
Abstract
Secondary vascular growth is a conserved mechanism that gives rise to vascular tissues produced via a single vascular cambium. Molecular mechanisms underlying this process are characterized mainly in model species with typical vascular architectures, while the genetics underlying ecologically-important atypical vascular architectures remain unexplored. We use developmental anatomy, comparative transcriptomics, molecular evolutionary analyses, and heterologous gene expression to address this knowledge gap, investigating how multiple ectopic cambia (EC) form in the woody vine Japanese wisteria. Anatomical studies show EC in Japanese wisteria arise from cortical parenchyma, and cambium-specific transcriptome comparisons reveal that genes acting as regulators of typical cambium development in model species are likewise associated with EC development. Gene trees of KNOX genes suggest that duplication events may contribute to EC formation, including a Fabaceae-specific duplication of KNAT2/6, which is detected as being under positive selection. We also demonstrate that KNOX genes from Japanese wisteria show canonical KNOX-like activity in heterologous functional assays, although no vascular aberrations were observed. Overall, these findings provide the first insights into the genetics of EC formation in “natural variants”, advancing our understanding of the molecular mechanisms regulating vascular variants in seed plants.
Similar content being viewed by others


Introduction
The vast majority of woody plants undergo a common type of secondary vascular growth that involves the production of a single meristematic niche called the vascular cambium, which is responsible for generating secondary xylem (wood) and secondary phloem (inner bark), thereby enabling the plants to expand in diameter1. With only a single vascular cambium, plants can live up to 5000 years (e.g., Bristlecone pines [Pinus longaeva D.K.Bailey] or build a sophisticated plumbing system to transport water upwards of 380 ft (e.g., Coastal Redwood [Sequoia sempervirens (D.Don) Endl.]2,3. However, some plants deviate from the canonical modality to create woody structures with unique growth patterns that result in novel anatomical formations, such as those found mainly within the stems of woody vines or “lianas.” Lianas reach the forest canopy by climbing over other plants rather than supporting their weight4. As they weave their way through dense forest understory canopy, the stems of lianas are frequently damaged either through the collapse of host trees or the regular wear and tear of twisting and twining5. The woody stems of many lianas exhibit “ectopic cambia.” Ectopic cambia is a convergently evolved vascular variant6 that may serve as a mechanism for lianas to repair their stems after injury while maintaining key vascular functions of transportation and support5. Ectopic cambia (EC) result from the development of multiple vascular cambia in atypical locations, after the first-formed and central vascular cambium is established7. In addition to their role in lianas, EC may also facilitate the success of self-supporting herbs, shrubs, and trees living in harsh conditions such as mangroves and arid environments7,8,9. Among lianas, EC have been identified in the stems of the temperate Wisteria vines10,11, potentially facilitating their success as invasive vines in the Eastern United States12.
To date, all anatomical studies show that EC arise through three steps: (1) de-differentiation of living parenchyma cells, (2) cell division to give rise to a new cambium, and (3) production and differentiation of localized vascular tissues from the new cambium layer8,10,13. While the vast majority of the literature on the natural diversity of EC is purely anatomical and descriptive, transgenic phenotypes in model species suggest that ECs can emerge by simply manipulating the expression of conserved genes responsible for vascular development7.
Recent investigations demonstrate that Arabidopsis thaliana (L.) Heynh and Populus spp. share a suite of genes in the regulatory pathway controlling the canonical model of secondary growth by a single vascular cambium1,14,15. Notably, transgenic lines that perturb the expression of conserved vascular genes produce “experimental mutants” with EC in stems and/or roots7. Various studies indicate that EC can appear as the result of the misexpression of members of the class III homeodomain-leucine zipper (HD-ZIP III) transcription factor: for example, EC are developed in Poplar trees stems through overexpression of PRE, an ortholog of the gene REVOLUTA (REV)16, while EC have been shown to develop in A. thaliana through the overexpression of AtHB717 and repression of AtHB418. ECs can also be formed following overexpression of WOX4, a member of the WOX family19, and ECs were observed in double mutants following the overexpression of WOX4 and repression of PETAL LOSS (PTL)20. Finally, regenerated cambium can be induced by girdling trees, allowing investigators to characterize how such cambia (here categorized as EC, given their de novo establishment) can function to stitch together a wounded vascular cambium21,22. KNOTTED1-like HOMEOBOX (KNOX) transcription factors––key regulators of vascular proliferation––often emerge as differentially expressed genes in cambium development studies17,18,20,23. Yet, their role is underexplored in the formation of EC development. Our current knowledge of EC formation arises from molecular manipulation or direct perturbation––either mechanical (i.e., girdling) or genetic (i.e. transgenics)––in model species that don’t naturally form EC. To understand how EC contribute to plant diversity in form and function, we must realize how EC are produced in systems in which they naturally occur (“natural variants”), particularly in lianas, where they have a key functional role in allowing plants to climb.
In this work, we seek to elucidate the developmental anatomy, transcriptomic profile, molecular evolution, and gene expression of molecular markers underlying cambium formation in a pair of vines in the Fabaceae family, the ectopic cambia-producing Japanese wisteria (Wisteria floribunda (Willd.) DC.) and the non-ectopic cambia-producing common bean (Phaseolus vulgaris L.). We confirm previous reports that ectopic cambia form within the cortex of Japanese wisteria in a haphazard fashion. To reveal the transcriptomic profile underlying EC formation, we leveraged tangential cryosectioning to isolate tissues of interest for RNA sequencing. Here, we report the differential expression of several conserved genes in species and cross-species analyses of cambium-specific samples, including known cambium markers and genes involved in epigenetic regulation. We identified KNOX genes as prospective candidates in EC development in Japanese wisteria, as multiple transcript clusters are differentially expressed in typical v. ectopic cambia contrasts. We explored the molecular evolution of KNOX genes across 45 species, spanning both gymnosperms and angiosperms, and used branch models to analyze the nonsynonymous to synonymous substitution ratio and detect positive selection in KNOX transcripts. Finally, we confirmed the conserved function of Japanese wisteria KNOX genes by heterologous expression in Arabidopsis thaliana, further supporting their role in plant development. Together, these results expand our understanding of both typical and atypical cambium development, while identifying promising contenders underpinning EC formation in woody plants.
Results
Common bean and Japanese wisteria exhibit typical cambium development; Japanese wisteria develops ectopic cambia during the later stages of stem growth
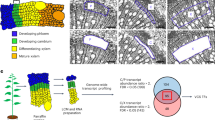
To characterize the origin of ectopic cambia (EC), we began by detailing the developmental anatomy of common bean and Japanese wisteria (Fig. 1a–j). Young stems of both species have the same organization of primary tissues, with vascular bundles arranged in a ring (Fig. 1b, f). In young stems (3rd to 5th internodes, see Fig. 1a, e), we find the initiation of the vascular cambium from the fascicular cambium within each bundle and the interfascicular cambium connecting the bundles (Fig. 1c, g). This continuous cambium gives rise to secondary xylem (wood) inward and secondary phloem (inner bark) outward (Fig. 1d, h). This stage marks the final step in the stem development of common bean (Fig. 1d), giving rise to the mature form; however, Japanese wisteria development continues with further elaborations (Fig. 1i) to reach maturity. In stems with at least 20 mm diameter, Japanese wisteria starts to produce additional cambia ectopically (Fig. 1j), with older stems displaying multiple layers of EC and its products, i.e., secondary xylem and secondary phloem (Fig. 1i). The first EC in Japanese wisteria arise from the cortex (Fig. 1j), where existing cortical cells undergo localized cell divisions, creating a mass of cells from which a new cambium will emerge to produce secondary xylem and phloem (Fig. 1j). These EC do not arise in a uniform concentric pattern but rather initiate asynchronously across the stem circumference forming discrete strands of secondary xylem and secondary phloem (Fig. 1i).

a–d Common bean. e-i Japanese wisteria. a, e Common bean and Japanese wisteria plants sampled for developmental anatomy. The dashed rectangle in “(e)” indicates the position of the image displayed in stage 4 (i). b, f Primary growth showing vascular bundles (dashed circle), formed by xylem inwards and phloem outwards, arranged in a single ring delimiting the pith. c, g Initiation of typical cambium from the fascicular cambium located within the bundles, and the interfascicular cambium between the bundles. d, h Secondary growth generated by the typical cambium (dashed yellow line), producing secondary xylem and secondary phloem. In (d), the bottom right inset shows details of the typical cambium extracted from the top right inset. i Mature stem of Japanese wisteria with EC (dashed purple line) forming multiple vascular increments in addition to the vascular cylinder produced by the typical cambium (dashed yellow line). j Detail of stem in advanced typical secondary growth derived from the typical cambium (dashed yellow line) and the emergence of EC (dashed purple line) in Japanese wisteria; the dashed box indicates the position of the inset (on the right). Gray arrow = pericyclic fibers. All figures are displayed in cross-section and indicate magnification. Anatomical analysis was performed on two samples from each species.
Transcriptomes of common bean and Japanese wisteria
Having isolated key developmental stages, we sought to identify which genes underlie the formation of EC. Transcriptomic profiles were made from the typical cambium of the common bean and Japanese wisteria and the EC of Japanese wisteria, with at least five biological replicates for each of the three tissues sampled. The tissues for transcriptomic analyses consisted of multiple sections of secondary xylem-vascular cambium-secondary phloem obtained through tangential cryosectioning (Fig. 2). Sequencing of 17 samples across cambium types and species yielded an average of 21,420,218 (128,521,310 total) and 23,468,348 (258,151,832 total) paired-end reads per sample for the common bean and Japanese wisteria, respectively. The transcriptome assemblies vary in BUSCO, mapping rate, and N50, but both assemblies yielded BUSCO scores and mapping rates over 80% with N50s greater than 1500 base pairs (bp) (Supplementary Table 1).

The inset shows a cross-section of a standard sample of secondary xylem–vascular cambium–secondary phloem taken through tangential cryosectioning (schematic at the bottom left) to produce the transcriptomes. Tissue collection ranged from approximately 40 to 100 sections per sample, with each section ~12 μm thick. This inset is approximately 700 μm in size and represents the average amount of tissue collected per sample (typically 2 mm wide). In the schematic, blue, tan, and pink represent the longitudinal positions of phloem, cambium, and xylem, respectively. Tangential cryosectioning was performed for six samples from each species. Drawings were created in Adobe Illustrator 2025.
Differential cluster expression and gene ontology enrichment
Comparison across species
Using the transcript clusters for common bean (42,827) and Japanese wisteria (74,960), 10,498 one-to-one orthologous transcript clusters were identified and passed filtering (Expressed > 0.5 counts per million) (Supplementary Data 1). The number of differentially expressed clusters varied dramatically between contrasts (Table 1). The most differentially expressed (DE) transcript clusters were identified in the “Japanese wisteria v. Common bean” contrast, representing overall differences between the two species (>6000). This was followed by the number of DE transcripts when comparing the “Typical Cambia: Japanese wisteria v. Common bean” (>5000). The third highest number of DE transcripts was found in the “Ectopic Cambia v. Typical Cambia of Japanese wisteria and Common bean” contrast (>2000) (Table 1). Lastly, we compared the “Japanese wisteria: Ectopic v. Typical cambia” contrast, which surprisingly yielded no DE transcript clusters in this analysis (see further analyses “Comparisons within Japanese wisteria” below). In annotating the DE transcript clusters with Trinotate24 against the UniProt database for Fabales (UniProt Taxon ID 72025)25, we found transcript clusters related to DNA methylation (e.g., methyltransferases, DNA methylase), phytohormones (e.g., indoleacetic acid, auxin response factors), cell cycle control (e.g., cyclins), cell differentiation (e.g., KNOX), and other cambial markers (e.g., PXY). The “Ectopic Cambia v. Typical Cambia of Japanese wisteria and Common bean” contrast is the only comparison that yielded enriched gene ontology terms based on identified DE clusters, and the GO terms are mostly related to RNA binding, splicing, and processing (Supplementary Table 2).
Comparison within Japanese wisteria
Given our focus on elucidating the genes related to EC, we further explored the “Japanese wisteria: Ectopic v. Typical cambia” contrast using differential expression for repeated measures (dream)26 to compare across cambium types with the individual as a random effect. This approach enabled us to isolate the most likely candidate genes associated with EC formation in Japanese wisteria. After filtering (Expressed > 0.5 counts per million in at least two samples), 46,225 transcript clusters were retained for differential transcript cluster expression analysis across the two Japanese wisteria cambium types using dream26, with 14 clusters (2▲ and 12▼) found to be differentially expressed (Table 2). The differentially expressed clusters include transcripts related to DNA methylation, cell expansion (e.g., expansin-like B1), and cell differentiation (e.g., KNOX), all of which were downregulated in the EC of Japanese wisteria (Fig. 3). Only two transcript clusters are upregulated, one of which includes the epigenetic gene Methyltransf_7 (Table 2). Interestingly, we found five differentially expressed KNOX genes across the four contrasts (Table 3). KNOX clusters identified as two potential KNOX genes are linked to typical cambium development in Japanese wisteria and common bean (e.g., STM, KNATM), while EC formation is more directly associated with the expression of KNOX2/6 (Table 3, Supplementary Fig. 1). These observations make KNOX genes of particular interest, given their defined role in vascular cambium proliferation in model systems.

The y-axis is the negative log10 of the Benjamini-Hochberg multiple testing burden false discovery rate adjusted p-value for a two-sided test, while the x-axis is the log fold change. The transcript clusters, colored by classification, are related to cambium development in other plants, and the group annotation is provided in the legend. The analysis was conducted with six biological replicates for the typical cambium of common bean and ectopic cambia (EC) of Japanese wisteria, and five replicates for the typical cambium of Japanese wisteria.
Identification, characterization, and phylogenetic analysis of KNOX genes analysis
To investigate the relationship between KNOX genes and EC evolution, we began by testing the hypothesis that the diversification of KNOX genes has led to EC. To achieve this aim, we broadened our analyses to encompass all nine members of the KNOX gene family (based on the A. thaliana genome) across 45 seed plant species, including gymnosperms, monocots, and eudicots (Fig. 4; Supplementary Fig. 2). Our sampling included species with and without EC (Supplementary Data 2, 3). Although retrieved through the tBLASTx search, we removed eight species due to their poor alignment, which potentially resulted in an erroneous phylogenetic placement (Supplementary Fig. 3).

The phylogenetic tree includes members of KNOX genes obtained from a tBLASTx search, encompassing seed plants from 20 families, 33 genera, and 38 species, including 11 species that develop ectopic cambia. KNOX are divided into three classes: KNOX1 (branches in green), KNOX2 (branches in blue), and KNATM (branches in magenta). These classes are further divided into clades corresponding to individual genes named after Arabidopsis thaliana orthologs, as indicated in bold (in black). All recovered transcript clusters annotated for KNOX genes are likewise highlighted in bold colors corresponding to their identified clades (KNOX1, KNOX2, or KNATM). The tree is rooted in a member of the BEL1 gene family. Circles indicate angiosperm (yellow), eudicots (blue), Poales (gray), and Fabaceae-specific (magenta) duplications.
The final sequence alignment contained 466 sequences. The maximum likelihood phylogeny grouped KNOX genes into three main clades that correspond to defined classes: KNOX1, KNOX2, and KNATM (Fig. 4). In this phylogeny rooted with a representative of BEL1 genes, KNOX2 diverges first, with the subsequent divergence of KNOX1 and KNATM as sister lineages. The three KNOX classes can be further divided into major clades named according to their putative A. thaliana ortholog (Fig. 4; Supplementary Fig. 2). Class KNOX1 includes three main clades corresponding to STM, KNAT1, and KNAT2/6 (Clusters “W2” and “1332” are phylogenetically placed within a clade comprised of KNAT2 and 6 Arabidopsis reference sequences, highlighting their similarity to both genes. Hereafter, we use the notation “KNAT2/6” to refer to these genes). Class KNOX2 is subdivided into two clades corresponding to KNAT3/4/5 and KNAT7.
Using our phylogenetic approach, we determined the orthology of each of the differentially expressed KNOX transcript clusters (Fig. 4). All DE clusters belong to class KNOX1: Two of them group with A. thaliana STM ortholog, one of which is nested within a clade derived from a Fabaceae-specific gene duplication containing the soybean SBH1 reference; two transcript-clusters group with KNAT2/6, one being associated with each of the major clades resulting from gene duplication following the divergence of angiosperms (Supplementary Fig. 2); and one transcript-cluster groups within KNATM (Fig. 4; Supplementary Fig. 2).
The number of KNOX gene copies recovered from each taxon varies due to duplication events within major clades. We recovered duplication events in most lineages with EC including at the base of the angiosperms for KNAT2/6 (indicated by sequences of Amborella trichopoda Baill. in Fig. 4), eudicots for STM, KNAT2/6, and KNAT3/4/5) (indicated by sequences of Vitis vinifera L. in Fig. 4), as well as within the Fabaceae which included duplications for both KNOX1 and KNOX2 genes (indicated by sequences of Wisteria floribunda or Spatholobus suberectus Dunn in Fig. 4). Duplication events are also observed in lineages without EC, including the conifers for KNAT1 and the Poales (monocots) for KNAT1 and KNAT3/4/5 (indicated by sequences of Zea mays L. or Triticum dicoccoides (Asch. & Graebn.) Schweinf. in Fig. 4).
We did not find an association between the number of KNOX genes and the presence/absence of EC across plants (Fig. 5). The A. thaliana genome contains nine KNOX, genes, but lineages with EC include both increases (e.g., Japanese wisteria with 16 copies and other species of Fabaceae with 17 copies) and decreases (e.g., Amaranthaceae species with five copies), including the possible absence of KNOX genes in the EC-producing gymnosperms Cycas panzhihuaensis L.Zhou & S.Y.Yang and Gnetum montanum Markgr. (Fig. 5). A Fabaceae-specific duplication in KNATM after the divergence of Cercis canadensis L. is apparent in this dataset (Fig. 5).

Copy numbers are inferred from the phylogeny in Fig. 4 and associated with duplication events. Squares indicate copy number, with filled squares indicating gene presence, empty squares inferring gene losses, and white crossed squares indicating gene absence. Whole genome duplications (WGD) inferred in Fig. 4 are indicated by colored circles; stars indicate inferred taxon and/or copy-specific duplication events. Expansion of some KNOX genes is evident after WGD events in angiosperms (yellow circle), eudicots (blue circle; gray box), and Fabales (magenta circle). Names of species with ectopic cambia (EC) are in bold. Black ellipses indicate variation in copy number in lineages with EC. All copies were recovered from the blast pipeline to build the phylogeny (Fig. 4), except for Momordica charantia L. KNATM (red asterisk), which was downloaded from Genbank.
Selection analyses
To test whether KNOX genes in lineages with EC are under positive selection (indicative of adaptive evolution), we compared PAML codon-based nested models (models 0 = one omega value for the tree, 1 = a unique omega per branch, 2 = foreground and background omega values)27. We tested these models in individual gene trees for members of the KNOX genes that were differentially expressed in the transcriptome analysis (i.e., STM, KNATM, and KNAT2/6). By comparing foreground (EC present) and background (EC absent or typical growth) branches (model 2), the only gene set that showed a significant difference was KNAT2/6 (p-value = 0.00406). To detect whether some branches along KNAT2/6 lineages were under positive selection, we employed branch model 1 on the KNAT2/6 phylogenetic rooted tree, which showed nine branches with omega values greater than 1 (ω > 1), therefore, indicating positive selection in specific lineages (Fig. 6). Out of the nine branches, only two have EC, both of which are sequences differentially expressed from two transcript clusters in the gene expression analysis (Wisteria_floribunda_Cluster_W2, ω = 999; Wisteria_floribunda_Cluster_1332, ω = 958.234). Taxa under positive selection belonging to species with typical growth include Dioscorea rotundata (ω = 999), Populus trichocarpa (ω = 964.749 and 999), and Z. mays (ω = 1.29156), including some Fabaceae such as Phaseolus vulgaris (ω = 999) and Prosopis alba (1.27446) (Fig. 6). Despite statistical tests not supporting differences in synonymous v. nonsynonymous changes for the KNATM, results indicate only two branches with higher omega values, both of which are species with EC from the Fabaceae, including Phaseolus lunatus (ω = 1.53725) and W. floribunda (ω = 1.15616), representing the KNATM sequence from the DGE analysis (Cluster_4072) (Supplementary Fig. 4a). Only one branch belonging to P. trichocarpa (ω = 999) is under positive selection in the rooted STM tree (Supplementary Fig. 4b).

Rooted tree fitted for branch models for the KNAT2/6 gene investigated under selection analysis. A unique omega value (dN/dS ratio, ω) is assigned per branch (model 1). Branches in red indicate ω > 1 (positive selection; indicated by arrows), and blue branches indicate ω < 1 (purifying selection).
KNOX genes from Japanese Wisteria show canonical KNOX-like activity in heterologous functional assays
Transcriptomic, phylogenetic, and selection analyses suggest that two KNAT2/6 orthologs (Cluster_W2 and Cluster_1332) are candidates to mediate EC formation. However, neither gene has been formally shown to possess KNOX activity. To directly test this, we turned to heterologous expression in Arabidopsis, as transgenesis has not yet been established in Japanese wisteria. Importantly, this approach has been used to demonstrate the functional conservation of KNOX genes from such evolutionarily distinct genera as Cardamine L., Liriodendron L., Solanum L., and Picea A.Dietr.28,29,30,31. Cluster_1332 was selected as a representative candidate, placed downstream of the strong UBIQUITIN10 promoter (pUBQ:W1332), and pUBQ:W1332 was transformed into wildtype Arabidopsis. The overexpression of Cluster_1332 resulted in strong phenotypes in most primary transformants grown under both long-day (~30 days) and short-day (~90 days) conditions. For instance, transgenic plants were significantly smaller than their wildtype Col-0 counterparts in both experiments (Fig. 7), particularly in short-day plants (Supplementary Fig. 5). The leaves of transformants showed apparent developmental abnormalities, including reduced petiole lengths, rumpled leaf blades, and prolonged growth of their distal serrations (Fig. 7). Vascular aberrations were not observed (Supplementary Fig. 5). However, in short-day plants, transformants showed delayed bolting (~30 days after Col-0), which was characterized by delayed vascular development, including the absence of fibers in the secondary xylem at ~90 days post-germination (Supplementary Fig. 5).

a, b Images of 17-day-old (a) and 23-day-old (b) Arabidopsis seedlings. Compared to wildtype (left), primary transformants carrying pUBQ:W1332 are smaller in stature, and have reduced petiole lengths, rumpled leaves, and prolonged growth of their distal serrations (right). Scale bars = 1 cm.
Discussion
KNOX genes are likely candidates for modulating EC formation in Japanese wisteria
Until recently, information on the molecular regulation of EC formation was obtained exclusively from experimental studies in Populus and A. thaliana, which do not naturally produce EC but develop them through the misexpression of conserved vascular development genes, as reviewed by ref. 7. Given these data and considering the conserved nature of vascular development across seed plants, we initially hypothesized that two developmental pathways lead to the formation of EC, i.e., misexpression of HD-ZIP III genes and WOX4, similar to the observations from experimental mutants7. Although these candidate genes and other markers involved in their regulatory network were annotated in the common bean and Japanese wisteria transcriptome, they were not present in the final analysis of differentially expressed genes due to either not being assigned as one-to-one orthologs across species, not being annotated in one or the other species (e.g., AtHB4, AtHB8, WOX4), not being annotated in both species (e.g., CLE41/42/44, WOX14), or being present in both species and not differentially expressed (e.g., REV). These results reflect differences stemming from both biological variation and methodological limitations (e.g., de novo transcriptomes, unavailable genome for Japanese wisteria). Despite that, we found evidence from various lines of inquiry that KNOX gene are key candidates in regulating EC in Japanese wisteria.
By placing our five differentially expressed KNOX transcript clusters into a phylogenetic context, we demonstrate that, although gene copy number varies across lineages, there is no correlation with the evolution of EC. The expansion of KNOX genes could be derived from different mechanisms, and the specific cause has not been identified in this study. For instance, both WGD and segmental duplications in multiple genes could explain the increase in copy number in eudicots, the Fabaceae32, and the studied grasses33. In addition, paleopolyploidization events have been reported for all sampled angiosperm genera/species34; however, most have gone through diploidization events (see Supplementary Data 4), indicating that associating copy number variation with WGD should be taken with caution. Differences in copy number within some lineages may also result from sampling transcriptomes (e.g., Ipomoea purpurea (L.) Roth, Maianthemum canadense Desf.) rather than genomes. Generating a KNOX gene phylogeny was also crucial for exploring signals in protein evolution; selection analyses indicate that KNOX genes (e.g., KNAT2/6) are under positive selection, as evidenced by two sequences from Japanese wisteria in the transcript clusters obtained through differential expression analysis. In addition to Japanese wisteria, however, contigs from other species with or without EC are also under positive selection. Cases such as Dioscorea, which form underground storage organs and potentially atypical vasculatures, may corroborate selective pressures in the evolution of copies of KNAT2/6 associated with vascular oddities. At the same time, copies of KNOX genes were not found in Cycas panzhihuaensis and Gnetum montanum, which represent two genera of gymnosperms known to form EC35,36. Therefore, more analyses are needed to explore the gene-phenotype relationship at different biological levels, from possible Fabaceae-specific mechanisms to other molecular mechanisms that flowering plants might use compared to gymnosperms.
KNOX1 genes (STM, KNAT1, and KNAT2/6) have been reported to act as major regulators of organ initiation, tissue proliferation, and meristem maintenance, including the formation of vascular cambium20,37,38,39. Given these results and our findings indicating that KNAT2/6 is a contender in EC formation, we overexpressed the Japanese wisteria KNAT2/6 in Arabidopsis thaliana to further elucidate their function. We observed that the transformed plants exhibited numerous aberrations. Significantly, these phenotypes closely resemble those seen in other heterologous KNOX activity assays28,29,30, as well as in Arabidopsis mutants known to accumulate KNAT2/6 in their leaves40,41,42. Despite these morphological observations, overexpression of the Japanese wisteria KNAT2/6 did not cause significant vascular alterations in Arabidopsis mutants. Our findings thus strongly support the idea that Cluster_1332 is a bona fide ortholog of KNAT2/6 with potential roles in EC formation, in addition to their participation in typical cambium development in model species43,44,45. However, the role of these genes in EC development is still not fully understood, just as their function in typical development remains unclear, as discussed further below.
Evidence for the participation of KNAT2/6 in vascular oddities may be noted from its differential expression in mutants of other genes46,47. For instance, higher transcript levels of KNAT6 in knat1 and pny (PENNYWISE, also known as BEL1-LIKE HOMEODOMAIN 9) mutants of A. thaliana have been reported. These mutants exhibit vascular defects ranging from increased cambial activity between the phloem and xylem cells46 to extra vascular bundles that enlarge and reduce the interfascicular space47. Previous research also indicates that cambium regulation in the stems and roots of A. thaliana may be influenced by functional redundancy20,43,45 and antagonistic relationships among KNOX genes, including KNAT1 and KNAT643,48. Notably, these interactions are also observed in other systems, such as leaf development49. Furthermore, KNOX proteins (e.g., KNAT1 and STM) are downstream of the TDIF-PXY-WOX4 signaling pathway, a conserved module responsible for cambium development in woody plants1,14,15, which is implicated in EC formation in experimental mutants in both Arabidopsis and poplar (see also ref. 7). These observations suggest that EC development in Japanese wisteria likely derives from modifications to the TDIF-PXY-WOX4 pathway, possibly leading to changes in the regulation of downstream KNOX genes and their interactions. Our findings that the transcript-cluster identified as KNAT2/6 is downregulated in the “Japanese wisteria: Ectopic v. Typical cambia” contrast concur with the downregulation of other KNOX genes in A. thaliana mutants with EC20. However, just as the mechanism by which KNAT6 functions in vascular development remains largely unclear in model species37,43,45,47, additional studies will be required to clarify how KNAT2/6 plays a role in EC development, as well as the involvement of other KNOX proteins (e.g., STM), which is also downregulated in the contrast comparing “Ectopic Cambia v. Typical Cambia of Japanese wisteria and Common bean”.
Both constitutive and injury-induced ectopic cambia converge on similar molecular processes
KNOX genes also emerge as important regulators underpinning the ability of perturbed Populus trees to form de novo cambia post-girdling21 (see Supplementary Data 5, Supplementary Methods 1 for a comparison of cambial markers across papers on vascular development). In girdled Populus, KNOX genes (e.g., KNAT1, KNAT3, KNAT6) are up-regulated in most stages (e.g., dedifferentiating xylem parenchyma, differentiating cambium). In addition, they are down-regulated in the final stage when a regenerated cambium is finally observed 12 days after girdling, as we observed in the developed EC of Japanese wisteria. Conversely, KNOX genes are up-regulated in experimental mutants in which ectopically-formed cambia are observed, such as Populus HB4-repressed (e.g., KNAT318) and the A. thaliana wox4 (e.g., KNAT1)20 mutants (Supplementary Data 5). Given the contrasting expression patterns comparing EC in natural variants and regenerated cambia in girdled Populus with that of genetically-induced mutants, KNOX genes seem to have been independently co-opted in the molecular regulation of de novo cambia in both constitutively formed (Wisteria vines) and injury-induced (girdled stems of Populus) conditions (see Supplementary Data 6 for a comparison between Japanese wisteria and girdled Populus). Interestingly, KNOX genes are also down-regulated in the cambial zone of unperturbed Populus23, supporting their reduced expression levels in later stages of cambium development. Future studies examining the expression of KNOX genes in EC formation will provide further insight into their role in vascular development in a variety of natural and experimental systems.
In addition to KNOX genes, other major cambium markers, such as phytohormones, are differentially expressed in constitutive and injury-induced EC, suggesting similar mechanisms among these phenomena (see Supplementary Data 5, 6 for gene families differentially expressed across studies). For instance, auxin-related genes are considered master regulators of vascular development because they induce the expression of other conserved transcription factors (e.g., WOX4, PXY, and AtHB8)50, which are implicated in the emergence of EC in experimental mutants51. In line with these findings, changes in gene expression patterns in auxin-related genes (e.g., ARF, AUX, PIN6, TIR; Supplementary Data 6) in both injury-22,52 and non-injury-induced systems (this study) provide additional evidence for shared mechanisms in de novo cambium formation. Another example comes from the plant hormone ethylene. Previous investigations suggest that ETHYLENE RESPONSE FACTORS (AP2/ERF) may be upregulated in the absence of PXY, defining an alternative pathway to the PXY/CLE41 module that regulates cell division in typical cambium development53. Notably, PXY/CLE41 (as well as TDIF and WOX4) are not retained as differentially expressed genes in the comparisons among cambium types in our study (e.g., "Ectopic Cambia v. Typical Cambia of Japanese wisteria and Common bean"). In contrast, AP2/ERF genes are differentially expressed in both Japanese wisteria and girdled Populus, along with other genes from the ethylene signaling cascade (e.g., EIN2 and EIN3) (Supplementary Data 6). These findings strengthen the evidence for alternative regulatory networks involved in both typical and atypical vascular development, including through the interaction of phytohormones and other short-range signals (e.g., peptides)54.
Lastly, constitutive and injury-induced EC share signatures of epigenetic regulation, an area in vascular biology that remains poorly understood1,55,56. We found that numerous methyltransferases were differentially expressed across cambium types, being upregulated in Japanese wisteria EC (Supplementary Data 6). EC in lianas such as Japanese wisteria is potentially triggered by external environmental factors, particularly mechanical constraints associated with their climbing habit. Findings from gene expression analysis with another liana, Bignonia magnifica W.Bull (Bignoniaceae), show that adding external support for the vines to climb triggers changes in the xylem anatomy from self-supporting to liana-like with associated changes in the transcription profiling of the cambium57; thus, external cues relate to anatomical changes in these vines (see Supplementary Data 7). The association between external factors, gene regulation, and atypical vascular architectures has also been reported in shallow underground storage organs, which also thicken through parenchyma proliferation, additional vascular bundles, or EC itself (e.g., beets)58. A notable similarity between underground storage organs and EC in Japanese wisteria is the role of expansin-encoding genes (associated with cell wall loosening) in cell division and proliferation. We found expansin downregulated in the Japanese wisteria EC, a finding also observed in the underground storage organs of Bomarea multiflora (L.f.) Mirb.59. However, their specific role is yet to be further elucidated, given that these genes are up-regulated or down-regulated in different vascular systems (e.g., types of underground storage organs or EC)58.
In conclusion, our work provides anatomical and molecular insights into the unusual emergence of EC formation from cortical parenchyma in Japanese wisteria vines. Amongst the top-most differentially expressed genes are numerous conserved genes involved in hormone signaling, cell division and differentiation, and epigenetic regulation. Our findings identify KNOX genes as key factors in modulating EC development in Japanese wisteria, supported by evidence of positive selection and conserved gene functions observed through heterologous expression in Arabidopsis thaliana. Although none of the HD-ZIP III genes (i.e., REV, AtHB7, AtHB4) that generate EC in experimental mutants were retained as differentially expressed genes in Japanese wisteria, we cannot rule out that this pathway represents an alternative route for the emergence of de novo cambia in natural systems. Future investigations leveraging spatial and time-specific transcriptomes will further elucidate the genetic mechanisms controlling EC and its similarities with de novo cambia in natural and induced vascular phenomena in plants (e.g., cambium regeneration). This knowledge will inform the contribution of conserved genetic pathways leading to wood formation to the repeated evolution of the climbing habit in plants.
Methods
Plants and stem sample collection
Wisteria floribunda (Japanese wisteria) plants were collected in two populations growing in Ithaca, NY, United States. We performed DNA barcoding using the chloroplast intergenic spacer gene “ndhJ-trnF” to confirm the identification of plants belonging to the species W. floribunda11. Phaseolus vulgaris (genotype L88-57; common bean) plants were grown at Cornell University Agricultural Experiment Station Guterman greenhouses (Ithaca, NY) under the following conditions: 14 h light, 75 °F daytime temperature, 65 °F nighttime temperature, and ambient humidity. Samples from these plants were used for developmental anatomy and RNAseq analyses (see“methods” below).
Histology and microscopy
Stem samples of wild plants of common bean and Japanese wisteria were hand-sectioned (with the aid of a razor blade) and processed to generate temporary slides or processed using embedding in polyethylene glycol, sectioning with rotary or sliding microtome, and staining with Safranin and Astra Blue to generate permanent slides60. Slides were imaged using a light microscope (Olympus BH2) equipped with a digital camera (AmScope MU1000) at New York University. Arabidopsis thaliana plants were either hand-sectioned or sectioned with a rotary microtome (Thermo Scientific HM 355S), stained with Toluidine Blue O, and slides were imaged using a light microscope (Olympus BX51) equipped with a Raspberry Pi High Quality at Florida International University.
RNA sequencing and data processing
Sampling and RNA isolation
Developed stem samples were cut into small blocks containing cambium, secondary xylem, and secondary phloem, which were sectioned (~12 µm thick) using a cryostat at −20 °C (tangential cryosectioning approach)23,61 to obtain the tissue for total RNA isolation with RNeasy Mini Kit (QIAGEN). Each sample used for tangential cryosectioning was roughly 6 × 2 × 4 mm. For RNA extraction, about 40–110 sections of each sample containing cambium, secondary xylem, and secondary phloem were used for each replicate (Fig. 2). Six biological replicates were used for the typical cambium of common bean and ectopic cambia (EC) of Japanese wisteria. Five replicates were used for the typical cambium of Japanese wisteria. The same adult plants used for comparative transcriptome analysis were also used for anatomical studies (see above). All sampling was performed at Cornell University.
RNA integrity and quantification
Total RNA was quantified using RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA), and RNA integrity was assessed using an AATI Fragment Analyzer (Agilent Technologies, CA, USA) at Cornell University. Purification, library preparation, and Illumina sequencing were performed by Novogene Co., Ltd (Davis, California). Quality control was performed on the RNA samples using Qubit® 2.0, and RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
Library preparation for transcriptome sequencing
RNA library for poly(A)-dependent RNA-seq was prepared using NEBNext Ultra II RNA Library Prep Kit for Illumina (New England BioLabs). The libraries were subjected to 150 bp paired-end sequencing using NovaSeq 6000 with the v1.5 S4 reagent kit. The library was checked with Qubit and real-time PCR for quantification and a bioanalyzer for size distribution detection.
Transcriptome assembly and annotation
Raw paired-end reads were trimmed for adapter sequences using an overlap stringency of three, a Phred score cut off of twenty, and a minimum sequence length of 50 base pairs (bp) using Trim Galore! version 0.6.662. De novo transcriptome assemblies were developed for P. vulgaris and W. floribunda. For each trimmed dataset, the range of potential kmer values was estimated using Kmergenie version 1.701663. De novo assemblies were generated following part of the TransPi pipeline that uses a multi-assembler approach, followed by merging assemblies, reducing complexity, and annotation64 (see Supplementary Methods 2 for additional details). Within the TransPi pipeline, the UniProt database for Fabales (UniProt Taxon ID 72025)25 was used for annotation (Supplementary Methods 2).
Differential expression and gene ontology enrichment
Trimmed reads were subset to 20 million reads, mapped to their respective transcriptomes with Hisat2 version 2.2.165, including all multiple alignments, and the mate-pair information of the resulting SAM files was fixed using Picard Tools version 2.27.5 FixMateInformation before being converted to BAM files with SAMtools version 1.1466. Corset version 1.0767 was used to group the mapped transcripts into transcript clusters and estimate read counts for each cluster separately for both transcriptomes. Each cluster was annotated using the Trinotate annotation of the longest sequence within the cluster. Clusters were retained if at least two samples in each species had a count of 0.5 or more per million. For the cross-species comparison, Orthofinder2 version 2.5.268 was used with our protein annotations from TransDecoder, and the proteomes of Vigna unguiculata (UniProt Taxon ID: 3917), Cajanus cajan (UniProt Taxon ID: 3821), Lotus japonicus69, Medicago truncatula70 and Mucuna pruriens (UniProt Taxon ID: 157652) from UniProt25 and Phytozome71 to identify orthologs between P. vulgaris and W. floribunda. We identified one-to-one orthologs at the transcript cluster level by removing any clusters with more than one ortholog group and any ortholog groups that contained sequences from more than one cluster. Differential cluster expression was tested using limma72 and voom73 comparing between species regardless of cambium type, between cambium types regardless of species, between the typical cambium of each species, and between the ectopic and typical cambium of Japanese wisteria (see Supplementary Methods 3 for additional details). Gene ontology enrichment was tested using the differentially expressed transcript clusters for each using GOseq74. To better leverage the repeated sampling within an individual, Japanese wisteria clusters were tested separately from the cross-species comparison using differential expression for repeated measures (dream26) with voom73 and variancePartition75 to compare across cambium types with individual as a random effect. Gene ontology enrichment for differentially expressed clusters was implemented in GOseq. For comparison with the cross-species analysis, ortholog groups were subsequently mapped back to the differentially expressed clusters identified by Dream. For all statistical analyses, p-values were adjusted for false discovery rates using a Benjamini-Hochberg procedure using two-sided tests.
Phylogenetic and selection analyses
To investigate the role of KNOX genes in EC formation across seed plants, we performed a phylogenetic analysis and selection analysis for this gene family. Candidate genes were identified from available genomes and transcriptomes using a BLAST pipeline that was previously used for investigating gene evolution across angiosperms76. Coding sequences (cds) were downloaded from Genbank or TreeGenes for 39 species, and raw RNA-seq files were downloaded from the SRA for six species (Supplementary Data 2.). In total, 45 species were included in the analysis, 11 containing EC. Raw sequencing files were cleaned with fastp version 0.23.377 and assembled with Trinity version 2.14.077,78 with the following flags: --max_memory 200 G --no_normalize_reads --min_contig_length 250 --trimmomatic. Reference sequences of candidate genes from A. thaliana were downloaded from Genbank, and a tBLASTx search was performed using blast version 2.1379. A maximum of 30 hits per taxon were retained if the matching length was 10 bp and an e-value of 0.0001. A fasta file containing all retrieved sequences was generated and checked for duplicates using the sRNAtoolbox80. Twenty-one sequences from different KNOX genes and taxa were downloaded from Genbank and included as additional references to identify specific clades (Supplementary Data 3). A species tree to depict this sampling (Fig. 4) was constructed using the R package V.PhyloMaker2 with scenario 3 for taxa binding81. Finally, the sequences of five transcript clusters identified as KNOX genes were included in the phylogenetic analysis. The resulting fasta file for phylogenetic reconstruction contained 466 sequences (Supplementary Methods 4, Supplementary Data 8 for additional details). These contigs were first aligned with MAFFT version 7.5282 with the following options: --auto --adjustdirectionaccurately --op 3 --leavegappyregion. The sequences were then realigned with the codon-aware aligner MACSE version 2.0783 and the alignSequences command. The resulting alignment was cleaned up with trimAL version 1.4.184 with the -gappyout option before converting premature stop codons to NNN while retaining the final stop codons with the exportAlignment command in MACSE, which were cleaned up with trimAL version 1.4.184. See Supplementary Dataset 9, 10. for aligned and unaligned sequences. Tree building was performed repeatedly to exclude poorly aligned and gappy contigs. Trees were generated with RaxML version 8.2.12 with the GTRGAMMA model of molecular evolution and 100 bootstrap replicates85.
For further investigation of specific candidate genes, a fasta file containing the nucleotide sequences of specific KNOX genes identified in the previous tree was aligned with MAFFT version 7.52, realigned with the codon-aware aligner MACSE version 2.07, and then cleaned with trimAL version 1.4.1. The inferred phylogeny and cleaned alignment were input into PAML version 10.627 for codeml analysis to explore signatures of selection. Both model = 0 (one omega value for the entire alignment), model = 1 (omega value for each branch), and model = 2 (foreground and background omega values) were conducted on both the rooted and unrooted trees. Branch-specific omega values were plotted on the phylogeny with the R package ggtree version 3.6.286.
Molecular cloning and plant transformation
A genomic fragment of Cluster_1332 (KNAT2/6) from the start codon to the stop codon was PCR amplified from Japanese wisteria genomic DNA as a 4936 bp band using W1332 F pUBQ10 OL and W1332 R pcr8 OL primers. A 4812 bp fragment comprised of pUBQ10 and the pCR/GW/TOPO vector was amplified from an existing template using pCR8 F W1332 OL and pUBQ10 pCR8 R W1332 OL primers. Fragments were assembled using Gibson assembly (NEBuilder® HiFi DNA Assembly Master Mix Cat. E2621L) and sequence validated using Plasmidsaurus. The resulting pUBQ10:W1332 cassette was then cloned into a modified pB7GW binary vector (VIB Ghent) using Gateway LR technology (Gateway™ LR Clonase™ II Enzyme mix Cat. 11791020) and transformed into the Col-0 ecotype of Arabidopsis thaliana using standard Agrobacterium-mediated floral dip protocols. Primary transformants were grown at 22 °C under long (16-h light, 8-h dark) and short (8-h light, 16-h dark) day conditions on soil for 4 weeks (~30 days) or 12 weeks (~90 days) post-germination, respectively. Plants were grown and imaged at the University of Pennsylvania. Cloning primers are listed in Supplementary Data 11.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets generated and/or analyzed during the current study are publicly available. The RNA sequencing data are available in SRA [SRR28389658], [SRR29802342-SRR29802358]. Other datasets generated in this study have been deposited in the GitHub database (Cunha-Neto et al.87; https://github.com/anthonysnead/Fabaceae-Ectopic_Cambia_Transcriptomics) and Figshare (Cunha-Neto et al.88; https://doi.org/10.6084/m9.figshare.26524381). Source data are provided with this paper.
Code availability
A custom R script was written for gene expression analyses and is available on GitHub (https://github.com/anthonysnead/Fabaceae-Ectopic_Cambia_Transcriptomics).
References
Hunziker, P. & Greb, T. Stem cells and differentiation in vascular tissues. Annu. Rev. Plant Biol. 75, 11.1–11.27 (2024).
Lanner, R. M. Why do trees live so long? Ageing Res. Rev. 1, 653–671 (2002).
Sillett, S. C. et al. Increasing wood production through old age in tall trees. For. Ecol. Manag. 259, 976–994 (2010).
Rowe, N. P. & Speck, T. The evolution of angiosperm lianescence: a perspective from xylem structure-function. in Ecology of lianas. 221–250 (JohnWiley & Sons, Chichester, 2015).
Fischer, J. B. & Ewers, F. W. Structural responses to stem injury in vines. in The Biology of Vines. 99–124 (Cambridge University Press, Cambridge, 1991).
Cunha-Neto, I. L. Vascular variants in seed plants—a developmental perspective. AoB PLANTS 15, 1–15 (2023).
Cunha-Neto, I. L. & Onyenedum, J. G. Ectopic cambia: connections between natural and experimental vascular mutants. Am. J. Bot. 110, e16246 (2023).
Terrazas, T., Aguilar-Rodríguez, S. & Ojanguren, C. T. Development of successive cambia, cambial activity, and their relationship to physiological traits in Ipomoea arborescens (Convolvulaceae) seedlings. Am. J. Bot. 98, 765–774 (2011).
Robert, E. M. R. et al. Successive cambia: a developmental oddity or an adaptive structure? PLoS ONE 6, e16558 (2011).
Nejapa, R., Cabanillas, P. A. & Pace, M. R. Cortical origin of the successive cambia in the stems of the charismatic temperate lianescent genus Wisteria (Fabaceae) and its systematic importance. Bot. J. Linn. Soc. 199, 667–677 (2022).
Cunha-Neto, I. L. & Onyenedum, J. G. How to climb and grow stronger: lessons from ornamental wisterias. Arnoldia 80, 38–47 (2023).
Trusty, J. L., Lockaby, B. G., Zipperer, W. C. & Goertzen, L. R. Horticulture, hybrid cultivars and exotic plant invasion: a case study of Wisteria (Fabaceae). Bot. J. Linn. Soc. 158, 593–601 (2008).
Tamaio, N., Vieira, R. C. & Angyalossy, V. Origin of successive cambia on the stem in three species of Menispermaceae. Rev. bras. Bot. 32, 839–848 (2009).
Turley, E. K. & Etchells, J. P. Laying it on thick: a study in secondary growth. J. Exp. Bot. 73, 665–679 (2022).
Groover, A. The vascular cambium revisited. IAWA J. 44, 531–538 (2023).
Robischon, M., Du, J., Miura, E. & Groover, A. The Populus class III HD ZIP, popREVOLUTA, influences cambium initiation and patterning of woody stems. Plant Physiol. 155, 1214–1225 (2011).
Zhu, Y., Song, D., Sun, J., Wang, X. & Li, L. PtrHB7, a class III HD-zip gene, plays a critical role in regulation of vascular cambium differentiation in Populus. Mol. Plant 6, 1331–1343 (2013).
Zhu, Y., Song, D., Xu, P., Sun, J. & Li, L. An HD-ZIP III gene, PtrHB4, is required for interfascicular cambium development in Populus. Plant Biotechnol. J. 16, 808–817 (2018).
Kucukoglu, M., Nilsson, J., Zheng, B., Chaabouni, S. & Nilsson, O. WUSCHEL - RELATED HOMEOBOX 4 (WOX 4) -like genes regulate cambial cell division activity and secondary growth in Populus trees. N. Phytol. 215, 642–657 (2017).
Zhang, J. et al. Transcriptional regulatory framework for vascular cambium development in Arabidopsis roots. Nat. Plants 5, 1033–1042 (2019).
Zhang, J. et al. Molecular features of secondary vascular tissue regeneration after bark girdling in Populus. N. Phytol. 192, 869–884 (2011).
Zhang, Y., Wang, X., Zhang, J. & He, X.-Q. Plant in situ tissue regeneration: dynamics, mechanisms and implications for forestry research. For. Res. 3, 3–8 (2023).
Schrader, J. et al. A high-resolution transcript profile across the wood-forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell 16, 2278–2292 (2004).
Bryant, D. M. et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776 (2017).
UniProt Consortium UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res 51, D523–D531 (2023).
Hoffman, G. E. & Roussos, P. Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics 37, 192–201 (2021).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Hay, A. & Tsiantis, M. KNOX genes: versatile regulators of plant development and diversity. Development 137, 3153–3165 (2010).
Ma, J., Mei, G., Liu, H. & Li, H. Overexpression of a novel LcKNOX transcription factor from liriodendron chinense induces lobed leaves in arabidopsis thaliana. Forests 11, 33 (2020).
Larsson, E., Sitbon, F. & von Arnold, S. Differential regulation of Knotted1-like genes during establishment of the shoot apical meristem in Norway spruce (Picea abies). Plant Cell Rep. 31, 1053–1060 (2012).
Shani, E. et al. Stage-specific regulation of solanum lycopersicum leaf maturation by class 1 KNOTTED1-LIKE HOMEOBOX proteins. Plant Cell 21, 3078–3092 (2009).
Zhuang, Y. et al. Phylogenomics of the genus Glycine sheds light on polyploid evolution and life-strategy transition. Nat. Plants 8, 233–244 (2022).
Landis, J. B. et al. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 105, 348–363 (2018).
Zhang, T. et al. Phylogenomic profiles of whole-genome duplications in Poaceae and landscape of differential duplicate retention and losses among major Poaceae lineages. Nat. Commun. 15, 3305 (2024).
Terrazas, T. Origin and activity of successive cambia in cycas (Cycadales). Am. J. Bot. 78, 1335–1344 (1991).
Carlquist, S. Wood anatomy of Gnetales in a functional, ecological, and evolutionary context. Aliso https://doi.org/10.5642/aliso.20123001.05 (2012).
Groover, A. T. et al. The Populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant Mol. Biol. 61, 917–932 (2006).
Du, J., Mansfield, S. D. & Groover, A. T. The Populus homeobox gene ARBORKNOX2 regulates cell differentiation during secondary growth. Plant J. 60, 1000–1014 (2009).
Liebsch, D. et al. Class I KNOX transcription factors promote differentiation of cambial derivatives into xylem fibers in the Arabidopsis hypocotyl. Development 141, 4311–4319 (2014).
Byrne, M. E. et al. Asymmetric leaves1 mediates leaf patterning and stem cell function in Arabidopsis. Nature 408, 967–971 (2000).
Semiarti, E. et al. The ASYMMETRIC LEAVES2 gene of Arabidopsis thaliana regulates the formation of a symmetric lamina, the establishment of venation and the repression of meristem-related homeobox genes in leaves. Development 128, 1771–1783 (2001).
Byrne, M. E., Simorowski, J. & Martienssen, R. A. ASYMMETRIC LEAVES1 reveals knox gene redundancy in Arabidopsis. Development 129, 1957–1965 (2002).
Woerlen, N. et al. Repression of BLADE-ON-PETIOLE genes by KNOX homeodomain protein BREVIPEDICELLUS is essential for differentiation of secondary xylem in Arabidopsis root. Planta 245, 1079–1090 (2017).
Kim, M.-H. et al. Wood transcriptome profiling identifies critical pathway genes of secondary wall biosynthesis and novel regulators for vascular cambium development in Populus. Genes 10, 690 (2019).
Zhao, Y. et al. KNAT 2/6b, a class I KNOX gene, impedes xylem differentiation by regulating NAC domain transcription factors in poplar. N. Phytol. 225, 1531–1544 (2020).
Smith, H. M. S. & Hake, S. The interaction of two homeobox genes, BREVIPEDICELLUS and PENNYWISE, regulates internode patterning in the Arabidopsis inflorescence. Plant Cell 15, 1717–1727 (2003).
Khan, M. et al. Antagonistic Interaction of BLADE-ON-PETIOLE1 and 2 with BREVIPEDICELLUS and PENNYWISE regulates Arabidopsis inflorescence architecture. Plant Physiol. 158, 946–960 (2012).
Ragni, L., Belles-Boix, E., Günl, M. & Pautot, V. Interaction of KNAT6 and KNAT2 with BREVIPEDICELLUS and PENNYWISE in Arabidopsis Inflorescences. Plant Cell 20, 888–900 (2008).
Champagne, C. E. M. et al. Compound leaf development and evolution in the legumes. Plant Cell 19, 3369–3378 (2007).
Johnson, L. A. & Douglas, C. J. Populus trichocarpa MONOPTEROS/AUXIN RESPONSE FACTOR5 (ARF5) genes: comparative structure, sub-functionalization, and Populus – Arabidopsis microsynteny. This article is one of a selection of papers published in the Special Issue on Poplar Research in Canada. Can. J. Bot. 85, 1058–1070 (2007).
Smetana, O. et al. High levels of auxin signalling define the stem-cell organizer of the vascular cambium. Nature 565, 485–489 (2019).
Chen, J. et al. Differential regulation of auxin and cytokinin during the secondary vascular tissue regeneration in Populus trees. N. Phytol. 224, 188–201 (2019).
Etchells, J. P., Provost, C. M. & Turner, S. R. Plant vascular cell division is maintained by an interaction between PXY and ethylene signalling. PLoS Genet 8, e1002997 (2012).
Wybouw, B., Zhang, X. & Mähönen, A. P. Vascular cambium stem cells: past, present and future. N. Phytol. 243, 851–865 (2024).
Dai, X. et al. Cell-type-specific PtrWOX4a and PtrVCS2 form a regulatory nexus with a histone modification system for stem cambium development in Populus trichocarpa. Nat. Plants 9, 96–111 (2023).
Groover, A. Roles for epigenetics in wood formation and stress response in trees–from basic biology to forest management. Front. Epigenet. Epigenom. https://doi.org/10.3389/freae.2025.1476499 (2025).
Lima, A. C. et al. Liana's attachment to supports leads to profound changes in xylem anatomy and transcriptional profile of cambium and differentiating xylem. Plant, Cell Environ. 47, 5172–5188 (2024).
Plunkert, M. L., Martínez-Gómez, J., Madrigal, Y., Hernández, A. I. & Tribble, C. M. Tuber, or not tuber: molecular and morphological basis of underground storage organ development. Curr. Opin. Plant Biol. 80, 102544 (2024).
Tribble, C. M., Martínez-Gómez, J., Alzate-Guarín, F., Rothfels, C. J. & Specht, C. D. Comparative transcriptomics of a monocotyledonous geophyte reveals shared molecular mechanisms of underground storage organ formation. Evol. Dev. 23, 155–173 (2021).
Barbosa, A. C. F., Pace, M. R., Witovisk, L. & Angyalossy, V. A new method to obtain good anatomical slides of heterogeneous plant parts. IAWA J. 31, 373–383 (2010).
Qiu, Z. et al. Genome-wide analysis reveals dynamic changes in expression of microRNAs during vascular cambium development in Chinese fir, Cunninghamia lanceolata. J. Exp. Bot. 66, 3041–3054 (2015).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Chikhi, R. & Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37 (2014).
Rivera-Vicéns, R. E., Garcia-Escudero, C. A., Conci, N., Eitel, M. & Wörheide, G. TransPi-a comprehensive Transcriptome Analysis Pipeline for de novo transcriptome assembly. Mol. Ecol. Resour. 22, 2070–2086 (2022).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Davidson, N. M. & Oshlack, A. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 15, 410 (2014).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Li, H., Jiang, F., Wu, P., Wang, K. & Cao, Y. A high-quality genome sequence of model legume lotus japonicus (MG-20) provides insights into the evolution of root nodule symbiosis. Genes 11, 483 (2020).
Tang, H. et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 15, 312 (2014).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14 (2010).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinforma. 17, 483 (2016).
Phillips, H. R., Landis, J. B. & Specht, C. D. Revisiting floral fusion: the evolution and molecular basis of a developmental innovation. J. Exp. Bot. 71, 3390–3404 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652 (2011).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 421 (2009).
Aparicio-Puerta, E. et al. sRNAbench and sRNAtoolbox 2022 update: accurate miRNA and sncRNA profiling for model and non-model organisms. Nucleic Acids Res. 50, W710–W717 (2022).
Jin, Y. & Qian, H. V. PhyloMaker2: an updated and enlarged R package that can generate very large phylogenies for vascular plants. Plant Divers. 44, 335–339 (2022).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Ranwez, V., Harispe, S., Delsuc, F. & Douzery, E. J. P. MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLOS ONE 6, e22594 (2011).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Cunha-Neto, I. L., Snead, A. A., Landis, J. B., Callery, N. I. & Husbands, A. Y. et al. Ectopic cambia in Japanese wisteria (Wisteria floribunda) vines are associated with the expression of conserved KNOX genes. GitHub https://doi.org/10.5281/zenodo.18200460 (2026).
Cunha-Neto, I. L., Snead, A. A., Landis, J. B., Callery, N. I. & Husbands, A. Y. et al. Ectopic cambia in Japanese wisteria (Wisteria floribunda) vines are associated with the expression of conserved KNOX genes. Figshare https://doi.org/10.6084/m9.figshare.26524381 (2026).
Acknowledgments
We thank undergraduate research technician Danielle C. Sonnenleiter (Cornell University) for assistance in data collection, past and current members of the Onyenedum lab for continuous feedback, especially Angelique A. Acevedo, for assistance in growing the bean plants, and Mariane S. S. Baena for insightful discussions. We also thank Jocelyn Rose’s lab at Cornell University for allowing access to the cryostat. We want to acknowledge the Arnold Arboretum of Harvard University for providing access to the living collections and financial support through a Sargent Award for Visiting Scholars (I.L.C.N.). This work was supported in part through the NYU IT High Performance Computing resources, services, and staff expertise, as well as the Boyce Thompson Institute’s Computational Biology Center and Cornell’s BioHPC. This work was funded by startup laboratory funds from Cornell University’s College of Agriculture and Life Sciences and New York University, and NSF 2401675 to J.G.O., and startup laboratory funds from Florida International University to I.L.C.N.
Author information
Authors and Affiliations
Contributions
Conceptualization: I.L.C.-N. J.G.O. Methodology: I.L.C.-N., A.A.S., J.B.L., N.I.C., A.Y.H., C.D.S, and J.G.O. Investigation: I.L.C.-N., J.B.L., A.A.S., N.I.C., A.Y.H., and J.G.O. Data curation: I.L.C.-N., A.A.S., and A.Y.H. Writing – Original Draft: I.L.C.-N., J.B.L., A.A.S., and J.G.O. Writing – Review & Editing: I.L.C.-N., A.A.S., J.B.L., N.I.C., A.Y.H., C.D.S., and J.G.O.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cunha-Neto, I.L., Snead, A.A., Landis, J.B. et al. Ectopic cambia in wisteria vines are associated with the expression of conserved KNOX genes. Nat Commun 17, 2190 (2026). https://doi.org/10.1038/s41467-026-68669-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68669-w
