
Abstract
Folding stability is crucial for the vast majority of proteins. Computational methods suggested to date for the absolute folding stability (ΔG) prediction, including those driven from protein structure prediction AIs, show clear limitations in reproducing quantitative experimental values. Here we present IFUM, a deep neural network that jointly estimates ΔG and the equilibrium ensemble of folded and unfolded states represented by residue-pair distance probability distributions. This joint learning considerably enhances prediction accuracy compared to learning ΔG alone. Trained on a dataset including Mega-scale small proteins, disordered proteins, and wild-type natural proteins, IFUM is robust to various protein types and can accurately predict complex mutational effects like insertions or deletions. Here, we show that IFUM effectively guides real-world design challenges, exhibiting strong correlation with experimental melting temperatures in protein engineering and outperforming AlphaFold-based metrics in de novo design selection.
Similar content being viewed by others


Introduction
A majority of proteins function by folding into unique three-dimensional structures1. Because naturally evolved proteins are often only marginally stable in their unfolding free energy (ΔGunfolding, or ΔG hereafter)2,3, a small shift in the folding stability can result in a crucial impact. For instance, structure-disrupting mutations with poorer ΔG can lead to protein misfolding and aggregation, which can eventually cause human diseases4,5 or poor protein expressions6. Thus, accurate ΔG quantification is highly demanded in many biotechnological applications, such as therapeutic development or biocatalysis7,8.
Measuring the ΔG of a protein experimentally presents significant challenges due to several factors. These measurements often require extrapolation from non-native conditions, such as high concentrations of denaturants or elevated temperatures, which can introduce inaccuracies9. Furthermore, the process is typically labor-intensive, necessitating the purification of each protein variant, even with the aid of automated methods. The resulting experimental data are also frequently inhomogeneous, stemming from variations in methods and experimental conditions across different studies, making direct comparisons and comprehensive analyses difficult9.
Computational estimation of ΔG, which can significantly reduce such experimental cost, has been studied from various perspectives. One of the most popular methods these days is to approximate it using the confidence estimate from structure prediction results. For instance, AlphaFold10,11 or ESMFold12 provide metrics called plDDT and pTM, which are confidence estimates of structural accuracy of predicted structures learned by separate modules. Although not explicitly learned for protein stability, it has been shown that those metrics can be effective for filtering out unstable designed proteins13, and then became a popular computational filter in various design studies13,14. However, these methods are not designed to estimate ΔG and therefore show limitations in providing a quantitative estimate. Similarly, the pseudolikelihood assigned to a sequence by protein language models (PLMs) or inverse folding models has been explored as a proxy for protein stability15. In parallel, considerable efforts have been made to build networks for the estimation of ΔΔG, the change in stability upon mutation (ΔGmutant – ΔGwildtype)16,17. However, despite their utility for protein engineering, these methods provide little insight into the absolute protein stability, and are generally limited to single point mutations.
We reasoned that the challenge of ΔG prediction originates from the ambiguity of the sequence-to-structure relationship at unfolded states. Because ΔG is by definition the gap between Gfolded and Gunfolded, to accurately quantify ΔG, it is very natural to consider the unfolded state along with the folded state. In fact, sequences not only influence the thermodynamic characteristics of folded states but also those of unfolded states; for instance, unfolded states of poly-Leu and poly-Lys peptides cannot be equal to each other. This sequence-dependence to unfolded states was either modeled using a linear model called “reference state energy”18, which was quite effective yet too simple to capture the complex nature of unfolded states, or was unmodeled and left as a null state regardless of protein sequences in many deep learning approaches15,19. We hypothesized that the key question in this endeavor should be how to explicitly abstract the unfolded state into a deep learning architecture to effectively represent the protein unfolded state.
In this work, we introduce IFUM (In silico evaluation of unfolding Free energy with Unfolded state ensemble Modeling, pronounced [ip͈ɯm]), a deep learning model that addresses the challenge of accurate ΔG prediction by explicitly abstracting the unfolded state ensemble. Conceptually speaking, IFUM makes two hypotheses to abstract the unfolded state representation: first, proteins fold via a two-state folding model20, and second, that the myriad conformations within the unfolded state ensemble can be effectively simplified and represented as an ensemble-averaged distance map using the Flory random coil model21. The Flory random coil model, a fundamental concept in polymer physics, describes an ideal polymer chain whose conformation is statistically averaged as a random walk, thus providing a simplified yet powerful framework for modeling the highly diverse and disordered ensemble of protein unfolded states22. By employing these principles, IFUM estimates ΔG and the relative thermodynamic preferences of a given sequence at the average structures of folded and unfolded states represented as residue-pairwise distance maps, which corresponds to the unfolding free energy definition: ΔG = –RT ln([U]/[F])20. IFUM demonstrates that this approximation works for accurately predicting ΔG for various types of proteins, and also for estimating ΔΔG from sequence deletions or insertions that previous ΔΔG-centric methods were not capable of. IFUM is freely available at github.com/HParklab/IFUM and through Colab.
Results
Overview of IFUM
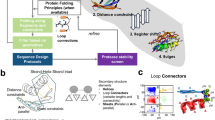
As highlighted above, the key concept of IFUM is to abstract the conformations of both folded and unfolded states of proteins. We use the residue-pair Cα distance distribution histogram (distogram) for this purpose. IFUM takes two types of inputs from pre-trained models: sequence-based and structure-based ones. The sequence embedding is obtained from ProtT523. The folded state structural embedding is generated using ESM-IF124 on the structure predicted by deep-learning based structure prediction tools such as ESMFold12. This structural embedding and the mean pairwise Cα distances (as a form of one-hot encoded distogram with 21 bins with a range of 2–42 Å) of the folded state structure are fed as structural inputs (Fig. 1a and Supplementary Fig. 1). No specific input was provided for the unfolded state because the Flory model only depends on the residue-pair sequence separation.

a Schematic illustration of the IFUM model. Sequence embeddings from ProtT5 (blue solid arrow; seq. rep.), structure embeddings from ESM-IF1 (red solid arrow; str. rep.), and a residue-pair Cα distogram (distance histogram) from ESMFold (red dashed arrow; folded state distogram) are fed into a main module with task-specific heads. b Equilibrium ensemble derivation. Energy landscape diagram illustrating the unfolded (U, GU) and folded (F, GF) states separated by ΔG, with protein schematics and formulas for ΔG and the folded state population α. c, d Derivation of the equilibrium ensemble distogram. c Distograms for the unfolded and folded states are weighted by (1−α) and α, respectively, and then summed. Example residue-pair (i,j) is highlighted. d Cα-Cα distogram for a residue pair (i,j). Bar charts show unfolded (orange) and folded (blue) distograms, weighted by conformational probabilities (1−α) and α, to give the equilibrium ensemble distogram (see “Methods”).
Using the sequence and the folded-state structural information, a transformer-based module jointly updates these embeddings and the distogram inspired by AlphaFold2’s Evoformer10. The network jointly estimates target objectives by applying separate heads: ΔG, ensemble distogram, and an auxiliary head for sequence recovery. The ΔG head predicts per-residue ΔG, a vector with the same length as a protein, which is summed up to predict the net ΔG of a protein. Protein ΔG labels are derived from experimental measurements (Mega-scale20) or set as less than 0.5 kcal/mol for disordered proteins (DisProt25). The distogram head predicts the probability distribution of distances for all residue pairs given the sequences and the estimated folded structure distances. For the ensemble distogram labels, mean residue distances at the folded state derived from the predicted structure and the unfolded state derived from the Flory model are one-hot encoded into a distogram form, with the ratio derived from the Boltzmann distribution (Fig. 1b–d). By doing so, the network can effectively learn to what extent the given sequence can accommodate a denatured structure with respect to the folded structure.
IFUM was trained on a dataset composed of two distinct sources: a subset of 648,650 proteins (30–80 amino acids in length) from the Mega-scale dataset20, which features ΔG values inferred from cDNA display proteolysis, and a set of 3219 disordered proteins from the DisProt database25 with at most 70 amino acids. For a rigorous test-only evaluation, we compiled several independent datasets, including (i) 6007 sequences from the CATH database26 with lengths up to 869 amino acids, (ii) 26 protein-engineered sequences with experimentally measured melting temperatures (Tm) (12 IFN-λ14, 8 IL-10, and 6 UGT76G18), (iii) a manually curated literature set of 57 unique wild-type sequences with experimental ΔG values; 40 from the S669 dataset27 (Supplementary Table 1) and 17 from Maxwell et al. 28, and iv) a manually curated literature set of 413 de novo designed sequences from five distinctive folds with their corresponding experimental expression data29,30,31,32,33,34,35. To ensure no data leakage, none of the sequences in these test sets shared more than the sequence identity of 0.30 with any data in the Mega-scale training set. Further details on the model architecture, inputs, ΔG labels, unfolded state ensemble modeling, and dataset curations are available in the “Methods”.
IFUM can predict unfolding free energy with a low mean error
We first tested the accuracy of IFUM predictions on the Mega-scale test set containing 86 wild-type and de novo designed domains. Testing on this set offers an objective estimation of the network’s precision limit, at least for small proteins, at ideal conditions free from experimental disparities (e.g., pH, temperature, solvent, etc). IFUM achieved an RMSE (root mean squared error) of 1.16 kcal/mol and a PCC (Pearson correlation coefficient) of 0.78 when both labels and predictions are clamped to the dataset’s experimental dynamic range, [–1, 5] kcal/mol20 (Fig. 2a). Detailed performance metrics for the training, validation, and test sets are provided in Supplementary Table 2.

a Scatter plot of IFUM-predicted ΔG (ΔGpred) versus experimental ΔG (ΔGexp) for the Mega-scale test set. ΔG values are clamped between –1 and 5 kcal/mol, the reported experimental dynamic range20. Color intensity indicates point density (bright: high density; dark: low density). The model achieved a Pearson correlation coefficient (PCC) of 0.78 and root mean square error (RMSE) of 1.16 kcal/mol on this dataset. b, c PCC and RMSE comparisons. b IFUM compared to IFUMbaseline, ESMtherm, ESM2 pseudo-perplexity (ESM2pppl), and protein length as a baseline (baseline-length) on the Mega-scale Common (see “Methods”). c IFUM compared to the IFUMbaseline model, trained without unfolded state modeling, and baseline-length on the Mega-scale test set. d Example predictions for HECTD1 CPH domain (PDB: 3DKM) mutants Q5L and V16D. Left: Overlay of ESMFold-predicted structures for Q5L (blue) and V16D (orange) mutants, showing their ΔGpred and ΔGexp values. Mutation sites (Q5L and V16D) are indicated with a dotted circle. Corresponding predicted equilibrium ensemble distograms are shown in a heatmap (orange: V16D, blue: Q5L). e Histogram comparing ΔGpred values between stable (ΔGexp ≥ 5 kcal/mol) and unstable (ΔGexp ≤ –1 kcal/mol) subsets of the Mega-scale test set. The x-axis labels denote specific ranges of ΔGpred values, where parentheses indicate an exclusive boundary and square brackets indicate an inclusive boundary. f A scatter plot of ΔGpred versus ΔGexp for 57 unique wild-type proteins from literature data. The marker size corresponds to protein length, and color corresponds to the ESMFold predicted Local Distance Difference Test (plDDT) score. The PCC ranges from 0.53 to 0.97 with a higher plDDT cutoff. The dashed line indicates a perfect correlation (y = x). g Histogram comparing ΔGpred values between CATH and DisProt datasets (two-sided Welch’s t-test p « 0.001). Source data are provided as a Source data file.
To see how this compares to other existing tools, we ran ESMtherm19, a fine-tuned version of ESM2 for ΔG prediction, and collected the ESM2 sequence pseudolikelihood12 (see “Methods”). For a fair comparison, we used the sequences that overlap between two test sets from IFUM and ESMtherm. This resulted in a common test set (containing 27 domains, noted as Mega-scale Common hereafter) of 5356 sequences from the original Mega-scale dataset. With this particular smaller test set (Mega-scale Common), IFUM achieved a PCC of 0.91 and an RMSE of 0.78 kcal/mol, outperforming ESMtherm, ESM2, and a sequence length-based baseline (PCC: 0.86, 0.44, and 0.11; RMSE: 1.40, N/A, and N/A, respectively) (Fig. 2b), with clamped ΔGpred and ΔGexp values (unclamped results in Supplementary Fig. 2). We stress that this common set is mainly taken for the purpose of comparison, and that the full-set result should better reflect the actual performance.
Training IFUM to predict unfolded state ensemble improves ΔG prediction
Then, we confirmed whether this improvement resulted from the incorporation of unfolded state ensemble modeling. Training a simpler version of IFUM that does not contain any unfolded-state-related aspects (IFUMbaseline), the PCC dropped from 0.78 to 0.70, and RMSE worsened from 1.16 to 1.39 kcal/mol when clamped (Fig. 2c and Supplementary Table 3). A simple baseline based solely on sequence length showed negligible correlation (PCC: 0.10). This low performance is expected because the dataset is dominated by mutational variants (e.g., point mutations) where stability changes significantly despite the sequence length remaining constant. Inspecting the equilibrium ensemble distogram prediction, it was highly consistent with the ΔG prediction; while stable proteins had ordered distograms resembling those from folded states, unstable ones had fuzzy distograms close to mixtures of folded and unfolded distograms (Fig. 2d). More examples showing the consistency between the model’s predicted equilibrium ensemble and ΔG are provided in Supplementary Fig. 3.
We further investigated ablation studies to reveal the importance of key components of the network (Supplementary Table 3). Removing the triangle multiplicative updates10 or replacing all differential self-attention layers36 with conventional self-attention significantly reduced performance. Critically, training IFUM without the equilibrium ensemble prediction objective resulted in a comparable performance decrease. This shows that, for the ΔG estimation, equilibrium ensemble modeling is as crucial as other well-established innovations in deep learning architectures.
IFUM is broadly applicable to various types of proteins
Given the promising prediction accuracy on small proteins at highly controlled conditions (PBS, pH 7.4, T = 298 K), we next evaluated IFUM’s performance on more realistic problems. First, we asked about its discriminative power in distinguishing unstable (ΔGexp ≤ –1 kcal/mol) versus stable (ΔGexp ≥ 5 kcal/mol) sequences within the well-controlled Mega-scale test set (Fig. 2e). The network was able to discriminate between those, giving a Welch’s t-test p-value « 0.001. The figure shows evaluation of the ΔGpred values clamped to the dataset’s dynamic range ([−1, 5] kcal/mol); the same analysis on unclamped ΔGpred values is provided in Supplementary Fig. 4.
We then tested its performance on labeled proteins of broader types, with sizes ranging from 33 to 375 residues. A set of 57 wild-type protein sequences was collected with their experimentally determined ΔG values27 (Fig. 2f, Supplementary Table 1; see “Methods”) from Maxwell et al. 28 and other literature. Across all 57 sequences, IFUM achieved a PCC of 0.53, which is poorer than that on the well-controlled data set. We found that the quality of folded state structure affects the prediction accuracy; restricting the analysis to the 45 sequences with plDDT > 80 improved the PCC to 0.66; with plDDT > 90 (10 sequences), PCC further increased to 0.97 (Supplementary Fig. 5A). With a more controlled subset from Maxwell et al. collected from similar experimental conditions, the correlation was much clearer (Supplementary Fig. 5C). A sequence-length-based baseline model, known to weakly correlate with the stability37, showed a low PCC of 0.38 (Supplementary Fig. 5D). These findings suggest that IFUM’s ΔG prediction accuracy is correlated with the confidence and quality of the input target folded state, with higher accuracy observed for proteins with well-defined structures. This is related to the ESMFold modeling accuracy, which was generally not an issue for the small proteins in training data; further analysis between ΔG prediction and folded state quality is in the Discussion.
Next, we moved on to a more challenging yet realistic problem, which was to apply the same discrimination test on semi-labeled wild-type natural proteins. This dataset includes 6,007 folded sequences from CATH, which contain structured domains from wild-type proteins, and 685 disordered proteins from DisProt (Fig. 2g; see “Methods”). The main purpose of this test was to check IFUM on a larger scale if (i) it returns any unexpected results for unseen types of proteins (e.g., larger than 100 residues), and (ii) it can discriminate well-folded proteins against intrinsically disordered proteins. Although these proteins do not have quantitative ΔGexp values, we expected IFUM should predict positively to CATH domains while negatively to disordered ones.
We found that the distributions of ΔGpred values reflected the differences in the characteristics in two distinct datasets, giving a Welch’s t-test p-value « 0.001, implying IFUM can generally distinguish ordered proteins from disordered ones. However, still a considerable portion of CATH proteins had ΔGpred < 1 kcal/mol. Investigating such underestimated CATH domains, we observed a higher prevalence of solvent-exposed hydrophobic residues required for forming obligate oligomers, stable domain interfaces, or embedding transmembrane domains (Supplementary Table 4 and Supplementary Fig. 6). This analysis underscores the need for cautious IFUM application depending on the expected protein context. On the other hand, there was also a good portion of predictions with excessively positive values (>25 kcal/mol), implying overestimation of stability. This will be further discussed in the Discussion.
IFUM can accurately predict ΔΔG for various types of mutants
IFUM exhibited strong predictive performance for ΔΔG, encompassing point/double and insertion/deletion (indel) mutants within the Mega-scale test set (Fig. 3a). Within the full Mega-scale test set, IFUM achieved a PCC of 0.81, 0.80, and 0.63 for point mutants, indels, and double mutants, respectively. To further benchmark this capability, we used the Mega-scale Common test set and compared IFUM against ThermoMPNN16 (ThermoMPNN-D38 for double mutants), ESMtherm, ESM2, FoldX39 and Rosetta FastRelax and Cartesian ddg protocols40,41 (Fig. 3b, c). For Mega-scale Common indels, a comparison with ThermoMPNN was not applicable, IFUM demonstrated a clear advantage with a PCC of 0.76 over ESMtherm, ESM2, FoldX, and Rosetta (0.66, 0.35, 0.22, and 0.01, respectively). For Mega-scale Common double mutants, IFUM achieved a PCC of 0.61, and the others 0.38, 0.70, 0.50, 0.50, and 0.78 (ThermoMPNN-D, ESMtherm, ESM2, FoldX, and Rosetta, respectively). Data points with ΔΔGexp < −10 kcal/mol or ΔΔGexp > 6 kcal/mol in Fig. 3a were excluded from the visualization for point, indel, and double mutants (n = 169, 5, and 71, respectively). The source data are given as a Source data file.

a Scatter plots of IFUM-predicted ΔΔG (ΔΔGpred) versus experimental ΔΔG (ΔΔGexp) for different mutation types within the point, insertion/deletion (indel), and double mutants in the Mega-scale test set. Color intensity represents point density (bright = high density). The model achieved a Pearson Correlation Coefficient (PCC) of 0.81, 0.80, and 0.63, respectively. b Scatter plots of ΔΔGpred versus ΔΔGexp for four case study proteins: Myoglobin, P53, S. nuclease, and T4 lysozyme. The dashed gray lines indicate the perfect correlation, and the dashed black lines are linear regression lines. The model achieved a PCC of 0.75, 0.63, 0.71, and 0.70, respectively. c Bar charts comparing Pearson correlation coefficients (PCC) for IFUM against ThermoMPNN, ESMtherm, ESM2pppl, FoldX, and Rosetta. Comparisons are shown for the Mega-scale Common (MS Common) indel subset (ΔG), MS Common double mutant subset (ΔΔG), and the four case study datasets (ΔΔG). Source data are provided as a Source data file.
Next, we evaluated the models on four case studies selected for single point mutation ΔΔG: Myoglobin42, P5343, S. nuclease44, and T4 lysozyme44. IFUM achieved PCC values of 0.75, 0.63, 0.71, and 0.70, respectively (Fig. 3b), comparable to the ThermoMPNN specifically trained for single point mutations (0.56, 0.72, 0.76, and 0.69, respectively). Other methods (ESMtherm and ESM2) showed relatively poor performance in reproducing point mutation effects. Specific PCC values are reported in Fig. 3c, more details on double mutants ΔΔG predictions are shown in Supplementary Fig. 7 and Supplementary Table 5. Overall, IFUM was the only method among tests that robustly worked across various types of mutations ranging from single point mutations to indels among these methods.
We then compared IFUM with a free energy perturbation (FEP) method45 on ΔΔG prediction. FEP is a physics-based method that offers rigorous mutational estimates with higher computational cost. For the comparison with a previous FEP work, we filtered the original FEP dataset to non-redundant 76 points (sequence identity <0.30 with the training set). The FEP+ method showed a better performance (RMSE of 1.06 kcal/mol) than IFUM (RMSE of 1.44 kcal/mol) (Supplementary Fig. 8). However, the computational cost differed by orders-of-magnitude: several hours for FEP + 45 and 30 s for IFUM (both in a single GPU). This demonstrates the IFUM’s utility for a scalable alternative tool for screening.
In the following paragraphs, we demonstrate the practical utility of IFUM on two important protein design or engineering problems.
Practical application 1. Protein stabilization engineering
It is quite common practice in protein engineering to introduce sequence length modifications or multiple-sequence substitutions. For instance, loops are often truncated for protein stabilization engineering8 but are elongated instead when new functionality needs to be introduced46. Although structure prediction confidence estimations are broadly used as guidelines (e.g., filtering) for these processes13, their quantitative contribution remains unclear. Here, we demonstrate that IFUM can be a good alternative to these structure prediction metrics.
We tested it on three protein engineering scenarios in which multiple sequence substitutions and/or sequence length modifications were introduced (Fig. 4a). For two targets (IFN-λ3 and IL-10), IFUM was blind tested in parallel with experimental human-driven engineering; for UGT76G1, IFUM was compared against previously reported data (per-target details in the following paragraphs). Computational metrics were compared to experimental melting temperature (Tm) values, which are known to well correlate with ΔG within the same protein domain47,48 (Supplementary Fig. 9). Tm is chosen here as it is a more affordable and broadly used measurement for protein engineering, although its difference with ΔG can introduce further noise to labels. Due to the less confident ESMFold models, Rosetta FastRelaxed AF3 models were used for the folded state structural input to the network.

a Scatter plots of experimental melting temperature (Tm) versus IFUM-predicted ΔG (ΔGpred) for IFN-λ, IL-10, and UGT76G1 sequence and backbone redesigns. The model achieved a Pearson Correlation Coefficient (PCC) of 0.75, 0.62, and 0.87, respectively. The marker size corresponds to protein length, and the marker color corresponds to the number of mutations relative to each wild-type protein. Markers with the labels indicate the wild-type proteins and IL-10M1. Rosetta FastRelaxed AF3 model structures were used in these scatter plots. The dashed lines are linear regression lines (measure of center), and the shaded regions indicate the 95% confidence interval (error bands) for each correlation, calculated via bootstrapping. b, c Performance on the in silico screening of designed proteins. b Composition of design datasets for five different protein folds, showing the number of computationally designed proteins that were experimentally found to express in E. coli in a soluble monomeric form (blue) or not (red). c Predictive performance, measured by the Area Under the Receiver Operating Characteristic curve (AUROC), for classifying non-expressing designs from expressing ones. The performance of IFUM’s predicted ΔGpred (blue) is compared with the predicted Local Distance Difference Test (plDDT) from ESMFold (orange) and AF3 (green). Source data are provided as a Source data file.
The first group consists of engineered IFN-λ3 proteins designed to enhance their protein fitness and stability. In particular, the flexible loop harboring the thrombin cleavage site was excised and replaced with a structurally inpainted α-helix, which effectively shielded the adjacent hydrophobic patch and conferred resistance to proteolytic degradation. Thermal shift assays were performed to determine the Tm of 9 engineered IFN-λ3 variants and, for comparison, 3 wild-type IFN-λ isoforms (IFN-λ1, IFN-λ2, and IFN-λ3)14. The wild-type IFN-λ1 and IFN-λ2 consist of 200 amino acids, and IFN-λ3 consist of 196 residues. For engineered IFN-λ3 variants, 12 residues were deleted from the wild-type IFN-λ3 sequence, and 12 residues were inserted, resulting in overall length preserved.
As a second example, we analyzed 8 engineered monomeric IL-10 variants. The wild-type human IL-10 consists of 178 amino acids per chain. In the designed group, 11 residues were deleted from the wild-type sequence, 15 residues (for 2 variants), and 20 residues (for 5 variants) were inserted, resulting in a net 4–9 residues addition. IL-10M146, a previously developed monomeric IL-10 containing a GS linker, was also included for comparison.
Finally, the wild-type UGT76G1 and 5 engineered UGT76G1s and their measured Tm were collected from previous work8. In this group of engineered proteins, 14–62 mutations were introduced to the original sequence length of 458 amino acids, including 11 deletions relative to the wild-type. See Supplementary Tables 6–8 for measured Tm and predicted ΔG of IFN-λ, IL-10, and UGT76G1, respectively. The final number of mutations per sequence was recalculated based on the respective sequence alignment with the corresponding wild-type.
Shown in Fig. 4a, IFUM prediction results showed a good positive correlation with the experimental Tm for all groups of engineered proteins. For IFN-λ, IL-10, and UGT76G1 proteins, PCC of 0.75, 0.62, and 0.87 were observed, respectively, although the results should be interpreted with caution due to the small sample sizes (p-values: 0.005, 0.104, and 0.023, respectively). For comparison, we selected the AF3 confidence metric (plDDT) (note that all the other ΔΔG prediction methods were not applicable due to length changes). Experimental Tm was much less consistent with the AF3 metric, which even negatively correlated with IL-10 and UGT76G1 groups (Supplementary Fig. 10). This highlights the broad generalization of IFUM over AF3 in guiding quite challenging stability engineering problems where both sequence length change and multiple sequence substitution occur simultaneously. Yet, despite the good correlation within a protein topology, caution should be taken on absolute ΔG values across three distinct families, e.g., prediction ranging from +6 to +10 for UGT, while from −1 to +3 for IFN-λ, even though they are in similar stability. This implies that as proteins get larger, IFUM can still be effective on predicting mutational effects, but less accurate for absolute stability estimation, which is further discussed in the limitation part of the Discussion.
Practical application 2. Screening de novo protein designs for soluble monomeric expression
A common final evaluation step in computational de novo protein design is to use a structure prediction model to prioritize candidate sequences13. Here, the discriminative power of IFUM, ESMFold, and AF3 is compared for their ability to identify designed proteins that express as soluble monomers in E. coli (Fig. 4b, c). Filtering with IFUM’s ΔGpred achieved consistently improved discrimination in AUROC (Area Under the Receiver-Operating Characteristics Curve) over the filtering based on plDDT scores from ESMFold and AF3 across all five different scaffold types: the Rossmann fold29,30, TIM barrel31, helical repeat32, beta barrel33, and jelly roll fold34,35. For this comparison, any sequences with sequence identity >0.30 to the training data were filtered out. This highlights that, similar to its application to protein engineering, IFUM can replace the routinely applied AlphaFold-based metrics in the de novo design campaigns to improve design success rates. Detailed performance metrics, including confusion matrices, precision, recall, and F1-scores, when using ΔGpred (>4 or >5 kcal/mol) and plDDT (>80 or >90) as thresholds for predicted stable proteins, are provided in Supplementary Fig. 11 and Supplementary Table 9.
Finally, to assess the computational scalability of IFUM, we measured the wall-clock time required to process the dataset of 1045 variants from Fig. 3b (Supplementary Table 10). IFUM processed the entire dataset in approximately 5 minutes in a single A6000 GPU (0.3 s per sample). This inference speed, comparable to that of FoldX, demonstrates that IFUM is well-suited for high-throughput screening applications where computational cost can be a limiting factor.
Discussion
To verify the contribution of the large-scale dataset to our model’s performance, we conducted an ablation study on the training set size with the fixed test set (n = 90,958). We trained IFUM using varying fractions of the Mega-scale dataset, ranging from a baseline of only wild-type and de novo proteins to 1%, 5%, 10%, 25%, 50%, 75%, and 100% of the full training set (Supplementary Fig. 12). Notably, the model’s performance (i.e., RMSE and PCC) on the Mega-scale test set did not plateau even when using the full 100% dataset. This absence of a plateau demonstrates that the current large-scale dataset is essential for IFUM’s accuracy. Furthermore, it suggests that the model has not yet reached its capacity, implying that performance could be improved even further with larger datasets in the future.
Our results suggest that explicitly considering the unfolded state ensemble contributes to improved accuracy in ΔG prediction. This aligns well with the thermodynamic basis of ΔG defined as the free energy difference between folded and unfolded states. We have shown this in two ways; first, many ΔΔG predictors relying solely on a single folded state (typically the wild-type structure) revealed their limitations on capturing more complex types of mutations16,17, as supported by our benchmark comparison results (Fig. 3c). Second, the observed performance decrement in IFUM when neglecting the equilibrium ensemble was as significant as that of removing key architectural components seen with ablations (Supplementary Table 3).
IFUM predicts ΔG and the equilibrium ensemble by integrating pre-trained sequence and structure embeddings from ProtT5 and ESM-IF1, along with structural input. Leveraging transfer learning from extensive datasets, including 2.1 billion and 45 million protein sequences in BFD and UniRef50 (used to train ProtT5), respectively, and 12 million AlphaFold2-predicted structures (used to train ESM-IF1), likely contributes to mitigating overfitting in IFUM, a strategy also seen in ThermoMPNN16. This pre-training approach may offer an advantage over methods like ESMtherm, which showed significant overfitting when fine-tuning the entire ESM2 model on a single dataset19. We confirmed this by observing a significant performance decrement in IFUM when removing the ProtT5 embedding and using one-hot encodings instead (Supplementary Table 3).
We then point out the impact of the input folded structure on the prediction results. Shown in Fig. 2f, for natural proteins up to 375 residues, IFUM predictions show much improved correlation with experimental data when the subset of high-confidence AF3 models (plDDT >90) were used as inputs (Supplementary Fig. 5A). A similar trend was observed in every group of engineered proteins, where replacing Rosetta FastRelaxed AF3 models with ESMFold models led to a significant decrease in correlation between measured Tm and predicted ΔG values (Supplementary Fig. 13). This dependence on input structure quality is likely arising from the nature of the network which assumes that the input structure representation correctly matches the actual folded state structure, while the ESMFold modeling accuracy especially does not satisfy this assumption for larger proteins (>200aas). Further analysis highlights the role of the quality of folded state structures tested in the literature set (n = 57). While unrealistically low ΔG values were predicted for wild-type proteins when using poor-quality ESMFold structures (indicated by low plDDT or pTM scores), substituting these with AF3 structures—more closely resembling the corresponding crystal structures (Supplementary Fig. 14)—resulted in more reasonable ΔG predictions (Supplementary Fig. 5b, c). We further investigated this point on a dataset from Cianferoni et al. 49, which contains 17 experimental ΔΔG values and 7 corresponding high-resolution crystal structures. Using ESMFold yielded an RMSE of 1.52 kcal/mol, but replacing 7 with their crystal structures (keeping ESMFold for the rest) improved the RMSE to 0.90 kcal/mol (Supplementary Fig. 15). These observations suggest that IFUM prediction is more reliable when supplied with more confident folded structural inputs.
Based on these observations, we tried re-training IFUM using the AF3 structures as inputs. However, testing the re-trained model on the Mega-scale test set with AF3 structural inputs did not improve the performance (RMSE: +0.1 kcal/mol, PCC: −0.05). This was because the ESMFold structures were at high quality50 for the small proteins in the training data, unlike the larger proteins in Fig. 2f or Fig. 4a for which ESMFold output significantly differed from AF3 outputs. From a practical point of view, we recommend using AF3 models as structural inputs if the ESMFold modeling quality is expected to be low (e.g., plDDT <80).
IFUM predicts the stability of a protein in the sum of per-residue contribution predictions. These residue-wise decomposed values can potentially guide the selection of high-priority residues for stability optimization. To test this idea, sequence redesign on de novo designed proteins51 using ProteinMPNN52 was performed, comparing two strategies: (1) full redesign and (2) selective redesign focusing only on residues with negative (i.e., destabilizing) contribution predicted by IFUM. As shown in the Supplementary Fig. 16, selective redesign improved IFUM ΔGpred over the original design while full redesign did not. While this result is promising, it might be simply a self-consistent outcome, and hence, we expect to experimentally validate this strategy in more depth in our future work.
While IFUM demonstrates promising performance in protein stability prediction, several inherent limitations warrant consideration. First, the primary training dataset, Mega-scale, possesses an experimental dynamic range of [–1, 5] kcal/mol, with protein sizes ranging up to 80 residues20. This restricted range inherent to the proteolysis-based experimental methodology may constrain IFUM’s capacity for accurate extrapolation beyond these boundaries, and may limit its performance for proteins with exceptional stability or with larger sizes. To assess the model’s extrapolation ability, we examined the ΔGpred distribution from the AlphaFold Human proteome database (Supplementary Fig. 17). This analysis clearly showed that (i) the model can predict values beyond the label range in the training data, [−1, 5], but (ii) begins to predict unexpectedly large values (>25 kcal/mol) when protein size exceeds 200 amino acids. This trend is also evident in Fig. 2f. Furthermore, we found that ΔGpred has shown higher correlation with the length of a protein than ΔGexp in wild-type proteins (Supplementary Fig. 5E). These findings indicate that predictions of absolute stability for proteins larger than 200 amino acids might be treated with caution.
Investigating the topology dependence, it was not prominent as long as proteins are small enough (Supplementary Fig. 18). However, we identified a systematic tendency to predict lower stability for high loop content proteins (Supplementary Fig. 19). This bias may stem from the inclusion of scrambled sequences and intrinsically disordered regions (IDRs) in the training set, which likely conditioned the model to associate extensive loop regions with instability. This tendency explains the lower stability predictions for IFN-λ proteins and their variants (IFN-λ1 loop content: 45%) revealed in the protein engineering study. Consequently, caution is advised when applying IFUM to proteins with significant disordered regions or high loop contents.
Second, IFUM’s framework is built on a simplified two-state folding model. The simplified and discrete approximation of ensemble representation in this hypothesis should differ from a more realistic continuous conformational distribution. While this approximation is widely used20, it may not fully capture the complexities of the folding landscapes particularly for larger or multi-domain proteins. It is reported that the denatured state structural ensemble is different from the Flory model and closer to partially-folded structures53. Hence, we believe that using more direct strategies like CALVADOS54 or other heuristics that can better represent the denatured ensemble may improve the model’s performance. We plan to explore this and other strategies in more depth in our future work.
Third, the network does not apply to membrane proteins and obligate oligomers because it was mainly trained on proteins that are water-soluble and monomeric. We intend to address this in further studies, specifically to predict proteins in their natural conditions.
Finally, as IFUM relies on transfer learning from large-scale pre-training, its performance is intrinsically linked to the quality and scope of these foundation models and datasets. While transfer learning likely mitigates overfitting, the model’s standalone utility without this pre-training remains to be fully established. These limitations listed above define key directions for the future model development toward enhancing the scope and accuracy of protein stability predictions.
We have shown IFUM’s strong performance on a broad range of test sets as well as its practical utility in protein de novo design and engineering, as demonstrated by the correlation between its ΔGpred and experimental Tm for engineered proteins with many mutations. Given these results, IFUM can provide guidance to various types of protein design or engineering processes, complementing standard checks like plDDT and PAE score based filtering13 (Fig. 4 and Supplementary Figs. 10, 13). In summary, IFUM offers a robust method for ΔG and ΔΔG prediction and proposes a new concept in the deep-learning architecture design. While these findings are encouraging, further research is needed to fully explore the model’s capabilities and limitations and to guide future development in protein stability engineering and design.
Methods
IFUM architecture
IFUM is a transformer-based model designed to predict multiple protein properties, mainly the unfolding free energy (ΔG) and the equilibrium ensemble of conformational states. A schematic description of the IFUM model can be found in Supplementary Fig. 1. The model takes three inputs: a sequence embedding of shape [N, 1024] generated by ProtT5 embedder, a one-hot encoded, binned pairwise distance map (distogram) of shape [N, N, 21] representing the folded structure (predicted by ESMFold unless otherwise stated), and a structure embedding of shape [N, 512] extracted from ESM-IF1, where N is the sequence length of target protein. The IFUM architecture comprises two main components: a main transformer module incorporating triangle multiplicative updates and a task-specific simple output multilayer perceptron (MLP).
Initially, IFUM reshapes the sequence embedding and the structure embedding, respectively, to a common dimension of [N, 21] using differential self-attention36. These reshaped embeddings are then concatenated into a tensor of shape [2, N, 21].
The main module is a modified version of AlphaFold2’s Evoformer, where triangle attention is removed due to its memory demand (but keeping triangle multiplicative updates) and all self-attention layers are replaced with differential self-attention. During the main blocks, the pairwise distogram and the concatenated embeddings are jointly updated through differential self-attention with pair bias and outer product mean as similar to the original Evoformer. IFUM’s main module comprises 11 of these main blocks.
Finally, an MLP with two hidden layers predicts the final output for each objective. Specifically, ΔG and sequence recovery are predicted from the main module-updated embeddings, while the equilibrium ensemble is predicted from the main module-updated pairwise distogram. IFUM predicts a protein’s ΔG by predicting per-residue ΔG contributions and then through an unweighted summation of these contributions. We observed that optimizing destabilizing sequences of a protein based on per-residue contribution results in a higher predicted ΔG than naively redesigning the whole sequence, suggesting potential usage of IFUM on de novo protein design (Supplementary Fig. 11). IFUM can also predict ΔΔG by subtracting the predicted wild-type ΔG from a predicted mutant ΔG, obtained from independent runs of the model.
Equilibrium ensemble modeling
The Flory random coil model is a well-established model for describing the unfolded state ensemble of proteins (Eq. 1).
This model provides a statistical description of the average distances expected between residues in a flexible, disordered chain. Therefore, we employed the Flory random coil model with fitted parameters55,56 (Eq. 2) to calculate the root-mean-square (RMS) pairwise Cα distances between residue indices i and j in the unfolded states (\({r}_{i,j}^{U}\)) of the protein:
We use these RMS pairwise distances as an explicit representation of the spatial extent of unfolded states to model their contribution to the conformational ensemble.
Protein structure prediction AIs often exhibit insensitivity to structure-disrupting mutations, sometimes predicting structures for mutated sequences that are nearly identical to the wild-type structure. Therefore, we used the ESMFold-predicted structure as a proxy for the target folded (near wild-type) state, recognizing the limitations of AI models in capturing mutational effects. Pairwise Cα distances between residue indices i and j can be directly calculated from this modeled structure.
Assuming a simple two-state equilibrium between folded and unfolded states, we constructed our model by incorporating the Flory random coil model for unfolded conformations and the ESMFold-predicted structure as a proxy for folded conformations. Within this model, the distance between any two residues was represented as a weighted average of their folded and unfolded state distances. This weighting factor, reflecting the equilibrium ratio between folded and unfolded states (Eq. 3), can be derived directly from the protein’s unfolding free energy, ΔG (Eq. 4, note that this is only used at the training stage):
Consequently, using this equilibrium ratio, we calculated the ratio of pairwise distances between residues i and j in folded and unfolded states (Eq. 5):
which is then represented as a mixed label distogram (probability values above at rF and rU bins, otherwise 0) with a range of [2.0, 42.0] Å and a bin width of 2 Å (Fig. 1b, c. α corresponds to p(\({r}_{i,j}^{F}\))). IFUM was trained to predict this equilibrium ensemble distogram.
Training configuration
IFUM was trained on a single A6000 48 GB GPU using an AdamW with a weight decay of 0.01 and a learning rate of 1 × 10−4. The loss function was a weighted combination of three components (Eq. 6, \(L\)):
which consists of a Gaussian negative log-likelihood loss for ΔG prediction \(({L}_{\varDelta G})\), cross-entropy loss for equilibrium ensemble modeling \(({L}_{{{\rm{ensemble}}}})\), and a cross-entropy loss for sequence reconstruction \(({L}_{{{\rm{seq}}}})\). During training, we used a batch size of 80, increasing it to 100 for validation and testing, and reducing it to 1 for test-only datasets. Data was shuffled at the start of each training epoch, and masks were carefully employed during calculation to enable batch processing of sequences with varying lengths. We acknowledge that we did not explicitly evaluate the impact of different mask sizes on prediction performance.
Dataset preparation
The Mega-scale dataset was first curated by removing data with deltaG values marked as unreliable (dG_ML = “—”). We then filtered out scrambled sequences with deltaG values greater than 0.5 kcal/mol to avoid potential artifacts caused by aggregation. This curation process resulted in a Mega-scale dataset containing 829,927 sequences, with deltaG values used as the training labels.
The DisProt dataset was curated by collecting sequences shorter than 70 amino acids to enrich for IDPs. Longer sequences in the raw dataset often represented stable proteins with flexible loops, not primarily IDPs. This curation yielded a DisProt dataset with 4705 sequences, and this size of curated subset is noteworthy as it is comparable to, or even larger than, training sets commonly employed in similar studies. Although the DisProt dataset lacks experimental ΔG labels, it is a database of proteins known to be disordered or unstable. Therefore, during the training of IFUM, we incorporated the DisProt dataset by modifying the loss function to encourage the model to predict ΔG values lower than 0.5 kcal/mol for these sequences. This approach implicitly guides the model to learn features associated with disordered proteins, even in the absence of explicit ΔG values for DisProt. We reasoned that by training IFUM to predict low ΔG values for sequences known to be disordered, the model would better capture the characteristics of unfolded or unstable protein states.
The CATH database (‘cath-dataset-nonredundant-S20’) was curated by first removing sequences with noncanonical amino acids or unknown amino acids or more than two segments (Supplementary Fig. 6G), then collecting sequences shorter than 900 amino acids due to computational limitations when using ESMFold for structure prediction (specifically, CUDA out-of-memory issues encountered). Finally, any CATH sequences with a higher sequence identity of 0.30 with the Mega-scale test set were filtered out. This curation resulted in a CATH dataset with 6007 sequences for testing purposes. Hydrophobicity was calculated by using SAP values following the protocol (‘cao_2021_protocol_guide’) from Cao et al.51.
The literature dataset was first curated by collecting sequences and their corresponding experimental ΔG values of wild-type proteins from the S669 dataset’s original 92 papers. The dataset was then filtered to include only entries with experimental pH values between 7.0 and 7.5 and temperatures between 293 and 328 K (Supplementary Table 1). Subsequently, we removed homooligomerizing sequences based on UniProt annotations57, as well as any sequences with greater than 0.30 sequence identity to those in the Mega-scale train set. This yielded a curated, test-only dataset containing 40 sequences and experimental ΔG values.
We also used the same four case studies, p53, myoglobin, staphylococcal nuclease, and T4 lysozyme, as previously employed by ThermoMPNN as test-only datasets.
We collected wild-type UGT76G1 and 7 redesigned sequences and their measured Tm values from Seong-Ryeong Go et al. 8. Seong-Ryeong Go et al. used Rosetta based protocol and redesigned the wild-type UGT76G1, enhancing the thermostability and enzymatic activity.
Finally, we collected 413 de novo designed sequences from five distinctive folds with their corresponding experimental expression data29,30,31,32,33,34,35.
Design workflow of IFN-λ3 and IL-10
To improve structural stability, native loop structures of wild-type IFN-λ3 and IL-10 were computationally redesigned using RFdiffusion58, respectively. Among the generated models, those with α-helical linkers were identified using the DSSP algorithm and were chosen for sequence design. For the selected backbones, ProteinMPNN generated amino acid sequences, while keeping the undesigned region fixed. Final sequences of those we measured the Tm will be available in our GitHub repository (github.com/HParklab/IFUM). See Jeongwon Yun et al. 14 for more details on protein expression and Tm measurement.
Dataset splitting
To ensure a robust evaluation and prevent data leakage between sets, we first grouped sequences by homology. This part was essential as many wild-type sequences were sequentially close to each other. We used the MMseqs2 easy-cluster59 to cluster the wild-type sequences of the Mega-scale and whole DisProt datasets with a minimum sequence identity cutoff of 0.30. Proteins were then randomly split by clusters into train and validation/test sets, so that all members in a cluster belong to the same set, to ultimately yield an 8:2 ratio of sequences in the final datasets. The validation and test sets were then split randomly in half. The exact values can be found in Supplementary Table 11.
A set of 5356 sequences, common to both the IFUM Mega-scale test set and the ESMtherm test-set-only domains, was assembled and used for IFUM and ESMtherm comparison (Figs. 2b and 3c; Mega-scale Common).
Ablation results
All modifications were trained on the same training sets with the same hyperparameters. The best model for each modification was selected based on the highest coefficient of determination (R2) on the validation set. All modifications were evaluated with the same test sets to calculate the performance metrics.
Other predictors
ThermoMPNN is a graph neural network, a transfer-learned version of ProteinMPNN, trained on the Mega-scale dataset to predict ΔΔG from point mutations. It is important to note that ThermoMPNN is designed for point mutations only and does not predict the effects of insertions or deletions, as it requires the wild-type and mutant protein sequences to be of the same length. We generated single mutants ΔΔG values for comparison using the Google Colab implementation of ThermoMPNN. We used ThermoMPNN-D to predict double mutants ΔΔG.
ESM2 is a PLM trained on the UniRef database to predict masked amino acids in protein sequences. It has demonstrated the ability to predict the functional effects of mutations without requiring any specific training (zero-shot prediction). To quantify this predictive power on predicting ΔG, we calculated the ESM2 pseudolikelihood for a sequence of length N by summing the log probabilities of predicting each residue \({r}_{i}\) when it is masked (Eq. 7):
We used esm2_t36_3B_UR50D to generate these pseudo-loglikelihood (plll) values for our comparisons.
ESMtherm is a PLM, a fine-tuned version of ESM2, trained on the Mega-scale dataset to predict ΔG of proteins. We used the original script provided in the ESMtherm GitHub repository to generate ΔG values for our comparisons.
FoldX calculates the ΔG value of a protein using an energy function. We used predicted structures and the FoldX Stability protocol (FoldX 5.0) to calculate the ΔG and ΔΔG values, including indel mutants.
Rosetta uses physical and empirical potentials to calculate the ΔG value. We used predicted structures and the FastRelax and Cartesian ddG protocols40,41 (ref2015) to calculate the ΔG and ΔΔG values, respectively, including indel mutants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data used in this study can be found in the original research publications: Mega-scale20, DisProt25, CATH26, S66927, UBA14, MyUb5, Myoglobin42, P5343, S. nuclease44, T4 lysozyme44, IFN-λ14, UGT76G18, Rossmann fold29,30, TIM barrel31, helical repeat32, beta barrel33, jelly roll fold34,35, Maxwell et al. 28, Duan et al. 45, and Cianferoni et al. 49. Every AF3 model structure used for our test sets was generated from the official AlphaFold3 server (www.alphafoldserver.com)11. Source data are provided with this paper.
Code availability
The code for IFUM is publicly available on GitHub (github.com/HParklab/IFUM) or Google Colab (https://colab.research.google.com/drive/14TbHFp-BXfiv0vrCSNxyIlqMDOWX-8nV?usp=sharing).
References
Shoichet, B. K., Baase, W. A., Kuroki, R. & Matthews, B. W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA 92, 452–456 (1995).
Dill, K. A. Dominant forces in protein folding. Biochemistry 29, 7133–7155 (1990).
Taverna, D. M. & Goldstein, R. A. Why are proteins marginally stable? Proteins Struct. Funct. Bioinform. 46, 105–109 (2002).
Wang, Q., Goh, A. M., Howley, P. M. & Walters, K. J. Ubiquitin recognition by the DNA repair protein hHR23a. Biochemistry 42, 13529–13535 (2003).
He, F. et al. Myosin VI contains a compact structural motif that binds to ubiquitin chains. Cell Rep. 14, 2683–2694 (2016).
Hargrove, J. L. & Schmidt, F. H. The role of mRNA and protein stability in gene expression. FASEB J. 3, 2360–2370 (1989).
Gumulya, Y. et al. Engineering highly functional thermostable proteins using ancestral sequence reconstruction. Nat. Catal. 1, 878–888 (2018).
Go, S.-R., Lee, S.-J., Ahn, W.-C., Park, K.-H. & Woo, E.-J. Enhancing the thermostability and activity of glycosyltransferase UGT76G1 via computational design. Commun. Chem. 6, 265 (2023).
Lindorff-Larsen, K. & Teilum, K. Linking thermodynamics and measurements of protein stability. Protein Eng. Des. Sel. 34, gzab002 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Bennett, N. R. et al. Improving de novo protein binder design with deep learning. Nat. Commun. 14, 2625 (2023).
Yun, J. et al. Computational design and glycoengineering of interferon-lambda for nasal prophylaxis against respiratory viruses. Adv. Sci. 20, e06764 (2025).
Cagiada, M., Ovchinnikov, S. & Lindorff-Larsen, K. Predicting absolute protein folding stability using generative models. Protein Sci. 34, e5233 (2025).
Dieckhaus, H., Brocidiacono, M., Randolph, N. Z. & Kuhlman, B. Transfer learning to leverage larger datasets for improved prediction of protein stability changes. Proc. Natl. Acad. Sci. USA 121, e2314853121 (2024).
Diaz, D. J. et al. Stability oracle: a structure-based graph-transformer framework for identifying stabilizing mutations. Nat. Commun. 15, 6170 (2024).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017).
Chu, S. K. S., Narang, K. & Siegel, J. B. Protein stability prediction by fine-tuning a protein language model on a mega-scale dataset. PLoS Comput. Biol. 20, e1012248 (2024).
Tsuboyama, K. et al. Mega-scale experimental analysis of protein folding stability in biology and design. Nature 620, 434–444 (2023).
Flory, P. J. Statistical Mechanics of Chain Molecules (Interscience Publishers, 1969).
Alston, J. J., Ginell, G. M., Soranno, A. & Holehouse, A. S. The analytical flory random coil is a simple-to-use reference model for unfolded and disordered proteins. J. Phys. Chem. B 127, 4746–4760 (2023).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2022).
Hsu, C. et al. Learning inverse folding from millions of predicted structures. Proceedings of the 39th International Conference on Machine Learning (eds Chaudhuri, K. et al.) 8946–8970 (PMLR, 2022).
Aspromonte, M. C. et al. DisProt in 2024: improving function annotation of intrinsically disordered proteins. Nucleic Acids Res. 52, D434–D441 (2024).
Dawson, N. L. et al. CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 45, D289–D295 (2017).
Pancotti, C. et al. Predicting protein stability changes upon single-point mutation: a thorough comparison of the available tools on a new dataset. Brief Bioinform. 23, bbab555 (2022).
Maxwell, K. L. et al. Protein folding: Defining a ‘standard’ set of experimental conditions and a preliminary kinetic data set of two-state proteins. Protein Sci. 14, 602–616 (2005).
Koga, N. et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2012).
Koga, N. et al. Role of backbone strain in de novo design of complex α/β protein structures. Nat. Commun. 12, 1–12 (2021).
Listov, D. et al. Complete computational design of high-efficiency Kemp elimination enzymes. Nature 643, 1421–1427 (2025).
Brunette, T. J. et al. Exploring the repeat protein universe through computational protein design. Nature 528, 580–584 (2015).
Dou, J. et al. De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018).
Marcos, E. et al. De novo design of a non-local β-sheet protein with high stability and accuracy. Nat. Struct. Mol. Biol. 25, 1028–1034 (2018).
Marcos, E. et al. Principles for designing proteins with cavities formed by curved β sheets. Science 355, 201–206 (2017).
Ye, T. et al. Differential transformer. arXiv [cs.CL] https://doi.org/10.48550/ARXIV.2410.05258. (2024).
Ghosh, K. & Dill, K. A. Computing protein stabilities from their chain lengths. Proc Natl Acad Sci USA 106, 10649–10654 (2009).
Dieckhaus, H. & Kuhlman, B. Protein stability models fail to capture epistatic interactions of double point mutations. Protein Sci. 34, e70003 (2025).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Park, H. et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J. Chem. Theory Comput. 12, 6201–6212 (2016).
Frenz, B. et al. Prediction of protein mutational free energy: benchmark and sampling improvements increase classification accuracy. Front. Bioeng. Biotechnol. 8, 558247 (2020).
Kepp, K. P. Towards a ‘Golden Standard’ for computing globin stability: Stability and structure sensitivity of myoglobin mutants. Biochim. Biophys. Acta 1854, 1239–1248 (2015).
Pires, D. E. V., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 (2014).
Musil, M. et al. FireProt 2.0: web-based platform for the fully automated design of thermostable proteins. Brief Bioinform. 25, bbad425 (2023).
Duan, J., Lupyan, D. & Wang, L. Improving the accuracy of protein thermostability predictions for single point mutations. Biophys. J. 119, 115–127 (2020).
Josephson, K. et al. Design and analysis of an engineered human interleukin-10 monomer. J. Biol. Chem. 275, 13552–13557 (2000).
Huang, P. et al. Evaluating protein engineering thermostability prediction tools using an independently generated dataset. ACS Omega 5, 6487–6493 (2020).
Watson, M. D., Monroe, J. & Raleigh, D. P. Size-dependent relationships between protein stability and thermal unfolding temperature have important implications for analysis of protein energetics and high-throughput assays of protein-ligand interactions. J. Phys. Chem. B 122, 5278–5285 (2018).
Cianferoni, D. et al. Artificial intelligence and first-principle methods in protein redesign: a marriage of convenience? Protein Sci 34, e70210 (2025).
Hýskova, A., Maršálková, E. & Šimeček, P. Balancing speed and precision in protein folding: a comparison of AlphaFold2, ESMFold, and OmegaFold. In ICML 2025 Generative AI and Biology (GenBio) Workshop (2025).
Cao, L. et al. Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022).
Dauparas, J. et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
Cortajarena, A. L. et al. Non-random-coil behavior as a consequence of extensive PPII structure in the denatured state. J. Mol. Biol. 382, 203–212 (2008).
Cao, F., von Bülow, S., Tesei, G. & Lindorff-Larsen, K. A coarse-grained model for disordered and multi-domain proteins. Protein Sci. 33, e5172 (2024).
Kohn, J. E. et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA 101, 12491–12496 (2004).
Raccosta, S. et al. Scaling concepts in serpin polymer physics. Materials 14, 2577 (2021).
UniProt Consortium UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Acknowledgements
We thank Prof. Junseock Koh, Byung-hyun Bae, Seongin Jung, Soohong Min, and Jinyoung Byun for insightful discussions. This work was supported by the National Research Foundation of Korea (NRF) grant (No. 2022R1C1C1007817 for H.P.), Korea-US Collaborative Research Fund (KUCRF) funded by the Ministry of Science and ICT and Ministry of Health & Welfare (grant number RS-2024-00467483 for H.P.), the Korea Institute of Science and Technology (KIST) Institutional Program (No. 2E33791 for H.P.), and Scale-up TIPS funded by the Ministry of SMEs and Startups (RS-2024-00467110 for H.M.K.).
Author information
Authors and Affiliations
Contributions
H.L., H.M.K., and H.P. conceived and designed the study. H.L. and H.P. developed the deep learning model. H.L. curated datasets. M.S. reviewed the code. Y.C., J.Y., and H.M.K. conducted protein expression and Tm measurement. H.L., H.P., and H.M.K. wrote the manuscript. Y.C. and M.S. edited the manuscript. H.L. and M.S. worked on the Google Colab implementation. All authors contributed to the discussion of the results and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68637-4
