplar34 and Zebrafish37. The test sets are completely independent of the training sets, with the majority of the reads coming from different biological samples. This allowed us to validate the performance of the basecallers across a diverse range of transcriptome data. For RNA004 sequencing, the basecallers (except RODAN that does not support RNA004 reads) were evaluated on the independent RNA004 sample of the HEK293T human cell line from Singapore Nanopore Expression Project41. More details of the datasets and experimental settings can be found in the “Methods” section, as well as in Tables S1–S3.
The basecalling performance was evaluated in terms of six metrics, including read accuracy, mismatch rate, insertion rate, deletion rate, read length (the length of a read excluding the left and right soft clips), and the number of unaligned reads. There is no available data on unaligned reads for Guppy in the RNA001/002 test datasets, which include only the reads successfully basecalled by Guppy and aligned to the reference sequence. On the RNA001/002 test datasets (Fig. 2a–f), Coral outperformed other basecallers across all evaluation metrics. Coral achieved an advancement in basecalling accuracy with an average median read accuracy of ~97%. The improvement is especially notable in the human, Yeast, and Arabidopsis datasets. The average median mismatch, insertion, and deletion rates of Coral were 0.96%, 0.69%, and 1.20%, respectively, markedly lower than those of other basecallers. Coral also yielded the fewest unaligned reads and the longest read lengths, which verifies that higher basecalling accuracy can facilitate better alignment to the reference sequence. On the HEK293T RNA004 sample (Fig. 2g), Coral maintained strong performance by achieving the highest median read accuracy of 99.52%. The results suggest that RNA004 reads can be basecalled more accurately than those from RNA001/002. Coral also achieved the lowest median mismatch, insertion, and deletion rates (0.10%, 0.00%, and 0.14%, respectively). Its identified read length and the proportion of unaligned reads were comparable to Dorado. The findings highlight Coral’s high accuracy and robustness on RNA004 chemistry.

a–d Heatmaps illustrating the median read accuracy, mismatch error rate, insertion error rate, and deletion error rate for five basecallers applied to the RNA001/002 test datasets. In these heatmaps, darker colors signify superior performance. The 'Avg' row represents the average value across all six test datasets. e, f Bar plots depicting the median read length and the number of unaligned reads tested on the RNA001/002 datasets. g Summary metrics for the HEK293T RNA004 sample, including median read accuracy, mismatch rate, insertion rate, deletion rate, median read length, and the proportion of unaligned reads. Statistics were derived from n = 100,000 reads for each RNA001/002 dataset and n = 13,027,670 reads for the HEK293T RNA004 dataset. Source data are provided as a Source Data file.
To rigorously assess Coral’s ability to generalize to previously unseen data, we also performed leave-one-out cross-validation experiments at both the species and chromosome levels. For the RNA001/002 datasets, this involved a leave-one-species-out approach where Coral, RODAN, and GCRTcall were trained on all but one species and subsequently evaluated on the held-out species’ test reads. A similar leave-one-chromosome-out validation was conducted for the human RNA004 dataset, with models trained after excluding all reads from the representative chromosome 22. Note that there are no training pipelines available for Guppy and Dorado, yet they were also included in the comparison by using the corresponding original official models, i.e., no leave-one-out training was done for Guppy and Dorado. In the leave-one-species-out evaluations on the RNA001/002 datasets, Coral achieved a robust average median read accuracy of 95.19% on the unseen species—a modest decrease of only 1.74% compared to the fully trained model—while consistently outperforming the other retrainable basecallers as well as Guppy and Dorado (Table S4). On the held-out chromosome evaluation on the human RNA004 dataset, Coral achieved a median read accuracy of 97.78%, higher than that of RODAN (92.44%) and GCRTcall (94.15%), and comparable to that of Dorado (98.13%) (Table S5). These findings indicate that Coral can avoid overfitting to reference-specific details and learn generalizable and robust mapping from raw electrical signals to nucleotide sequences, making it a reliable tool for analyzing novel species or genomic regions.
Given that homopolymer sequencing errors are a significant type of error in nanopore sequencing26, we investigated the accuracy and the proportions of insertion, mismatch, and deletion of the basecallers on calling homopolymers of varying lengths on the RNA001/002 human dataset (Fig. 3a). Coral is more accurate than the other basecallers for all homopolymer lengths, and it works better in identifying shorter homopolymers. Since short homopolymers (lengths of 2 and 3) constitute a significant proportion (90.64%) of nanopore DRS data (Fig. 3b), Coral can effectively refrain the primary homopolymer errors. Similar results can also be observed on other species (Figs. S1 and S2) and the HEK293T RNA004 dataset (Fig. S3a, b).

a Accuracy, proportions of insertion, mismatch, and deletion errors for homopolymers of varying lengths in the human dataset, derived from the n = 100,000 input reads. Homopolymer accuracy refers to the frequency of homopolymers being correctly basecalled. Error proportions reflect the frequency of insertion, deletion, or mismatch errors occurring within these regions. b Distribution of homopolymer lengths in the human dataset (n = 100,000 reads). c Read length distribution of Coral outputs across all six test datasets (n = 100,000 reads per dataset). d Read accuracy stratified by read length intervals in the human dataset (n = 100,000 reads). e The average four-base match accuracy, mismatch/deletion error of C/U at the single nucleotide level across all six test datasets (n = 100,000 reads per dataset). f Accuracy of k-mer predictions (lengths 5 to 11) by each basecaller in the human dataset (n = 100,000 reads). g Scatter plot showing the accuracy of next-base predictions given a correct 5-mer context in the human dataset (n = 100,000 reads). Circle markers are colored yellow, green, orange, blue, and red for Guppy, RODAN, GCRTcall, Dorado, and Coral, respectively. For all box-and-whiskers plots in a, d, f, the center line represents the median, the edges of the box denote the first and third quartiles, thus enclosing the interquartile range (IQR), and the whiskers extend to the most extreme data points within 1.5 × IQR from the edges of the box. Data points beyond the whiskers are plotted individually as potential outliers. Source data are provided as a Source Data file.
To evaluate the performance of Coral with respect to different read lengths, we assessed the basecalling accuracy as a function of read length. Since the majority of read lengths fall within the range of (0, 2000] (Fig. 3c), the basecalling accuracy of the five basecallers on the RNA001/002 datasets (Figs. 3d and S4) and the HEK293T RNA004 dataset (Fig. S3c, d) is plotted across different read length intervals within (0, 2000]. It can be seen that the accuracy of all the basecallers decreases as the read length increases, whereas Coral obtained higher accuracy than the others across all length intervals.
We further explored the match accuracies of the basecallers at the individual base level (Fig. 3e). The bases ‘C’ and ‘U’ are more likely to be wrongly identified, and the mismatch errors between them (‘C’ to ‘U’ and ‘U’ to ‘C’) constituted the highest proportion of mismatch errors. The deletion errors of ‘C’ and ‘U’ also account for the majority of deletion errors. This is potentially due to their current signal similarity and longer dwelling time, a finding consistent with the previous study16. Similar results are also observed on the HEK293T RNA004 dataset, and Coral reduced the C/U mismatch error rate from ~1% on RNA001/002 reads to around 0.25% on RNA004 reads (Fig. S3e).
Effectiveness of modeling contextual dependencies
On top of signal context dependencies as considered in most CTC-based basecallers, the encoder-decoder architecture of Coral is also intended to capture the contextual dependencies between output bases. Accurate context information is essential for effective decoding in Coral, as error contexts can lead to noisy contextual representations, which are detrimental to subsequent decoding processes. To investigate the impact of k-mer contextual information on basecalling accuracy, we show the accuracy of the decoded k-mers across varying lengths (5 to 11 bases) (Figs. 3f, S3f, and S5) and the accuracy on predicting the next base given a correct 5-mer context (Figs. 3g and S3g). Coral consistently demonstrated superior accuracy. The advantage increases as the length of k-mers increases, i.e., Coral achieved significantly higher accuracy (>0.8) compared to other basecallers (<0.5) in 9-mer, 10-mer, and 11-mer contexts (Fig. S6). Further investigation into the impact of erroneous 5-mer contexts revealed that while an incorrectly predicted 5-mer context increases the error rate of the immediately subsequent base prediction compared to predictions from correct 5-mer contexts (Fig. S7a), this detrimental impact progressively diminishes after ~4 subsequent bases (Fig. S7b). These findings collectively highlight Coral’s superior utilization of dual contextual information for robust decoding and its resilience to contextual errors.
To evaluate the contributions of key components for modeling contextual dependencies, we performed a series of ablation experiments using the Coral framework on the RNA001/002 and RNA004 test datasets, with detailed architectures and parameter sizes provided in Tables S6 and S11. We analyzed the relationship between model scale and basecalling performance across all ablated models to disentangle the effects of architecture and size (Figs. S10 and S11). The results confirm two key findings. First, comparing models at a similar parameter scale highlights that architectural choices are critical. For instance, Coral-L3 (356 MB) significantly outperforms both Coral-CTC (292 MB, where the original Transformer decoder of Coral was replaced by a single linear layer trained with the CTC loss) and Coral-LSTM (309 MB, where LSTM is short for long short-term memory network), affirming the advantage of the auto-regressive Transformer decoder (Table S7). Likewise, comparing Coral-L3 with the LSTM-Transformer (351 MB) highlights the superiority of the CNN-Transformer encoder (Table S8). Comparison between the two modes of Coral, i.e., the normal mode Coral (predicting a single base per step, 548 MB) and the fast mode Coral-FAST (predicting k bases per step, 554 MB), revealed that the impact of symbol prediction granularity, with Coral yielding slightly better performance (Table S9). Second, the performance of Coral scales with model size, as demonstrated by accuracy gains from Coral-S (44 MB) and Coral-M (115 MB) to the larger Coral-Lx variants (356–548 MB, where x = 3, 6, and 12 represents the number of decoder layers) (Table S10). The performance gain becomes moderate when the model size reaches a certain scale, likely due to limitations in training data and the inherent complexity of the task—a trend consistent with neural scaling laws. A similar trend in performance scaling with respect to model size was also observed on the RNA004 dataset (Table S12). Compared with the official Dorado models, Coral also achieved superior performance at comparable model scales on both the RNA002 and RNA004 datasets (Figs. S10 and S11). Overall, the ablation studies provide a robust justification of the current Coral design, demonstrating that the full Transformer encoder-decoder architecture, when appropriately scaled, is critical for achieving state-of-the-art basecalling performance.
Transcript isoform discovery and quantification
To verify the effects of improved basecalling accuracy on the downstream transcript isoform discovery, we applied the five basecallers to nanopore DRS data of three human cell lines, namely A549, HCT116, and NA12878/GM12878, on which high-quality annotated references are available, and for A549 and HCT116 matched deep-coverage Illumina short-read RNA-seq data are also available to enable in silico validation (Table S15). The average median basecalling accuracy of Coral exceeded that of Guppy, RODAN, GCRTcall, and Dorado by 6.87%, 5.23%, 4.33%, and 4.32%, respectively, across the three datasets (Table S16). Coral and Guppy exhibited comparable read lengths, whereas RODAN and Dorado showed a shorter read length, especially on the NA12878 dataset.
Based on the basecalling results, ESPRESSO42, a robust computational tool designed for error-prone long sequencing reads, was utilized to detect the splice junctions (SJs) and transcript isoforms. We investigated the de novo SJs identification, supported by at least two reads with perfect alignments around canonical splice sites. Coral’s outputs achieved comparable precision and outperformed the other basecallers in terms of true positive counts, recall rate, and F1 scores (Fig. 4a). The mean perfect reads count (MPRC) indicates that Coral’s outputs support SJs with a higher number of perfectly aligned reads compared to other basecallers.

a Radar plots comparing de novo splice junction detection performance using ESPRESSO across human cell lines (A549: n = 1,254,612 reads; HCT116: n = 987,488 reads; NA12878: n = 8,540,683 reads). Metrics include precision, recall, true positive rate (TPR), true negative rate (TNR), and mean perfect read coverage (MPRC) at splice junctions. All values are normalized to the best-performing value, shown in parentheses. b Bar plots showing the number of reads classified into transcript categories: FSM, ISM, SEX, NIC, NNC, and NCD. c Upset plot (bottom) showing the intersection size of annotated transcripts discovered by Illumina (using Salmon) and the specific basecallers indicated by linked filled dots below each bar in the A549 dataset. Intersection sets smaller than 300 elements are not displayed. The total number of annotated transcripts identified by different basecallers is shown in parentheses, with each method distinguished by a unique color. Strip plot (upper) showing the abundance distribution of each basecaller across different annotated transcript sets. d Upset plot (top-left panel) showing the intersection size of SIRV transcripts discovered by the specific basecallers indicated by linked filled dots below each bar. The total number of SIRV transcripts identified by different basecallers is shown in the left-hand horizontal bars. Scatter plots (remaining panels) showing the correlation between transcript expression estimates obtained from Coral/Guppy/RODAN/GCRTcall/Dorado reads and the known SIRV input concentrations. The analysis was performed on n = 68 SIRV transcripts. The solid line represents the linear regression fit, and the shaded band indicates the 95% confidence interval (CI) of the regression estimate. Pearson’s correlation coefficient (r) and Spearman’s rank correlation coefficient (ρ) are provided for each basecaller. The expression is quantified as abundance (read count). Source data are provided as a Source Data file.
We further compared the ESPRESSO results against the well-studied human transcript annotations to discover the transcript isoforms. Based on the confidence and configuration of SJs, ESPRESSO categorizes reads into full splice match (FSM), incomplete splice match (ISM), novel in catalog (NIC), novel not in catalog (NNC), single-exon (SEX), and not completely determined (NCD)43. With Coral’s basecalling outputs, ESPRESSO identified the highest number of FSM reads across all datasets, indicating a superior ability to accurately capture and represent known transcript structures (Fig. 4b). Compared to Guppy, Coral identified comparable ISM reads while maintaining the fewest NCD reads that signify reads with low-confidence SJs. RODAN exhibited a higher read accuracy than Guppy (Table S16), yet it yielded the fewest FSM reads, and the highest number of ISM reads in most datasets. This discrepancy may stem from RODAN’s shorter read alignments, which limit its ability to detect complete SJs, and a similar pattern was also observed with Dorado on the NA12878 dataset.
Coral also led to the highest number of annotated transcript isoforms across all three datasets (Figs. 4c and S12). Transcripts commonly discovered by all basecallers exhibited higher expression levels compared to those detected by only one or two basecallers (Figs. 4c and S12), as low-abundance transcripts are generally more difficult to detect. Among these low-abundance annotated transcripts, Coral exhibited the highest sensitivity by identifying the largest number of such transcripts. Furthermore, among the transcripts uniquely detected by each basecaller, Coral consistently identified the largest number of low-abundance transcripts, and these Coral-specific isoforms were further corroborated by supporting evidence in the Illumina short-read data (2219 in the A549 dataset, 1814 in the HCT116 dataset), significantly outperforming the other basecallers (Figs. 4c and S12). Quantification of these Coral-specific isoforms also showed the strongest concordance with the Illumina data (Fig. S13). Coral was also demonstrated to identify the most annotated transcripts across all read proportions (Fig. S14). As the read proportion increases, Coral’s advantage over other basecallers expands.
To further benchmark the transcript identification and quantification accuracy, we performed an additional experiment using the publicly available HEK293T dataset42 spiked with 25 ng of SIRV-Set 1-E2 RNA (Lexogen, lot no. 001418), which comprises 68 artificial transcripts with known sequences and precisely defined concentrations spanning two orders of magnitude. The detected transcripts from different basecallers were then compared with the known SIRV transcripts, and we assessed the correlation between the estimated transcript abundances and the known input concentrations (fmol/ul). The results (Figs. 4d, S15, and S16) highlight the superior performance of Coral in both transcript detection and quantification accuracy. Coral and Dorado successfully identified the largest number of SIRV transcripts, followed by Guppy and GCRTcall, while RODAN detected the fewest SIRV transcripts. Coral also achieved the highest Pearson and Spearman correlation coefficients for abundance estimation compared to the other basecallers.
For the annotated transcripts uniquely detected by Coral, the majority of assigned reads by Coral were classified as FSM or ISM, whereas the same reads in Guppy, RODAN, GCRTcall, and Dorado were mainly classified as NCD or ISM (Fig. 5a). The results indicated that Coral can significantly bolster the reliability of transcript annotation. On the ten most abundant annotated transcripts uniquely discovered by Coral, the read accuracy (by transcriptome alignment) was consistently higher than that of other basecallers (Tables S17–S19). We then inspected the reads assigned to the most abundant annotated transcripts that were identified exclusively by Coral, including TM4SF1-201 in A549, CHCHD10-202 in HCT116, and HLA-A-203 in NA12878 (Fig. 5b). On these transcripts, the reads basecalled by Coral were primarily classified as FSM, whereas the corresponding reads basecalled by other basecallers were mainly categorized as NCD. Coral also achieved a significantly higher proportion of perfectly aligned reads at the splice junctions of these transcript models, further confirming these findings (Fig. S17). These results demonstrated Coral’s superior capability to maintain the integrity of SJs, thus significantly enhancing the reliability of transcript identification and quantification.

a Classification of reads assigned to the annotated transcripts identified by Coral but missed by other basecallers across three human cell lines. b Classification of the reads assigned to the most abundant transcript isoform discovered only by Coral across three datasets. c The detection status and proportion of spanning reads of 12 specific MCF-7 cell fusion genes (validated by RT-PCR and Sanger sequencing) using different basecallers. This analysis was performed on n = 7,230,921 reads. d Upset plot showing the intersection size of the genes with allele-specific expression (ASE) identified by the specific basecallers indicated by linked filled dots below each bar in the NA12878 dataset. The total number of ASE genes identified by different basecallers is shown in the left-hand horizontal bars. Intersection sets smaller than 9 elements are not displayed in the Upset plot. e Parental allele assignment for the genes HLA-C, GSTP1, CRYZ based on basecalling by Guppy, RODAN, GCRTcall, Dorado, and Coral. The number of reads assigned to maternal (M) and paternal (P) alleles at the heterozygous SNP with the highest total coverage of each gene is shown. Source data are provided as a Source Data file.
We further investigated the biological roles of the uniquely identified transcripts. In the NA12878 dataset, the transcripts uniquely identified by Coral exhibited the highest abundances, with a predominant association with immune regulation. Specifically, the transcripts ENST00000376809.1044, ENST00000376806.945, and ENST00000706900.1, which were identified with abundances of 4656.61, 2330.06, and 564.65, respectively, correspond to different isoforms of the HLA-A gene encoding a major histocompatibility complex class I molecule. The HLA system plays an important role in the occurrence and outcome of infectious diseases, including those caused by the malaria parasite, the human immunodeficiency virus, and the severe acute respiratory syndrome coronavirus. Additionally, ENST00000216802.1046 identified with abundance 1942.10 corresponds to PSME2, encoding PA28 protein which can enhance the generation of antigenic peptides by inducing dual substrate cleavages by the 20S proteasome47. In contrast, transcripts uniquely identified by other basecallers, although also involved in immune regulation, exhibited lower expression levels, such as ENST00000375224.1 (Guppy)48, ENST00000372874.9 (RODAN)49, ENST00000316757.4 (GCRTcall)50, and ENST00000244204.11 (Dorado)51, with abundances of 128.72, 459.67, 68.20, and 93.99, respectively.
In addition to detecting annotated transcripts, identifying fusion genes is also critical, as they serve as key diagnostic markers and therapeutic targets in cancer cells, and long-read transcriptome sequencing enables the detection of fusion genes along with their full-length transcripts52. To further assess Coral’s performance in detecting fusion genes, we used JAFFAL52 to identify fusion genes from an ONT DRS replicate of the MCF-7 cancer cell line41 and compared the results to previously validated fusion events in the Mitelman database53. Coral demonstrated a median basecalling accuracy improvement of 9.25%, 7.68%, 6.18%, and 4.91% over Guppy, RODAN, GCRTcall, and Dorado, respectively (Table S16). Coral identified 44 known fusion genes, exceeding Guppy’s 29, RODAN’s 20, GCRTcall’s 33, and Dorado’s 36 (Fig. S18). For validation against experimentally confirmed events, we utilized findings from ref. 41, which experimentally validated 12 specific fusion genes within the same MCF-7 sample using RT-PCR followed by Sanger sequencing. Coral successfully identified all 12 (100%) of the experimentally validated fusion genes, which outperformed other basecallers. Guppy and Dorado both detected 11 out of 12 (91.7%), while RODAN detected the fewest (Fig. 5c). RODAN’s lower detection rate may be due to its significantly shorter read lengths (Table S16). Furthermore, Coral generated the highest number of spanning reads aligning across these validated fusion gene breakpoints, indicating robust detection sensitivity. These findings highlight that Coral’s improved read accuracy enhances the detection of fusion genes in DRS.
Allele-specific expression analysis
ASE refers to the preferential transcription of RNA from either the paternal or maternal allele of a gene. RNA-seq can provide quantitative estimates of ASE for genes with transcribed polymorphisms. To investigate the effects of the basecallers on ASE, we used HapCUT2 to identify reads with allele-informative variants in the NA12878 dataset. Reads were required to contain at least two heterozygous SNPs, with no less than 75% of heterozygous SNPs consistent with parental alleles, leveraging the NA12878 small variant truth set for accurate parental origin assignment. A binomial test was used to test for statistically significant allele-specific analysis, and the P values were corrected for multiple hypothesis testing using the Benjamini–Hochberg procedure, with a false discovery rate (FDR) of 0.01. Coral identified 269 significant ASE genes, surpassing Guppy’s 165, RODAN’s 147, GCRTcall’s 152, and Dorado’s 116 (Fig. 5d). On the ASE genes identified by all basecallers, i.e., GSTP1, CRYZ, and HLA-C, the number of reads assigned to parental alleles at various heterozygous SNP sites clearly showed that Coral exhibited stronger parental allele specificity (Fig. 5e).
Haplotype phasing
The splicing of RNA transcripts as they mature from pre-mRNAs to mRNAs provides an opportunity to mitigate the problem of short-read spans by bringing together exons across large genomic distances, thus enabling the recognition of heterozygous alleles from the same chromosomal copy. RNA-seq enhances this capability by directly capturing splicing events and providing full-length transcript data, which is crucial for accurate haplotype phasing. To evaluate the performance of Coral in haplotype phasing, we analyzed whole-cell nanopore DRS data from the GM12878 cell line. The data were basecalled, followed by SNP calling with Clair3-RNA54, and haplotype phasing with HapCUT255. The phased haplotype blocks were compared against the gold-standard GIAB validation phases.
The SNP identified across all human chromosomes showcased Coral’s basecalled sequences achieved superior identification precision, recall, and F1 scores (Figs. 6a and S19). In the subsequent haplotyping analysis, the basecalling of Coral also exhibited fewer phase switch errors and lower Hamming errors compared to the other basecallers. With Coral, the switch error and Hamming error rates were reduced by up to 78.8% and 76%, respectively, while simultaneously up to 36% more SNPs were also phased, which is critical for maintaining the integrity of haplotype blocks across the genome. Dorado achieved the lower F1 score in SNP identification and phased fewer SNPs than other basecallers, which may stem from Dorado’s low read accuracy (91.4%) and its significantly shorter read length in the GM12878/NA12878 dataset (Table S16). We further assessed the impact of phased SNPs on phasing genes by linking all heterozygous SNPs within these genes. Coral delivered more complete phasing results, which include notable examples that have clinical associations with diseases, e.g., DDR156, PTK2B57, FRA10AC158, CTSC59, and TNFRSF13B60 (Fig. 6c).

a Performance on SNP identification and haplotype phasing over all chromosomes in GM12878. This analysis was performed on n = 8,540,683 reads. Precision, Recall, and F1 scores were calculated by comparing the chromosome position, allele, and genotype between unphased SNPs against the GIAB gold standard. Switch error (SE) and Hamming error (HE) rates were calculated by comparing the haplotype differences between the phased blocks and the standard at the identical heterozygous variants. To align with other metrics, the original SE and HE were normalized as SEmin/SE and HEmin/HE such that larger values indicate better performance. Best scores per metric are noted in parentheses. In the radar chart, larger areas indicate better overall performance. b F1 scores of SNP identification and haplotyping error rate on simulated datasets with varying accuracy. c Haplotype phasing of reads basecalled by different basecallers across five disease-associated genes. Solid lines in different colors represent different phased blocks, with circles of the same color within each block indicating SNP sites that were correctly phased. SNPs that were incorrectly phased (either due to phasing errors or incorrect allele compared to the ground truth) are represented by black empty circles. d F1 scores of discovered transcripts using simulated datasets with varying accuracy. e F1 scores of detected ASE genes using the simulated datasets with varying accuracy (FDR = 0.01). Simulated accuracy is capped at 96% due to PBSIM3 limitations. The simulated result with an accuracy exceeding 96% was extrapolated based on the observed trend. In b, d, e, the solid lines represent the mean performance, and the error bands (shaded regions) indicate the 95% confidence intervals (95% CI), derived from n = 7 independent simulation experiments performed at each accuracy level. Source data are provided as a Source Data file.
Discussion
Nanopore DRS has revolutionized transcriptome research by enabling the direct sequencing of RNA molecules, facilitating the reconstruction of full-length transcripts, quantifying transcript abundance with high strand specificity, and providing high-resolution detection of allele-specific variants. Nevertheless, long-read sequencing tools still lag behind short-read counterparts in terms of accuracy14. Basecalling accuracy of nanopore DRS should be substantially improved to better support the downstream transcriptome analyzes and promote its broader acceptance5.
To access the extent to which the downstream transcriptome analyzes benefit from improvements in basecalling accuracy, we conducted simulation experiments to evaluate the impact of sequencing accuracy on transcript isoform discovery, ASE detection, SNP detection, and haplotype phasing (Fig. 6b, d, e). The simulations for transcript and SNP discovery were conducted using Badread61 across a range of read accuracies (89% to 99%) with 30× sequencing coverage. Simulations for ASE detection were performed using PBSIM362. It is observed that increasing sequencing accuracy significantly improved transcript discovery, ASE detection, SNP identification, and haplotype phasing, highlighting the tangible benefits of enhanced basecalling precision for downstream analyzes. Notably, as basecalling accuracy approached 95%, all task performance showed a sharp increase, while switch error and Hamming error rates showed a sharp decline, followed by a stabilization phase. Beyond this threshold, further improvements in sequencing accuracy led to diminishing gains. Coral has approached the optimal performance at this threshold, capturing the critical information required for accurate transcript identification, ASE detection, SNP detection, and haplotype phasing. The plateau effect observed beyond 95% suggests that Coral can successfully extract the majority of relevant transcriptomic and genomic insights. These findings establish Coral as a benchmark for accuracy in nanopore DRS applications.
Computational efficiency is another critical concern of nanopore basecalling tools. We analyze the theoretical decoding complexities of the basecallers, including the CTC-based methods (i.e., Guppy, RODAN, GCRTcall, and Dorado) and the auto-regressive decoder-based Carol variants, i.e., Coral, Coral-FAST, and Coral-LSTM (Coral with LSTM-based decoder) (Table S14). The complexity of CTC-based methods scales primarily with the length of signal features T. While the computational complexity of Coral variants inherently scales with sequence length L, leading to increased computational demand, architectural refinements in Coral can mitigate this. Specifically, the Transformer variants (Coral and Coral-FAST) employ efficient cross-attention and standard beam search, offering lower theoretical complexity than the LSTM-based variant (Coral-LSTM). Coral-FAST further reduces complexity by scaling with L/k (Table S14). Empirically, these refinements yield significant gains: Coral-FAST achieves an ~4× speed-up over Coral-LSTM while also improving accuracy (Fig. S8). Compared to Coral, Coral-FAST also demonstrates a roughly 4× speed-up on RNA004 data with only a negligible reduction in median accuracy (Fig. S9), showcasing an effective trade-off between precision and efficiency. More details about computational efficiency are presented in the Supplementary Note 1.
Coral is an open-source DRS basecalling tool built upon an encoder-decoder framework. It provides users with access to both the code and pre-trained models, enabling them to train or fine-tune Coral on their own datasets and design new architectures tailored to specific applications. Coral’s design represents a deliberate trade-off that prioritizes accuracy over raw computational speed. This capability is particularly crucial for novel research applications, such as sequencing non-standard sample types or accurately identifying modified bases, where a proprietary, pre-trained model may be suboptimal. As a transparent tool, Coral dismantles the “black box” nature of commercial basecallers, empowering the scientific community to scrutinize, innovate, and extend the basecalling architecture itself across various chemistries and sequencing platforms.
In summary, we have developed Coral, an advanced basecalling tool tailored for error-prone nanopore DRS, which has been integrated into transcriptome analysis to uncover a high proportion of transcript isoforms and refine haplotype and ASE detection. Leveraging advancements in RNA sequencing technology, Coral is expected to facilitate the study of RNA modifications at single-nucleotide resolution, crucial for a comprehensive understanding of the epitranscriptome and its regulatory role in gene expression. By revealing a greater diversity of transcript isoforms, Coral could provide profound insights into gene expression’s regulatory mechanisms. Notwithstanding rigorous computational and orthogonal validation, the reliance of this study on public datasets means direct experimental validation remains absent. Pursuing such validation constitutes a primary aim of our subsequent research. The implementation of Coral can also be further optimized in specific application scenarios, i.e., model quantization, knowledge distillation, and hardware acceleration can be applied to significantly reduce the computational overhead.
Methods
Encoder-decoder architecture
Coral employs a Transformer-based encoder-decoder architecture tailored for DRS basecalling. This architecture comprises two primary components: an encoder that processes raw electrical signals to extract contextual features, and a decoder that interprets these features to generate nucleotide sequences. Given an input raw signal represented as a one-dimensional vector \({{{\bf{x}}}}\in {{\mathbb{R}}}^{T}\) with T indicating the number of sampled values, Coral outputs the corresponding ribonucleotide sequence u ∈ ΣL, where L is the length of the ribonucleotide sequence and Σ = {A, C, G, U} is the alphabet of bases.
Encoder
The encoder of Coral comprises convolution modules followed by Transformer network (Figs. 1b and S20), designed to process the input signals and generate rich contextual representations. Initially, the raw ionic current signals are processed by five convolutional modules. Each convolutional module consists of a one-dimensional convolutional network, batch normalization operation63, and Swish activation function64. The sizes of the convolutional kernels in these modules are in the sequence of 5, 5, 9, 9, and 5, while the corresponding channel numbers expand in the order of 64, 64, 128, 128, and 512. The convolution modules perform a 12-fold down-sampling on the input signal, resulting in an output feature sequence of length t = T/12. The down-sampling of the raw signal not only effectively extracts its local features but also substantially shortens the sequence length, thereby reducing the computational complexity and overhead of the self-attention mechanism in the subsequent Transformer encoder. Following convolutional processing, the encoder integrates 18 Transformer layers. Each Transformer encoder layer comprises a Multi-Head Self-Attention (MHSA) module and a Multi-Layer Perceptron (MLP) module, both with residual connections and layer normalization. Given an input signal feature sequence \({{{\bf{h}}}}\in {{\mathbb{R}}}^{t\times d}\), where d is the feature dimension (d = 512), the MHSA mechanism first projects the input features into queries \({{{{\bf{q}}}}}_{h}^{(e)}\), keys \({{{{\bf{k}}}}}_{h}^{(e)}\), and values \({{{{\bf{v}}}}}_{h}^{(e)}\) for each attention head e ∈ {1, …, E}, where E is the number of heads. Specifically:
where \({{{{\bf{Q}}}}}_{h}^{(e)},{{{{\bf{K}}}}}_{h}^{(e)},{{{{\bf{V}}}}}_{h}^{(e)}\in {{\mathbb{R}}}^{d\times {d}_{e}}\) denote the learnable projection matrices that transform the signal feature sequence into queries, keys, and values, respectively. de is the head dimension (de = 64). Additionally, rotary positional embeddings65 are then applied to the queries \({{{{\bf{q}}}}}_{h}^{(e)}\) and keys \({{{{\bf{k}}}}}_{h}^{(e)}\) to incorporate relative positional information. Afterward, the self-attention weight \({{{{\boldsymbol{\alpha }}}}}_{h}^{(e)}\) and the output representations \({{{{\bf{o}}}}}_{h}^{(e)}\) are computed for each head:
The outputs from all attention heads are concatenated and projected to the original dimension:
where \({{{{\bf{W}}}}}_{O}\in {{\mathbb{R}}}^{(E\cdot {d}_{e})\times d}\) is a learnable output projection matrix. The MLP module contains a Swish Gated Linear Unit (SwiGLU)66 followed by a linear projection layer. The SwiGLU introduces nonlinearity, dimension expansion, and feature selection into the encoder layer. Following Transformer layers, the resultant features are subsequently up-sampled since decoder requires an output sequence from the encoder that is longer than the target ribonucleotide sequence to ensure a stable and accurate signal-to-sequence alignment, while a 12-fold down-sampling risks shortening the sequence excessively. Specifically, the up-sampling model is achieved through a linear projection. The feature sequence from the encoder \({{{\bf{h}}}}\in {{\mathbb{R}}}^{t\times d}\), is transformed using a learnable matrix \({{{\bf{U}}}}\in {{\mathbb{R}}}^{d\times 2d}\) to produce an expanded sequence of doubled length while maintaining the same feature dimension d, resulting in an overall sixfold down-sampling relative to the raw signal (final feature length t = T/6). This sixfold resolution provides finer granularity for decoding and adequately covers the dwell time of most signal events (Fig. S22). To validate the down-then-up sampling strategy, we trained an ablated Coral-L3 model (i.e., Coral with three decoder layers) by removing the up-sampling step and directly applying sixfold down-sampling followed by Transformer layers in the encoder. The ablated Coral-L3 was compared to the standard Coral-L3 model on the HEK293T RNA004 dataset. As shown in Table S13, the standard Coral-L3 attained the same median accuracy while requiring substantially fewer computational resources in terms of GFLOPs and GPU memory usage. The results indicate that 12-fold down-sampling is adequate for Transformer-based contextual modeling and yields significantly higher efficiency.
Decoder
The decoder of Coral employs a Transformer network (Fig. 1c) to extract contextual dependencies between bases and learn the alignment between signal and base sequence. The nucleotide sequence is modeled as a sequential prediction task, where each base is predicted conditioned upon previously decoded bases. To optimize decoding efficiency, the sequence u = (u1, u2, …, uL) is partitioned into adjustable-length units, or bins, denoted as \(y=\left({y}_{0},{y}_{1},{y}_{2},\ldots,{y}_{l},{y}_{l+1}\right)\), where y0 = 〈sos〉 marks the start of the sequence; yl+1 = 〈eos〉 denotes the end of the sequence. The binning strategy adapts to throughput requirements: single-nucleotide bins for high-accuracy decoding (default mode) and multi-nucleotide bins for high-throughput decoding (fast mode). The binning process is defined as follows: yi = u(i−1)*k+1u(i−1)*k+2…ui*k (\(i\in \left[1,l\right]\)) represents a k-bin; The total number of bins l is calculated as: \(l=\left\lfloor \frac{L-k}{k}\right\rfloor+1\). In the fast mode, we use k = 5 for RNA001/002 kit and k = 6 for RNA004 kit.
Sequence representations \(y=\left({{y}_{0},y}_{1},{y}_{2},\ldots,{y}_{l}\right)\) are first embedded into a high-dimensional space via an embedding layer. Specifically, each symbol is mapped to a vector using a learnable lookup table, resulting in a sequence of embeddings \({{{\bf{s}}}}\in {{\mathbb{R}}}^{l\times d}\). These embeddings are then passed through N stacked decoder blocks. Each block includes two causal self-attention Transformer layers (Fig. S21a) and one fusion layer containing a cross-attention Transformer layer (Fig. S21b). In the causal self-attention layers, a causal mask \({{{{\bf{m}}}}}_{s}\in {{\mathbb{R}}}^{l\times l}\) is introduced to prevent the sequence representation si from attending to any future positions j > i. The causal mask is defined as:
In the cross-attention layer, the signal context representation \({{{\bf{h}}}}\in {{\mathbb{R}}}^{t\times d}\) serves as the source of keys and values, while the sequence representation \({{{\bf{s}}}}\in {{\mathbb{R}}}^{l\times d}\) acts as the source of queries. The cross-attention weight \({{{{\boldsymbol{\alpha }}}}}_{c}^{(e)}\in {{\mathbb{R}}}^{l\times t}\) for each head e ∈ {1, …, E} is computed follows the same procedure as that used for self-attention weight \({{{{\boldsymbol{\alpha }}}}}_{h}^{(e)}\) in the encoder. The expected alignment position of yi for head e ∈ {1, …, E} is then given by:
where \({{{{\boldsymbol{\alpha }}}}}_{c}^{(e)}(i,j)\) denotes the probability that symbol yi aligns to signal representations at position j. Although the alignment position β is an average across all positions, the model quickly learns to assign meaningful weights only to relevant parts of the signal. As shown in Fig. S23, as training progresses, the cross-attention matrix αc of the final cross-attention layer (closest to the output logits) becomes sharply focused and nearly diagonal. The probabilities for distant and unrelated signal positions become vanishingly small (close to zero). Consequently, the contribution of unrelated positions to the weighted average is negligible, and the calculated β value accurately reflects the true, localized alignment position.
Finally, a linear layer with Softmax activation produces a probability distribution over nucleotide symbols. In our implementation, we use 4 decoder blocks (12 layers) for RNA001/002 kit, and 5 decoder blocks (15 layers) for RNA004 kit. The Transformer decoder layers adopt an architecture similar to that of the Transformer encoder layers, with distinction in the normalization strategy: encoder layers utilize Post-LayerNorm (Post-LN, places layer norm outside of the residual blocks), whereas decoder layers employ a Pre-LayerNorm (Pre-LN, places layer norm inside of residual blocks), which has been shown to enhance training stability67.
Training and inference details
Datasets
The training dataset for the RNA001/002 sequencing kit was selected from Arabidopsis wild type (SRA accession SRX14052225)23, Homo sapiens (NA12878 RNA project BHAM_Run1)7, Epinano synthetic constructs data (SRA accession SRX5177013)30, C. elegans (ENA accession ERX3290234)31, E. coli (SRA accession SRX8525119)32, Mouse (ENA accession ERX12326526)33, Yeast (SRA accessions SRX8120016 and SRX17942557)3536, Poplar (SRA accessions SRX9290560 and SRX9290561)34, and Zebrafish (ENA accession ERX9536682)37. The majority of biological samples used for training differ from those in the test sets, except Zebrafish, for which only one public DRS sample is currently available; in this case, training was performed using reads distinct from those in the test dataset. The sequencing reads were first basecalled using Guppy, then aligned to the corresponding transcriptome reference to filter low-quality reads, and finally established signal-to-sequence mappings using the resquiggle command from Tombo v1.5.168. Subsequently, 4 million high-accuracy chunks (accuracy≥96%) with 4096 signal samples were randomly selected from the dataset for training.
The training dataset for the RNA004 sequencing kit was created from a publicly available Universal Human Reference RNA sample (ENA accession ERX12369345)38. This RNA004 sample was basecalled using the Dorado v0.8.322 “rna004_130bps_sup@v5.1.0" model and aligned to the human transcriptome reference. We then performed the signal mapping refinement (re-squiggling) using the remora v3.3.069, and extracted 12 million high-quality signal chunks (12,000 samples per chunk, alignment accuracy≥98%) for training. More details and data links of the training dataset are provided in Table S1.
The test dataset for the RNA001/002 sequencing kit was comprised of six species, including H. sapiens (Human, NA12878 RNA project HOPKINS_RUN1)7, Arabidopsis thaliana (Arabidopsis, ENA accession ERX3766448)39, M. musculus (Mouse, ENA accession ERX3444723)40, S. cerevisiae S288C (Yeast, SRA accession SRX8120036)36, Populus trichocarpa (Poplar, SRA accession SRX9347134)34, and D. rerio (Zebrafish, ENA accession ERX9536682)37. The test dataset was generated through a series of pre-processing steps. Initially, the sequencing reads were basecalled with Guppy. Subsequently, these reads were aligned to their respective transcriptome references, and reads aligned to the mitochondrial genome were discarded. From the remaining aligned reads in each sample, 100,000 were randomly chosen for inclusion in the test dataset. For the performance evaluation of RNA004, we used a complete RNA004 sample from the HEK293T human cell line provided by the Singapore Nanopore Expression Project (Sample ID: SGNex_Hek293T_directRNA_replicate5_run1, accessed on [2024-11-21] at registry.opendata.aws/sg-nex-data)41. Detailed sequencing configurations and data links of the test dataset are listed in Table S2.
Optimization and loss functions
The model parameters θ are estimated by minimizing the loss function with labeled data. We use the cross-entropy loss function \({{{{\mathcal{F}}}}}_{{{{\rm{CE}}}}}\left(x,y;\theta \right)\) as follows:
In normal mode, Coral is initially trained to predict a single base per step using only the cross-entropy loss. Upon completion, the best-performing checkpoint is used to initialize all parameters except for the cross-attention layers, which are randomly initialized. The model is then retrained using the cross-entropy loss combined with additional monotonic alignment regularization loss \({{{{\mathcal{F}}}}}_{{{{\rm{RE}}}}}\)70, which penalizes non-monotonic alignments and excessively small alignment gaps between adjacent symbols, following the formulation:
Here, ϵ is a hyperparameter representing the minimum allowed alignment gap between adjacent symbols. In our implementation, we set ϵ = k, i.e., ϵ = 1 for the normal mode training and ϵ = 5 or 6 for the fast mode training. The regularization loss is applied exclusively to the final cross-attention layer (closest to the output logits) to preserve the model’s capacity for soft alignment in intermediate decoder layers while enforcing a near-diagonal, monotonic pattern in the final alignment.
After training the Coral model with the monotonic alignment constraint, we again load its best checkpoint to initialize all parameters, except for the decoder embedding layer and output layer, which are randomly initialized due to the different vocabulary size in the Coral-FAST model compared to the Coral model. The Coral-FAST model, which predicts k bases per auto-regressive step, is then trained with the same monotonic alignment regularization loss. This pre-training strategy is crucial for ensuring training stability. The significantly larger output vocabulary of Coral-FAST makes optimization from a random initialization challenging and can lead to unstable gradients. In contrast, initializing Coral-FAST with weights from a pre-trained Coral model provided a much more stable and effective starting point for optimization, leading to a smoother gradient norm and faster convergence of the loss value (Fig. S24).
Signal file pre-processing
To enable the basecalling of the models, the input FAST5/POD5 files should undergo pre-processing as follows:
-
1.
Convert the discrete 16-bit signals stored in FAST5 into current values through digital-to-analog conversion. This conversion requires some metadata (offset, range, and digitisation) recorded in FAST5. For POD5 files, the raw current values can be directly read from the file using the official API.
-
2.
Remove the open-pore signal at the beginning of the sequencing that exceeds the normal signal level.
-
3.
Normalize the signal using the Median Absolute Deviation:
$${{{\rm{signal}}}}=\frac{{{{\rm{signal}}}}-{{{\rm{median}}}}({{{\rm{signal}}}})}{{{{\rm{median}}}}\left(\left|{{{\rm{signal}}}}-{{{\rm{median}}}}\left({{{\rm{signal}}}}\right)\right|\right)}$$(8) -
4.
Segment the original signal into fixed-length chunks. In our implementation, the chunk length is 4096 for RNA001/002, and 6000 for RNA004. The chunks are decoding independently. Since the ONT nanopore DRS proceeds from the 3′ to 5′ direction, the decoding results should be concatenated to one complete nucleotide sequence, which is then reversed to align with the natural orientation.
Model inference
The standard beam search algorithm is applied to determine the most probable nucleotide sequence, with several optimization strategies to accelerate the decoding process, including key-value caching (KV Cache), FlashAttention71, and a two-stage decoding scheme. KV caching avoids redundant computation by storing the keys and values of previously decoded tokens in the decoder’s transformer layers. FlashAttention is an I/O-aware algorithm that provides an accelerated attention implementation, significantly improving the speed and memory efficiency of Transformer models. Additionally, we adopt a two-stage decoding approach: an initial fast decoding pass with a reduced number of auto-regressive steps for most sequences, followed by a second pass using a longer decoding window for remaining unfinished sequences. The beam search decoding process proceeds as follows:
-
1.
The input signal is encoded using the CNN-Transformer encoder to produce the signal feature sequence.
-
2.
Decoding begins with a start-of-sequence input symbol 〈sos〉. The cross-attention mask is initialized as \({{{{\bf{m}}}}}_{c}(0,j)=0\left(j\in [1,t]\right)\). During the subsequent decoding process, this mask is added to the attention logits (before the softmax operation) of the final cross-attention layer to guide alignment-based decoding.
-
3.
At decoding step i, the decoder predicts the probability distribution of the next output symbol based on previously decoded symbols and the signal features. Simultaneously, it estimates the alignment position of the output symbol from the final cross-attention layer, denoted as \({\beta }_{i}^{*}\), following the formulation:
$${\beta }_{i}^{*}={\min }_{e\in \{1,\ldots,E\}}{\beta }_{i}^{(e)}$$(9) -
4.
From all expanded sequences, the top-B hypotheses (beam size B) with the highest cumulative log-probabilities are selected. Decoding terminates when any of the following conditions are met by the best hypothesis:
-
An end-of-sequence symbol 〈eos〉 is generated.
-
The decoding length reaches the predefined maximum sequence length.
-
The alignment position satisfies \({\beta }_{i}^{*}\ge t\), where t is the length of signal features.
-
-
5.
If none of the stopping criteria are met, the cross-attention mask mc(i, j) is updated based on the current alignment position \({\beta }_{i}^{*}\), following the formulation:
$${{{{\bf{m}}}}}_{c}(i,j)=\left\{\begin{array}{ll}0 & \,{{{\rm{if}}}}\,j\ge \lfloor {\beta }_{i}^{*}\rfloor \\ -\infty & \,{{{\rm{if}}}}\,j < \lfloor {\beta }_{i}^{*}\rfloor \end{array}\right.$$(10)This masking approach prevents the sequence representation from attending to any signal representations at positions \(j < \lfloor {\beta }_{i}^{*}\rfloor\) for the next symbol decoding, which enforces monotonic decoding and effectively filters out low-contribution noise from earlier or irrelevant positions, without sacrificing contextual representation.
-
6.
Repeat from Step 3 to predict the next output symbol.
Benchmark performance comparison
In the comparison experiments, RODAN and GCRTcall were run using their default configuration. Guppy was executed using the RNA model rna_r9.4.1_70bps_hac.cfg with the option “–disable_qscore_filtering” to deactivate the default quality control. Dorado was executed using the RNA002 model “rna002_70bps_hac@v3” or the RNA004 model “rna004_130bps_sup@v5.1.0” according to the sequencing kit. The download links and parameter configurations of Guppy, RODAN, GCRTcall, Dorado, and Coral are summarized in Table S3. After basecalling, the reads were aligned to the respective transcriptome reference using minimap2 (v2.17-r941)72 with the options “-axmap-ont–secondary=no–eqx” to disable secondary alignments. For the RNA004 sequencing kit, we use minimap2 (v2.28-r1209) with the options “-axlr:hq–for-only–secondary=no–eqx" for more accurate transcriptome alignment. The unaligned reads and the supplementary alignments were further excluded from the evaluation. The read accuracy is used as the primary metric for evaluating the basecalling performance:
where Nmatch, Nmis, Nins, and Ndel, extracted from the CIGAR string, represent the numbers of matching bases, mismatches, insertions, and deletions of a read, respectively. The error (mismatch, insertion, and deletion) rate per read can be defined as:
Downstream analyses pipelines
Several publicly available datasets were involved in human transcriptome analysis, including one A549 replicate, one HCT116 replicate, and one MCF-7 replicate from the Singapore Nanopore Expression Project41, 20 runs from the NA12878 RNA project7, and three HEK293T replicates spiked with 25 ng of SIRV-Set 1-E2 RNA from ESPRESSO42. The data links of the human dataset are provided in Table S15. To assess the basecalling performance, we aligned the basecalled reads to the human reference transcriptome from GENCODE (v45) using minimap2 (v2.17-r941) with the options “-axmap-ont–secondary=no–eqx”.
The transcriptome analyzes were carried out with ESPRESSO (v1.5.0)42. Before running the ESPRESSO, the basecalled reads were aligned to the human reference genome GRCh38 using minimap2 (v2.17-r941)72 with the options “-axsplice-uf-k14–secondary=no". We apply “–splice-flank=no" option to let minimap2 only model GT..AG splicing signal for SIRV splice alignment. After the spliced alignment, samtools (v1.10)73 was used to create sorted and filtered BAM files as input for ESPRESSO. ESPRESSO works in three steps, i.e., high-confidence SJ identification ("S”), SJ correction and recovery("C”), and transcript discovery and quantification ("Q”). The human transcript annotations from GENCODE (v45) were considered during the final step of ESPRESSO. The output files from ESPRESSO, including an updated GTF annotation, abundance of detected isoforms, and compatible isoforms of each read, were used for further statistical analysis. We used JAFFAL (v2.3)52 with default options to detect fusion genes from basecalled reads of cancer cell samples.
The NA12878 DRS dataset was used for allele-specific analysis. We obtained genotype information for NA12878 from existing phased Illumina platinum genome data (https://platinum-genomes.s3-eu-central-1.amazonaws.com/2017-1.0/hg38/small_variants/NA12878/NA12878.vcf.gz). We used bcftools (v1.8)74 to filter variants that are heterozygous in NA12878. We used the extractHAIRS utility of the HapCUT2 (v1.3.4)55 to identify aligned reads with allele-informative variants. We followed the script (https://github.com/nanopore-wgs-consortium/NA12878/blob/master/nanopore-human-transcriptome/scripts/ase.py) provided by ref. 7 for allelic assignment. Finally, we performed a binomial test and adjusted the P values using the Benjamini–Hochberg procedure with a FDR of 0.01 to identify genes with significant ASE. To simulate ASE, we selected hundreds of transcripts containing at least two heterozygous variants from chromosome 1. Parental-specific expression was randomly assigned to each transcript. Using the “trans" mode of PBSIM3 (v3.0.4)62, we generated sequencing reads with varying accuracy, following predefined expression profiles. We then applied the same ASE detection method, with a FDR of 0.01, to analyze the simulated reads.
The NA12878 DRS dataset was used for haplotype phasing. We employed Clair3-RNA (v0.0.1)54 to call SNPs and HAPCUT2 (v1.3.4)55 for haplotype phasing. The phased VCF was compared to the GIAB HG001 benchmark (v3.3.2) using the compare command from whatshap (v2.0)75.
Thousands of transcripts from chromosome 1 were used to establish a transcriptome reference for our transcript discovery simulation experiments. To perform simulation for SNP discovery and haplotype phasing, we also generated a diploid transcriptome reference that includes variation data from the GIAB HG001 benchmark (v3.3.2) using g2gtools (v2.0.0) (https://github.com/churchill-lab/g2gtools). The simulated nanopore sequencing reads varying the accuracies from 89% to 99% were generated using Badread (v0.4.1)61.
Implementation and environment configuration
All experiments were conducted on a Linux server equipped with two 10-core Intel®Xeon®Silver 4210 CPUs (2.20 GHz), four NVIDIA GeForce RTX 3090 GPUs, and 128 GB of system RAM. The operating system was Ubuntu 22.04.3 LTS. The implementation was developed using Python 3.10 and PyTorch 2.6.0 with CUDA 11.8 for GPU acceleration. Coral was trained using gradient descent with the AdamW optimizer76, an initial learning rate of 0.0002, and a batch size of 128. The model was trained for 10 epochs and converged (achieving the peak validation accuracy) at the 6th epoch. More details of the training configuration are listed in Table S20.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA sequencing data used in this study are available in the Zenodo database under [https://doi.org/10.5281/zenodo.4557005] and [https://doi.org/10.5281/zenodo.11632496], and in the SRA/ENA databases and AWS Open Data registry under the accession codes and links listed in Supplementary Tables S1, S2, and S15. The processed data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Code availability
The code package for this study, along with a guide to use the code, is available on GitHub at [https://github.com/BioinfoSZU/Coral] and is also available on Zenodo under [https://doi.org/10.5281/zenodo.18153247]77.
References
Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656 (2019).
Hansen, K. D., Brenner, S. E. & Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 38, e131–e131 (2010).
Kovaka, S., Ou, S., Jenike, K. M. & Schatz, M. C. Approaching complete genomes, transcriptomes and epi-omes with accurate long-read sequencing. Nat. Methods 20, 12–16 (2023).
Garalde, D. R. et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206 (2018).
Jain, M., Abu-Shumays, R., Olsen, H. E. & Akeson, M. Advances in nanopore direct RNA sequencing. Nat. Methods 19, 1160–1164 (2022).
Soneson, C. et al. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 10, 3359 (2019).
Workman, R. E. et al. Nanopore native RNA sequencing of a human poly (A) transcriptome. Nat. Methods 16, 1297–1305 (2019).
Gunter, H. M. et al. mRNA vaccine quality analysis using RNA sequencing. Nat. Commun. 14, 5663 (2023).
Chan, A., Naarmann-de Vries, I. S., Scheitl, C. P., Höbartner, C. & Dieterich, C. Detecting m6A at single-molecular resolution via direct RNA sequencing and realistic training data. Nat. Commun. 15, 3323 (2024).
Baek, A. et al. Single-molecule epitranscriptomic analysis of full-length HIV-1 RNAs reveals functional roles of site-specific m6As. Nat. Microbiol. 9, 1340–1355 (2024).
Wu, Y. et al. Transfer learning enables identification of multiple types of RNA modifications using nanopore direct RNA sequencing. Nat. Commun. 15, 4049 (2024).
Glinos, D. A. et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353–359 (2022).
Berger, E. et al. Improved haplotype inference by exploiting long-range linking and allelic imbalance in RNA-seq datasets. Nat. Commun. 11, 4662 (2020).
Pardo-Palacios, F. J. et al. Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat. Methods 21, 1349–1363 (2024).
Wang, J. et al. Direct RNA sequencing coupled with adaptive sampling enriches RNAs of interest in the transcriptome. Nat. Commun. 15, 481 (2024).
Liu-Wei, W. et al. Sequencing accuracy and systematic errors of nanopore direct RNA sequencing. BMC Genomics 25, 528 (2024).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 1–10 (2019).
Teng, H. et al. Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience 7, giy037 (2018).
Zeng, J. et al. Causalcall: Nanopore basecalling using a temporal convolutional network. Front. Genet. 10, 1332 (2020).
Huang, N., Nie, F., Ni, P., Luo, F. & Wang, J. SACall: a neural network basecaller for Oxford Nanopore sequencing data based on self-attention mechanism. IEEE/ACM Trans. Comput. Biol. Bioinf. 19, 614–623 (2020).
Graves, A., Fernández, S., Gomez, F. & Schmidhuber, J. Connectionist temporal classification: labelling unsegmented sequence data with recurrent neural networks. In Proc. 23rd International Conference on Machine Learning 369–376 (Association for Computing Machinery, New York, NY, USA, 2006).
Oxford Nanopore Technologies. Dorado. https://github.com/nanoporetech/dorado.
Neumann, D., Reddy, A. S. & Ben-Hur, A. RODAN: a fully convolutional architecture for basecalling nanopore RNA sequencing data. BMC Bioinf. 23, 142 (2022).
Li, Q., Sun, C., Wang, D. & Lou, J. GCRTcall: a Transformer based basecaller for nanopore RNA sequencing enhanced by gated convolution and relative position embedding via joint loss training. Front. Genet. 15, 1443532 (2024).
Vaswani, A. et al. Attention is all you need. In Proc. Advances in Neural Information Processing Systems 30 (Curran Associates, Inc., Red Hook, NY, USA, 2017).
Pagès-Gallego, M. & de Ridder, J. Comprehensive benchmark and architectural analysis of deep learning models for nanopore sequencing basecalling. Genome Biol. 24, 71 (2023).
Dorey, A. & Howorka, S. Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics. Nat. Chem. 16, 314–334 (2024).
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365 (2021).
Oxford Nanopore Technologies. Guppy. https://nanoporetech.com/community.
Liu, H. et al. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 10, 4079 (2019).
Roach, N. P. et al. The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res. 30, 299–312 (2020).
Grünberger, F. et al. Nanopore sequencing of RNA and cDNA molecules in Escherichia coli. RNA 28, 400–417 (2022).
Krawczyk, P. S. et al. Re-adenylation by TENT5A enhances efficacy of SARS-CoV-2 mRNA vaccines. Nature 641, 984–992 (2025).
Gao, Y. et al. Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing. Genome Biol. 22, 1–17 (2021).
DeMario, S., Xu, K., He, K. & Chanfreau, G. F. Nanoblot: an R-package for visualization of RNA isoforms from long-read RNA-sequencing data. RNA 29, 1099–1107 (2023).
Jenjaroenpun, P. et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 49, e7–e7 (2021).
Begik, O. et al. Nano3P-seq: transcriptome-wide analysis of gene expression and tail dynamics using end-capture nanopore cDNA sequencing. Nat. Methods 20, 75–85 (2023).
Samarakoon, H. et al. Flexible and efficient handling of nanopore sequencing signal data with slow5tools. Genome Biol. 24, 69 (2023).
Parker, M. T. et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. eLife 9, e49658 (2020).
Bilska, A. et al. Immunoglobulin expression and the humoral immune response is regulated by the non-canonical poly(A) polymerase TENT5C. Nat. Commun. 11, 2032 (2020).
Chen, Y. et al. A systematic benchmark of Nanopore long-read RNA sequencing for transcript-level analysis in human cell lines. Nat. Methods 22, 801–812 (2025).
Gao, Y. et al. ESPRESSO: robust discovery and quantification of transcript isoforms from error-prone long-read RNA-seq data. Sci. Adv. 9, eabq5072 (2023).
Tardaguila, M. et al. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 28, 396–411 (2018).
Parham, P. & Ohta, T. Population biology of antigen presentation by MHC class I molecules. Science 272, 67–74 (1996).
Neefjes, J., Jongsma, M. L., Paul, P. & Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11, 823–836 (2011).
Ma, C., Slaughter, C. A. & DeMartino, G. N. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J. Biol. Chem. 267, 10515–10523 (1992).
Voges, D., Zwickl, P. & Baumeister, W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 (1999).
Liu, Q. et al. IKZF1 and UBR4 gene variants drive autoimmunity and Th2 polarization in IgG4-related disease. J. Clin. Invest. 134, e178692 (2024).
Gao, Z. -w et al. The roles of adenosine deaminase in autoimmune diseases. Autoimmun. Rev. 20, 102709 (2021).
Hara, T. et al. Deletion of the Mint3/Apba3 gene in mice abrogates macrophage functions and increases resistance to lipopolysaccharide-induced septic shock. J. Biol. Chem. 286, 32542–32551 (2011).
Nataf, S., Guillen, M. & Pays, L. The immunometabolic gene N-acetylglucosamine kinase is uniquely involved in the heritability of multiple sclerosis severity. Int. J. Mol. Sci. 25, 3803 (2024).
Davidson, N. M. et al. JAFFAL: detecting fusion genes with long-read transcriptome sequencing. Genome Biol. 23, 10 (2022).
Mitelman F, J. B. & F, M. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. https://mitelmandatabase.isb-cgc.org (2024).
Zheng, Z. et al. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat. Comput. Sci. 2, 797–803 (2022).
Edge, P., Bafna, V. & Bansal, V. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 27, 801–812 (2017).
Roig, B. et al. The discoidin domain receptor 1 as a novel susceptibility gene for schizophrenia. Mol. Psychiatry 12, 833–841 (2007).
Yan, Y. et al. Genetic association of FERMT2, HLA-DRB1, CD2AP, and PTK2B polymorphisms with Alzheimer’s disease risk in the southern Chinese population. Front. Aging Neurosci. 12, 16 (2020).
von Elsner, L. et al. Biallelic FRA10AC1 variants cause a neurodevelopmental disorder with growth retardation. Brain 145, 1551–1563 (2022).
Chitsamankhun, C. et al. Cathepsin C in health and disease: from structural insights to therapeutic prospects. J. Transl. Med. 22, 777 (2024).
Salzer, U. et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 113, 1967–1976 (2009).
Wick, R. R. Badread: simulation of error-prone long reads. J. Open Source Softw. 4, 1316 (2019).
Ono, Y., Hamada, M. & Asai, K. PBSIM3: a simulator for all types of PacBio and ONT long reads. NAR Genomics Bioinf. 4, lqac092 (2022).
Ioffe, S. & Szegedy, C. Batch normalization: accelerating deep network training by reducing internal covariate shift. In Proc. 32nd International Conference on Machine Learning 448–456 (JMLR.org, 2015).
Ramachandran, P., Zoph, B. & Le, Q. V. Searching for activation functions. In Proc. 6th International Conference on Learning Representations (OpenReview.net, 2018).
Su, J. et al. Roformer: enhanced transformer with rotary position embedding. Neurocomputing 568, 127063 (2024).
Shazeer, N. Glu variants improve transformer. Preprint at https://arxiv.org/abs/2002.05202 (2020).
Liu, L., Liu, X., Gao, J., Chen, W. & Han, J. Understanding the difficulty of training Transformers. In Proc. Empirical Methods in Natural Language Processing 5747–5763 (Association for Computational Linguistics, 2020).
Oxford Nanopore Technologies. Tombo. https://github.com/nanoporetech/tombo.
Oxford Nanopore Technologies. Remora. https://github.com/nanoporetech/remora.
Rios, A., Amrhein, C., Aepli, N. & Sennrich, R. On biasing Transformer attention towards monotonicity. In Proc. North American Chapter of the Association for Computational Linguistics: Human Language Technologies 4474–4488 (Association for Computational Linguistics, 2021).
Dao, T., Fu, D., Ermon, S., Rudra, A. & Ré, C. Flashattention: Fast and memory-efficient exact attention with io-awareness. Adv. Neural Inf. Process. Syst. 35, 16344–16359 (2022).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Patterson, M. et al. WhatsHap: weighted haplotype assembly for future-generation sequencing reads. J. Comput. Biol. 22, 498–509 (2015).
Loshchilov, I. & Hutter, F. Decoupled weight decay regularization. In Proc. 7th International Conference on Learning Representations (OpenReview.net, 2019).
Xie, S. Coral: a dual context-aware basecaller for nanopore direct rna sequencing - code. https://doi.org/10.5281/zenodo.18153247 (2026).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2022YFF1202104 and 2019YFA0707003), National Natural Science Foundation of China (62471310, 32401256, and 62571401), and the Agricultural Science and Technology Innovation Program (CAAS-ZDRW202503).
Author information
Authors and Affiliations
Contributions
S.X and Z.Z. conceived the idea of Coral. J.R. and Z.Z. coordinated and supervised the project. S.X. designed and implemented the Coral algorithm. S.X. and L.D. performed experiments, analyzed data, and drafted the manuscript. L.L. and Y.Y. contributed to the design and implementation of the Coral method. Y.S., J.L., L.L., J.R., and Z.Z. provided critical comments on algorithm evaluations and improved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jianxin Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, S., Ding, L., Yu, Y. et al. A dual context-aware basecaller for nanopore direct RNA sequencing. Nat Commun 17, 1851 (2026). https://doi.org/10.1038/s41467-026-68566-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68566-2
