
Abstract
G proteins and arrestins are key transducers for G protein-coupled receptor (GPCR) signaling, mediating distinct downstream pathways. Recent evidence suggests that G proteins and β-arrestins (βarrs) can directly or functionally interact. However, the molecular details and functional consequences of Gα–βarr interactions remain poorly understood. Here, we quantify the binding affinities between βarr1 and Gαs or Gαi1 in various activation states using microscale thermophoresis (MST). βarr1 in the active conformational ensemble state favors binding, whereas Gα activation status is less determinant. Hydrogen/deuterium exchange mass spectrometry reveals distinct conformational changes between Gαs versus Gαi1 upon βarr1 binding, suggesting differential binding mechanism between Gαs–βarr1 and Gαi1–βarr1 complexes. Both the Ras-like domain and the α-helical domain of Gα contribute to complex formation. Functionally, a BODIPY-FL–GTPγS assay shows that βarr1 does not alter GDP/GTP turnover of Gαs or Gαi1, whereas β-strand XX (βXX) release assays demonstrate that Gαs enhances βarr1 C-tail release. Together, these results propose molecular mechanism of the interaction and asymmetric functional coupling within Gα–βarr complexes and uncover a previously underappreciated layer of GPCR signal transduction.
Similar content being viewed by others


Introduction
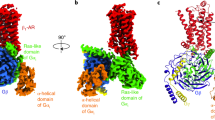
G proteins and arrestins are the two major downstream signaling molecules of G protein-coupled receptors (GPCRs)1,2,3. Activated GPCRs interact with the Gα subunit of the heterotrimeric G protein, releasing guanosine diphosphate (GDP) from Gα. The empty Gα is rapidly occupied with guanosine triphosphate (GTP), activating the G proteins (Fig. 1a)4,5,6. Based on sequence homology and signaling properties, Gα subunits are classified into four subfamilies: Gαs, Gαi/o, Gαq/11, and Gα12/134.

a Schematic diagram of G protein-coupled receptor (GPCR) signal transduction. Agonist-induced receptor activation initiates two major signaling pathways. On one hand, the activated receptor recruits and activates heterotrimeric G proteins, triggering guanosine diphosphate (GDP)/guanosine triphosphate (GTP) exchange on the Gα subunit, followed by Gα and Gβγ dissociation, leading to distinct G protein-mediated downstream signaling. On the other hand, the activated receptor recruits GPCR kinase (GRK), which phosphorylates the receptor. Subsequently, the phosphorylated receptor promotes βarr recruitment and activation, resulting in receptor desensitization, internalization, or βarr-mediated signaling. b Structural features of basal βarr1 (gray, PDB: 1G4M). The auto-inhibitory C-terminal tail is highlighted in red. The polar core is indicated by an orange circle. The three-element interaction comprising β-strand XX (red), the αI, and β-strand I (blue) is marked in a blue circle, with hydrophobic side chains shown in yellow. c Structural superimposition of basal βarr1 (gray, PDB: 1G4M) and V2Rpp-bound active βarr1 (wheat, PDB: 4JQI). V2Rpp is shown as a green ribbon. Structural rearrangement upon activation, including loop rearrangement and C-terminal tail release, is highlighted in dark orange. Activation-induced interdomain rotation is shown as a black curved arrow. d–o Microscale thermophoresis (MST)-based affinity measurement between βarr1 and Gαs or Gαi1 in different activation states. The difference in normalized fluorescence ΔFnorm[‰] of labeled βarr1 is plotted as a function of the concentration of Gαs or Gαi1. Data points represent the mean values ± SEM from 3–4 independent experiments. Curve fitting was performed using GraphPad Prism 10. The dissociation constant is obtained using the Kd mode of MO.Affinity Analysis software (v2.3) and presented as the best-fit Kd ± Kd confidence, corresponding to a 68% confidence level (~1 standard deviation). MST experiments were performed at 20% excitation power and medium MST power.
The activated GPCRs are also phosphorylated and interact with arrestins; the receptor-bound arrestins desensitize the G protein-mediated signaling7,8, regulate GPCR internalization, and/or initiate G protein-independent signal transduction (Fig. 1a)8,9,10,11. Arrestins are composed of four subtypes, arrestin1–4; arrestin1 and 4 are primarily expressed in the visual system, while arrestin 2 and 3 (also called β-arrestin1 and 2 [βarr1 and βarr2], respectively) are expressed ubiquitously7,10,12.
Most studies have focused on the interaction between GPCRs and their downstream signaling molecules; however, emerging evidence suggests that G proteins and arrestins can physically or functionally interact13,14,15,16, βarr1 directly interacts with Gαs16, GPCRs can form a “mega-complex” comprising both G proteins and βarrs15, and receptor-mediated Gαi–βarr complexes facilitate extracellular signal-regulated kinase 1/2 (ERK1/2) activation14. Despite these findings, the precise functional and structural mechanisms of βarr–Gα interactions remain poorly defined.
Herein, we studied the functional and structural interaction between βarr and Gα. As previous reports suggest that βarr1 can directly or functionally interact with Gαs or Gαi113,14,15,16, we selected βarr1 as a representative βarr and investigated its interaction with Gαs or Gαi1. We measured the binding affinities between βarr1 and Gαs or Gαi1 under various activation states using microscale thermophoresis (MST). The functional implications of these interactions were investigated using BODIPY-FL–GTPγS for Gα activation analyses and using the house-developed β-strand XX (βXX) release assay and an intramolecular bioluminescence resonance energy transfer (BRET) sensor for βarr1 activation analyses. The structural mechanism and binding interfaces between βarr1 and Gαs or Gαi1 interactions were analyzed using hydrogen/deuterium exchange mass spectrometry (HDX-MS). Together, these approaches provide a comprehensive framework for understanding βArr–Gα interaction at the molecular level.
Results
Interaction between different activation states of βarr1 and Gα
To test whether βarr1 physically interacts with Gαs or Gαi1, we measured the binding affinity (Kd) between purified βarr1 and purified Gαs or Gαi1 using MST. To assess how Gα activation state influences these interactions, we prepared Gα in GDP-bound (inactive) or GTPγS-bound (active) conformations.
The arrestin activation status can be modulated by introducing mutations or adding phosphorylated GPCRs. Basal state arrestins are stabilized by the interactions between the N- and C-domains, specifically through the interaction between βI, αI, and C-terminal βXX (three-elements interaction, Fig. 1b, blue circle) as well as the ionic interactions between the residues in βIII, βX, gate loop, and C-tail (polar core interaction, Fig. 1b, orange circle)17,18. Upon phosphorylated GPCR binding to the N-domain of arrestins, βXX is released from the N-domain, and the polar core is disrupted (Fig. 1c). Disruption of the interdomain interactions induces domain rotation and conformational changes of the loops between the two domains (such as finger, middle, lariat, and middle loops; Fig. 1c)19,20. These structural changes are canonical features of arrestin activation.
However, arrestin activation does not follow a simple binary switch between basal and active states. Instead, arrestins undergo dynamic conformational states that are finely tuned by the activating receptor, its ligand, and the receptor’s phosphorylation pattern. These inputs shape globally and locally distinct active states that selectively recruit downstream partners, as proposed in the barcode or flute models of arrestin signaling21,22,23,24.
Truncation of the C-terminus, including βXX, transforms arrestins into an “active conformational ensemble state” by mimicking βXX release but without occupying the phosphorylated receptor-binding pockets. This active conformational ensemble state is relieved from autoinhibition but not locked into a specific active conformation and therefore has been widely used to study the mechanism of arrestin activation19,25,26,27.
In this study, we used full-length βarr1 to represent the basal state, C-terminally truncated βarr1 (βarr1_ΔC) to represent the active conformational ensemble state rather than a single active state, and V2Rpp-bound βarr1 (V2Rpp, a phosphopeptide from the vasopressin type-2 receptor) as one specific receptor-tail–stabilized active conformer13,19,28.
When the interaction between the basal state βarr1 and GDP-bound or GTPγS-bound Gαs was analyzed using MST, the interactions were insignificant (Fig. 1d,e). Similarly, GTPγS-bound Gαi1 did not significantly interact with the basal state βarr1 (Fig. 1g). However, the GDP-bound Gαi1 interacted with basal state βarr1 (Kd = 290.48 ± 111.99 nM) with relatively low MST response amplitude (approximately 5‰) (Fig. 1f). When the interaction between the V2Rpp-bound βarr1 and GDP-bound or GTPγS-bound Gα was analyzed using MST, the interactions were insignificant for all tested conditions (Fig. 1h–k). Conversely, βarr1_ΔC significantly interacted with all tested Gα proteins (GDP-bound Gαs, GTPγS-bound Gαs, GDP-bound Gαi1, and GTPγS-bound Gαi1) with similar affinities (Kd: 80.68–161.92 nM; Fig. 1l–o).
These results indicate that Gαs and Gαi1, whether in their inactive or active states, preferentially interact with the active conformational ensemble state βarr1 (βarr1_ΔC) but not with basal or V2Rpp-specific active states. V2Rpp occupancy at phosphate-binding pockets may favor a particular active-like conformer that does not engage Gα efficiently. On the contrary, C-tail truncation permits interconversion among multiple active-like sub-states, increasing the probability of populating a Gα-compatible conformation. Of note, GDP-bound Gαi1 also engages basal βarr1, albeit with significantly lower affinity than with βarr1_ΔC (Supplementary Fig. 1b).
HDX analyses upon complex formation between βarr1_ΔC and Gαs or Gαi1
To identify interaction interfaces and conformational changes associated with βarr1–Gα complex formation, we performed HDX-MS. HDX-MS probes the conformational dynamics of proteins by measuring the exchange rate between the amide hydrogens in the proteins and deuterium in the solvent29,30. This method is particularly effective in detecting protein–ligand interfaces and conformational changes, even in systems with weak or transient interactions31,32. Upon complex formation, solvent accessibility typically decreases at the protein–protein interface, reducing HDX rate. Additionally, an altered HDX rate indicates altered backbone flexibility or changes in secondary structure stability.
Given the stronger binding affinity of βarr1_ΔC, we co-incubated purified βarr1_ΔC with GTPγS-bound Gαs or Gαi1, followed by HDX at various durations (10, 100, 1000, and 10,000 s) in D₂O buffer. Control samples included βarr1_ΔC, GTPγS–Gαs, and GTPγS–Gαi1 alone. After HDX, the samples were digested with pepsin, and peptide-level deuterium uptake was analyzed. The peptic peptides used for HDX-MS analyses are summarized in Supplementary Fig. 2, and the detailed information about the HDX-MS conditions and the peptic peptides is described in Supplementary Data 1 and 2, respectively.
Upon βarr1_ΔC binding, Gαs exhibited altered HDX in specific regions. Notably, the αD/αE loop showed enhanced deuterium uptake (Fig. 2a, red; Supplementary Fig. 3a, peptide 169–176), suggesting increased conformational flexibility in this region. In contrast, HDX decreased in multiple regions, including the αA/αB loop, αE/αF loop, switch I, and αG/α4 loop (Fig. 2a, blue; Supplementary Fig. 3a, peptides 108–118, 190–197, 201–208, and 316–331), indicating reduced solvent accessibility or increased structural stabilization upon complex formation.

a Changes in the HDX profile of GTPγS-bound Gαs co-incubated with C-terminal truncated βarr1 (βarr1_ΔC). Regions with reduced and increased HDX are colored in blue and red, respectively, on the Gαs structure (PDB: 6EG8). The deuterium uptake plots of selected peptides with HDX changes are shown in Supplementary Fig. 3a. b Changes in the HDX profile of GTPγS-bound Gαi1 upon co-incubation with βarr1_ΔC. Regions with reduced HDX are colored in blue on the Gαi1 structure (PDB: 1GP2). The deuterium uptake plots of selected peptides with HDX changes are shown in Supplementary Fig. 3b. c Changes in the HDX profile of βarr1_ΔC upon co-incubation with GTPγS-bound Gαs. Regions with reduced and increased HDX are colored in blue and red, respectively, on the basal state βarr1 structure (PDB: 1G4M). The deuterium uptake plots of selected peptides with HDX changes are shown in Supplementary Fig. 3c. d Changes in the HDX profile of βarr1_ΔC co-incubation with GTPγS-bound Gαi1. Regions with reduced HDX are colored in blue on the basal state βarr1 structure (PDB: 1G4M). The deuterium uptake plots of selected peptides with HDX changes are shown in Supplementary Fig. 3d. a–d White regions represent areas with no significant difference in HDX. Peptide coverage is shown in Supplementary Fig. 2. e Structural alignment of guanosine diphosphate (GDP)-bound Gαs (gray, PDB: 6EG8) with nucleotide-free Gαs (light pink for Ras-like domain [RD] and magenta for α-helical domain [AHD], PDB: 3SN6), illustrating the receptor-induced opening of the AHD (magenta) of Gα subunit. The receptor (β2-adrenergic receptor) is shown in green (PDB: 3SN6).
Co-incubation with βarr1_ΔC decreased the HDX of Gαi1 in the αA/αB loop extending through αB, αD/αE loop, switch II, α3 helix, and adjacent α3/β5 loop, and the C-terminal part of α5 (Fig. 2b, blue; Supplementary Fig. 3b), suggesting that these regions may participate in complex formation or undergo conformational stabilization.
We next assessed the conformational changes in βarr1_ΔC upon complex formation. Binding to GTPγS–Gαs-induced minimal HDX changes in βarr1_ΔC. A small increase in HDX was observed in the βIV/βV loop (Fig. 2c, red; Supplementary Fig. 3c, peptide 43–54), suggesting localized flexibility, while a minor decrease in the lariat loop (Fig. 2c, blue; Supplementary Fig. 3c, peptide 282–289) indicated slight structural stabilization, although the effect on the lariat loop was modest (<0.2 Da). A similar pattern was observed upon binding to GTPγS–Gαi1, where modest HDX reductions were detected in the finger loop (Fig. 2d, blue; Supplementary Fig. 3d, peptide 69–75) and lariat loop (Fig. 2d, blue; Supplementary Fig. 3d, peptide 279–289), again with low magnitude changes (<0.2 Da).
Collectively, the HDX-MS results indicate that complex formation with βarr1_ΔC distinctly changes deuterium uptake across multiple regions of both Gαs and Gαi. Conversely, βarr1_ΔC exhibited only modest changes in HDX upon binding to either Gαs or Gαi1, with localized effects observed in loop regions, such as the βIV/βV, finger, and lariat loops. While the magnitude of these changes was small, they may reflect Gα binding-associated subtle conformational rearrangements and warrant further structural investigation.
Domain-level contributions of Gαs and Gαi1 to βarr1 binding
While HDX-MS provides valuable insights into regions affected by βarr1–Gα complex formation, the observed changes may reflect either direct contact surfaces or allosteric rearrangements. To directly assess the structural domains of Gα subunits contributing to βarr1 binding, we dissected the interaction using individual domains of Gαs and Gαi1. Gα subunits are composed of a Ras-like GTPase domain (RD) and an α-helical domain (AHD), which together coordinate nucleotide binding and hydrolysis (Fig. 2e). Nucleotide exchange is closely linked to conformational rearrangements between these two domains, particularly AHD displacement.
Our HDX-MS data showed βarr1_ΔC-induced HDX changes in both RD and AHD regions of Gαs and Gαi1 (Fig. 2a, b), suggesting that both domains might contribute to the interaction interface. To test this, we employed truncated constructs: miniGα variants (lacking the AHD)33 and isolated AHD domains (residues 68–192 for GαsAHD and 61–183 for Gαi1AHD) (Supplementary Fig. 4). We then quantified their binding to βarr1_ΔC using MST.
The miniGαs construct displayed a significantly reduced binding affinity to βarr1_ΔC (Kd = 463.96 ± 140.49 nM) than the full-length GDP- or GTPγS-bound Gαs (Fig. 3a; Supplementary Fig. 1a). Conversely, the isolated GαsAHD retained strong binding to βarr1_ΔC (Kd = 47.24 ± 14.98 nM), comparable to the full-length Gαs under both nucleotide states (Fig. 3b). These findings suggest that the Gαs AHD plays a dominant role in mediating the interaction with βarr1_ΔC, whereas the Gαs RD is less critical.

a–d Microscale thermophoresis (MST)-based affinity measurement between βarr1_ΔC and miniGα or GαAHD. The change in normalized fluorescence ΔFnorm[‰] of labeled βarr1_ΔC is plotted as a function of the concentration of miniGαs (a), GαsAHD (b), miniGαi1 (c), or Gαi1AHD (d), as shown in dose–response curves. Data points represent the mean values ± SEM from 3–4 independent experiments. The dissociation constant is obtained using the Kd mode of MO.Affinity Analysis software (v2.3) and presented as the best-fit Kd ± Kd confidence, corresponding to a 68% confidence level (~1 standard deviation). MST experiments were performed at 20% excitation power and medium MST power. Changes in the HDX profile of miniGαs (e) or GαsAHD (f) upon co-incubation with C-terminal truncated βarr1 (βarr1_ΔC). Regions with reduced HDX are colored in blue on the Gαs structure (PDB: 6EG8). The deuterium uptake plots of selected peptides with HDX changes are shown in Supplementary Fig. 5a and 5b. g Regions consistently affected in GTPγS-bound Gαs (Fig. 2a) and miniGαs (Fig. 3e) or GαsAHD (Fig. 3f) upon co-incubation with βarr1_ΔC are shown in green on the Gαs structure (PDB: 6EG8). Changes in the HDX profile of miniGαi1 (h) or Gαi1AHD (i) upon co-incubation with βarr1_ΔC. Regions with reduced HDX are colored in blue on the structure of Gαi1 (PDB: 1GP2). The deuterium uptake plots of selected peptides with HDX changes are shown in Supplementary Fig. 5c, d. j Regions consistently affected in GTPγS-bound Gαi1 (Fig. 2b) and miniGαi1 (Fig. 4h) or Gαi1AHD (Fig. 4i) upon co-incubation with βarr1_ΔC are shown in green. e, f, h, and i White regions represent areas with no significant difference in HDX. Peptide coverage is shown in Supplementary Fig. 2.
However, both miniGαi1 and Gαi1AHD constructs exhibited Kd values comparable to that of the full-length GDP- or GTPγS-bound Gαi1 (Kd = 107.97 ± 27.08 and 62.54 ± 17.99 nM, respectively; Fig. 3c,d; Supplementary Fig. 1b). These data indicate that both the RD and AHD of Gαi1 contribute to βarr1 binding, and neither domain alone is solely responsible for the interaction.
HDX-MS analysis of βarr1 interaction interfaces within individual Gα domains
Building on our domain-level binding analysis, we next used HDX-MS to further map the potential interaction interfaces between βarr1_ΔC and the RD or AHD domains of Gαs and Gαi1 (Supplementary Data 3).
Upon co-incubation with βarr1_ΔC, miniGαs exhibited decreased deuterium uptake in the αG/α4 loop extending into α4 (Fig. 3e and Supplementary Fig. 5a), suggesting a conformational stabilization or reduction in solvent accessibility in this region. For the isolated AHD (GαsAHD), reduced HDX was observed in the αA helix, αA/αB loop, αD/αE loop, and αE/αF loop (Fig. 3f and Supplementary Fig. 5b; peptides 68–99, 110-119, 169–176, and 190–198). Notably, the αG/α4 loop in RD and αA/αB and αE/αF loops in AHD were also protected upon βarr1_ΔC binding to full-length Gαs (Fig. 2a and Supplementary Fig. 3a), identifying these regions as strong candidates for the direct βarr1 binding interface (Fig. 3g, green-highlighted). Other regions affected in full-length Gαs, such as switch I and the αD/αE loop (Fig. 2a), were not consistently observed across the constructs and may reflect allosteric conformational changes rather than direct contacts.
Co-incubating βarr1_ΔC with miniGαi1 reduced HDX in the C-terminal part of α5 (Fig. 3h and Supplementary Fig. 5c). Gαi1AHD displayed protection in the αA/αB loop extending into αB, αD/αE loop, and αE/αF loop (Fig. 3i and Supplementary Fig. 5d). Among these, the C-terminal part of α5 in the RD and the αA/αB through αB and αD/αE regions in the AHD were similarly protected in full-length Gαi1 upon βarr1_ΔC interaction (Fig. 2b and Supplementary Fig. 3b), reinforcing their likely involvement in complex formation (Fig. 3j, green-highlighted). Regions, such as switch II, α3, and α3/β5 loop, altered in the full-length Gαi1 complex (Fig. 2b), were not consistently affected in domain-specific constructs and may reflect allosteric effects.
Contrary to the HDX profiles of Gα domains, those of βarr1_ΔC remained largely unchanged upon co-incubation with miniGα or AHD constructs (Supplementary Data 3). The absence of significant protection in βarr1_ΔC may be because the binding occurs through side chains without affecting the backbone dynamics, which may not be sufficient to induce measurable HDX level changes within βarr1. Further structural characterization, such as crosslinking-mass spectrometry or cryo-EM, may be needed to define βarr1’s contact surfaces at higher resolution.
Collectively, the HDX-MS results indicate that although both RDs and AHDs are involved in both βarr1–Gαs and βarr1–Gαi1 complex formation, the binding interfaces (compare Fig. 3g and 3j) and allosteric conformational changes (compare Fig. 2a and 2b) might differ between these complexes.
βarr1 does not alter the activation status of Gα subunits
Having established that βarr1_ΔC directly binds both Gαs and Gαi1 through interfaces spanning RD and AHD, we next asked whether this interaction influences G protein activation. To assess the GDP/GTP exchange rate, we employed BODIPY-FL–GTPγS, a BODIPY fluorescence-labeled GTP analog (Fig. 4a). In solution, BODIPY fluorescence is quenched by the guanine ring; however, this quenching is relieved upon incorporation into Gα34,35. Thus, an increased fluorescence corresponds to GDP release and BODIPY-FL–GTPγS binding and serves as a proxy for Gα activation.

a Chemical structure of BODIPY-FL–GTPγS. The effect of βarr1_ΔC on GDP/GTP turnover of Gαs (b) and Gαi1 (c) was monitored by BODIPY-FL–GTPγS assay. Data are presented as mean ± SD from three independent experiments.
We measured fluorescence changes over time upon incubating BODIPY-FL–GTPγS with purified Gαs or Gαi1 in the presence or absence of βarr1_ΔC. In both cases, GTPγS incorporation rate and extent were unaffected by βarr1_ΔC (Fig. 4b, c), indicating that βarr1 binding does not significantly alter the nucleotide exchange activity of either Gα isoform under these conditions. These results suggest that while βarr1_ΔC engages both Gαs and Gαi1 directly, the interaction does not modulate their activation status, at least in terms of GDP/GTP turnover.
Gαs–βarr1 interactions modulate βarr1 activation status
A previous study has suggested that the interaction of Gαi1 with βarr1 regulates ERK1/2 activation14. Building on our observation of direct binding between βarr1_ΔC and Gαs or Gαi1 (Fig. 1l–o), we investigated whether these interactions affect βarr1 activation status. As βarr1 activation is associated with the displacement of its C-terminal βXX, we assessed this structural hallmark using a fluorescence-based βXX release assay36. This method leverages the environment-sensitive nature of bimane fluorescence and its quenching by nearby aromatic residues, such as tryptophan37.
In our βXX release assay system, residue 385 (adjacent to βXX) was labeled with bimane, and a tryptophan was introduced at residue 5 (Fig. 5a, yellow and cyan circles, respectively). In the basal state, proximity between these residues quenches bimane fluorescence (Fig. 5a, upper panel). Upon βXX release, the increased separation between residues 5 and 385 reduces the quenching effect (Fig. 5a, lower panel), resulting in an increase in fluorescence intensity accompanied by a slight red shift in the emission spectrum (Supplementary Fig. 6).

a Schematic illustration of the fluorescence-based βarr1 β-strand XX (βXX) release assay. In the basal state, βarr1, Gly5, and Asp385, highlighted by yellow and cyan circles, respectively, are in proximity; Gly5 is switched to Trp, and Asp385 is switched to Cys and labeled with Bimane. In the basal state, the Bimane fluorescence is quenched by nearby tryptophan. Upon activation, βXX is released, and the distance between these residues increases, reducing the quenching effect and resulting in enhanced fluorescence. Effects of pre or post co-incubation of GTPγS-bound Gα (b), miniGα (c), and GαAHD (d) on bimane fluorescence changes of βarr1 with or without V2Rpp. Statistical significance was assessed using one-way ANOVA followed by Tukey’s post-hoc test (*P < 0.05) comparing all tested conditions. For simplicity, the statistical significance was noted only for the pairs with functional importance. Data are presented as mean ± SD from 6–7 independent experiments. Exact P-values in (b) are as follows: vehicle vs. V2Rpp, 4.018 × 10⁻¹⁰; V2Rpp vs. V2Rpp+Gαs(post), 0.000773; V2Rpp vs. V2Rpp+Gαs(pre), 0.001929 (n = 6–7). Exact P-values for vehicle vs. V2Rpp in (c) and (d) are 0.000037 and 0.001466, respectively (n = 6).
Co-incubation with V2Rpp increased bimane fluorescence, reflecting βXX release upon V2Rpp-binding (Fig. 5b, gray solid bars). Conversely, GTPγS-bound Gαi1 or Gαs alone did not alter bimane fluorescence (Fig. 5b, green and red empty bars), suggesting that neither Gα directly activates βarr1 in the absence of receptor input in this in vitro system.
Interestingly, GTPγS-bound Gαs, but not Gαi1, enhanced V2Rpp-induced βXX release when added before or after V2Rpp co-incubation (Fig. 5b, red solid bars), indicating that active Gαs can potentiate receptor-mediated βarr1 activation. These findings suggest a cooperative mechanism where Gαs amplifies the conformational activation of βarr1 in response to receptor-derived phosphopeptides, while Gαi1 does not share this capability.
Both RD and AHD of Gαs are required for potentiating βarr1 activation
As βarr1_ΔC directly interacts with both miniGαs and GαsAHD (Fig. 3), we tested which domain is responsible for the potentiation of V2Rpp-induced βXX release. Neither miniGαs nor GαsAHD alone enhanced V2Rpp-induced βXX release (Fig. 5c, d). These findings indicate that while both domains contribute to βarr1 binding, with AHD playing the predominant role, they are individually insufficient to facilitate βarr1 activation. Instead, full-length Gαs requires concerted action between RD and AHD to promote βarr1 conformational response to V2R-derived signals.
Validation of the physical and functional interaction between Gαs and βarr1 in the cell
To validate the functional interaction between Gαs and βarr1, we first examined whether these proteins associate in cells using the proximity ligation assay (PLA) (Fig. 6a). HEK293S cells were transfected with V2R, and PLA signals representing endogenous Gαs–βarr1 complex formation were measured in the presence or absence of arginine vasopressin (AVP) stimulation. Upon AVP treatment, we observed a significant increase in PLA puncta, with 26.27 ± 3.20 puncta per nucleus, compared to untreated cells (P < 0.0001) (Fig. 6b, c). These results indicate that endogenous Gαs and βarr1 interact in cells, and that this interaction is enhanced upon activation. As our MST analyses showed that the activation state of Gαs does not affect the interaction with βarr1 (Fig. 1l, m), the AVP-dependent increase in complex formation is most likely explained by activation of βarr1 rather than by changes in Gαs.

a Schematic illustration of the PLA-based Gαs–βarr1 interaction monitoring system. b Representative PLA images showing the interaction between Gαs and βarr1 under the indicated conditions (NT = non-treated; +AVP = Arginine Vasopressin-treated). PLA puncta are shown in gray, and Hoechst-stained nuclei in cyan. Scale bars: 10 μm. c Quantification of PLA signals. Data represent pooled counts from 42 (NT) and 44 (AVP) randomly selected fields of view across 3 independent experiments. Bars indicate mean ± SEM. Scatter plots show individual data points. *P < 0.0001; statistical analysis performed using unpaired two-tailed Student’s t-test. The exact P-value is 0.000001191. d Schematic illustration of BRET-based βarr1 β-strand XX (βXX) release sensing system. e Basal BRET ratio in the absence or presence of Gαs is shown as violin plots with the median line (left panel); corresponding luminescence intensities are presented as mean ± SD with individual points from 27 wells per condition (right panel); unpaired two-tailed Student’s t-test, *P < 0.001. The exact P-values for basal BRET ratio and luminescence intensity are 3.486 × 10⁻14 and 0.3768, respectively. f BRET values measured 10 min after AVP (100 nM) in the absence or presence of Gαs, shown as violin plots with the median line; data are from 9 wells per condition; one-way ANOVA with Tukey’s post-hoc test, *P < 0.0001. Exact P-values are as follows: Gαs–/–AVP vs. Gαs–/+AVP, 3.259 × 10⁻9; Gαs–/–AVP vs. Gαs+/–AVP, 0.000015; Gαs–/+AVP vs. Gαs+/+AVP, 0.000131; Gαs+/–AVP vs. Gαs+/+AVP, 2.499 × 10⁻10. g Proposed model of Gαs–βarr1 interaction and its functional outcome.
Notably, our MST with purified proteins showed that Gαs does not interact with V2Rpp-bound βarr1 (Fig. 1h, i), suggesting a discrepancy between in vitro and cellular observations. This difference can likely be attributed to the distinct conformational states of βarr1 induced in each system. In cells, receptor activation leads to dual engagement of βarr1 through both the phosphorylated C-terminal tail and the receptor core38, whereas V2Rpp alone promotes only tail engagement, which may not fully stabilize the Gαs-interacting conformation. In addition, β-arrestins can remain signaling-competent after receptor dissociation, sustaining an active-like ensemble that can engage other downstream signaling molecules39,40. This receptor-free, signaling-competent ensemble likely resembles βarr1_ΔC, providing a basis for its interaction with Gαs in cells.
As a next step, we investigated whether the interaction affects the activation status of βarr1 using an intramolecular BRET sensor (Fig. 6d). In this sensor, the basal (βXX–engaged) conformation yields a higher BRET signal, whereas βXX release reduces BRET. Gαs overexpression significantly decreased the BRET signal, indicating that Gαs alone can promote βarr1 activation (Fig. 6e). As expected, AVP reduced the BRET signal as V2R phosphorylated C-tail engages at the N-domain and replacing βXX (Fig. 6e). Co-expression of Gαs potentiated this AVP-induced βXX release (Fig. 6e).
By contrast, in our bimane fluorescence quenching assay with purified proteins, Gαs alone did not trigger βXX release (Fig. 5b). This discrepancy likely reflects cell-specific factors that prime βarr1 in vivo. For example, overexpressed V2R can exhibit basal activity, generating a subpopulation of phosphorylated receptors that biases βarr1 toward activation. In addition, plasma-membrane lipids such as phosphatidylinositol 4,5-bisphosphate (PIP2) can allosterically favor pre-active arrestin ensembles even in the absence of receptor engagement36,41,42,43. Thus, cellular βarr1 likely exists as an ensemble of basal and active-like states, with Gαs overexpression shifting the equilibrium toward βXX release.
In summary, convergent evidence from cell-based and in vitro assays supports a model in which Gαs preferentially engages the active conformational ensemble state of βarr1 and promotes or stabilizes the βXX-released conformation (Fig. 6g) while the precise βarr1 sub-state favored by Gαs remains to be defined.
Conformational changes of full-length βarr1 or Gαs upon interaction
To explore the structural mechanism by which Gαs facilitates βarr1 activation (Figs. 5b and 6e, f), we performed HDX-MS on full-length βarr1 in the absence or presence of V2Rpp, with or without GTPγS-bound Gαs co-incubation. After 60 min co-incubation, HDX was initiated by transferring samples into D₂O. The samples were pepsin-digested and analyzed by mass spectrometry (Supplementary Data 4).
When GTPγS-bound Gαs was co-incubated, full-length βarr1 either with or without V2Rpp did not show HDX changes. Similarly, we could not detect HDX changes in Gαs. The lack of Gαs-induced HDX changes in full-length βarr1 either with or without V2Rpp can be explained by the lack of stable binding between Gαs and the basal or V2Rpp-bound state βarr1 (Fig. 1d, e, h, i).
Nevertheless, Gαs facilitated βXX release from V2Rpp-bound state in in vitro (Fig. 5b) and both basal and V2R-activated βarr1 in cells (Fig. 6e, f). We interpret this as a transient, low-occupancy allosteric effect. Gαs briefly engages βarr1 when the protein samples active conformational ensembles, thereby promoting βXX release, but dissociates as βarr1 transitions to subsequent conformations (Fig. 6g). In the in vitro setting, V2Rpp may populate only a small fraction of such Gαs-competent ensembles, yielding functional potentiation without sufficient complex lifetime or abundance for detection by MST or HDX-MS. In cells, receptor activity and membrane factors likely enrich these active-like ensembles even within subpopulations of basal or V2R-activated βarr1, enabling the observed effect.
Discussion
In this study, we provide structural and functional insights into the direct interaction between βarr1 and two G protein α-subunits, Gαs and Gαi1. By quantifying binding affinities across different activation states, we demonstrated that βarr1 engages both Gαs and Gαi1, with the activation state of Gα exerting limited influence on binding affinity (Fig. 1d–k). Conversely, the active conformational ensemble state of βarr1 (βarr1_ΔC) exhibited markedly enhanced binding compared to its basal or V2Rpp-bound state, although GDP-bound Gαi1 retained some ability to associate with inactive βarr1. Domain-level binding assays (Fig. 3) and HDX-MS (Figs. 2 and 3) revealed that both the RD and AHD contribute to βarr1 interaction in both Gαs and Gαi1, albeit likely through isoform-specific structural interfaces.
Functionally, βarr1 binding did not influence GDP/GTP exchange in Gαs or Gαi1 (Fig. 4), suggesting that βarr1 does not act as a guanine nucleotide exchange factor (GEF) for G proteins. Gαs, however, not Gαi1, potentiated βXX release (Figs. 5 and 6), suggesting a role for Gαs in facilitating βarr1 activation. The difference between Gαs and Gαi1 in potentiating βXX release from βarr1 may be owing to different binding modes inferred by distinct HDX-MS profile changes.
These findings complement and expand on the results of a companion manuscript by Lee et al., which used both cellular and purified systems to characterize βarr–Gαi1 interaction. They reported βarr1–Gαi1 binding independently of Gαi1 activation state and identified the AHD as the primary βarr1 interaction site. Notably, in that study, βarr1 interaction with Gαs was not observed in cells, whereas we found compatible binding between βarr1 and Gαs (Fig. 6b, c). As prior studies have also reported βarr1–Gαs interactions in cellular systems16, these differences may arise from varying experimental conditions and potentially the transient nature of the interaction. Together, our findings and those of Lee et al. support a model where βarr1 engages Gα subunits in a conformation- and isoform-specific manner, with distinct structural and functional consequences.
Previous studies have shown that Gα and β-arrestin can co-localize at both the plasma membrane and endosomes13,14,15, suggesting that they may cooperate around these locations. The lack of GEF activity from βarr1, combined with its ability to bind G proteins, suggests that the interaction may serve spatial or allosteric regulatory functions rather than catalytic roles. Recently, the von Zastrow group reported that active Gαi is detected at the endosome independent of the receptor activation at the endosomes; the authors proposed that the receptor-activated Gαi at the plasma membrane is translocated to the endosomes44. It might be possible that βarr-Gαi interaction transports Gαi to the endosomes.
The potentiation of βXX release by Gαs could reflect a form of allosteric priming, wherein Gαs promotes βarr1 transition into a more active conformation. A similar modulatory effect has been observed with PIP₂, which enhances βXX conformational dynamics and V2Rpp binding22,36,43. Notably, our findings raise the possibility that Gα induces a distinct βarr1 conformation—distinct from classical “active” or “basal” states—thereby contributing to signaling diversity. Moreover, arrestins can remain active or signaling-competent state even after dissociation from the receptor39,40, potentially corresponding to the βXX-released “active conformational ensemble state”. It is possible that Gαs or Gαi1 can engage with βarr1 in this active conformational ensemble state within cells, promoting distinct conformational rearrangements, as captured by HDX-MS (Fig. 2c, d), which may underlie divergent downstream signaling profiles. This hypothesis also aligns with the “barcode” theory of arrestins, which posits that discrete arrestin conformations can encode specific downstream signaling outcomes45.
Mapping of potential binding interfaces via HDX-MS and domain dissection identified candidate contact regions. In Gαs, βarr1 may interact with the αG/α4 loop in the RD and αA/αB and αE/αF loops in the AHD (Fig. 3e, f), with AHD having a prominent role for the interaction (Supplementary Fig. 1a). For Gαi1, the C-terminal α5 helix (RD) and αA/αB and αD/αE loops (AHD) were implicated (Fig. 3h, i). On the βarr1 side, the lariat loop (Gαs and Gαi1), βIV/βV loop (Gαs), and finger loop (Gαi1) emerged as potential Gα contact regions (Fig. 2d, e). However, assembling a comprehensive βarr1–Gα complex structural model based on these binding interfaces remains challenging. Notably, full-length βarr1 did not exhibit measurable HDX changes upon Gαs binding in the presence or absence of V2Rpp, possibly reflecting transient interactions or localized structural effects below HDX-MS detection thresholds. Therefore, more sophisticated analysis is needed for understanding the complex structures; a high-resolution understanding of stabilized sub-states will be required to define the precise βarr1 sub-states and Gα-selective interfaces. Despite these limitations, the differential impact of Gαs and Gαi1 on βarr1 activation (Fig. 5b), along with different HDX-MS change profiles, suggests that distinct complex architectures or interaction dynamics underlie their divergent functions.
In conclusion, our data provide evidence for direct, conformation-sensitive interactions between βarr1 and Gαs/Gαi1, identify domain-level contact points, and reveal isoform-specific functional outcomes. These findings lay a foundation for future structural and cellular studies aimed at elucidating the physiological relevance of Gα–βarr1 complexes and their roles in GPCR signaling dynamics.
Methods
Gα, GαAHD, and miniGα expression and purification
As described previously33,46,47,48, the cDNA encoding human Gαi1 containing an N-terminal His-tag and TEV cleavage site was constructed in pET21a. Human Gαs cDNA with an N-terminal His-tag and an HRV 3C protease cleavage site was inserted into pET28a. Recombinant GαsAHD and Gαi1AHD with an N-terminal His-tag followed by a thrombin cleavage site, and a C-terminal FLAG tag were synthesized and constructed in pET28a. Recombinant miniGαs and miniGαi1 containing an N-terminal His-tag and TEV cleavage site were cloned into pET21a.
All constructs were transformed into Escherichia coli LOBSTR cells and grown in Terrific Broth (TB) medium in the presence of an antibiotic at 37 °C until OD600 reached 0.6–0.8. Protein expression was induced with 0.03 mM IPTG at 16 °C for 24 h for full-length Gα proteins, 0.5 mM IPTG at 37 °C for 4 h for AHD constructs, and 0.5 mM IPTG at 16 °C for 24 h for miniGα proteins. Cell pellets were harvested and resuspended in lysis buffer (20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 2 mM MgCl2, 20 µM GDP, 1:1000 protease inhibitor cocktail, 2.5 µg/mL leupeptin, 10 µg/mL benzamidine, 100 µM TCEP) in three-fold cell pellet volume. Lysozyme was added at 3 mg/mL and incubated at room temperature (22–25 °C) for 30 min, followed by incubation with 10 µg/mL DNase I for another 30 min. Lysates were collected by centrifugation at 18,000 × g for 30 min at 4 °C, supplemented with 20 mM imidazole, and loaded on an Ni-IDA column equilibrated with lysis buffer containing 20 mM imidazole. The resin was washed with wash buffer (20 mM HEPES pH 7.4, 300 mM NaCl, 2 mM MgCl2, 20 µM GDP, 1:1000 protease inhibitor cocktail, 2.5 µg/mL leupeptin, 10 µg/mL benzamidine, 100 µM TCEP, and 20 mM imidazole). The proteins were eluted with elution buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl2, 20 µM GDP, 1:1000 protease inhibitor cocktail, 2.5 µg/mL leupeptin, 10 µg/mL benzamidine, 100 µM TCEP, 200 mM imidazole) and further purified using a Superdex 200 (10/300) column with ÄKTA FPLC. For GTPγS-bound Gα preparation, 20 μM GTPγS was added instead of GDP during the wash and elution steps of Ni-IDA purification. Subsequently, 1 mM GTPγS was added to the Ni-IDA eluate and incubated on ice for 1 h, followed by gel filtration in buffer containing 20 μM GTPγS.
βarr1 expression and purification
Rat βarr1 cDNA with an N-terminal His-tag and a thrombin cleavage site was cloned into the pET28a vector. A C-terminal truncated variant (βarr1_ΔC, residues 1–365) was generated by introducing a stop codon at residue 365 via site-directed mutagenesis. A cysteine-free rat βarr1 variant (C59V/C125S/C140I/C150V/C242V/C251V/C269S) containing an N-terminal His-tag and a thrombin cleavage site was cloned into the pET28b vector. A site-directed mutant (G5W/D385C) was subsequently generated from this construct.
βarr1 constructs were expressed in E. coli BL21(DE3) cells. Cultures were grown in Luria Bertani (LB) medium containing antibiotics at 37 °C and induced with 0.03 mM IPTG when OD600 reached 0.6, followed by 24 h incubation at 16 °C. After induction, the cells were harvested, centrifuged, and resuspended in the lysis buffer (20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 1:1000 protease inhibitor cocktail, 2.5 µg/mL leupeptin, 10 µg/mL benzamidine, 100 µM TCEP) in five-fold cell pellet volume. Lysozyme was added at 3 mg/mL and incubated at room temperature for 30 min, followed by incubation with 10 µg/mL DNase I for another 30 min. The lysates were collected by centrifugation at 18,000 × g for 30 min at 4 °C. The supernatant was supplemented with 20 mM imidazole and loaded on a Ni-IDA column pre-equilibrated with lysis buffer containing 20 mM imidazole. The resin was washed with wash buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 1:1000 protease inhibitor cocktail, 2.5 µg/mL leupeptin, 10 µg/mL benzamidine, 100 µM TCEP, and 20 mM imidazole). The proteins were eluted with elution buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1:1000 protease inhibitor cocktail, 2.5 µg/mL leupeptin, 10 µg/mL benzamidine, 100 µM TCEP, 200 mM imidazole). The Ni-IDA purified βarr1 and βarr1_ΔC proteins were further purified using a HiTrap Q column or an SP column (GE Healthcare), respectively, and subsequently buffer-exchanged using a Zeba desalting column to reduce salt concentration for further use. For the cysteine-free βarr1 G5W/D385C variant, Ni-IDA purified proteins were further purified by size-exclusion chromatography using a Superdex 200 Increase 10/300 GL column on an ÄKTA FPLC system.
Synthetic peptide
The V2Rpp peptide (ARGRpTPPpSLGPQDEpSCpTpTApSpSpSLAKDTSS), derived from the phosphorylated C-terminus of the human V2 vasopressin receptor, was synthesized by the Tufts University Core Facility.
Microscale thermophoresis (MST)
βarr1 was labeled using the Monolith NT™ Protein Labeling Kit RED-MALEIMIDE 2nd Generation (NanoTemper) according to the manufacturer’s instructions. Labeled βarr1 (100 nM) was mixed with serially diluted unlabeled proteins (Gα, GαAHD, or miniGα) in interaction buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 2 mM MgCl₂, 100 μM TCEP, 0.05% Tween-20, and 20 μM GDP or GTPγS. For V2Rpp-bound βarr1, labeled βarr1 was pre-incubated with a 10-fold molar excess of V2Rpp for 30 min at room temperature to pre-form the V2Rpp–βarr1 complex. This complex was then titrated with unlabeled Gα, Gα_AHD, or miniGα in the same interaction buffer supplemented with 30 μM V2Rpp to maintain the peptide-bound state during the binding assay. After incubating for 15 min at room temperature, the samples were loaded into premium capillaries (NanoTemper). MST measurements were performed on a Monolith NT.115 (NanoTemper) using 20% excitation power and medium MST power at 25 °C. Data were collected from the MST-on phase (5 s), and best-fit Kd values were calculated using MO.Affinity Analysis software (v2.3) with a defined 68% confidence interval (~1 standard deviation) for uncertainty. All measurements with a signal-to-noise ratio of ≥5, as well as response amplitude ≥ 5, were accepted as binding events.
HDX-MS
To form βarr1–Gα complexes, βarr1 and G protein (Gα, GαAHD, or miniGα) were mixed at 1:1 (40 μM: 40 μM) and 1:2 (40 μM: 80 μM) molar ratios, followed by incubation for 40 min at room temperature. Hydrogen/deuterium exchange was initiated by mixing 5 μL G protein, βarr1, and complex samples with 25 μL D2O buffer (20 mM HEPES pD 7.4, 150 mM NaCl, 2 mM MgCl₂, 20 μM GTPγS or GDP, 100 μM TCEP, 10% glycerol in D2O) and incubating on ice for 10, 100, 1000, or 10,000 s. The deuterated samples were quenched by adding 30 μL ice-cold quench buffer (60 mM NaH2PO4, pH 2.01, 10% glycerol, 20 mM TCEP) at each time point. Non-deuterated control samples were prepared in parallel using H2O buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl₂, 20 μM GTPγS or GDP, 100 μM TCEP, 10% glycerol in H2O), followed by quenching and freezing as above.
The quenched samples were sent for digestion by passing through an immobilized pepsin column (2.1×30 mm²; Life Technologies, Carlsbad, CA, USA) at a 100 µL/min flow rate with 0.05% formic acid in H2O at 12 °C. The peptide fragments were subsequently collected on a C18 VanGuard trap column (1.7 µm × 30 mm; Waters, Milford, MA, USA) and desalted with 0.05% formic acid in H2O. Peptic peptides were then separated using ultra-pressure liquid chromatography (UPLC) on an ACQUITY UPLC C18 column (1.7 µm, 1.0 × 100 mm²; Waters) at 40 µL/min with an acetonitrile gradient created by two pumps: mobile phase A (0.15% formic acid in H2O) and B (0.15% formic acid in acetonitrile). The gradient started at 8% B and increased to 85% B over 8.5 min. To minimize the back-exchange of deuterium to hydrogen, the sample, solvents, trap, and UPLC column were all maintained at pH 2.5 and 0.5 °C during analysis. Mass spectrometry analyses were performed using a Xevo G2-XS QTof (Waters) as established previously49,50. The peptides were identified in the ND samples using the ProteinLynx Global Server 2.4 (Waters). To process the HDX-MS data, the amount of deuterium in each peptide was determined by measuring the centroid of the isotopic distribution using DynamX 3.0 (Waters). The average back-exchange in our system was approximately 30–40%. Back-exchange correction was not performed, as the proteins exhibited aggregation in fully deuterated samples, and the analyses focused on comparative assessments between different conformational states.
BODIPY-FL–GTPγS assay
Nucleotide exchange function of Gα was assessed by monitoring the binding of BODIPY-FL–GTPγS (Invitrogen, Waltham, MA, USA) to GDP-bound Gα proteins. BODIPY-FL–GTPγS (final concentration: 500 nM) was prepared in imaging buffer containing 20 mM HEPES, pH 7.4, 150 mM NaCl, 10 mM MgCl₂, 100 μM TCEP. Fluorescence baseline was recorded for 120 s using a Synergy Neo or Neo2 plate reader (BioTek, Winooski, VT, USA) in a 96-well black plate, with excitation at 485 nm (3 nm bandwidth) and emission at 510 nm (20 nm bandwidth). After baseline acquisition, 1.5 μM GDP-bound Gα or Gα–βarr1 complex was added to the wells, and fluorescence was recorded every 10 s for 80 min. All experiments were performed at room temperature and repeated in triplicate. The resulting kinetics spectra were plotted using GraphPad Prism 8.0.
β-arrestin βXX release assay
Cys-free βarr1 G5W/D385C (final concentration: 20 μM) was prepared in 20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl₂, and labeled by co-incubation with a 10-fold molar excess of bromobimane for 1 h on ice. Excess dye was removed using a Zeba spin desalting column. The labeled βarr1 was then diluted to 5 μM final concentration and incubated with 15 μM Gα, GαAHD, or miniGα for 1 h at room temperature in the presence or absence of pre-incubated 50 μM V2Rpp. Fluorescence measurements were performed in a MicroFluor 96-well plate, with excitation at 390 nm and emission spectra recorded from 420 to 600 nm in 1-nm increments using a Synergy Neo or Neo2 plate reader (BioTek, Winooski, VT, USA). Fluorescence changes were quantified by measuring the emission intensity at the peak wavelength and normalized as a fold change relative to the vehicle-treated control.
Proximity ligation assay (PLA)
PLA experiment was done using the Duolink® in situ detection reagent green kit (Cat#DUO92008, Sigma-Aldrich) following the provided protocol. Briefly, cells were fixed and permeabilized before incubation with primary antibodies (rabbit anti-Gαs, #ab235959, Abcam; mouse anti-βarr1 (ZA005), Cat#39-5000, Invitrogen) for 1 h at RT, followed by incubation with the provided oligonucleotide-conjugated secondary antibodies for 30 min at 37 °C with washes after each step. Ligation and amplification were performed by incubating cells with the provided ligase, polymerase, and detection solutions. PLA images were acquired on a Leica Thunder imager (Leica), equipped with a 40x/1.3NA objective and a Leica K8 sCMOS (Leica) controlled by LAS X software (Leica). The DAPI channel was acquired using a 391/32-nm excitation filter, a DFT51010 filter cube (11525418, Leica), a 415-nm dichroic mirror, and a 435/30-nm emission filter. PLA signal was acquired using a 479/33-nm excitation filter, a DFT51010 filter cube (11525418, Leica), a 500-nm dichroic mirror, and a 519/25-nm emission filter. PLA fluorescence images were analyzed using FIJI. Raw fluorescence images were corrected by background subtraction and converted to grayscale. Images of PLA puncta and cell nuclei were then adjusted for brightness and converted to binary mode. PLA puncta and nuclei were counted using the analyze particle plugin in FIJI based on the size criteria 0.3–10 pixel2 for PLA puncta and 30-infinite pixel2 for nuclei.
Bioluminescence resonance energy transfer (BRET) assay
A BRET-based β-arrestin1 conformational biosensor was generated by fusing mCitrine and NanoLuc at the N- and C-terminus of rat β-arrestin1, respectively, as described previously51. Upon activation, conformational changes of β-arrestin can be monitored by detecting changes in BRET ratios in living cells. For the BRET assay, ΔGαs cells were seeded at a density of 4 × 105 cells per well in 6-well plates and co-transfected with plasmids encoding the β-arrestin1 BRET sensor, human V2R, and human Gαs using Lipofectamine™ 2000 (Invitrogen, #11668019) according to the manufacturer’s instructions. After 6 h, the transfected cells were harvested and replated into white 96-well plates (4 × 104 cells per well; Greiner, #655083) for 24 h. On the day of the assay, culture medium was replaced with HBSS buffer (pH 7.4) containing 20 mM HEPES, and NanoLuc substrate (Promega, #N1662) was added at a 1:1000 dilution. BRET signals were recorded using a CLARIOstar plate reader (BMG) in monochromator mode, with donor emission collected at 440–480 nm and acceptor emission at 515–555 nm. Before measurement, cells were dark-adapted in the reader for 5 min, followed by five baseline readings. After AVP addition, BRET changes were monitored for 20 min. The BRET ratio was calculated as the value of the acceptor channel divided by that of the donor channel.
Statistics and reproducibility
For HDX-MS analysis, statistical significance was determined using a two-tailed unpaired Student’s t-test (P < 0.05) combined with a mass difference >0.2 Da to assess the differences between samples with and without the binding partner. For the C-tail release assay, a one-way ANOVA followed by Tukey’s post-hoc test was applied to analyze the differences among multiple conditions, with statistical significance set at P < 0.05. For the PLA assay, a two-tailed unpaired Student’s t-test was used to assess the differences in PLA signals with or without AVP treatment, and the statistical significance was set at P < 0.0001. For BRET assays, a two-tailed unpaired Student’s t-test was used to compare BRET signals in the absence or presence of Gαs, whereas a one-way ANOVA followed by Tukey’s post-hoc test was performed to analyze BRET responses across conditions with or without AVP treatment in the absence or presence of Gαs, with statistical significance set at P < 0.0001. More than three independent experiments were conducted for each dataset.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this manuscript are available in the Source Data File. A reporting summary for this Article is available as a Supplementary Information file. Data for sequence analysis in Supplementary Fig. 4 are available from the GPCR database (gpcrdb.org). HDX-MS data have been deposited to ProteomeXchange Consortium via PRIDE partner repository with the set identifier PXD065019. The following accession codes are used in the study: 1G4M, 4JQI, 1GP2, 6EG8, 3SN6. Further information and requests for reagents should be directed to and fulfilled by the corresponding author, Ka Young Chung (kychung2@skku.edu). Source data files are provided in this paper. Source data are provided with this paper.
References
Shukla, A. K., Xiao, K. & Lefkowitz, R. J. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469 (2011).
Hilger, D., Masureel, M. & Kobilka, B. K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 (2018).
Weis, W. I. & Kobilka, B. K. The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018).
Syrovatkina, V., Alegre, K. O., Dey, R. & Huang, X. Y. Regulation, signaling, and physiological functions of G-proteins. J. Mol. Biol. 428, 3850–3868 (2016).
Mahoney, J. P. & Sunahara, R. K. Mechanistic insights into GPCR-G protein interactions. Curr. Opin. Struct. Biol. 41, 247–254 (2016).
Milligan, G. & Kostenis, E. Heterotrimeric G-proteins: a short history. Br. J. Pharmacol. 147, S46–S55 (2006).
Wilden, U., Hall, S. W. & Kuhn, H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc. Natl. Acad. Sci. USA 83, 1174–1178 (1986).
Benovic, J. L. et al. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. USA 84, 8879–8882 (1987).
Smith, J. S. & Rajagopal, S. The beta-arrestins: multifunctional regulators of G protein-coupled receptors. J. Biol. Chem. 291, 8969–8977 (2016).
Lohse, M. J., Benovic, J. L., Codina, J., Caron, M. G. & Lefkowitz, R. J. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550 (1990).
DeFea, K. A. Beta-arrestins as regulators of signal termination and transduction: how do they determine what to scaffold? Cell. Signal. 23, 621–629 (2011).
Attramadal, H. et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 267, 17882–17890 (1992).
Daly, C. et al. beta-Arrestin-dependent and -independent endosomal G protein activation by the vasopressin type 2 receptor. eLife https://doi.org/10.7554/eLife.87754 (2023).
Smith, J. S. et al. Noncanonical scaffolding of G(alphai) and beta-arrestin by G protein-coupled receptors. Science https://doi.org/10.1126/science.aay1833 (2021).
Thomsen, A. R. B. et al. GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919 (2016).
Li, B., Wang, C., Zhou, Z., Zhao, J. & Pei, G. beta-Arrestin-1 directly interacts with Galphas and regulates its function. FEBS Lett. 587, 410–416 (2013).
Granzin, J. et al. X-ray crystal structure of arrestin from bovine rod outer segments. Nature 391, 918–921 (1998).
Hirsch, J. A., Schubert, C., Gurevich, V. V. & Sigler, P. B. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell 97, 257–269 (1999).
Shukla, A. K. et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141 (2013).
Kang, Y. et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015).
Liu, Q. et al. DeSiphering receptor core-induced and ligand-dependent conformational changes in arrestin via genetic encoded trimethylsilyl 1H-NMR probe. Nat. Commun. 11, 4857 (2020).
Zhai, R. et al. Distinct activation mechanisms of beta-arrestin-1 revealed by (19)F NMR spectroscopy. Nat. Commun. 14, 7865 (2023).
Maharana, J. et al. Molecular insights into atypical modes of beta-arrestin interaction with seven transmembrane receptors. Science 383, 101–108 (2024).
He, Q. T. et al. Structural studies of phosphorylation-dependent interactions between the V2R receptor and arrestin-2. Nat. Commun. 12, 2396 (2021).
Pulvermuller, A. et al. Functional differences in the interaction of arrestin and its splice variant, p44, with rhodopsin. Biochemistry 36, 9253–9260 (1997).
Kim, Y. J. et al. Crystal structure of pre-activated arrestin p44. Nature 497, 142–146 (2013).
Kim, D. K., Yun, Y., Kim, H. R., Seo, M. D. & Chung, K. Y. Different conformational dynamics of various active states of beta-arrestin1 analyzed by hydrogen/deuterium exchange mass spectrometry. J. Struct. Biol. 190, 250–259 (2015).
Latorraca, N. R. et al. How GPCR phosphorylation patterns orchestrate arrestin-mediated signaling. Cell 183, 1813–1825.e1818 (2020).
Hvidt, A. & Nielsen, S. O. in Advances in Protein Chemistry Vol. 21 (eds Anfinsen, C. B. et al.) 287–386 (Academic Press, 1966).
Wales, T. E. & Engen, J. R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 (2006).
Qu, C. et al. Scaffolding mechanism of arrestin-2 in the cRaf/MEK1/ERK signaling cascade. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2026491118 (2021).
Ham, D., Inoue, A., Xu, J., Du, Y. & Chung, K. Y. Molecular mechanism of muscarinic acetylcholine receptor M3 interaction with Gq. Commun. Biol. 7, 362 (2024).
Nehme, R. et al. Mini-G proteins: novel tools for studying GPCRs in their active conformation. PLoS ONE 12, e0175642 (2017).
McEwen, D. P., Gee, K. R., Kang, H. C. & Neubig, R. R. Fluorescent BODIPY-GTP analogs: real-time measurement of nucleotide binding to G proteins. Anal. Biochem. 291, 109–117 (2001).
Tontson, L., Babina, A., Vosumaa, T., Kopanchuk, S. & Rinken, A. Characterization of heterotrimeric nucleotide-depleted Galpha(i)-proteins by Bodipy-FL-GTPgammaS fluorescence anisotropy. Arch. Biochem. Biophys. 524, 93–98 (2012).
Kim, K. & Chung, K. Y. Molecular mechanism of beta-arrestin-2 pre-activation by phosphatidylinositol 4,5-bisphosphate. EMBO Rep. https://doi.org/10.1038/s44319-024-00239-x (2024).
Jones Brunette, A. M. & Farrens, D. L. Distance mapping in proteins using fluorescence spectroscopy: tyrosine, like tryptophan, quenches bimane fluorescence in a distance-dependent manner. Biochemistry 53, 6290–6301 (2014).
Bous, J. et al. Structure of the vasopressin hormone-V2 receptor-beta-arrestin1 ternary complex. Sci. Adv. 8, eabo7761 (2022).
Ranjan, R., Gupta, P. & Shukla, A. K. GPCR Signaling: beta-arrestins Kiss and Remember. Curr. Biol. 26, R285–R288 (2016).
Eichel, K., Jullie, D. & von Zastrow, M. beta-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 18, 303–310 (2016).
Eichel, K. et al. Catalytic activation of beta-arrestin by GPCRs. Nature 557, 381–386 (2018).
Grimes, J. et al. Plasma membrane preassociation drives beta-arrestin coupling to receptors and activation. Cell 186, 2238–2255.e2220 (2023).
Janetzko, J. et al. Membrane phosphoinositides regulate GPCR-beta-arrestin complex assembly and dynamics. Cell 185, 4560–4573.e4519 (2022).
Fisher, N. M. & von Zastrow, M. Opioid receptors reveal a discrete cellular mechanism of endosomal G protein activation. Proc. Natl. Acad. Sci. USA 122, e2420623122 (2025).
Nobles, K. N. et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci. Signal 4, ra51 (2011).
Ahn, D. et al. Galphas slow conformational transition upon GTP binding and a novel Galphas regulator. iScience 26, 106603 (2023).
Ham, D. et al. Conformational switch that induces GDP release from Gi. J. Struct. Biol. 213, 107694 (2021).
Ham, D., Ahn, D., Chung, C. & Chung, K. Y. Isolation and conformational analysis of the Galpha alpha-helical domain. Biochem. Biophys. Res. Commun. 685, 149153 (2023).
Du, Y. et al. Assembly of a GPCR-G protein complex. Cell 177, 1232–1242 e1211 (2019).
Kim, H. R. et al. Structural mechanism underlying primary and secondary coupling between GPCRs and the Gi/o family. Nat. Commun. 11, 3160 (2020).
Charest, P. G., Terrillon, S. & Bouvier, M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep. 6, 334–340 (2005).
Acknowledgements
We thank Asuka Inoue (Kyoto University) for kindly providing the ΔGαs cell line. This study was supported by grants from the Ministry of Science and ICT (RS-2019-NR040057, RS-2021-NR059713, and RS-2025-23323103 to K.Y.C.). S.K. was supported by the Brain Korea 21 FOUR program “Education and Research Center for Fostering Pharmaceutical Researchers towards Leading Innovative Growth” at the School of Pharmacy, Sungkyunkwan University. Y.K. was supported by grants of the Korea Basic Science Institute (National research Facilities and Equipment Center), National Research Foundation of Korea (NRF) (RS-2024-00340059 and RS-2024-00398706), the Korea Health Technology R&D Project (the Korea Health Industry Development Institute, KHIDI) (RS-2024-00512909), and the Gwangju Institute of Science and Technology (GIST) research fund (Future-leading Specialized Research project, 2025). J.S was supported by the National Research Foundation of Korea (NRF) (RS-2023-00227950, RS-2024-00407331, RS-2024-00338426).
Author information
Authors and Affiliations
Contributions
L.D. prepared βarr1 and Gα proteins and performed MST, HDX-MS, BODIPY-FL–GTPγS assay, and β-arrestin βXX release assay for all samples. H.K. and J.S. performed the BRET assay. Y.S. and Y.K. performed the PLA assay. D.A. assisted in Gα purification and BODIPY-FL–GTPγS assay. S.K. and J.H. performed the structural analysis. K.Y.C. initiated and supervised the project. K.Y.C. and L.D. analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
L.D. and K.Y.C. have a patent pending for βarr C-tail release assay (application number: 10-2024-0091925). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, L., Kim, H., Suh, Y. et al. Functional and structural insights into interactions between β-Arrestin 1 and Gαs or Gαi1. Nat Commun 17, 1879 (2026). https://doi.org/10.1038/s41467-026-68690-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68690-z
