
Abstract
Hard ticks depend upon an ability to precisely and dynamically regulate their saliva to successfully evade host haemostatic and immune defences during extended blood feeding. Although pilocarpine, an exogenous muscarinic acetylcholine receptor (mAChR) agonist, can stimulate salivation experimentally, the endogenous control of saliva secretion by acetylcholine remains poorly understood. Here, we identify and characterise two pharmacologically distinct mAChRs (type A and B) in the genome of the medically important tick Ixodes ricinus. Molecular dynamics simulations and targeted mutagenesis reveal that type B mAChRs exhibit an atypical muscarinic profile, suggesting unconventional receptor signalling. Combining immunolabelling, in vivo pharmacology, and proteomics, we show that specific central neurons interact with distinct salivary gland regions via mAChR type-specific axons, coordinating fluid and protein secretion through separate acini and likely acting upstream of a neuropeptide-dependent cascade. This previously unrecognised mechanism of neural control offers new insights into how ticks modulate their saliva advancing our understanding of vector-host interactions, with potential implications for disrupting pathogen transmission.
Similar content being viewed by others


Introduction
Hard ticks can secrete saliva into vertebrate hosts over days or weeks, depending on tick species and developmental stage1. Factors within tick saliva prevent host blood coagulation, suppress host defences, remove excess fluids and ions, and thus maintain homeostasis2,3,4. Salivary molecules are critical in facilitating pathogen transmission and are produced by specialised cells in the salivary glands (SGs)2,5.
Female tick SG contains three acinus types (I, II, and III), each with distinct regulation and functions6,7,8,9,10. Salivation can be induced experimentally by catecholaminergic and cholinergic agents11,12,13,14, enabling the collection of large saliva volumes. While the role of dopamine is well established15,16,17, the physiological relevance of cholinomimetics remains unclear.
Currently, in ticks, the muscarinic agonist pilocarpine is known to evoke strong, sustained salivary secretion in vivo, accompanied by elevated activity in SG-innervating nerves11,18. This response can be blocked by atropine11, or by severing connections between the synganglion and SGs19, indicating that axonal mAChRs mediate saliva secretion via a non-cholinergic secretomotor mechanism20.
Here, we identified mAChRs in the axons of peptidergic neurons projecting to type II and III SG acini21,22,23, revealing the long-sought cholinoceptive pathway4,20 controlling tick salivation. We characterised two pharmacologically distinct mAChRs in I. ricinus using molecular dynamics, heterologous expression, and receptor-specific antibodies. We demonstrated in vivo that coordinated activation of both receptors is required for complete salivary secretion, including its dynamic and proteomic output. We also identified additional neurosecretory cells (NSC) expressing mAChRs, indicating broader cholinergic sensitivity within the synganglion. These findings reveal an integrated cholinergic control system in ticks, linking the synganglion and SGs to coordinate secretion.
Results
Ir-mAChR-A and -B act in an excitatory manner and exhibit distinct muscarinic profiles in heterologous systems
Genome and transcriptome searches identified one type A and one type B mAChR in hard ticks. Phylogenetic analyses placed Ir-mAChR-A as an ortholog to mammalian receptors, while Ir-mAChR-B clustered within the protostome-specific clade24 (Fig. 1a). The insect-specific type C receptor25 was absent in ticks.

a Circular cladogram showing the phylogenetic relationships of type A and type B mAChRs across the animal kingdom; tick taxa are highlighted (see Supplementary Dataset 1 for protein sequences). b Mean inward ion currents evoked by 0.5 µM drug stimulation of Ir-mAChR-A and -B expressed in Xenopus oocytes. For Ir-mAChR-A, n = 19, 14, 18, and 15 from six independent oocyte batches (N = 6); for Ir-mAChR-B, n = 8, 9, 8, and 8 from seven batches (N = 7). Multiple recordings were sometimes obtained from a single oocyte. The box plots show the median (central line), interquartile range (bounds of the box), and the full range from minimum to maximum values (whiskers). c Representative current traces recorded upon administration of various drugs. Dose-response curves of calcium mobilisation mediated via Ir-mAChR-B activation in CHO cells without (d) or with (e) Gα15(16) expression. f Dose-response curves of cAMP elevation via Ir-mAChR-B activation in HEK cells. Heat map showing agonistic (g) and antagonistic (h) activity of cholinergic agents on Ir-mAChR-A and -B in CHO/Gα15(16) aequorin assay. Data in (d–f) are from three independent transfections (N = 3); (g, h) show means of two biological replicates (N = 2), normalised to the ACh-evoked response (100%) (Supplementary Fig. 1g). Statistics: (d–f) nonlinear regression. Data are mean ± SD. Source data are available in the Source Data file.
In Xenopus oocytes, 0.5 µM acetylcholine (ACh) induced inward currents via both Ir-mAChR-A (−4.62 ± 3.8 µA) and Ir-mAChR-B (−0.63 ± −0.27 µA) (Fig. 1b, c). Muscarine produced similar responses (−2.79 ± 2.12 µA and 0.16 ± 0.09 µA, respectively), confirming both receptors as common agonist targets. By contrast, the mammalian non-subtype-selective muscarinic agonists methacholine and oxotremorine activated Ir-mAChR-A (−3.69 ± 2.86 µA and −4.15 ± 4.20 µA)26,27 but not Ir-mAChR-B.
We previously showed that Ir-mAChR-A couples to the Gq/11 family of proteins in Chinese hamster ovary (CHO) cells, whereas Ir-mAChR-B could not be functionally expressed8. Here, codon optimisation with a Kozak sequence enabled Ir-mAChR-B expression in CHO cells, allowing analysis with aequorin and GloSensor™ assays (Fig. 1d–f). Aequorin assays revealed robust ACh- (EC50 0.68 μM) and muscarine- (EC50 6.6 μM) induced calcium mobilisation consistent with Gq/11 activation (Fig. 1d and Supplementary Fig. 1a). Co-expression of Ir-mAChR-B with human Gα15(16) enhanced sensitivity ∼4.6 times for ACh (EC50 0.15 μM) and ∼2.3 for muscarine (EC50 2.1 μM) (Fig. 1e and Supplementary Fig. 1b). GloSensor assays in human embryonic kidney (HEK293) cells further showed that Ir-mAChR-B couples to the Gs pathway to elevate cAMP (ACh, EC50 2.7 μM) (Fig. 1f and Supplementary Fig. 1c), while also mediating partial inhibition of forskolin-stimulated cAMP (Supplementary Fig. 1d–f).
A screen of 37 cholinergic drugs with known affinity for mammalian mAChR subtypes (M1–M5), revealed common activators and blockers for both Ir-mAChR types, but type-specific ligands only for Ir-mAChR-A (Fig. 1g, h). Notably, pilocarpine selectively activated Ir-mAChR-A while the archetypal anticholinergic drug, atropine, inhibited only Ir-mAChR-A, consistent with reports that atropine antagonises type A mAChRs8,28,29—including all five mammalian mAChR subtypes30—but shows no28,29,31 or little32 antagonistic effects on invertebrate-specific type B muscarinic receptors.
Active-site mutations in Ir-mAChR-A and -B destabilise receptor conformations and alter their interactions with atropine
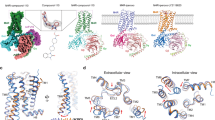
Both Ir-mAChR types share the conserved G protein-coupled receptor (GPCR) seven transmembrane (TM) fold and are structurally similar to the human M1 receptor (Hs-M1, PDB 6WJC)33. Their N-terminal extracellular regions are disordered, with Hs-M1 shorter than Ir-mAChR-A, and considerably shorter than Ir-mAChR-B (Supplementary Fig. 2). Amino termini are critical for both GPCR export from the Golgi and membrane integration. Specifically, a conserved Y–S pair proximal to TM134 is present in Ir-mAChR-A (Y42-S43) but absent in Ir-mAChR-B, which instead carries a W77–Q78 pair conserved in Hs-M1 (W23-Q24, Supplementary Fig. 3a).
Comparison with the Hs-M1 active site that binds atropine33, showed full conservation in Ir-mAChR-A but distinct substitutions in Ir-mAChR-B (Fig. 2a–c). To test their effects in silico, the four functionally divergent Ir-mAChR-B residues were individually introduced into the Hs-M1 model, and atropine coordination was analysed by classical molecular dynamics simulations (Fig. 2d). Simulations revealed that the most divergent substitution, A193I, caused the largest deviation of atropine (4.1 ± 0.6 Å, vs. 2.0 ± 0.4 Å in Hs-M1 WT). The Y106H substitution also induced large fluctuations (3.5 ± 0.3 Å). The remaining substitutions, S109C (3.1 ± 0.4 Å) and N382H (2.6 ± 0.5 Å), likewise produced higher deviations than the Hs-M1 WT (Fig. 2d). After ~300 ns, A193I and Y106H showed opposite shifts in atropine coordination.

a Structural alignment of the resolved Hs-M1 (grey), and predicted Ir-mAChR-A (black) and -B (cyan) structures showing atropine (spheres: red, oxygen; blue, nitrogen; green, carbon) and key binding residues (sticks). Upper inset: sequence alignment of Hs-M1 atropine-binding residues with Ir-mAChR-A and -B (asterisk, identical; colon, strongly similar; period, weakly similar). Lower inset: Hs-M1 atropine-binding residues (grey) indicating substitutions between Ir-mAChR-A/Hs-M1 and -B (black). b A bird’s-eye view (90° rotation of a) of superimposed Hs-M1, Ir-mAChR-A and -B showing residues coordinating atropine in the binding site. c Sequence alignment of human M1–M5 receptors and tick mAChR-A and -B highlighting residues in TM3, TM5, and TM6 that determine atropine selectivity. Species: Ir, I. ricinus; Is, I. scapularis; Rm, Rhipicephalus microplus; Rs, Rhipicephalus sanguineus; Da, Dermacentor andersoni; Ds, Dermacentor silvarum. Full sequences are provided in Supplementary Dataset 1. d 500-ns molecular dynamics trajectories showing RMSD of atropine (y-axis) across single Hs-M1, reflecting Ir-mAChR-B substitutions, and the Hs-M1Ir4B (e). WT, wild type. f Fragments of Ir-mAChR-A and -B sequences showing mutation sites (green and yellow, respectively) used to generate Ir-mAChR-A4B and -B4A mutant variants. g, h Aequorin assays comparing responses of WT and Ir-mAChR-A4B and -B4A to 45 μM agonists; ACh responses were normalised to (100%) in each of the three biological replicates. Effect of atropine pre-treatment on 30 μM muscarine- (i) and arecoline- (j) induced responses of WT and Ir-mAChR-A4B or -B4A in CHO/Gα15(16) aequorin assay. (g–j) Data are from three independent transfections (N = 3). Statistics: (i, j) nonlinear regression. Data are mean ± SD. Source data are available in the Source Data file.
Molecular dynamics simulations of the Hs-M1Ir4B variant, which incorporates all four Ir-mAChR-B–to–Hs-M1 substitutions (Fig. 2a–c) produced an average atropine deviation of 3.3 ± 0.3 Å after 15 ns compared to the Hs-M1 WT (2.1 ± 0.3 Å) (Fig. 2e). The Hs-M1Ir4B also produced structural changes in transmembrane helices and loops (Supplementary Fig. 3b). Residues 132–134 of intracellular loop 2 (IL2) showed the largest displacement, deviating up to 7.0 ± 0.6 Å after 200 ns vs 3.0 ± 0.5 Å in the Hs-M1 WT. By 450 ns, additional deviations appeared in residues 164–166 of TM4 and 179–193 of extracellular loop 2-TM5, which includes the A193I mutation (Supplementary Fig. 3b).
In silico analyses predicted that replacing the four conserved active-site residues of Ir-mAChR-A with those of Ir-mAChR-B (Y128H, S131C, A216I, and N510H) would destabilise atropine binding and reduce affinity, whereas introducing the reciprocal substitutions in Ir-mAChR-B (H160Y, C163S, I247A, and H765N) were predicted to enhance affinity. To test these predictions, we generated the corresponding constructs encoding Ir-mAChR-A4B and Ir-mAChR-B4A (Fig. 2f and Supplementary Fig. 2 and 3c), and compared their properties in CHO cells using nine cholinergic agonists at 45 μM (Fig. 2g, h), the maximal dose for Ir-mAChR-B in our assays (Fig. 1e). All ligands activated the Ir-mAChR-A WT, with similar potency to ACh (equivalent to 100%). Muscarine activation of the Ir-mAChR-A4B approximately doubled the response compared to ACh, whereas carbachol, arecoline, aceclidine, and methacholine elicited responses less than half of the ACh response (<50%), and pilocarpine and bethanechol were almost inactive (<10%) (Fig. 2g). In contrast, both muscarine and arecoline elicited activation of the Ir-mAChR-B4A with potency equal to or greater than that of ACh. Strikingly, methacholine and oxotremorine—previously selective for Ir-mAChR-A (Fig. 1b, g)—elicited responses in the Ir-mAChR-B4A nearly equivalent to ACh (~80–120%) (Fig. 2h).
We next examined dose-dependent inhibitory effects of atropine on WT and mutant Ir-mAChRs stimulated with muscarine or arecoline (Fig. 2i, j, and Supplementary Fig. 3d–h), the most potent activators of each mutant (Fig. 2g, h). In the Ir-mAChR-A4B mutant, atropine antagonism was completely abolished, whereas higher atropine doses paradoxically potentiated muscarine responses. In contrast, the reciprocal Ir-mAChR-B4A mutant enabled partial dose-dependent inhibition by atropine.
mAChR-A and -B expressing neurons and their axonal projections in the synganglion and associated nerve trunks
To investigate muscarinic regulation of neural pathways from the synganglion to the SGs11,19, we used I. ricinus type-specific antibodies to localise mAChR-A and -B in the synganglia of females (Fig. 3a–l). Both antibodies labelled distinct neuronal populations, some with axons projecting through different nerve trunks, while others remained confined within the synganglion.

a–l Immunolocalisation of Ir-mAChR-A and -B in neurons and axons of the female synganglion. Z-stack projections from dorsal (a, g), middle (b, h), and ventral (c, i) regions show lateral PcLNS1,2 and medial PcMNS1-5 NSC in the anterior protocerebrum, with their dorso-lateral surface axons (arrowheads). d Immunoreactivity (IR) in lateral nerve (LN) axons and the lateral segmental organ (LSO) of a three-day-fed female. e Lack of IR in the LSO of an unfed female, but positive labelling (arrows) in SG nerve 3 (SgN3) originating from OsSG1,2 neurons. f, l Schematic drawings of IR distribution (green); in panel l, only representative neurons are annotated. j IR detected in the axon (arrows) of SG nerve 1 (SgN1), a branch of the palpal nerve (PaN), from the PcSG neuron. k IR-positive axons (arrows) exiting the synganglion (S) via the ano-myosomal (An-myN) and genital (GenN) nerves. DAPI, blue. PgSh, periganglionic sheath; Pl-myN, postlatero-myosomal nerve; PspN, paraspiracular nerve. m Double labelling with anti-Ir-mAChR-A and -B in PcLNS1,2 and PcMNS1-5 NSC and their dorso-lateral axons. Open arrowheads, Ir-mAChR-A-positive axons (red); filled arrowheads, Ir-mAChR-B-positive axons (green). SG-innervating neurons OsSG1,2 and PcSG are indicated. n Double labelling with anti-Ir-mAChR-A and -B plus anti-MS or anti-FMRFa highlights PcMNS1-5 and PcLNS1,2 and their dorso-lateral axons. Filled arrowheads, PcMNS1-5 axons; open arrowheads indicate PcLNS1,2 axons; yellow, co-localisation. See schematics in (f) and (l) and Supplementary Fig. 6d. o TEM micrographs showing double immunogold labelling (see insets in n) in axons within the dorso-lateral perineurium (Pn, pink). Colour-coded arrows indicate IR. NL neurilemma (brown), Nb neuronal cell bodies (yellow). Supplementary Fig. 6a provides details. p SEM of a sagittal section of a 48 h-fed nymph; inset (right) shows TEM of a comparable region. A aorta, MG midgut, E oesophagus, SG salivary gland, Np neuropile, PgSh periganglionic sheath, PgS periganglionic sinus, Ax axon, Nb neuronal cell bodies. q Double labelling with anti-Ir-mAChR-A and anti-MS. Z-stack projection showing internal axonal networks of PcLNS1,2 (green; open arrowheads) and PcMNS1-5 (red; filled arrowheads), arborising into distinct lobes (see schematic in Fig. 4d). Validation of anti-Ir-mAChR-A and -B antibodies is shown in Supplementary Fig. 4a–c. For neuron nomenclature, see the “Methods” section. Scale bars: 50 µm (a–d, g–i, m, n, p, q); 10 µm (e, j, k insets of d, m and n); 2 µm (o); 1 µm (insets of o and p).
The cells with the strongest Ir-mAChR-A-positive reaction were protocerebral lateral NSC (PcLNS1,2), which formed dense axonal arborisation on the dorso-lateral synganglion surface (Fig. 3a, b, f), and opistosomal SG-innervating neurons (OsSG1,2) projecting to SG nerves 2–4 (Fig. 3a, f). Additionally, Ir-mAChR-A immunoreactivity was observed in lateral segmental organs in a three-day-fed female (Fig. 3d, f). In contrast, Ir-mAChR-B antibodies identified medial protocerebral NSC (PcMNS1-5) with arborisations on the dorso-lateral synganglion (Fig. 3h, i, l), as well as protocerebral SG-innervating neurons (PcSG) projecting to the SG nerve 1 (Fig. 3h, j, l). These data show that Ir-mAChR-A and -B are expressed in distinct neuronal populations, including NSC and SG-innervating neurons, pointing to type-specific muscarinic circuits in tick neural regulation.
Lateral and medial peptidergic NSC express distinct mAChR types
Double immunolabelling with Ir-mAChR-A and -B antibodies distinguished lateral and medial NSC, each extending prominent axons that arborise at the synganglion surface (Fig. 3m). In a Rhipicephalus appendiculatus study, antibodies against myosuppressin (MS) and FMRFamide neuropeptides recognised medial and lateral NSC22, corresponding to those identified here, thus confirming their peptidergic identity. Double immunolabelling of Ir-mAChR-A and -B with MS35 and FMRFamide36 antibodies verified the cholinoceptive nature of these NSC in I. ricinus (Fig. 3n and Supplementary Movie 1). Immunogold labelling revealed that axon terminals of PcLNS1,2 and PcMNS1-5 are positioned within the dorso-lateral perineurium (Fig. 3o), beneath the acellular neurilemma, which faces the haemolymph-filled periganglionic sinus (Fig. 3p). Other axons originating from these NSC extended towards internal synganglion lobes, arborising into fine terminals across medial and lateral regions (Fig. 3q). Because whole-mount confocal imaging lacks membrane resolution, the predominantly intracellular mAChR signal in axons—confirmed by ultrastructural analysis10—is interpreted as reflecting receptor trafficking. Most labelled profiles likely represent axonal shafts rather than synaptic terminals, where membrane receptors are concentrated.
In insects, both FMRFamide and MS belong to the FMRFamide-related peptide family, defined by a C-terminal RFamide sequence37,38. Hard ticks appear to encode a single ortholog that is structurally intermediate between insect FMRFamide and MS, with a C-terminal I/L-L/MHFamide motif, recently designated FMRFamide_MS-like (FMRFa_MS-L)39. We molecularly characterised the I. ricinus FMRFa_MS-L transcript and generated an antibody against one of its mature peptides (Supplementary Fig. 5a and 6b–d). Co-localisation of anti-FMRFa_MS-L and anti-Ir-mAChR-A in PcLNS1,2 cells and axons indicated that insect FMRFa antibody likely recognises FMRFa_MS-L in these neurons (Fig. 4a and Supplementary Fig. 6d). By contrast, the anti-MS antibody, raised against the silkworm MS N-terminal35, likely cross-reacts with an unknown protein in PcMNS1-5 cells (Fig. 3n, o, q and Supplementary Fig. 6c, d, f). Instead, the neuropeptide leucokinin40 was labelled in PcMNS1-5 cells and axons (Fig. 4b, c and Supplementary Fig. 5b and 6e). A schematic summary presenting NSC labelling patterns is shown in Fig. 4d.

a, b Double labelling shows co-localisation (yellow) of anti-Ir-mAChR-A with anti-FMRFa_MS-L in PcLNS1,2 neurons and of anti-Ir-mAChR-B with anti-leucokinin in PcMNS1-5 neurons, including their dorso-lateral axons (open and filled arrowheads, respectively). c Sagittal section of the synganglion immunolabelled with anti-leucokinin, showing the medial region at the PcMNS1-5 level and a deeper plane through the pedal lobes (Pd); arrowheads mark internal axonal projections from PcMNS1-5. Dotted lines outline Pd boundaries. d Schematic lateral view of the synganglion summarising medial (red) and lateral (green) NSC and their projections based on 3D confocal z-stacks and serial sections. Ch cheliceral lobe, Pa palpal lobe, Pd1–4 pedal lobes, Pc protocerebrum, Os opistosomal lobe, E oesophagus, MG midgut. IR immunoreactivity. Insets: structure of the dorso-lateral surface showing multiple axon terminals (green, red, grey) within the perineurium (Pn). PgSh, periganglionic sheath; PgS, periganglionic sinus; NL, neurilemma; Nb, neuronal cell bodies. e TEM micrographs showing double immunogold labelling with anti-Ir-mAChR-A (15 nm gold, blue) and anti-FMRFa (10 nm gold, red) in the dorso-lateral perineurium; inset shows contrast-enhanced detail. Schematic illustrates labelled axons (1–10), perineurium (11, 12), and the region of neuronal cell bodies (13). f Quantification of immunogold-labelled nanoparticles from (e). Bars represent Ir-mAChR-A (blue) and FMRFa (red) labelling. The raw counting data are provided in the Source Data file. g Quantification of fluorescence intensities in dorso-lateral axonal arborisation from PcMNS1-5 and PcLNS1,2 cells labelled with anti-leucokinin and anti-FMRFa, respectively, in uninjected, water-, or ACh-injected unfed females. Methodological details are in Supplementary Fig. 7. Anti-leucokinin and anti-FMRFa intensities: N = 6, 5, 4, 6, 5 synganglia; for anti-leucokinin, n = 12, 10, 8, 13, 11 lateral sides of synganglia or subdivisions; for anti-FMRFa, n = 12, 10, 12, 11 lateral sides or subdivisions (subdivisions used to avoid nonspecific signal). Only statistically significant comparisons (p < 0.05) are indicated. Validation of anti-Ir-FMRFa_MS-L and anti-leucokinin antibodies is shown in Supplementary Fig. 5. Statistics: g One-way ANOVA (two-sided) followed by Tukey’s multiple-comparison post hoc test. Data are mean ± SD. Source data are available in the Source Data file. Scale bars: 50 µm (a–c); 10 µm (insets in a, b); 1 µm (e).
We next examined PcLNS1,2 axon terminals and adjacent regions on the dorsal synganglion surface using double immunogold labelling with anti-Ir-mAChR-A and anti-FMRFa (Fig. 4e, f). FMRFa-labelled nanoparticles were abundant in the perineurium, but sparse near neuronal somata, suggesting axon-derived peptides are trafficked towards the neurilemma and then released into the haemolymph. Low levels of FMRFa-labelled nanoparticles were also observed in neighbouring axons that possibly originate from PcMNS1-5 or other neurons41, (Fig. 4e), which may reflect peptide transfer, uptake, or a technical artefact.
Finally, immunostaining suggested that cholinoceptive axons from PcLNS1,2 and PcMNS1-5 cells in the dorsal perineurium (summarised in Fig. 4d) may release neuropeptides upon ACh stimulation. To test this, we injected unfed female ticks with 100 nL water or 100 nL of 10 μM ACh and monitored fluorescence in axons double-labelled with anti-leucokinin (PcMNS1-5) and anti-FMRFa (PcLNS1,2) (Fig. 4g and Supplementary Fig. 7a–c). Water injection produced a gradual fluorescence increase over 30 min, whereas ACh elicited a significantly attenuated response. We interpret fluorescence increases as likely reflecting enhanced peptide synthesis and axonal transport triggered by haemolymph pressure, whereas the reduction following ACh injection may reflect mAChR-dependent neuropeptide release. These findings indicate that PcLNS1,2 and PcMNS1-5 axons release neuropeptides into the haemolymph via mAChR-mediated mechanisms, linking cholinergic signalling to systemic endocrine and osmoregulatory processes.
Peptidergic axons in type II and III SG acini express distinct mAChR types, whereas epithelial mAChRs are localised to type I acini
Tracing immunoreactive axonal projections from the synganglion to SGs revealed two pathways: (i) Ir-mAChR-A–positive axons from OsSG1,2 neurons exclusively innervating type II acini (Fig. 5a); and (ii) Ir-mAChR-B–positive axons from PcSG neurons targeting both type II and III acini (Fig. 5b). The peptidergic identity of these neurons and projections21,23 was confirmed by double labelling with antibodies against the insect neuropeptides orcokinin42 and SIFamide43 (Fig. 5a, b). SIFa-positive axons also reacted with anti-FMRFa and anti-FMRFa_MS-L antibodies, likely reflecting cross-reactivity at the conserved C-terminal Phe-NH₂ motif (Supplementary Fig. 5a and 8a–c), though this requires further validation.

Double immunolabelling of anti-Ir-mAChR-A with anti-orcokinin (a) and anti-Ir-mAChR-B with anti-SIFa (b) in synganglial neurons and their axonal projections to the SG of an unfed female. Co-localisation of anti-Ir-mAChR-A with anti-orcokinin marks OsSG1,2 neurons (yellow) and their axon terminals (yellow, arrowheads) targeting type II acini, while anti-Ir-mAChR-B co-localises with anti-SIFa in PcSG cells (yellow) and their axon terminals (yellow, arrowheads) innervating type II and III acini. c Double immunolabelling of SG acini from an unfed female with anti-Ir-mAChR-A (red, open arrowheads) and anti-Ir-mAChR-B (green, filled arrowheads) showing that type II acini receive both inputs, whereas type III acini are labelled exclusively with anti-Ir-mAChR-B. d Association of anti-Ir-mAChR-A- and anti-Ir-mAChR-B-positive axon terminals (green, arrowheads) with a single myoepithelial cell visualised with anti–βIII-tubulin (red, arrows) across feeding stages, showing acinar enlargement from unfed to fully engorged females. e Number of female SGs positive for anti-Ir-mAChR-A and anti-Ir-mAChR-B immunoreactivity in axon terminals in type II and III acini. Tick numbers (n) are shown in the figure. f Double labelling of type I acini with anti-Ir-mAChR-A or anti-Ir-mAChR-B with anti-ChAT shows scattered Ir-mAChR-type-positive immunoreactivity (green, arrowheads) in acinar cells adjacent to ChAT-positive axon terminals (red, arrows). DAPI, blue. A schematic of SG-innervating neurons and their axons is shown in Fig. 6a, b. Validation of anti-Ir-mAChR-A and anti-Ir-mAChR-B antibodies is provided in Supplementary Fig. 4. Scale bars, 50 µm.
Immunostaining showed that type II acini are the only site where Ir-mAChR-A and -B expressing axons form close synaptic varicosities (Fig. 5c; summarised in Fig. 6a, b). Within type II acini, Ir-mAChR-A terminals extended apically, while Ir-mAChR-B terminals remained basally restricted (Supplementary Fig. 8d, e). Examination across feeding stages (Fig. 5d) revealed more persistent Ir-mAChR-B immunoreactivity in type II and III acini, compared with less consistent Ir-mAChR-A labelling, which was confined to type II acini (Fig. 5e). Consistent with previous findings10 and confirmed here (Fig. 5d and Supplementary Fig. 8d), anti–βIII-tubulin staining demonstrated that axon terminals from PcSG or OsSG1,2 neurons made close contact with a single myoepithelial cell in both type II and III acini. These data indicate that Ir-mAChR-A and -B axons converge in type II acini but segregate spatially, with Ir-mAChR-B demonstrating broader persistence across acini and feeding stages, pointing to complementary receptor-type–specific roles in regulating myoepithelial cell function and secretion dynamics.

a Schematic overview depicting muscarinic cholinoceptive NSC, cholinoceptive neurons innervating type II and III acini, and cholinergic neurons innervating type I acini (unfed female). SgN1-5, salivary nerves; PaN, palpal nerve; LN, lateral nerve; LSO, lateral segmental organ; OsN, opistosomal nerve; MD, midgut. Previously reported immunoreactivity (IR) for pigment dispersing factor (PDF), myoinhibitory peptide (MIP), elevenin and orcokinin neuropeptides in axons entering acini is indicated21,22,88. b Simplified diagram highlighting major muscarinic cholinoceptive neurons—including NSC and SG-innervating neurons—and cholinergic neurons projecting to the SG (5 day-fed female). PcMNS and PcLNS neurons extend axons toward the haemocoel-facing surface, while additional projections arborise internally and may contact or modulate SG-innervating axons. Inset: magnified view of synaptic varicosities within a type II acinus where neuropeptides accumulate. Arrows indicate saliva flow direction in the main duct. mAChR-A, -B and ChAT-expressing neurons are colour-coded (A - green, B - red and ChAT - blue). (a) and (b) include 3D schematics of individual acini adapted and modified from Šimo et al.16 and Mateos-Hernández et al.8, reused under the Creative Commons Attribution 4.0 International License (CC-BY 4.0; https://creativecommons.org/licenses/by/4.0/). Quantification of free choline and ACh in SGs (c) and synganglia (d) from unfed, 1–6-day-fed, and fully engorged (FE) females. Negative ACh values indicate undetectable levels, after subtracting free choline from total choline (free choline + ACh). The inset in (c) presents a dot plot of free choline and ACh levels in type I and type II/III acini isolated from a five-day-fed female. Because tissue is inevitably lost during acini separation, these measurements are not directly proportional to overall SG content. e Relative mRNA levels of chat, vacht, machr-a and -b in SG and synganglion from unfed, 1-, 3-, and 5-day-fed, and FE females. Expression in unfed ticks was normalised to 1; p-values above bars indicate significant differences versus the unfed stage. Due to high variability among biological replicates, most comparisons in (c–e) were not statistically significant (p ≥ 0.05). c–e Each data point represents a pooled organ sample from multiple females, obtained from three independent biological replicates (N = 3) for all unfed and fed stages, except D5 in some subpanels of (e), which includes four replicates (N = 4). Statistics: c–e two-tailed Student’s unpaired t-test. Data are mean ± SD. Source data are provided in the Source Data file.
Type I acini, which have not been shown to produce saliva7, were previously identified as targets of synaptic cholinergic signals8. In this study, both anti-Ir-mAChR-A and -B antibodies labelled type I acini epithelial cells positioned adjacent to cholinergic axon terminals as detected with an antibody against choline acetyltransferase (ChAT, Fig. 5f and Supplementary Fig. 4d). Thus, Ir-mAChR-A and -B are also expressed in type I acini epithelia, suggesting neuronal cholinergic modulation of non-secretory acini.
SGs are a major pool of non-neuronal ACh
To investigate whether cholinoceptive axon terminals in saliva-producing type II and III acini (Figs. 5a–e and 6a, b) are exposed to paracrine ACh signalling, we quantified free choline and ACh in SGs during feeding (Fig. 6c). SGs contained variable free choline concentrations in the low nanogram range across feeding stages. ACh displayed greater variability, with detectable increases on days 3 and 6 of feeding. Free choline was detected exclusively in type II and III acini across three independent replicates, while all measurements in type I acini were below the detection limit (Fig. 6c), suggesting an epithelial origin rather than release from cholinergic axons innervating type I acini (Figs. 5f and 6a, b). In the synganglion, both free choline and ACh were present at all timepoints, though at consistently lower levels than in the SGs and with variable ACh detection among replicates (Fig. 6d).
Additional evidence for endogenous ACh synthesis in I. ricinus SGs comes from the transcripts encoding ChAT, which mediates ACh biosynthesis, and vesicular ACh transporter (VAChT, which packages ACh into secretory vesicles) (Fig. 6e and Supplementary Fig. 9). Given the absence of anterograde vacht and chat mRNA transport in cholinergic axons innervating type I acini8, their detection in SG tissue suggests local synthesis. However, despite extensive attempts, the ChAT protein was unable to be visualised in SGs by immunolabelling of whole-mount or sectioned tissues at any feeding stage, leaving the precise identity of ACh-producing cells in type II and III acini unresolved. machr-a and machr-b transcripts were detected at low levels in SGs (Ct >36 and 39, respectively, Fig. 6e), likely reflecting expression in epithelial cells of the less abundant type I acini, which receive neuronal ACh input (Figs. 5f, 6a, b). In synganglia, chat, vacht, and machr-a were consistently detected at all timepoints, whereas the machr-b transcript was barely detectable throughout feeding (Fig. 6e).
These data support local ACh synthesis in the SGs, where it may act in a paracrine manner to modulate neuropeptide release from cholinoceptive axon terminals. ACh injection into unfed females did not alter anti-SIFa fluorescence in Ir-mAChR-B-expressing axon terminals located within type II and III acini (Supplementary Fig. 10), suggesting that exogenous ACh either failed to reach the receptors or inhibited peptide release.
In vivo effects of muscarinic agonists and antagonists on SG secretion
To assess the physiological roles of axonal Ir-mAChR-A and -B in regulating saliva secretion, we injected pharmacological discriminators (Fig. 1g, h) into five-day-fed I. ricinus females (Fig. 7a). These included common agonists (ACh, muscarine), Ir-mAChR-A-selective agonists (methacholine, pilocarpine), Ir-mAChR-A-selective antagonists (scopolamine, atropine), or a broadly acting antagonist (propiverine) (Fig. 7b–j). Using a previously established in vivo dose of ~500 µmol·kg−1 of body weight in a pilot experiment11, ACh and pilocarpine produced no or only weak secretion. Increasing the dose tenfold to 5 mmol·kg−1 of body weight elicited robust secretion with both compounds (Supplementary Fig. 11a, b). These results established a dosing framework for testing muscarinic agonists and antagonists in vivo.

a Experimental setup showing the anterior part of a partially fed I. ricinus female salivating into a glass capillary attached to the hypostome. The arrow indicates the saliva level. b–e Cumulative salivary secretion rates over 60 min following injection of muscarinic agonists or 5-min pre-treatment with a muscarinic antagonist followed by agonist injection. n values indicate individual female ticks analysed: b n = 14, 9, 8, 8; c n = 9, 8, 8, 8; d n = 10, 8, 8, 8; and e n = 8, 9, 8, 8. f–j Truncated violin plots (solid line: median; dotted line: quartiles; bounds: data limits) showing total saliva volume secreted over 60 min by females treated with the indicated compounds. Each dot, square, diamond, or triangle represents one tick (see also Supplementary Fig. 11c). k Eight muscarinic drug combinations used to induce saliva secretion were analysed for protein content (l) and composition (m, n). Conditions 1–6 correspond to treatments shown in (b–e); conditions 7 and 8 (asterisk) refer to additional treatments shown in Supplementary Fig. 11d, e. l Comparison of protein content in saliva samples 1–6 collected from induced salivation experiments (k). m Principal component (PC) analysis of saliva from 5-day-fed females, showing clustering by treatment condition; colours and numbers correspond to those in (k). n UpSet plot showing protein intersections among the eight experimental conditions (see also Supplementary Fig. 12 and Supplementary Dataset 2). l–m Saliva analyses were performed in three technical replicates (n = 3). Statistics: b–e, l data are mean ± SD; f–j and I one-way ANOVA (two-sided) with Tukey’s post hoc test for multiple comparisons; p-values shown above the bars. Source data are provided in the Source Data file.
Injection of ACh and muscarine elicited immediate secretion, where ACh-stimulated activity plateaued within 30 min, whereas muscarine resulted in continuously increased secretion throughout the 60-min test period (Fig. 7b, c). Pilocarpine and methacholine also triggered rapid responses that plateaued at 30 min, with reduced secretion for the next 30 min (Fig. 7d, e). Notably, pilocarpine—widely used to induce tick salivation11—produced only ~50% of the maximal saliva volume induced by ACh, muscarine, or methacholine (Fig. 7b–f; Supplementary Fig. 11c). The Ir-mAChR-A-selective antagonist scopolamine partially inhibited ACh- and muscarine-stimulated secretion ( ~ 60%) (Fig. 7b, c, g, h), while virtually abolishing pilocarpine- and methacholine-induced responses (Fig. 7d, e, i, j). This suggests that any residual secretion under scopolamine/ACh or scopolamine/muscarine conditions reflects Ir-mAChR-B activity. Atropine fully blocked responses to all agonists, while propiverine nearly abolished ACh-, muscarine-, and pilocarpine-induced secretion, but demonstrated less effective inhibition of methacholine-induced responses (Fig. 7b–e, g–j). Together, these results demonstrate that Ir-mAChR-A and -B contribute to salivation with distinct effects on secretion volume and rate.
Notably, injection of atropine or propiverine—but not scopolamine—caused paralysis-like symptoms, including stretching and shaking of the forelegs, and reduced overall leg movement. Whether these effects reflect toxicity at the administered doses, and whether this influenced salivary secretion, remains unclear.
Impact of mAChR-A and -B activity on saliva protein quantity and composition
Saliva was collected under four pharmacological conditions designed to mimic or inhibit Ir-mAChR-mediated responses (Fig. 7k). Protein content was quantified for three of these conditions, and all four were analysed by principal component analysis (PCA) (Fig. 7l, m).
Saliva induced by ACh or muscarine contained comparable protein concentrations (~0.7 and 0.5 μg/μL, respectively). Pilocarpine-induced saliva exhibited higher protein levels (~1 μg/μL), whereas methacholine-induced saliva showed markedly lower protein (~0.26 μg/μL). Strikingly, scopolamine pre-treatment followed by ACh or muscarine injection resulted in significantly elevated protein concentrations (~1.5 and ~2 μg/μL) compared with controls without scopolamine treatment (Fig. 7l).
To assess the impact of receptor-type–specific activity on the saliva proteome, we performed PCA of 218 proteins, including 37 host-derived proteins (Fig. 7m, see also Supplementary Dataset 2). Saliva induced by each drug formed distinct clusters. ACh- and muscarine-induced samples did not clearly separate from pilocarpine- and methacholine-induced samples, which overlapped near the origin. In contrast, ACh- and muscarine-induced saliva following scopolamine or propiverine pre-treatment showed clear separation from all other groups. Muscarine alone separated from ACh along PC2, and for both agonists, this divergence became more pronounced after scopolamine pre-treatment. Protein identities varied across conditions (Fig. 7k, n and Supplementary Fig. 12 and Supplementary Dataset 2). Unique sets were detected in saliva induced by ACh (17 proteins), methacholine (15), and pilocarpine (9). In scopolamine-pre-treated ticks, subsequent ACh and muscarine injection yielded 15 and 9 unique proteins, respectively. These findings suggest that muscarinic inputs do not generate stimulus-specific secretomes, but rather shift the relative abundance of salivary proteins, reflecting flexible regulation across acinar cell types. Importantly, abundant host proteins, including haemoglobin, albumin, and serum transferrin, and immune effectors such as neutrophilic granule protein and protein S100, were consistently detected across conditions (Supplementary Dataset 2). Their recurrent presence indicates that vertebrate haemostatic and immune factors are not contaminants but integral components of the saliva proteome, underscoring their dynamic role at the host-parasite interface.
Discussion
Functional and pharmacological studies on invertebrate mAChRs progress rather slowly compared to research on their mammalian counterparts. As yet, only a few studies have investigated these receptors in nematodes and arthropods, but their specific physiological functions in invertebrates remain largely unknown. Central to our findings is the anatomico-physiological model of two distinct axonal mAChR types (A and B) mediating salivary secretion in I. ricinus females. In addition to their sequence, the selective affinity of Ir-mAChR-A and -B to a number of classical muscarinic drugs supports a recent classification that all deuterostome mAChRs—including mammalian M1–M5—belong to the type A group, while in protostomes, additional pharmacologically distinct type B mAChRs are present24,29.
Ir-mAChR-A and Ir-mAChR-B induced ionic currents of different amplitudes in Xenopus oocytes, but the identity of the ion channels and the mechanisms linking receptor activation to channel opening remain unknown. These differences likely reflect intrinsic receptor properties, such as ligand-binding affinity and G protein coupling efficiency, assessed within a simplified oocyte membrane environment. In contrast, similar Ir-mAChR-B (this study) and Ir-mAChR-A (in Mateos-Hernández et al.8) EC₅₀ values measured in CHO cells are influenced by endogenous signalling pathways, membrane composition, and/or regulatory proteins, which buffer intrinsic receptor differences. Together, these results highlight the need to further investigate how mAChRs and other GPCRs couple to those ion channels playing key roles in tick SG physiology44,45.
Gq/11 pathway activation by Ir-mAChR-B in CHO cells mirrors signalling previously described for GAR-1 and GAR-2 receptors in the T. spiralis nematode29. cAMP production was also stimulated in HEK cells, indicating engagement of a second distinct cascade; however, parallel assays revealed cAMP inhibition at lower ligand concentrations, consistent with concentration-dependent bidirectional regulation of adenylate cyclase. These findings challenge the prevailing view that all mAChR-B receptors couple exclusively to Gi proteins and act solely as inhibitory receptors24. Indeed, previous studies have reported that Gi or Gq/11-coupled mAChRs may, under certain conditions, activate adenylyl cyclase and elevate cAMP46, and that varying ligand concentrations can bias between Gs and Gi in other GPCR systems47. These observations underscore the need for detailed analyses of mAChR-B signalling under different physiological contexts, which is essential to defining the full functional repertoire of these receptors.
Immunolabelling revealed an extensive network of cholinoceptive peptidergic axon terminals at the latero-dorsal synganglion perineurium (summarised in Fig. 4d). Although neurosecretory axons at this region were described in the 1980s48, our study is the first to trace their origin to lateral and medial NSC. By morphology and location, these cells likely correspond to insect protocerebral NSC, which are also cholinoceptive49. In ticks, the barrierless perineurium/neurilemma complex may allow direct exposure of these axons to hormonal ACh. In turn, their activation could release neuropeptide hormones into the haemolymph, and/or initiate signalling cascades through internal peptidergic axons across synganglion lobes. Additional support for a hormonal ACh pathway comes from Ir-mAChR-A detection on haemolymph-surrounded lateral segmental organs, although the physiological relevance of this system remains unclear. We speculate that protocerebral cholinoceptive NSC in tick are part of the previously suggested sensory pathway20, responsive to haemolymph volume and acting directly or indirectly on SG-innervating neurons. Furthermore, nicotinic acetylcholine receptors (nAChRs) are expressed in synganglion50,51, but their precise localisation and role in tick neurophysiology remain unresolved.
Our data support the existence of the long-postulated cholinoceptive secretomotor nerves that extend to the SG4,20. Unlike in vertebrates, neuron-neuron communication rarely occurs at the level of cell somata in insects52, instead taking place along axons or their terminals—a pattern that likely also applies to ticks. However, our prior patch-clamp study detected ACh-, muscarine-, and nicotine-sensitive sites at the somata of neurons resembling PcSG cells23,53. Therefore, we suggest that cholinergic signal clusters may diffuse to the somata of these SG-innervating neurons20 or act directly on their axo-axonic contacts within or near the synganglion to mediate their activation. The presence of mAChR-A and -B at peptidergic axon terminals in SG acini further support their role as presynaptic heteroreceptors, capable of modulating neuropeptide release as shown in mammals54. In this case, ACh signals would likely originate from adjacent SG cells. Although we confirmed substantial ACh levels in I. ricinus SGs, we were unable to localise cholinergic cells, possibly due to rapid ChAT protein turnover55 consistent with scattered ACh signal and variable chat and vacht transcript levels across individuals. To date, no evidence supports expression of nAChRs in tick SG50. Thus, mAChRs remain the primary candidates for mediating cholinergic signalling in this tissue.
The reported inability of ACh to stimulate fluid secretion in hard ticks11 likely reflects the long-standing reliance on the exogenous cholinomimetic pilocarpine. In contrast, we found that injection of ACh or muscarine triggered robust secretion in I. ricinus, with approximately twice the efficacy of pilocarpine. Our pharmacological data suggest that pilocarpine’s reduced efficacy arises from its inability to activate Ir-mAChR-B and from its partial agonism at Ir-mAChR-A (this study and Mateos-Hernández et al.8). By comparison, methacholine—a potent Ir-mAChR-A–specific agonist—induced secretion volumes similar to those evoked by ACh and muscarine.
In 1982, Kaufman and Harris19 demonstrated that severing lateral SG-innervating nerves 2 and 3 in Amblyomma hebrareum reduced carbachol-induced salivation, whereas severing the palpal nerve caused only a minor reduction in saliva volume. At the time, however, the receptor specificity of carbachol action was unknown. Here, we show that carbachol activates both Ir-mAChR types, acting as a full agonist at Ir-mAChR-A and a partial agonist at Ir-mAChR-B. Consistent with this mechanistic insight in I. ricinus, selective stimulation of mAChR-A immunolocalised to axon terminals innervating type II acini via SG nerves 2–4, triggered robust secretion, with saliva volumes comparable to those observed when both receptor types were activated. In contrast, blocking mAChR-A and stimulating salivation with a common agonist markedly reduced secretion, revealing a strong dependence on mAChR-A signalling. Under these conditions, secretion would therefore be mediated solely by mAChR-B, which we detected in an axonal branch of the palpal nerve projecting to type II and III acini. Together, these results suggest a predominant role for type A mAChR in driving fluid secretion.
Our model of axonal mAChR type-specific regulation of tick SG secretion aligns closely with Kaufman and Harris’ findings19, and highlights the need to dissect how mAChR-A signalling via OsSG1,2 neurons that innervate type II acini drives robust fluid secretion. Although mAChR-A stimulation elicits high saliva volumes, the fluid likely originates from the large-lumen type III acini, because type II acini—with their small lumens and high abundance of granular cells—are generally specialised for protein and molecule secretion6,7. The intermittent presence of mAChR-A protein in type II axon terminals further suggests that it may act as a rate-limiting factor in cholinergic control of fluid output. In contrast, mAChR-B–positive axons innervate the basal regions of both type II and III acini, most evident during late feeding, suggesting readiness to regulate secretion and drive acinar emptying through myoepithelial cell contractions that also control the basal valve10. mAChR-A-positive axons also enter type II acini via the basal region, suggesting a role in valve regulation. However, unlike mAChR-B, mAChR-A axons extend further toward apical regions, suggesting additional functions such as specific cell targeting or modulating apical secretory mechanisms, consistent with earlier observations of neuropeptide-positive axonal projections10.
In this model, mAChR-A-positive axons may initiate secretion by stimulating type II acini and simultaneously transmit neuropeptide signals via axo-axonic synapses within the type II acini to adjacent mAChR-B-positive axons. Such interaction could coordinate activation of type II and type III acini, coupling protein release with fluid flushing to move contents into the main salivary duct (see Fig. 6b). Although mAChR-A stimulation alone produces saliva volumes comparable to combined mAChR-A and -B activation, the protein concentrations were significantly lower, suggesting that mAChR-A alone cannot generate the full saliva cocktail. In contrast, mAChR-B activity yielded saliva enriched in proteins, but with reduced fluid volume, likely reflecting a primary role in ejecting pre-accumulated material rather than shaping the protein composition of saliva. It remains unresolved whether proteins predominantly derive from type II acini and fluids from type III acini under these experimental conditions. It also remains unclear whether mAChR-A and -B act independently or are co-activated during feeding. Nonetheless, our findings suggest that constitutive mAChR-B activity enables full mAChR-A signalling and coordinated secretion of protein and fluid components, positioning mAChR-B as a permissive regulator of the complete secretory response.
Although saliva volume and protein concentration can be broadly associated with the activity of type II or III acini, we were unable to directly assign specific proteins to individual acinus types. Moreover, the distinct saliva proteomic profiles elicited by drugs that either activate or inhibit the same type of mAChR suggest that multiple, context-dependent modes of action are involved. Resolving these discrepancies will require acinus-specific proteomics in combination with other omics approaches to define the contribution of different structural subunits. In parallel, the roles of additional modulators such as neuropeptides, dopamine, or others9, and their potential interactions must be investigated to fully understand the dynamic regulation of tick SG secretion.
The localisation of both Ir-mAChR-A and -B to type acini I was expected, given the presence of cholinergic innervation previously reported for these structures8. This pathway has been implicated in tick rehydration through water uptake8, as well as in the resorption of ions via type I acini from hyperosmotic primary saliva produced by upstream type II and III acini56. Whether this ion resorption is also mediated by muscarinic signalling—via mAChR-A or -B—and whether it operates in synchrony with the secretory activities of type II and III acini, remain open questions.
Given the highly versatile functions of tick SGs2, their performance depends on the coordinated activity of three distinct acinus types, each under specialised neural control. Among these, two sets of peptidergic neurons9—expressing different types of mAChRs—govern saliva production through the innervation of hundreds of acini. Our study reveals how cholinergic signalling through these neurons orchestrates acinar output. Further, it offers new insights into the neural regulation of tick salivation with potential implications for future pharmacological and vector control strategies. In this context, the invertebrate-specific Ir-mAChR-B emerges as a promising candidate for selective intervention. More broadly, our study highlights the evolutionary distinctiveness of this cholinergic regulatory system among blood-feeding arthropods, underscoring its central role in enabling ticks to adapt to their specialised parasitic lifestyle.
Methods
Ticks and experimental animals
All tick feeding procedures followed established protocols57,58. Animals used in France were approved by the ComEth Anses/ENVA/UPEC Ethics Committee for Animal Experimentation (permit No. APAFIS #35511–2022022111197802 v2). In the Czech Republic, procedures complied with the Animal Protection Law of the Czech Republic No. 246/1992 Sb. (Regulation 419/2012) and ethics approval No. 13/2021-P. Animals were maintained ad libitum in an accredited facility (Ministry of Agriculture No. 4253/2019-MZE-17214 and 1643/2019-MZE-17214). I. ricinus adults were obtained from the tick rearing facility of UMR BIPAR (Maisons-Alfort, France) and the Institute of Parasitology (České Budějovice, Czech Republic). Ticks were maintained in 100 mL plastic vials with sterile wood shavings at 22 °C, 90% relative humidity, and 11 h/13 h light-dark cycle (PHCbi, Japan). For adult female feeding stages, up to 80 tick pairs were fed on 11–12-week-old New Zealand White female rabbits (Charles River). To obtain 5-day-fed females for in vivo salivation experiments, two adult pairs were placed on a 5–7-week-old Oncins France 1 female mouse (Charles River).
Phylogenetic analyses
BLAST searches using previously identified mAChRs from I. scapularis (XP_002403135.1, XP_002416160.3), D. melanogaster (AFJ23965.1, AGE13748.1), and GAR-1-3 from Trichinella spiralis (KRY43096.1, OR220883, KRY27749.1)8,28,29, were performed against various animal genomes, transcriptomes, and protein databases in NCBI (http://www.ncbi.nlm.nih.gov/). For phylogenetic analysis, the transmembrane regions of putative mAChRs were aligned using ClustalW in MEGA11, and a neighbour-joining tree was constructed with 500 bootstrap replications59. A circular phylogenetic tree was coloured and annotated using Adobe Photoshop 26.3.0 and Adobe Illustrator 29.3 (Adobe Inc.).
Oocyte injection and voltage-clamp recordings
Defolliculated Xenopus laevis oocytes were obtained from TEFOR Paris-Saclay University, France and incubated in standard oocyte saline (SOS: 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM HEPES pH 7.5) supplemented with penicillin (100 U·mL−1), streptomycin (100 mg·mL−1), gentamycin (50 mg·mL−1) and sodium pyruvate (2.5 mM)60,61. Oocytes were enzymatically treated with Ca2+-free SOS solution containing collagenase IA (2 mg·mL−1) and trypsin inhibitor (0.8 mg·mL−1) (Sigma, France) and manually defolliculated.
Oocytes were microinjected with 50.6 nL of cRNA (10–20 ng/oocyte) coding for Ir-mAChR-A or -B (GenBank: GIXL01002966.1 and PP921913) using a Nanoliter 2020 injector (World Precision Instrument). Recombinant pcDNA3.1+ plasmids containing Ir-mAChR-A or -B open reading frames8 were linearised with PmeI (New England Biolabs, USA), and cRNAs were transcribed in vitro using the T7 mMESSAGE mMACHINE kit (Ambion, USA). G protein-coupled inwardly rectifying potassium channel (GIRK)61 cRNA was not co-injected. Oocytes were maintained at 18 °C and recorded 3–9 days post-injection.
Electrophysiological recordings were performed with the Roboocyte 2 system (Multi Channel Systems, Germany). Membrane currents were recorded using two 3 M KCl-filled microelectrodes (0.2 and 5 MΩ)61. Oocytes were voltage-clamped at −60 mV and perfused with SOS at 3 mL.min−162. Drugs were applied for 10 s followed by 30 min washout63,64. Oocytes lacking Ir-mAChR-A or -B expression were excluded. Negative controls (0.5 µM ACh, muscarine, oxotremorine, or methacholine on non-injected oocytes) produced no inward currents (Supplementary Fig. 13a). For drugs used, see Supplementary Table 1.
Heterologous expression of Ir-mAChR-B in mammalian cell lines and functional assays
The full-length Ir-mAChR-B open reading frame (PP921913) was codon- optimised for expression in CHO cells and chemically synthesised (Biomatik, Canada). The sequence was inserted into a pcDNA3.1(+) vector (Invitrogen), with the GCCGCCACC Kozak consensus sequence preceding the start codon. The receptor was expressed in CHO-K1 cells (85051005, Sigma, France) together with the aequorin reporter65, with or without wild-type human Gα15(16) (cDNA Resource Center, Bloomsburg University of Pennsylvania), to monitor intracellular calcium mobilisation-induced bioluminescence upon receptor activation66. Bioluminescence was measured using Fluostar Omega (BMG Labtech) or Glomax Discover (Promega) microplate readers. Half maximum activation (EC50) values were calculated using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA).
To assess cAMP modulation upon Ir-mAChR-B stimulation, we used the non-lytic GloSensor™ cAMP assay system (Promega)16. HEK293 (85120602, Sigma, France) were co-transfected with CHO-codon-optimised Ir-mAChR-B in pcDNA3.1(+) plasmid and the GloSensor-expressing plasmid (Promega). For the activation assay, cells were directly exposed to varying ACh concentrations and luminescence was recorded. For the inhibition assay, cells were pre-treated for 5 min at RT with different ACh doses followed by 10 μM forskolin. Luminescence was recorded every ~1 min for 25 min, and values were normalised to the maximal response (at 15 min for inhibition and 5 min for activation) after removing background.
Mock transfections using only aequorin/Gα15(16) (calcium assay)8 and GloSensor (cAMP assay; Supplementary Fig. 13b) served as negative controls. Detailed transfection and culture procedures are described in Šimo et al.16.
Pharmacology of Ir-mAChR-A and -B
Agonistic and antagonistic effects of cholinergic agents on Ir-mAChR-A and -B were tested in CHO-K1 cells co-expressing each receptor with the aequorin reporter and Gα15(16). Ir-mAChR-A was expressed using the Ir-mAChR-A/pcDNA3/Zeo(+) construct from Mateos-Hernández et al.8, and Ir-mAChR-B using the Ir-mAChR-B/pcDNA3.1(+) construct described above.
For agonist assays, cells were exposed to 0.5 or 5 μM ligand, and luminescence was recorded immediately. For antagonist assay, cells were pre-treated with 5 or 50 μM antagonist for 5 min at RT, followed by 3 μM ACh with luminescence immediately recorded. For drugs used, see Supplementary Table 1.
Protein structure prediction and preparation
Transmembrane regions and loop orientations were predicted using DeepTMHMM67. I. ricinus mAChR protein models were generated in Phyre268 using intensive mode, which aligns the query to remote homologues. Disordered regions, including N-termini (unless stated) and IL3, were truncated. Predicted structures and the Hs-M1-atropine complex33 were prepared and optimised in Maestro69. Hydrogen atoms were replaced, steric clashes resolved by local minimisation, and hydrogen-bond networks optimised using the Protein Preparation Wizard70. In silico mutations of the Hs-M1-atropine complex33 were performed in Maestro69. The Hs-M1 was solvated in a 12 Å orthorhombic TIP3P water box71,72, neutralised and supplemented with 0.15 M NaCl for molecular dynamics preparation.
Classical molecular dynamics
Protein–ligand complexes and ions were parameterised using the CHARMM36 force field73, and atropine parameters were generated with SwissParam74. Simulations were performed on a GPU-accelerated workstation using Desmond75, which included multiple equilibration steps followed by a production run under isotropic NPT conditions, employing a Nose-Hoover thermostat76 and a Martyna-Tobias-Klein barostat77. Simulations were conducted at 300 K with a RESPA integrator78 and a 2-fs inner time step. Images were prepared in Maestro69, and trajectory analyses were performed using Visual Molecular Dynamics79.
Generation of Ir-mAChR-A and -B mutants and functional assays
Based on molecular dynamic simulations (see “Results” section: Active-site mutations in Ir-mAChR-A and -B destabilise receptor conformations and alter their interactions with atropine), mutant forms of Ir-mAChR-A and -B were manually designed (Supplementary Fig. 2). Sequences were codon-optimised for expression in CHO cells, chemically synthesised (Biomatik, Canada), and cloned into pcDNA3.1(+) (Invitrogen) with a Kozak sequence preceding the start codon. Functional assays were performed in CHO cells using the aequorin reporter and Gα15(16) as described in the “Methods” section (Pharmacology of Ir-mAChR-A and -B, Mateos-Hernández et al.8 and Ning et al.29). Dose-response curves were generated with GraphPad Prism 9 (GraphPad).
Whole-mount immunofluorescence and antibodies
Whole-mount staining followed previously established protocols for tick tissues22,80. Primary antibodies and dilutions were listed in Supplementary Table 2. Antibodies against Ir-mAChR-A or -B were generated in rabbits using carboxy-terminal peptide antigens conjugated to keyhole limpet haemocyanin, followed by affinity purification (Lifetein, USA). The affinity-purified anti-I. ricinus FMRFa_MS-L antibody was produced in guinea pigs using an antigen corresponding to the sixth mature peptide of the precursor protein (Leiftein, USA, Supplementary Fig. 6b, c). Secondary antibodies included goat anti-rabbit Alexa 488, goat anti-rabbit Alexa 594, goat anti-mouse Alexa 594 and goat anti-guinea pig Alexa 488 (Life Technologies) diluted 1:1000 in 0.5% Triton X-100 in PBS (PBST). Secondary antibodies (Life Technologies) included goat anti-rabbit Alexa 488, goat anti-rabbit Alexa 594, goat anti-mouse Alexa 594, and goat anti-guinea pig Alexa 488, all diluted 1:1000 in PBST. For double staining of Ir-mAChR-B with Ir-mAChR-A, SIFamide, or leucokinin (rabbit antisera), tissues were first incubated with the primary antibody, followed by goat anti-rabbit Alexa 594, then incubated overnight in PBST containing anti-Ir-mAChR-B directly labelled with NHS-fluorescein (ThermoFisher).
Negative controls (Supplementary Fig. 4, 5) included antibody pre-adsorption with 250 μg.mL−1 antigen for 3 h at RT or a replacement with pre-immune serum. Anti-leucokinin antibody40 (raised against cockroach leucokinin) was pre-adsorbed with I. ricinus leucokinin peptide (Synpeptide Co., Ltd, China). Immunocytochemistry of CHO cells transfected with ir-machr-a or -b compared to mock-transfected cells was used to verify Ir-mAChR-A and -B antibody specificity (Supplementary Fig. 4a, b). Stained tissue or cells were mounted in Prolong Antifade Diamond Mountant with DAPI (Life Technologies) and imaged using a Leica DMI8 confocal microscope (Leica Microsystems) with z-stacking. Z-stack composites were adjusted in Adobe Photoshop (Adobe Inc.).
Immunostaining of sectioned tissue
Immunohistochemical staining of sectioned I. ricinus nymphs followed a standard paraplast embedding protocol. Legs and posterior idiosoma were removed before fixation in Bouin’s solution for 3 h at RT. Samples were dehydrated and cleared through graded 70%, 96%, and 100% ethanol and chloroform, then embedded in melted paraplast (Leica, Biosystems). Sections (12 μm) were cut using a semi-automated Histocore Multicut microtome (Leica, Biosystems). Xylene-deparaplasted sections were rehydrated through graded 96% and 70% ethanol and distilled water, then blocked for 20 min with normal goat serum (Sigma, France). Sections were incubated overnight with anti-leucokinin (1:1000), followed by a 6 h incubation with HRP-labelled anti-rabbit antibody (New England Biolabs, Canada) at RT. Immunoreactivity was visualised using 3,3′-diaminobenzidine (Sigma, France) and H2O2 in Tris-HCl buffer (pH 7.6) under a dissection microscope. Images were acquired with a UC90 camera mounted on an Olympus BX53 microscope. Contrast and brightness were adjusted in Adobe Photoshop (Adobe Inc.).
Nomenclature of the neuropeptidergic cells
Neuronal nomenclature for I. ricinus synganglion followed Šimo et al.81. The first two letters indicate the neuron’s position within a specific lobe: protocerebral (Pc), pedal 1–4 (Pd1–4), or opisthosomal (Os). Subsequent letters indicate anatomical orientation: dorsal (D), ventral (V), anterior (A), posterior (P), medial (M), lateral (L), or interneurons (IN). Neurons projecting axons to the synganglion surface—a putative neurohaemal site—were designated as neurosecretory cells (NS). Neurons projecting axons into SG were designated as SG neurons. Numbers following the letter code indicate the neuron pair number.
Molecular identification of I. ricinus FMRFa_MS-L neuropeptide transcript
The putative I. ricinus FMFRa-L_MS prepropeptide (GenBank: GIXL01007324) was identified by BLAST search using the Rhipicephalus microplus FMFRa-L_MS prepropeptide (GenBank: XP_037272417.1) against the I. ricinus NCBI Bioproject PRJNA657487. A 1324 bp fragment was amplified using primers 5′-AAGATGGTGAAGTGATTAGGTA-3′ and 5′-ATTCGAAGACAGCTTCCTCGT-3′, cloned into the pGEM®-T Easy vector (Promega), and sequenced (Eurofins Scientific, France). The confirmed sequence was then used to design and generate an antibody against the precursor’s mature peptide 6.
Immunogold TEM
Synganglia from unfed female I. ricinus were fixed in 4% formaldehyde with 0.1% glutaraldehyde in 0.1 M HEPES for 1 h at RT. After washing in HEPES buffer, specimens were embedded into 15% gelatine, cryoprotected in 2.3 M sucrose for 72 h at 4 °C and frozen by plunging into liquid nitrogen. Ultrathin cryosections were cut at −100 °C, and collected using 1.15 M sucrose/1% methylcellulose (25 cp, Sigma). Sections were incubated 1 h at RT in 1% fish skin gelatin (FSG), then incubated with primary antibodies diluted 1:40 in FSG 1 h at RT, washed, then incubated 1 h with gold-conjugated secondary probes (BBI; 1:40 in FSG; see Supplementary Table 2). After HEPES washes, sections were post-fixed for 5 min in 1% glutaraldehyde diluted in 0.1 M HEPES, rinsed in distilled H2O, and contrasted/embedded with 2% methylcellulose and 3% aqueous uranyl acetate (9:1). Samples were examined using a JEOL 1400 TEM. Control sections were processed without the primary antibody. See Supplementary Table 2 for a list of antibodies used, their dilutions, and the corresponding secondary antibodies.
Electron tomography was used to localise immunogold nanoparticles in specimens double-labelled with antibodies against FMRFamide and mAChR-A. Anti-FMRFamide antibody was selected over anti-FMRFa_MS-L due to its specific recognition of PcLNS1,2 dorso-lateral axons, whereas the latter also labels surface-adjacent neurons (Figs. 4a, 3n and Supplementary Fig. 6d, f), potentially interfering with nanoparticle quantification near neuronal cell bodies. Tomography data were acquired by JEOL 2100 F TEM equipped with the Gatan K2 Summit direct electron detector controlled by SerialEM. Tilt series were collected from ±60° at 1° increments, with each image captured as a 2 × 2 map. Reconstruction and model generation were performed using Imodmop (IMOD package). Black-and-white (B/W) masks delineating individual cell areas were created by converting 3D model contours from tomograms using Imodmop (IMOD package). Coordinates of gold nanoparticle centres in reconstructed tomograms were detected using Findbeads3d (IMOD package). Nanoparticles within cell-specific B/W masks were counted in Matlab, and statistical distributions were generated. Detailed nanoparticle counts are provided in the Source Data.
Serial block-face scanning electron microscopy
I. ricinus nymphs were fed on mice for 48 h. After removing the dorsal cuticle, the body was fixed in 2.5% glutaraldehyde in phosphate buffer for 24 h at 4 °C. Samples were stained sequentially (RT unless stated otherwise) with 2% OsO4 for 2 h, 3% ferrocyanide for 2 h, 1% thiocarbohydrazide for 1.5 h, 2% OsO4 for 3 h, and 1% uranyl acetate overnight at 4 °C. After each staining step (except the first osmium staining), the samples were washed three times for 15 min. Samples were dehydrated in an acetone series and infiltrated with 33%, 50%, 66% acetone/resin mixtures for 2 h each, then infiltrated with 100% Hard Resin Plus 812 (EMS) overnight and polymerised at 62 °C for 48 h. Resin-embedded blocks were trimmed and imaged using MAPS software on an Apreo SEM with VolumeScope (Thermo Fisher Scientific). Serial images were acquired at 3 keV, 1.6 nA, 50 Pa with 20 nm resolution, 140 nm slice thickness, and 1 µs dwell time per pixel. Image data were processed using TrakEM282, Microscopy Image Browser83, and Amira (Thermo Fisher Scientific).
Quantitative real-time reverse transcriptase PCR (qRT-PCR)
Ten dissected synganglia and ten pairs of SG per time point were pooled for RNA extraction using the RNeasy Micro Kit (Qiagen). qRT-PCR was performed on a LightCycler 480 II (Roche) with the LightCycler 480 SYBR Green I Master (Roche). The ribosomal protein S4 (GenBank: DQ066214) served as the reference gene84. Gene-specific primers were as described in Mateos-Hernández et al.8. Transcripts were quantified using the ΔΔCt method, corrected by amplification efficiency and expressed as fold differences85 from three biological replicates. Amplicon sizes were verified by agarose gel electrophoresis, and chat and vacht amplicons from SG cDNA were sequenced (Eurofins; Supplementary Fig. 9).
Acetylcholine detection assay
Pools of ten synganglia and ten pairs of SG from unfed, 1–6-day-fed, and replete I. ricinus females were dissected in ice-cold PBS, immediately frozen in liquid nitrogen and stored at −80 °C. Choline and ACh levels in tissue homogenates were quantified using the Choline/Acetylcholine Assay Kit (Abcam, Cat. No. ab65345) following the manufacturer’s instructions. Each pooled sample was analysed in duplicate on 96-well plates and read using a Glomax Discover microplate reader (Promega). Assays were conducted in three independent biological replicates from separate feeding experiments.
In vivo fluid secretion assay
Five-day mouse-fed I. ricinus females were secured ventral side up on water-resistant double-sided tape (Scotch 3M), and their hypostomes were inserted into calibrated capillary micropipettes (volume 2 or 3 μL, length 55 mm, Drummond MicroCaps) for saliva collection. Ticks were injected ventrally near the third leg pair with cholinomimetic agents using a Nano-Injector pump (Drummond) connected to a Micro 4 controller (World Precision Instruments). All agonists and antagonists were administered at 5 mmol·kg−1 of tick body weight, an effective dose for I. ricinus (Supplementary Fig. 11a, b) and a tenfold higher dose than that reported for Amblyomma hebraeum by Kaufman11. In specific tests, lower doses (1.25 mmol·kg−1) of the antagonist propiverine were used. Drugs were dissolved in sterile water with injection volumes ranging from 50–200 nL·mg−1 body weight. For agonist assays, saliva secretion was monitored for one hour post-injection by marking the saliva level in the micropipette at various timepoints under a Nikon SMZ800 stereomicroscope. For antagonist assays, ticks were first injected with the antagonist, rested for 5 min, and then injected with the agonist. Negative controls (water injections, 50–200 nL·mg−1 body weight) did not elicit secretion. For each treatment condition, saliva from individual ticks (Fig. 7k) was pooled and subsequently processed for proteomic analyses.
Proteomic analyses
Saliva from each experimental group was directly subjected to in-solution digestion without prior protein extraction, as described in Kozelková et al.86. Samples were reduced with 10 mM 1,4-dithiothreitol (DTT) at 56 °C for 45 min and alkylated with 55 mM iodoacetamide at RT in the dark for 20 min. Alkylation was subsequently quenched with 50 mM DTT. Proteins were digested using trypsin (Pierce Trypsin Protease, MS Grade; Thermo Fisher Scientific, #90057) at a ratio of 50:1 (protein:trypsin) overnight at 37 °C. Digestion was terminated by the addition of formic acid to a final concentration of 2.5% (v/v). NanoLC–MS/MS analysis was performed as described in Kozelková et al. (2023)86. Briefly, peptides obtained after overnight trypsin digestion were purified using StageTip C18 solid-phase extraction discs (Rappsilber et al., 2007). Purified peptides were dissolved in 30 µl of 3% acetonitrile/0.1% formic acid. Peptide separation was carried out on an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific) online-coupled to a timsTOF Pro mass spectrometer (Bruker Daltonics). An aliquot of 2 µL of peptide solution was loaded onto an Acclaim PepMap 100 C18 trapping column (300 µm i.d., 5 mm length, 5 µm particles, 100 Å pore size; Thermo Fisher Scientific, #160454) at a flow rate of 2.5 µL min⁻¹ with 2% acetonitrile/0.1% formic acid for 2 min. Peptides were then eluted onto an Acclaim PepMap 100 C18 analytical column (75 µm i.d., 150 mm length, 2 µm particles, 100 Å pore size; Thermo Fisher Scientific, #164534) and separated using a 48-min linear gradient from 5% to 35% acetonitrile/0.1% formic acid at a constant flow rate of 0.3 µL min⁻¹. Column temperature was maintained at 35 °C. Data were acquired in PASEF mode with positive ion polarity. Electrospray ionisation was performed using a CaptiveSpray source (Bruker Daltonics) with a capillary voltage of 1500 V, dry gas flow of 3 L min⁻¹, and a dry temperature of 180 °C. Ions were accumulated for 100 ms, and 10 PASEF MS/MS scans were recorded per topN acquisition cycle. The ion mobility range (1/K₀) was set from 0.6 to 1.6 Vs cm⁻², and mass spectra were acquired over an m/z range of 100–1700. Polygon filtering was applied to exclude low-m/z singly charged ions. Target intensity was set to 20,000 to enable repeated precursor selection for PASEF MS/MS, after which precursors were dynamically excluded for 0.4 min. Collision energies ranged from 20 to 59 eV and were adjusted in five equally spaced steps across the ion mobility range. Protein identification and quantification were performed using MaxQuant software (v2.4.2.0) with the Andromeda search engine against the I. ricinus (UniProt, 21.08.2023; 61,426 entries) and mouse (UniProt, 27.11.2023; 87,808 entries) databases. Default MaxQuant parameters were applied as described in Kozelková et al.86, including the TIMS-DDA search type and Bruker TIMS instrument settings. Trypsin/P was specified as the digestion enzyme with up to two missed cleavages allowed. Carbamidomethylation of cysteine was set as a fixed modification, while N-terminal protein acetylation and methionine oxidation were included as variable modifications. Minimum and maximum peptide lengths were set to 8 and 25 amino acids, respectively. Precursor mass tolerance was set to 20 ppm for the first search, and 10 ppm for the main search, and MS/MS fragment ion tolerance was set to 25 ppm. Peptide spectrum matches (PSMs) and protein identifications were filtered using a target–decoy strategy with a false discovery rate (FDR) of 1%. Label-free quantification (LFQ) was performed using the MaxQuant-integrated algorithm with a minimum ratio count of 1. Protein tables generated by MaxQuant were analysed using Perseus software (v1.6.14.0). Experimental conditions were analysed using one biological replicate, each measured in three technical replicates. Results were filtered to remove reverse hits, contaminants, and proteins identified only by modified peptides. Proteins identified by a single peptide, with a score <40, or detected in fewer than two technical replicates were excluded. LFQ intensity values were log₂-transformed, and principal component analysis (PCA) was performed using Perseus.
Statistics and reproducibility
Statistical analyses were performed using GraphPad Prism 9. Data are presented as mean±SD. Box plots show the median and interquartile range (IQR), with whiskers indicating minimum and maximum inward ion current values for Ir-mAChR-A and -B stimulation; all individual data points are shown. Truncated violin plots were used for total saliva secretion, displaying the median and IQR. Drug potency in bioluminescence assays was determined by nonlinear regression, and half-maximal effective concentrations (EC₅₀) were calculated where applicable. Sample sizes (N, n) and statistical tests are specified in the figure legends, except where multiple n values are indicated directly in the figure. All p-values are reported with uniform precision to four decimal places, and values smaller than 0.0001 are denoted as p < 0.0001, with p-values shown in the figures.
For imaging-based analyses, multiple samples, sections, and fields of view were examined, and representative images were selected based on optimal tissue preservation, orientation, and signal quality.
Figure 3a–c, e, g–k show synganglia from 4 to 5 independent ticks; staining was reproduced in ≥4 independent experiments and yielded similar labelling patterns. Figure 3d shows five synganglia from two independent staining experiments, with comparable lateral nerve staining patterns in all specimens and lateral segmental organs positivity in three of them. Figures 3m, n, q, and 4a, b are based on ≥6 synganglia across two independent experiments, each producing similar results. Figure 4c is based on four synganglia processed in a single experiment and showing consistent staining. Figure 5a, b are based on three synganglia and three paired SGs, reproduced in two and ≥3 independent biological replicates, respectively, each yielding similar results. Figure 5c comprises two independent experiments, each containing three paired SG, all yielding similar results. Figure 5d is based on >15 ticks per developmental stage across >3 independent biological replicates (three unfed and at least three different tick-rabbit feeding experiments); numbers of positive/negative SGs for individual females are shown in Fig. 5e. Figure 5f is based on 10 independent SG pairs across two experiments, all yielding similar results. Reproducibility and sample sizes (n) for immunostaining in the Supplementary Figs. are provided in the corresponding legends.
SEM data in Fig. 3p originate from two independent experiments and show comparable synganglion and associated structural morphologies. TEM immunogold data in Fig. 3o derive from four independent ticks, with multiple ultrathin sections examined per replicate and similar ultrastructural and staining features observed. Figure 4e is based on two synganglia showing consistent features across multiple sections; quantification was performed on one representative axon-rich section, with counting details provided in the Source Data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analysed in this study are included in the article and the supplementary information files. Mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE87 repository under dataset identifier PXD055362 (https://proteomecentral.proteomexchange.org). Source data are provided with this paper.
References
Kaufman, W. R. Gluttony and sex in female ixodid ticks: how do they compare to other blood-sucking arthropods? J. Insect Physiol. 53, 264–273 (2007).
Šimo, L., Kazimirova, M., Richardson, J. & Bonnet, S. I. The essential role of tick salivary glands and saliva in tick feeding and pathogen transmission. Front. Cell. Infect. Microbiol. 7, 281 (2017).
Mans, B. J. Chemical equilibrium at the tick–host feeding interface: a critical examination of biological relevance in hematophagous behavior. Front. Physiol. 10, 530 (2019).
Šimo, L. 50 Years since Kaufman and Phillips’ groundbreaking trilogy elucidating ion and water homeostasis in ixodid ticks. Pathogens 12, 385 (2023).
Nuttall, P. A. Tick saliva and its role in pathogen transmission. Wien. Klin. Wochenschr. 135, 165–176 (2023).
Binnington, K. C. Sequential changes in salivary gland structure during attachment and feeding of the cattle tick, Boophilus microplus. Int. J. Parasitol. 8, 97–115 (1978).
Kim, D., Šimo, L., Vancová, M., Urban, J. & Park, Y. Neural and endocrine regulation of osmoregulatory organs in tick: Recent discoveries and implications. Gen. Comp. Endocrinol. 278, 42–49 (2019).
Mateos-Hernández, L. et al. Cholinergic axons regulate type I acini in salivary glands of Ixodes ricinus and Ixodes scapularis ticks. Sci. Rep. 10, 16054 (2020).
Šimo, L., Žitňan, D. & Park, Y. Neural control of salivary glands in ixodid ticks. J. Insect Physiol. 58, 459–466 (2012).
Vancová, M. et al. Ultrastructural mapping of salivary gland innervation in the tick Ixodes ricinus. Sci. Rep. 9, 6860 (2019).
Kaufman, W. R. Actions of some transmitters and their antagonists on salivary secretion in a tick. Am. J. Physiol. 235, R76–R81 (1978).
Kaufman, W. R. & Phillips, J. E. Ion and water balance in the ixodid tick Dermacentor andersoni: II. Mechanism and control of salivary secretion. J. Exp. Biol. 58, 537–547 (1973).
Kaufman, W. R. The influence of adrenergic agonists and their antagonists on isolated salivary glands of ixodid ticks. Eur. J. Pharmacol. 45, 61–68 (1977).
Tatchell, R. J. A modified method for obtaining tick oral secretion. J. Parasitol. 53, 1106–1107 (1967).
Kim, D., Šimo, L. & Park, Y. Orchestration of salivary secretion mediated by two different dopamine receptors in the blacklegged tick Ixodes scapularis. J. Exp. Biol. 217, 3656–3663 (2014).
Šimo, L., Koči, J., Žitňan, D. & Park, Y. Evidence for D1 dopamine receptor activation by a paracrine signal of dopamine in tick salivary glands. PLoS ONE 6, e16158 (2011).
Šimo, L., Koči, J., Kim, D. & Park, Y. Invertebrate specific D1-like dopamine receptor in control of salivary glands in the black-legged tick Ixodes scapularis. J. Comp. Neurol. 522, 2038–2052 (2014).
Binnington, K. C. & Rice, M. J. A technique for recording efferent neurone activity from normal and poisoned cattle ticks [boophilus Microplus (canestrini)]. Aust. J. Entomol. 21, 161–166 (1982).
Kaufman, W. R. & Harris, R. A. Neural pathways mediating salivary fluid secretion in the ixodid tick Amblyomma hebraeum. Can. J. Zool. 61, 1976–1980 (1983).
Kaufman, W. R., Aeschlimann, A. A. & Diehl, P. A. Regulation of body volume by salivation in a tick challenged with fluid loads. Am. J. Physiol. 238, R102–112 (1980).
Roller, L. et al. Orcokinin-like immunoreactivity in central neurons innervating the salivary glands and hindgut of ixodid ticks. Cell Tissue Res. 360, 209–222 (2015).
Šimo, L., Slovák, M., Park, Y. & Zitnan, D. Identification of a complex peptidergic neuroendocrine network in the hard tick, Rhipicephalus appendiculatus. Cell Tissue Res. 335, 639–655 (2009).
Šimo, L., Žitňan, D. & Park, Y. Two novel neuropeptides in innervation of the salivary glands of the black-legged tick, ixodes scapularis: myoinhibitory peptide and SIFamide. J. Comp. Neurol. 517, 551–563 (2009).
Ren, G. R., Folke, J., Hauser, F., Li, S. & Grimmelikhuijzen, C. J. P. The A- and B-type muscarinic acetylcholine receptors from Drosophila melanogaster couple to different second messenger pathways. Biochem. Biophys. Res. Commun. 462, 358–364 (2015).
Xia, R.-Y. et al. A new family of insect muscarinic acetylcholine receptors. Insect Mol. Biol. 25, 362–369 (2016).
Birdsall, N. J., Burgen, A. S. & Hulme, E. C. The binding of agonists to brain muscarinic receptors. Mol. Pharm. 14, 723–736 (1978).
Meyer, E. M. & Otero, D. H. Pharmacological and ionic characterizations of the muscarinic receptors modulating [3H]acetylcholine release from rat cortical synaptosomes. J. Neurosci. 5, 1202–1207 (1985).
Collin, C. et al. Two types of muscarinic acetylcholine receptors in Drosophila and other arthropods. Cell. Mol. Life Sci. 70, 3231–3242 (2013).
Nìng, C., Heckmann, A., Mateos-Hernández, L., Karadjian, G. & Šimo, L. Functional characterization of three G protein-coupled acetylcholine receptors in parasitic nematode Trichinella spiralis. Int. J. Parasitol. Drugs Drug Resist. 23, 130–139 (2023).
Rang, H. P., Dale, M. M., Ritter, J. M. & Moore, P. K. Pharmacology 5th edn 797 pp. (Churchill Livingstone, 2003).
Lee, Y. S. et al. Characterization of GAR-2, a novel G protein-linked acetylcholine receptor from Caenorhabditis elegans. J. Neurochem. 75, 1800–1809 (2000).
Kimber, M. J. et al. Identification of an Ascaris G protein-coupled acetylcholine receptor with atypical muscarinic pharmacology. Int. J. Parasitol. 39, 1215–1222 (2009).
Maeda, S. et al. Structure and selectivity engineering of the M1 muscarinic receptor toxin complex. Science 369, 161–167 (2020).
Dong, C. & Wu, G. Regulation of anterograde transport of alpha2-adrenergic receptors by the N termini at multiple intracellular compartments. J. Biol. Chem. 281, 38543–38554 (2006).
Yamanaka, N. et al. Regulation of insect steroid hormone biosynthesis by innervating peptidergic neurons. Proc. Natl. Acad. Sci. USA 103, 8622–8627 (2006).
Grimmelikhuijzen, C. J. & Spencer, A. N. FMRFamide immunoreactivity in the nervous system of the medusa Polyorchis penicillatus. J. Comp. Neurol. 230, 361–371 (1984).
Nichols, R. Signaling pathways and physiological functions of Drosophila melanogaster FMRFamide-related peptides. Annu. Rev. Entomol. 48, 485–503 (2003).
Leander, M. et al. Cardiac contractility structure-activity relationship and ligand-receptor interactions; the discovery of unique and novel molecular switches in myosuppressin signaling. PLoS ONE 10, e0120492 (2015).
Waldman, J. et al. Neuropeptides in Rhipicephalus microplus and other hard ticks. Ticks Tick. Borne Dis. 13, 101910 (2022).
Chen, Y., Veenstra, J. A., Davis, N. T. & Hagedorn, H. H. A comparative study of leucokinin-immunoreactive neurons in insects. Cell Tissue Res. 276, 69–83 (1994).
Medla, M. et al. Identification and expression of short neuropeptide F and its receptors in the tick Ixodes ricinus. J. Insect Physiol. 147, 104524 (2023).
Yamanaka, N. et al. Bombyx orcokinins are brain-gut peptides involved in the neuronal regulation of ecdysteroidogenesis. J. Comp. Neurol. 519, 238–246 (2011).
Terhzaz, S., Rosay, P., Goodwin, S. F. & Veenstra, J. A. The neuropeptide SIFamide modulates sexual behavior in Drosophila. Biochem. Biophys. Res. Commun. 352, 305–310 (2007).
Li, Z., Macaluso, K. R., Foil, L. D. & Swale, D. R. Inward rectifier potassium (Kir) channels mediate salivary gland function and blood feeding in the lone star tick, Amblyomma americanum. PLoS Negl. Trop. Dis. 13, e0007153 (2019).
Li, Z. et al. ATP-sensitive inward rectifier potassium channels regulate secretion of pro-feeding salivary proteins in the lone star tick (Amblyomma americanum). Int. J. Biol. Macromol. 253, 126545 (2023).
Nathanson, N. M. A multiplicity of muscarinic mechanisms: enough signaling pathways to take your breath away. Proc. Natl. Acad. Sci. USA 97, 6245–6247 (2000).
Daaka, Y., Luttrell, L. M. & Lefkowitz, R. J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91 (1997).
Binnington, K. C. Ultrastructural identification of neurohaemal sites in a tick: evidence that the dorsal complex may be a true endocrine gland. Tissue Cell 15, 317–327 (1983).
Lapied, B., Le Corronc, H. & Hue, B. Sensitive nicotinic and mixed nicotinic-muscarinic receptors in insect neurosecretory cells. Brain Res 533, 132–136 (1990).
Lees, K. et al. Functional characterisation of a nicotinic acetylcholine receptor α subunit from the brown dog tick, Rhipicephalus sanguineus. Int. J. Parasitol. 44, 75–81 (2014).
Le Mauff, A. et al. Nicotinic acetylcholine receptors in the synganglion of the tick Ixodes ricinus: functional characterization using membrane microtransplantation. Int. J. Parasitol. Drugs Drug Resist. 14, 144–151 (2020).
Ito, K. et al. A systematic nomenclature for the insect brain. Neuron 81, 755–765 (2014).
Boussaine, K. et al. Isolation and electrophysiological recording of Ixodes ricinus synganglion neurons. J. Pharmacol. Toxicol. Methods 124, 107473 (2023).
Trendelenburg, A.-U., Gomeza, J., Klebroff, W., Zhou, H. & Wess, J. Heterogeneity of presynaptic muscarinic receptors mediating inhibition of sympathetic transmitter release: a study with M2- and M4-receptor-deficient mice. Br. J. Pharmacol. 138, 469–480 (2003).
Salvaterra, P. M. & McCaman, R. E. Choline acetyltransferase and acetylcholine levels in Drosophila melanogaster: a study using two temperature-sensitive mutants. J. Neurosci. 5, 903–910 (1985).
Kim, D., Urban, J., Boyle, D. L. & Park, Y. Multiple functions of Na/K-ATPase in dopamine-induced salivation of the Blacklegged tick, Ixodes scapularis. Sci. Rep. 6, 21047 (2016).
Mateos-Hernández, L., Rakotobe, S., Defaye, B., Cabezas-Cruz, A. & Šimo, L. A capsule-based model for immature hard tick stages infestation on laboratory mice. J. Vis. Exp. https://doi.org/10.3791/61430 (2020).
Almazán, C. et al. A versatile model of hard tick infestation on laboratory rabbits. J. Vis. Exp. https://doi.org/10.3791/57994 (2018).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Cieslikiewicz-Bouet, M. et al. Functional characterization of multifunctional ligands targeting acetylcholinesterase and alpha 7 nicotinic acetylcholine receptor. Biochem Pharma 177, 114010 (2020).
Cartereau, A., Martin, C. & Thany, S. H. Neonicotinoid insecticides differently modulate acetycholine-induced currents on mammalian α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 175, 1987–1998 (2018).
Papke, R. L. et al. Persistent activation of α7 nicotinic ACh receptors associated with stable induction of different desensitized states. Br. J. Pharmacol. 175, 1838–1854 (2018).
Papke, R. L. & Porter Papke, J. K. Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br. J. Pharmacol. 137, 49–61 (2002).
Gulsevin, A. et al. Allosteric agonism of α7 nicotinic acetylcholine receptors: receptor modulation outside the orthosteric site. Mol. Pharm. 95, 606–614 (2019).
Vernon, W. I. & Printen, J. A. Assay for intracellular calcium using a codon-optimized aequorin. BioTechniques 33, 730–734 (2002).
Offermanns, S. & Simon, M. I. Gα15 and Gα16 couple a wide variety of receptors to phospholipase C (∗). J. Biol. Chem. 270, 15175–15180 (1995).
Hallgren, J. et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural network. Preprint at https://doi.org/10.1101/2022.04.08.487609 (2022).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Schrödinger Release 2022-1: Maestro (Schrödinger. LLC, 2021).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Mahoney, M. W. & Jorgensen, W. L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 112, 8910–8922 (2000).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Huang, J. & MacKerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
Zoete, V., Cuendet, M. A., Grosdidier, A. & Michielin, O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 (2011).
Bowers, K. J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proc. 2006 ACM/IEEE Conference on Supercomputing 84–es (Association for Computing Machinery, New York, NY, USA, 2006).
Evans, D. J. & Holian, B. L. The nose–Hoover thermostat. J. Chem. Phys. 83, 4069–4074 (1985).
Martyna, G. J., Tobias, D. J. & Klein, M. L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 (1994).
Tuckerman, M., Berne, B. J. & Martyna, G. J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 97, 1990–2001 (1992).
Humphrey, W., Dalke, A. & Schulten, K. V. M. D. Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Šimo, L., Koči, J. & Park, Y. Receptors for the neuropeptides, myoinhibitory peptide and SIFamide, in control of the salivary glands of the blacklegged tick Ixodes scapularis. Insect Biochem. Mol. Biol. 43, 376–387 (2013).
Šimo, Daniel, S., E., Park, Y. & Žitňan, D. The Nervous and Sensory Systems: Structure, Function, Proteomics and Genomics. In Biology of Ticks (eds. Sonenshine, D. E. & Roe, R. M.) vol. 1 309–367 (Oxford Univ. Press, New York, 2014).
Cardona, A. et al. TrakEM2 software for neural circuit reconstruction. PLoS ONE 7, e38011 (2012).
Belevich, I., Joensuu, M., Kumar, D., Vihinen, H. & Jokitalo, E. Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biol. 14, e1002340 (2016).
Koči, J., Šimo, L. & Park, Y. Validation of internal reference genes for real-time quantitative polymerase chain reaction studies in the tick, Ixodes scapularis (Acari: Ixodidae). J. Med. Entomol. 50, 79–84 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408 (2001).
Kozelková, T. et al. Insight into the dynamics of the ixodes ricinus nymphal midgut proteome. Mol. Cell. Proteom. 22, 100663 (2023).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Kim, D., Šimo, L. & Park, Y. Molecular characterization of neuropeptide elevenin and two elevenin receptors, IsElevR1 and IsElevR2, from the blacklegged tick, Ixodes scapularis. Insect Biochem. Mol. Biol. 101, 66–75 (2018).
Acknowledgements
This study was supported by the French National Research Agency (ANR-21-CE14-0012, project AxoTick; L.Š). UMR BIPAR was supported by the French Government’s Investissement d’Avenir programme, Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases (ANR-10-LABX-62-IBEID; institutional support). Additional support was provided by the Czech Science Foundation (22-18424M; J.P.), (22-30920S; R.Š.), (21-08826S; P.K.), and the Ministry of Health of the Czech Republic (NU20-05-00396 R.Š.). The study was also supported by a grant from the Centre-Val de Loire Region, under the Electro-CELL, Appel à projet d’intérêt regional (APR IR 21060LBL; S.H.T.). We acknowledge the BC CAS core facility LEM supported by MEYS CR (LM2023050 Czech-BioImaging and OP VVV CZ.02.1.01/0.0/0.0/18_046/0016045; M.V.). We are grateful to Jan Veenstra from the University of Bordeaux, France, for providing the anti-SIFamide and anti-leucokinin antibodies.
Author information
Authors and Affiliations
Contributions
L. Šimo designed the study and supervised all work. C.N., L.M.-H., S.R., L.A.-D., L. Šofranková, K.B., A.C., E.T., H.F., V.U., T.K., F.D., O.H., R.Š., J.T., T.B., M.T., M.V., J.P., S.H.T., and L. Šimo performed the experiments and analysed and interpreted the data. J.J.V. performed the molecular dynamics analyses and wrote this part of the manuscript. N.H., M.S., H.A., P.K., and J.P. provided resources. L. Šimo prepared the final figures and wrote the manuscript. All authors revised and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Philip Ahring and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nìng, C., Valdés, J.J., Mateos-Hernández, L. et al. Two types of axonal muscarinic acetylcholine receptors mediate formation of saliva cocktail in the tick Ixodes ricinus. Nat Commun 17, 2867 (2026). https://doi.org/10.1038/s41467-026-68654-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68654-3
