
Abstract
For industrial water electrolysis, the development of active and stable catalysts for the oxygen evolution reaction remains a challenge. Here, we report a heterostructure catalyst composed of NiFe layered double hydroxide nanosheets anchored on pyramidal Fe2(MoO4)3 to activate lattice oxygen for efficient and durable oxygen evolution. Our investigation reveals that oxygen vacancies within the NiFe layered double hydroxide and the internal electrical field at the material interface optimize the electronic states, allowing oxygen atoms within the crystal lattice to participate directly in the reaction. The resulting heterostructured NiFe LDH/FeMoO catalysts possess high oxygen evolution reaction activity in 1 M KOH electrolyte with a low overpotential of 316 mV at 2 A cm−2 and maintain long-term stability over 3,000 h. Furthermore, integrating this anode into a solar-powered electrolyzer yields a high solar-to-hydrogen efficiency of 20.15%. This work provides a promising strategy for designing stable catalysts and advancing the integration of renewable energy with water electrolysis to produce clean hydrogen at scale.
Similar content being viewed by others


Introduction
Anion exchange membrane water electrolysis (AEMWE) has emerged as a promising technology for sustainable production of hydrogen, combining the advantages of low-cost alkaline water electrolysis (AWE) and the high current density capability of proton exchange membrane water electrolysis (PEMWE)1,2. However, its overall efficiency is fundamentally constrained by the sluggish reaction kinetics of the oxygen evolution reaction (OER) at the anode, which involves a multi-step four-electron transfer process and requires high overpotentials3. Conventional OER electrocatalysts acting through the adsorbate evolution mechanism (AEM) are limited by a linear scaling relationship between *OH and *OOH intermediates, requiring a theoretical overpotential of more than 0.37 V4,5,6. In contrast, the lattice oxygen mechanism (LOM), wherein lattice oxygen directly participates in O–O bond formation, bypasses this scaling constraint and enables substantially enhanced intrinsic activity7,8,9. Electrocatalysts designed to activate the LOM thus offer a viable pathway to reduce OER overpotentials and unlock the full performance potential of AEMWE systems.
NiFe-based hydroxides, especially NiFe layered double hydroxide (LDH), are promising OER electrocatalysts in alkaline conditions due to their low cost, high intrinsic activity, and flexible structure10,11. Recent efforts have focused on activating the LOM in NiFe LDH catalysts in order to overcome the activity limitations imposed by the conventional AEM12,13. Strategies such as heteroatom doping and heterostructure have been shown to be effective in modulating the covalence of metal-oxygen and enabling lattice oxygen redox14,15,16. However, activation of the LOM often undermines long-term durability of catalysts, as repeated lattice-oxygen redox and vacancy cycling progressively destabilize the structure and degrade the catalytic performance17,18,19. To address this issue, strategies have been reported to either prevent the direct consumption of lattice oxygen or enable the reversible regeneration to reinforce the structural integrity of electrocatalysts20,21,22. For instance, the introduction of oxygen vacancies in the catalyst can further trigger the lattice oxygen-mediated mechanism-oxygen vacancy site mechanism (LOM-OVSM), enabling the cycling of reversible oxygen species20,21. Coupling NiFe LDH with Ni4Mo alloy has been introduced to act as an oxygen pump, continuously replenishing lattice oxygen and mitigating vacancy-induced degradation22.
Beyond the structural instability induced by lattice-oxygen activation, the dissolution of Fe poses a severe challenge for NiFe-based catalysts in AEMWE. In alkaline OER, Fe is leached predominantly as ferrate (FeO42−), a chemically irreversible process that extracts lattice oxygen and disrupts the redox cycle, thereby further aggravating the lattice-oxygen instability23,24,25. This ferrate formation is accelerated under harsh industrial conditions, including high current densities (≥2 A cm−2) and concentrated alkaline electrolytes (30 wt.% KOH), which lead to the loss of active-site and rapid degradation of the performance26. Currently, most LOM activation strategies focus on increasing redox activity, but often overlook the chemical stability of metallic components under these industrial operating conditions. Therefore, the balance between high LOM-driven activity and robust stability at high current densities remains a key challenge in the design of OER electrocatalysts.
Here, we show a heterostructure electrocatalyst consisting of Fe2(MoO4)3 with a pyramid structure wrapped in amorphous NiFe LDH nanosheets (named NiFe LDH/FeMoO) for use as an efficient and stable OER. The NiFe LDH/FeMoO electrode produces oxygen with a low overpotential of only 316 mV at 2 A cm−2 in 1 M KOH. Remarkably, the NiFe LDH/FeMoO electrode can operate stably at 2 A cm−2 for 3000 h with a low operating voltage degradation rate of only 4.5 μV h−1. Using NiFe LDH/FeMoO anode and Y-NiMo/MoO2−x cathode in an AEMWE, an industrial-level performance of 2 A cm−2 at 2.0 V can be achieved. A photovoltaic (PV)-AEMWE system with high solar-to-hydrogen efficiency (20.15%) under practical current densities has been further demonstrated. This work provides fundamental insights into the design of robust OER catalysts and offers a practical strategy for advancing integrated solar-to-hydrogen conversion systems.
Results
Synthesis and characterization of the NiFe LDH/FeMoO electrocatalyst
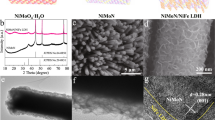
The NiFe LDH/FeMoO electrode was synthesized via a facile one-step water bath process. Specifically, clean nickel foam (NF) was used as substrate, immersed in a precursor solution containing sources of Ni, Fe, and Mo for 6 h at 90 °C (Supplementary Fig. 1). In this heterostructure, NiFe LDH is amorphous due to a competitive growth mechanism during the synthesis27. For comparison, NiFe LDH was synthesized by removing the source of Mo from the precursor solution under the same conditions (Supplementary Fig. 2). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) elucidate the microstructure of the NiFe LDH/FeMoO heterostructure. The NiFe LDH/FeMoO heterostructure formed by incorporation of the NiFe LDH nanosheets and FeMoO pyramid structure is evenly distributed on the NF substrate (Fig. 1a and Supplementary Fig. 3). In the heterostructure, FeMoO serves as a support with a pyramidal edge length of approximately 800 ± 25 nm, and NiFe LDH is characterized by thin nanosheets with a rough surface (Supplementary Fig. 4). The heterostructure configuration is adjustable according to the reaction time (Supplementary Fig. 5). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and elemental mapping (Fig. 1b, Supplementary Figs. 6 and 7) show a uniform distribution of Ni, Fe, and O across the nanosheets, while Fe, Mo, and O are concentrated in the pyramidal structure. These results confirm that the encapsulation of the pyramidal FeMoO with NiFe LDH nanosheets results in a well-defined heterostructure. The high-resolution TEM (HRTEM) image shows the distinct lattice fringes with a spacing of 0.207 nm, corresponding to the (121) plane of FeMoO (Fig. 1c). The adjacent amorphous regions are assigned to the NiFe LDH. As shown in Supplementary Fig. 8, the selected area electron diffraction (SAED) pattern of the nanosheet region displays broad and diffuse features. This is consistent with the amorphous nature of NiFe LDH, where weak rings are indexed to the NiFe LDH (110) and (012) planes. Figure 1d shows the X-ray diffraction (XRD) patterns of NiFe LDH and NiFe LDH/FeMoO. For the NiFe LDH/FeMoO heterostructure, three diffraction peaks are observed at 10.7°, 27.0°, and 29.4°, corresponding to the planes of (013), (028), and (051) for Fe2(MoO4)3 crystal, respectively. The lack of detectable NiFe LDH diffraction peaks confirms its low crystallinity as observed in HRTEM and SAED. Even after a longer reaction time, only an increase in intensity of the diffraction peaks attributable to FeMoO is observed (Supplementary Fig. 9). This is probably due to the amorphous nature of NiFe LDH, which makes it susceptible to ion diffusion limitations, whereas the crystalline structure of FeMoO allows for a continuous epitaxial growth. Their structural properties were further verified with Raman spectroscopy (Supplementary Fig. 10).

a SEM image of NiFe LDH/FeMoO heterostructure. b Elemental mapping of the NiFe LDH/FeMoO heterostructure. c HRTEM image of NiFe LDH/FeMoO heterostructure. The boundary between the NiFe LDH and FeMoO phases is marked with a yellow line. d XRD patterns of NiFe LDH and NiFe LDH/FeMoO. e Normalized Ni K edge XANES spectra of Ni foil, NiO, NiFe LDH, and NiFe LDH/FeMoO. The inset shows magnified XANES spectra. f Normalized Fe K edge XANES spectra of Fe foil, Fe2O3, NiFe LDH, and NiFe LDH/FeMoO. The inset shows magnified XANES spectra. g Ni K edge FT-EXAFS spectra for Ni foil, NiO, NiFe LDH, and NiFe LDH/FeMoO. R represents the radial distance. h Fe K edge FT-EXAFS spectra for Fe foil, Fe2O3, NiFe LDH, and NiFe LDH/FeMoO. i High-resolution XPS spectra of O 1 s for NiFe LDH and NiFe LDH/FeMoO.
The coordination and electronic structure of the NiFe LDH/FeMoO electrocatalyst were characterized by X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS). The normalized Ni K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 1e) show that the absorption edge energy of NiFe LDH and NiFe LDH/FeMoO is higher than that of Ni foil (Ni0) and NiO (Ni2+), indicating that the average oxidation state of Ni is greater than +2. Moreover, a slight shift of the absorption edge of NiFe LDH/FeMoO in comparison with NiFe LDH towards the higher energy region is consistent with the XPS results (Supplementary Fig. 11a). This can be attributed to the transfer of charge from NiFe LDH to FeMoO in the heterostructure, which reduces the electron density around Ni and increases its oxidation state. In the normalized Fe K-edge XANES spectra (Fig. 1f), the absorption edge energy of NiFe LDH and NiFe LDH/FeMoO is significantly higher than that of Fe foil (Fe0) and Fe2O3 (Fe3+), implying that the average oxidation state of Fe is greater than +3. Interestingly, in contrast to Ni, the Fe absorption edge of NiFe LDH/FeMoO is shifted to a lower energy relative to NiFe LDH, consistent with the negative shift in XPS Fe 2p binding energy (Supplementary Fig. 11b). The opposite shifts observed for Ni and Fe indicate a charge redistribution across the heterostructure interface, generating an intrinsic electric field directed from NiFe LDH to FeMoO. The high-resolution Mo 3d XPS spectra (Supplementary Fig. 11c) reveal two main peaks at ~235.3 eV and ~232.2 eV, which can be assigned to the Mo 3d5/2 and Mo 3d3/2 levels of Mo6+ ion in the MoO42− units, confirming the presence of the Fe2(MoO4)3 compound in the heterostructure28. The coordination structure of the NiFe LDH/FeMoO heterostructure was further investigated using Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectroscopy. As depicted in Fig. 1g, the Ni K-edge FT-EXAFS spectra show two prominent peaks for NiFe LDH/FeMoO at 1.65 and 2.79 Å, respectively, corresponding to the Ni–O and Ni–M (M = Ni, Fe, Mo) coordination. Similarly, the Fe K-edge FT-EXAFS analysis (Fig. 1h) reveals Fe–O and Fe–M coordination at 1.62 and 2.85 Å, respectively. Compared to NiFe LDH, NiFe LDH/FeMoO heterostructure exhibits a decreased peak intensity of Ni–M and Fe–M coordination. This observation explains the introduction of lattice disorder during the formation of the heterostructure of amorphous nature of NiFe LDH29. Meanwhile, the significant attenuation of peak intensity of Ni–O and Fe–O coordination in NiFe LDH/FeMoO likely originates from the generation of oxygen vacancy19. Oxygen vacancy characteristics were systematically investigated through high-resolution O 1s XPS. As shown in Fig. 1i, deconvolution of the O 1s XPS signal reveals four distinct components corresponding to the lattice oxygen (M–O, 529.6 eV), hydroxyl groups (M–OH, 531.0 eV), oxygen vacancies (Ov, 532.0 eV), and adsorbed water molecules (O–O, 532.8 eV)30. Quantitative analysis based on the peak area shows that the NiFe LDH/FeMoO heterostructure possesses a remarkably high oxygen vacancy content of 27.9%, which is significantly higher than that of the well-crystallized NiFe LDH (11.8%) (Supplementary Table 1). Electron paramagnetic resonance (EPR) analysis also suggests an increase in the concentration of oxygen vacancies in the NiFe LDH/FeMoO heterostructure (Supplementary Fig. 14)31.
Electrocatalytic performance for oxygen evolution
The electrocatalytic OER performance of the NiFe LDH/FeMoO electrode was systematically assessed in an alkaline solution (1 M KOH) using a standard three-electrode configuration. The reference electrode was calibrated with a reversible hydrogen electrode (RHE). Electrocatalysts such as NiFe LDH and commercial IrO2 were also tested for comparison. Figure 2a displays the linear sweep voltammetry (LSV) polarization curves of different electrocatalysts (90% iR compensation). The polarization curves without iR compensation are shown in Supplementary Fig. 16a. The NiFe LDH/FeMoO electrode exhibits notable OER performance requiring a low overpotential of 289 at 1 A cm−2 and 316 mV at 2 A cm−2, respectively (Fig. 2b). This is much lower than for the NiFe LDH (335 and 399 mV) and the IrO2 electrode (444 and 577 mV). The NiFe LDH/FeMoO electrode requires a low overpotential of 336 mV to achieve a high current density of 3 A cm−2. Supplementary Table 2 compares the overpotentials at various current densities of NiFe LDH/FeMoO with those of recently reported NiFe LDH-based electrocatalysts.

a Polarization curves with 90% iR compensation in 1 M KOH at a scan rate of 5 mV s−1. R was 1.1 ± 0.1 Ω. b Corresponding overpotentials at 1, 2, and 3 A cm−2 of NiFe LDH/FeMoO, NiFe LDH, and IrO2. c Δη/Δlog|j| values for different current density ranges. d The resistance of the interfacial charge transfer (Rct) at various potential for NiFe LDH and NiFe LDH/FeMoO. e The turnover frequency (TOF) values of catalysts at an overpotential of 300 mV (the black dotted line). f Polarization curve of NiFe LDH/FeMoO in alkaline electrolytes with different pH. g Polarization curves with 90% iR compensation of NiFe LDH/FeMoO in 1 M KOH and 1 M TMAOH, respectively. R was 1.1 ± 0.1 Ω. The inset shows corresponding Tafel plot. h The 34O2/32O2 ratio over four cyclic voltammetry (CV) cycles. i The stability test of NiFe LDH/FeMoO and NiFe LDH at 2 A cm−2 in 1 M KOH.
The NiFe LDH/FeMoO electrode has a smaller Tafel slope (48.9 mV dec−1) than that of NiFe LDH (70.7 mV dec−1), suggesting that the former has a more rapid OER kinetics (Supplementary Fig. 16b). This result also points out a possible change in the OER mechanism. The performance of the catalyst at high current densities is influenced by activity and mass transfer32. The slope of the overpotential versus current density was analyzed to determine the relative contribution of these factors. As shown in Fig. 2c, the NiFe LDH/FeMoO heterostructure exhibits a moderate increase in Δη/Δlog|j| from 53.0 to 113.6 mV dec−1 over the current density range of 0.1-3 A cm−2, in stark contrast to NiFe LDH (56.9 to 386.4 mV dec−1). This indicates that the NiFe LDH/FeMoO heterostructure enhances reaction kinetics and facilitates rapid mass transfer at high current densities. The thin, rough NiFe LDH nanosheets shorten the pathways for mass transfer, while the pyramidal FeMoO protrusions prevent the formation of gas bubbles and promote the recovery of electrolytes. This is further supported by contact angle measurements (Supplementary Fig. 17).
The resistance of the interfacial charge transfer (Rct) of the OER was determined by electrochemical impedance spectroscopy (EIS) (Supplementary Figs. 18 and 19), and the results are listed in Supplementary Tables 3 and 4. The NiFe LDH/FeMoO electrode shows a lower Rct than that of NiFe LDH at all tested potentials (Fig. 2d). This enhancement reflects the good interfacial charge‐transfer kinetics of NiFe LDH/FeMoO heterostructure. The electrochemical surface area (ECSA) of the NiFe LDH/FeMoO electrode was estimated to be 58.7 cm−2 with cyclic voltammetry (Supplementary Fig. 20), which is higher than that of the flat NiFe LDH (48.8 cm−2). The intrinsic activity of electrocatalysts was also compared through the ECSA-normalized polarization curves in Supplementary Fig. 21, showing that the NiFe LDH/FeMoO electrode maintains the lowest overpotential for the OER. In addition, the turnover frequency (TOF) of NiFe LDH and NiFe LDH/FeMoO was assessed to allow a clear comparison of their catalytic ability to the OER (Fig. 2e and Supplementary Fig. 22). At an overpotential of 300 mV, the NiFe LDH/FeMoO achieves a TOF of 0.140 s−1, which surpasses the NiFe LDH (0.056 s−1).
The NiFe LDH/FeMoO heterostructure shows extraordinary OER activity of lower overpotential and Tafel slope, suggesting a significant change in the reaction kinetics. Figure 2f shows the OER polarization curves of NiFe LDH/FeMoO in electrolytes with different pH levels, suggesting a strong pH dependence for the OER activity of NiFe LDH/FeMoO. Compared to the AEM pathway, the LOM pathway generates superoxide (O2−) and peroxide (O22−) anionic species, which serve as compelling indicators to determine whether the OER mechanism follows the LOM pathway4,33. These species were probed using tetramethylammonium cations (TMA⁺), which have a strong interaction with oxygen species. Figure 2g shows that the OER performance of the NiFe LDH/FeMoO heterostructure is significantly reduced in 1 M TMAOH electrolyte, as shown by increased polarization voltage and Tafel slope, whereas NiFe LDH is less affected (Supplementary Fig. 25).
To further investigate the OER mechanisms in NiFe LDH and NiFe LDH/FeMoO, differential electrochemical mass spectrometry (DEMS) was employed to monitor the oxygen signals generated during the OER process (Supplementary Fig. 26). In traditional AEM, 18O-labeled catalysts should only produce 32O2 during OER in a H216O electrolyte. Therefore, the release of 34O2 reflects the involvement of lattice oxygen34. The DEMS measurements showed a strong 34O2 signal and a weaker 36O2 signal, in addition to the normal 32O2 signal. This result indicates that the 18O-labeled lattice oxygen is activated and participates in the formation of 18O16O and 18O18O (Supplementary Fig. 27). Figure 2h shows the 34O2/32O2 ratio over four cyclic voltammetry (CV) cycles, which was used to quantify the contribution of the LOM35. The 34O2/32O2 ratio for NiFe LDH/FeMoO was approximately twice that for NiFe LDH, providing direct evidence that lattice oxygen in NiFe LDH/FeMoO participates more actively in the OER process via the LOM pathway. Furthermore, the gradual decrease in the 34O2/32O2 ratio in NiFe LDH/FeMoO over multiple cycles indicates that the 18O-labeled lattice oxygen was gradually consumed and replaced by 16O from the electrolyte36.
Apart from activity, electrocatalyst durability is a critical parameter for industrial applications, particularly under large current densities. As shown in Fig. 2i, the NiFe LDH/FeMoO heterostructure exhibits long-term stability, with a degradation rate of merely ~4.5 μV h−1 for the operating voltage over 3000 h of continuous operation at 2 A cm−2. Extrapolating this degradation rate suggests an increase of only ~88 mV in overpotential after 2 years of continuous operation. In contrast, NiFe LDH can only maintain stable performance for approximately 300 h before undergoing a significant performance decline. SEM and TEM images reveal that the specific architecture of the heterostructure, consisting of thin NiFe LDH nanosheets that wrap around the FeMoO pyramids, remains intact even after prolonged testing (Supplementary Figs. 28 and 29). In contrast, the NiFe LDH structure is susceptible to nanosheet agglomeration and structural collapse. These results highlight the industrial application potential of the NiFe LDH/FeMoO heterostructure.
Mechanistic insights of activity enhancement
In-situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was used to monitor the intermediates and to elucidate the mechanism of the reaction during oxygen evolution on NiFe LDH and NiFe LDH/FeMoO. The electrocatalysts were drop-cast onto a gold-coated silicon prism and tested in an electrochemical cell (Supplementary Figs. 30 and 31). ATR-SEIRAS spectra of NiFe LDH and NiFe LDH/FeMoO electrocatalysts were collected at different applied potentials (0.8-2.0 V), and the results are shown in Fig. 3a and b. The observed absorption peaks at ~1640 cm−1 correspond to the H–O–H bending mode (δH–O–H)of interfacial water molecules at open circuit potential (OCP)37. With the increase in applied potential, a new absorption at ~1110 cm−1 is observed, which is assigned to the *OOH intermediate38,39,40. This result supports the involvement of an AEM pathway for the OER process. Meanwhile, an absorption peak at ~1230 cm−1 corresponding to the *OO intermediate is observed, indicating the LOM pathway for OER41,42. The coverage of *OO and *OOH intermediates gradually increases as the potential increases, leading to a corresponding enhancement in peak intensity. Notably, NiFe LDH exhibits a more pronounced *OOH peak, indicating that the OER process predominantly follows the AEM pathway. In contrast, the NiFe LDH/FeMoO heterostructure shows a significantly stronger *OO signal than *OOH, suggesting greater involvement of the LOM pathway30.

In-situ ATR-SEIRAS spectra at open-circuit voltage, various applied potentials (from 0.8 to 2.0 V vs. RHE) for a NiFe LDH and b NiFe LDH/FeMoO. c Differential charge density diagram of the NiFe LDH/FeMoO heterostructure. The yellow and green regions represent charge accumulation and depletion, respectively. Projected density of states (PDOS) of d O 2p and e Ni 3d orbitals in the NiFe LDH and NiFe LDH/FeMoO. UHB represents the Upper Hubbard Band, and LHB represents the Lower Hubbard Band. f Schematic diagrams of the energy band structures induced by Mott-Hubbard splitting of NiFe LDH and NiFe LDH/FeMoO. U represents the energy gap, and Ef represents the Fermi level. g Gibbs free energy diagrams of the adsorbate evolution mechanism (AEM) pathway and lattice oxygen-mediated mechanism-oxygen vacancy site mechanism (LOM-OVSM) pathway. h Schematic diagrams of the reaction mechanism for NiFe LDH/FeMoO.
Density functional theory (DFT) calculations were performed to provide insights into the catalytic enhancement of the NiFe LDH/FeMoO heterostructure. A heterostructure model was constructed with a top layer of NiFe LDH rich in oxygen-vacancies and a bottom layer of Fe2(MoO4)3 (Supplementary Fig. 33a) according to the experimental results. NiFe LDH without oxygen vacancy was also constructed and assessed for comparison (Supplementary Fig. 33b). Differential charge density of NiFe LDH/FeMoO heterostructure was evaluated. Bader charge analysis was then performed along the Z axis, which is defined as the direction perpendicular to the interface, extending from the NiFe LDH layer toward the FeMoO substrate. A pronounced charge redistribution is revealed at the interface between NiFe LDH and FeMoO via differential charge density analysis (Fig. 3c). Specifically, electrons are depleted in the NiFe LDH layer but accumulated in the FeMoO layer. This electron density gradient suggests a net charge migration from NiFe LDH to FeMoO, consistent with the XANES measurements. This electron redistribution indicates the generation of an electric field within the heterostructure interface, thereby forming a space charge region that modulates the electronic structure of the catalyst.
The projected density of states (DOS) was analyzed to investigate the changes in the electronic states of the NiFe LDH/FeMoO heterostructure. Supplementary Fig. 34 presents the DOS of NiFe LDH and NiFe LDH/FeMoO heterostructure. A significant overlap between the d-band of metal and the 2p band of oxygen for NiFe LDH/FeMoO, indicating enhanced covalency between the metal centers and oxygen ligands. This improved covalency promotes the electron delocalization in the NiFe LDH/FeMoO heterostructure, which provides the basis for the lattice oxygen participation in the OER process15. Moreover, the partial density of states (PDOS) analysis shows that the O 2p band center (εp) shifts from −3.67 eV in NiFe LDH to −3.22 eV in the NiFe LDH/FeMoO heterostructure (Fig. 3d). This upward shift can be attributed to the abundant oxygen vacancies present in NiFe LDH, which elevate the O 2p states closer to the Fermi level and facilitate electron delocalization from oxygen centers. The increased proximity between the O 2p band and the Fermi level enhances the electronic coupling with adsorbed species under anodic potentials, thereby accelerating lattice oxygen oxidation and promoting a shift in the OER pathway toward the LOM43. According to molecular orbital theory, the hybridization between O 2p and Ni 3d orbitals leads to the formation of metal-oxygen (M–O) antibonding bands, which are split into an unoccupied upper Hubbard band (UHB) and an occupied lower Hubbard band (LHB)44,45. The calculated positions of the LHB and UHB centers, along with their corresponding energy gaps (U), for NiFe LDH and NiFe LDH/FeMoO are summarized in Fig. 3e and Supplementary Table 5. Notably, the NiFe LDH/FeMoO heterostructure exhibits a larger energy gap U value of 6.65 eV compared to 6.07 eV for NiFe LDH, which can be ascribed to the intrinsic electric field at the heterointerface that depletes electron density within the NiFe LDH layer, thereby reducing local dielectric screening46. The increased U value induces a downward shift of the LHB, as illustrated in Fig. 3f, significantly facilitating electron removal from oxygen sites under anodic polarization47. Importantly, the LHB center in NiFe LDH/FeMoO is located below the O 2p band center (Fig. 3f). Consequently, the combined effect of the downward shift of the LHB and the upward shift of the O 2p band center reduces the overlap between the metal 3d and oxygen 2p orbitals, thereby weakening the M–O bond strength. The presence of weaker M–O bonds lowers the activation energy for lattice oxygen release12,48. Together, these insights demonstrate that the cooperative regulation of oxygen vacancies and interfacial electric fields fundamentally reprograms the electronic structure of catalyst, thereby triggering the LOM pathway and enabling comparable OER performance.
The Gibbs free energies for OER via the AEM and LOM were calculated for the NiFe LDH/FeMoO heterostructure to validate the reaction mechanism (Supplementary Figs. 35–37 and Supplementary Table 6). As illustrated in Fig. 3g, the rate-determining step (RDS) for OER via the AEM pathway on NiFe LDH/FeMoO involves the evolution of *O to *OOH with a thermodynamic barrier of 0.58 eV. In contrast, the RDS for the LOM pathway shifts to the transformation of *O–OH to *O–O, reducing the barrier to 0.46 eV. These results indicate that the NiFe LDH/FeMoO heterostructure enhances OER kinetics by favoring the LOM pathway. As illustrated in Fig. 3h, the OER process via the LOM pathway begins with the adsorption of OH− anions onto lattice oxygen through nucleophilic attack. This leads to direct O–O coupling and the formation of O2, which then desorbs from the electrocatalyst surface. The generated oxygen vacancy is subsequently replenished by OH−, regenerating active sites for continuous catalysis.
Unraveling the mechanisms of stability improvement
According to the above reaction mechanism, strong adsorption affinity for OH− can effectively repair the oxygen vacancies generated along the LOM pathway, thereby suppressing the propagation of lattice defects and minimizing subsequent structural degradation. As shown in Supplementary Fig. 39a, b, the broad absorption band observed in the 2800–3800 cm−1 range in the in-situ ATR-SEIRAS corresponds to the O–H stretching vibrations (νO–H) of interfacial water molecules37. The intensity of the νO–H vibration increases progressively with the applied potential, indicating increasing adsorption of water molecules at the charged interface. Furthermore, the variation in the intensity of this band under different applied potentials was compared between NiFe LDH and NiFe LDH/FeMoO (Supplementary Fig. 39c). The NiFe LDH/FeMoO heterostructure exhibits a significantly stronger signal, indicating a higher surface coverage of *OH intermediates that facilitates the regeneration of lattice oxygen22,49. Laviron analysis was further conducted to investigate the adsorption behavior of OH− on the electrocatalyst surfaces50,51. As shown in Supplementary Fig. 41, steady-state redox currents associated with OH− transfer exhibit a linear correlation with the square root of the scan rate, indicating diffusion-limited adsorption behavior52. The calculated rate constant (ks) for NiFe LDH/FeMoO is 0.14 s−1, significantly higher than that of NiFe LDH (0.11 s−1) (Fig. 4a). This supports stronger OH− adsorption affinity on the NiFe LDH/FeMoO surface.

a Laviron analysis of NiFe LDH/FeMoO and NiFe LDH. E represents electrode potential, and E1/2 represents half-wave potential. In-situ Raman spectra of b NiFe LDH/FeMoO, and c NiFe LDH. d The ionic concentration in the electrolyte after chronopotentiometric tests every 4 h. e The UV-Vis spectrum of the electrolyte after 30 h of chronopotentiometric testing. f Schematic diagram of the improvement of the stability of NiFe LDH/FeMoO.
In-situ Raman spectroscopy was performed to further understand the evolution of the local structure of the catalyst during the OER process, which was collected within the potential range from the OCP to 1.41 V for the NiFe LDH and NiFe LDH/FeMoO electrodes (Supplementary Figs. 42 and 43). As shown in Fig. 4b, vibration peaks at 451 and 521 cm−1, which are attributed to Ni–OH and Ni–O in NiFe LDH/FeMoO, were consistently observed until the applied potential reached 1.32 V. As the potential increased to 1.35 V, two peaks appeared at 476 and 557 cm−1 corresponding to the Eg bending and A1g stretching vibration modes of Ni–O in γ-NiOOH53,54. A distinct peak at ~1060 cm−1 was observed, which can be assigned to the vibrations of interlayer carbonate species in the NiFe LDH55. In addition, the peaks at 780 cm−1 and 904 cm−1, originating from the asymmetric and symmetric stretching modes of the Mo–O bonds in the MoO4 units, were also detected56. With the applied potential increased from OCP to 1.41 V, the peak at 904 cm−1 remained clearly discernible, indicating the good structural stability of the pyramidal framework. In contrast, the peak at 780 cm−1 gradually decreased in intensity and eventually disappeared. This is likely due to the intrinsically weaker signal of FeMoO located beneath the NiFe LDH overlayer, combined with signal interference caused by gas bubble formation during electrolysis57.
In addition, as the voltage increased from the OCP to 1.41 V, the peaks of vibration corresponding to Mo–O bonding in FeMoO were always clearly visible, indicating the notable structural stability of its pyramid structure. Similarly, the nearly identical transformation process has been observed for NiFe LDH (Fig. 4c). It should be noted that the phase-transition potentials for NiFe LDH and NiFe LDH/FeMoO were 1.29 and 1.35 V, respectively. These results suggest that the nickel redox process of the NiFe LDH/FeMoO heterostructure is delayed by the aid of lattice oxygen oxidation12. Although elevated metal oxidation states are often observed in LOM-active catalysts, lattice-oxygen participation does not necessarily require highly oxidized metal centers15,58. In the NiFe LDH/FeMoO heterostructure, interfacial charge redistribution and oxygen-vacancy-induced modulation of the O 2p band enhance oxygen-hole character while suppressing excessive oxidation of Ni and Fe sites. Therefore, compared to NiFe LDH, NiFe LDH/FeMoO can be more easily stabilized during the OER process.
To further elucidate the underlying stabilization mechanism, a chronopotentiometric test was conducted on NiFe LDH/FeMoO and NiFe LDH in 10 M KOH under a high current density of 4 A cm−2 to accelerate the catalytic degradation. As shown in Supplementary Fig. 45, NiFe LDH/FeMoO electrode maintained its initial performance within 30 h, whereas the activity of NiFe LDH decreases to 96% of its initial value. The electrolyte was then examined every 4 h and the concentrations of Ni and Fe ions was measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES). As depicted in Fig. 4d, in NiFe LDH/FeMoO, Fe experienced slight dissolution in the early stage of the test and then remained stable at approximately 0.03 mg L−1, whereas in NiFe LDH, the leaching of Fe gradually increased to 0.29 mg L−1 along with the declined activity. Ni shows almost no dissolution in NiFe LDH/FeMoO and NiFe LDH (Fig. 4d), because Ni-based compounds are thermodynamically relatively stable under OER conditions59. To determine the form in which Fe elements are dissolved in the electrolyte from the catalyst, the electrolyte was examined with an ultraviolet-visible (UV-Vis) spectrophotometer after a 30-h chronopotentiometric test60. As shown in Fig. 4e, a signal peak at approximately 500 nm appears in the case of NiFe LDH, which corresponds to the d-d electron transition of the highly oxidized FeO42−61. In contrast, no such signal was observed for NiFe LDH/FeMoO, indicating the suppression of Fe over-oxidation. This is attributed to the formation of fast electron transport channels by the intrinsic electric field at the heterostructure interface, which effectively prevents the excessive oxidation and dissolution of iron species. Collectively, the good stability of NiFe LDH under industrial conditions can be primarily ascribed to three synergistic effects (Fig. 4f). Firstly, the pyramidal FeMoO framework provides robust mechanical support for NiFe LDH nanosheets, mitigating bubble-induced detachment and enhancing electrolyte diffusion kinetics. Secondly, the enhanced OH− chemisorption facilitates rapid replenishment of lattice oxygen vacancies, maintaining structural integrity during prolonged OER operation. Thirdly, the built-in electric field at the heterostructure interface accelerates charge transfer, suppressing over-oxidation and subsequent dissolution of active metal species.
Performance in AEMWE and integrated PV-AEMWE systems
To further assess the practical feasibility of the catalysts, we constructed an AEMWE cell utilizing a self-supported NiFe LDH/FeMoO anode and a Y-NiMo/MoO2−x62 cathode, as illustrated in Fig. 5a. A conventional AEMWE was also prepared using commercial catalysts of IrO2 and Pt/C loaded onto NF as anode and cathode for comparison. As shown in Fig. 5b, the electrolyzer based on NiFe LDH/FeMoO||NiMo/MoO2−x requires 1.7 V to reach a current density of 1 A cm−2 and 2.0 V for 2 A cm−2 at 60 °C, respectively, which was supplemented with a 3 M KOH solution at a flow rate of 380 mL min−1. In contrast, the IrO2||Pt/C electrolyzer requires a 1.9 V to reach a current density of 1 A cm−2. This performance, summarized in Supplementary Table 7, surpasses most state-of-the-art AEMWEs. In addition, the NiFe LDH/FeMoO||Y-NiMo/MoO2−x electrolyzer demonstrates durability over a period of 100 h at a current density of 1 A cm−2 with minimal voltage fluctuations (Fig. 5c). In contrast, the IrO2||Pt/C electrolyzer exhibits a rapid decline in performance under the same testing conditions.

a Schematic diagram of the designed AEMWE system. b j-V curves of AEMWE operated at 60 °C in 3 M KOH. c The durability of electrolyzer measured at 1 A cm−2 in 3 M KOH. d Schematics of the PV-AEMWE system. e The intersection points of the j-V characteristics of the perovskite/Si tandem solar cell measured under simulated AM1.5 G illumination (100 mW cm−2) and the polarization curves of the electrolysis cell. f Chronopotentiometric curves during PV-AEMWE operation and practical solar-to-hydrogen (STH) efficiency.
The integration of solar cell with electrolyzer has emerged as a promising strategy for achieving solar-water-splitting and cost-effective production of renewable hydrogen. As illustrated in Fig. 5d, a solar-water-splitting system was constructed using a monolithic perovskite/Si tandem solar cell (TSC) with high output voltage as the power source for an electrolyzer. The device exhibited notable photovoltaic characteristics with a VOC of ~1.82 V and a PCE exceeding 23.6% (Supplementary Figs. 46 and 47, and Supplementary Table 8)63. The predicted operating point of the integrated solar water splitting system, defined as the intersection of the j-V curves of the tandem solar cell and the electrolysis cell, yields a current density of 16.6 mA cm−2 at 1.43 V, corresponding to a solar-to-hydrogen (STH) efficiency of 20.41% (Fig. 5e). More importantly, this intersection point is very close to the maximum power point of the perovskite/Si tandem solar cell (16.19 mA cm−2 at 1.486 V), indicating that the energy loss during the conversion of electrical energy to chemical energy in this system is relatively small. In addition, the gas evolution of the PV-AEMWE system was measured by online gas chromatography (Supplementary Fig. 47a). As shown in Supplementary Fig. 47b, the yields of H2 and O2 in 30 h are close to a ratio of 2:1, being 9.16 and 4.64 mmol, respectively. Figure 5f presents the water splitting current generated by the PV-AEMWE system and the corresponding STH efficiency under continuous AM1.5G illumination. Encouragingly, the PV-AEMWE system exhibited an comparable STH efficiency of 20.15% under practical operation and maintained 97.1% of its initial current density after continuous operation for over 140 h, confirming its good durability (Supplementary Fig. 47d). Compared with recently reported photovoltaic-electrochemical (PV-EC) water-splitting devices, the present system exhibits notable performance in terms of both STH efficiency and operational stability (Supplementary Table 9). Additionally, the PV-EC systems based on perovskite/Si tandem cells consistently outperform single-junction Si or perovskite devices in terms of STH efficiency, highlighting the great potential of tandem perovskite photovoltaics to further increase photocurrent output and push the efficiency of integrated solar water splitting towards its theoretical limit.
Discussion
A NiFe LDH/FeMoO heterostructure has been designed and synthesized for the efficient and stable OER catalyst in alkaline electrolytes. A one-step water bath method allows the formation of amorphous NiFe LDH nanosheets encapsulating pyramid FeMoO, which introduces abundant oxygen vacancies and intrinsic electric field. The heterostructure catalyst achieves a high current density of 2 A cm−2 at a low overpotential of 316 mV in 1 M KOH. Mechanistic investigations, supported by in-situ spectroscopy and DFT calculations, confirm that the LOM pathway dominates OER, which can be assigned to the upshifted O 2p band center and downshifted Ni 3d lower Hubbard band, reducing the reaction energy barrier. Benefiting from the stable support of the pyramid in the heterostructure and the effective suppression of iron dissolution by the intrinsic electric field, notable stability exceeding 3000 h under high current density is ensured. Additionally, the practical feasibility of NiFe LDH/FeMoO as the anode was validated in an AEMWE system employing Y-NiMo/MoO2−x as the cathode. The AEMWE system achieves a high current density of 2 A cm−2 at just 2.0 V. We further demonstrated a PV-AEMWE system with a solar-to-hydrogen theoretical efficiency of 20.4% by integrating perovskite/Si tandem solar cell. This work offers a universal route to design OER catalysts combining lattice-oxygen activation with industrial-scale durability for sustainable hydrogen production.
Methods
Materials
Nickel nitrate (Ni(NO3)2·6H2O, ≥99%), chloroauric acid (HAuCl4·xH2O, Au ≥47.5%), and iridium oxide (IrO2, Ir ≥84.5%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Ferric nitrate (Fe(NO3)3·9H2O, 98%), yttrium nitrate (Y(NO3)3, 99.99%), ammonium fluoride (NH4F, 99%), and potassium hydroxide (KOH, 95%) were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, 99%), platinum on carbon (Pt/C, 20%), urea (CH4N2O, 99%), Nafion 117 (5%), and nickel foam (NF, 99.9%) were purchased from Beijing InnoChem Science & Technology Co., Ltd. Sodium sulfite (Na2SO3, ≥97%), sodium thiosulfate (Na2S2O3, 98.5%), hydrochloric acid (HCl, 36–38%) were purchased from Sinopharm Chemical Reagent Co.Ltd. All chemicals were used as received without purification.
Synthesis of NiFe LDH/FeMoO and NiFe LDH
Firstly, 500 mL precursor solutions were prepared with deionized water as solvent, containing nickel nitrate (2.9079 g), ferric nitrate, ammonium molybdate, urea (9.0090 g), and ammonium fluoride (1.3890 g). To regulate the Fe content, ferric nitrate concentrations were set to 2, 4, 6, and 8 mM while keeping other solute concentrations constant. Similarly, ammonium molybdate concentrations were adjusted to 13, 14, 15, and 16 mM to tune the Mo content under the same conditions. Nickel foam (NF, thickness: 1.5 mm, 5 × 5 cm2) was pre-treated by ultrasonic cleaning in 3 M hydrochloric acid for 15 min to remove surface impurities, followed by sequential ultrasonic rinsing in deionized water and absolute ethanol (15 min each). Subsequently, 150 mL of the precursor solution was transferred into a polytetrafluoroethylene beaker containing the cleaned NF, and the hydrothermal reaction was conducted at 90 °C for 6 h. After reaction, the as-synthesized NiFe LDH/FeMoO were collected, thoroughly rinsed with deionized water and absolute ethanol, and dried in a vacuum oven at 60 °C for 12 h. For comparison, NiFe LDH catalysts were synthesized via the same protocol but with the molybdenum source omitted from the precursor solution.
Preparation of Y-NiMo/MoO2−x
The preparation of Y-NiMo/MoO2−x was adapted from our group’s previous work62. Specifically, 0.6979 g of Ni(NO3)2·6H2O, 0.0184 g of Y(NO3)3·6H2O, and 0.7415 g of (NH4)6Mo7O24·4H2O were dissolved in deionized water and stirred until homogeneous. The solution was then transferred to a Teflon-lined stainless-steel autoclave, along with a piece of cleaned NF, followed by hydrothermal reaction at 150 °C for 6 h to form Y-NiMoO4. Subsequently, the as-prepared Y-NiMoO4 precursor was subjected to reduction in a tube furnace under an Ar/H2 atmosphere at 500 °C for 30 min, yielding Y-NiMo/MoO2−x.
Preparation of IrO2 and Pt/C catalysts on NF
The mass loading of NiFe LDH/FeMoO on NF was approximately 2.2 mg cm−2, calculated from the weight increase after the hydrothermal reaction. For reference, IrO2 and Pt/C catalysts were deposited on NF via the drop-casting method. Briefly, 22 mg commercial IrO2 catalyst was dispersed in a mixture of 90 μL 5 wt% Nafion, 260 μL ethanol, and 650 μL deionized water to form a homogeneous slurry. A 100 μL aliquot of this slurry was drop-cast onto cleaned NF and air-dried at room temperature. The commercial Pt/C catalyst was loaded on NF following the same procedure as IrO2.
Characterization
Scanning electron microscopy (SEM) was conducted on a JEOL-JSM IT800. Transmission electron microscopy (TEM) was performed on a JEOL-JEM 2100 F coupled with energy-dispersive spectroscopy (EDS). X-ray diffraction (XRD) was performed using a PANalytical X’pert PRO with Cu Kα radiation. Raman spectra acquired using a Horiba iHR320 spectrometer with a 532 nm He-Ne laser. X-ray absorption fine structure (XAFS) spectra were acquired at the Shanghai Synchrotron Radiation Facility (beamline BL14W1, China), with transmission mode employed for Ni, Fe, and Mo K-edges. X-ray photoelectron spectroscopy (XPS) was analyzed by a Thermo Fisher-ESCALAB 250Xi. Electron paramagnetic resonance (EPR) spectroscopy was recorded on a Bruker A300 instrument at −150 °C under a microwave frequency of 9.83 GHz.
Electrochemical measurements
All electrochemical measurements were conducted using a three-electrode configuration on an Autolab PGSTA302N workstation coupled with a BOOSTER10A current amplifier. The working electrode was the catalyst to be tested with a testing area of 1 cm2, the reference electrode was a Hg/HgO (1 M KOH, pH = 13.9 ± 0.1) electrode, and the counter electrode was a platinum plate. Calibration of the Hg/HgO reference electrode against the Reversible Hydrogen Electrode (RHE) was performed using a platinum wire in an electrolyte saturated with high-purity hydrogen. 56.11 g of KOH was weighed and transferred into a 1000 mL volumetric flask, followed by dilution to the graduation mark with deionized water. The obtained electrolyte (1 M KOH) was then transferred to a polytetrafluoroethylene (PTFE) bottle for storage. The experiments were performed under ambient conditions with the temperature maintained at 25 °C (room temperature). Before the linear sweep voltammetry (LSV) test, the working electrode was subjected to 30 cycles of CV activation treatment. The scanning range was from 0.8 to 1.1 V at a scan rate of 50 mV s−1. The polarization curves were measured using a slow-scan mode at 5 mV s−1. The obtained potential data were corrected by 90% iR compensation and calibrated to the RHE scale in accordance with the Nernst equation, as expressed by: ERHE = EHg/HgO + 0.0591 × pH + 0.098. Electrochemical impedance spectroscopy (EIS) analysis was carried out at different potentials ranging from 1.48 to 1.58 V vs. RHE. The frequency scanning range covered from 0.1 Hz to 100 kHz with an AC amplitude of 10 mV. The solution resistance is determinable through EIS analysis. The electrochemically active surface area (ECSA) was estimated via the double-layer capacitance (Cdl) method. Specifically, CV scans were recorded in the non-Faradaic region (0.924-1.024 V vs. RHE) at various rates, where Cdl was determined from the slope of the charging current vs. scan rate. ECSA was calculated as ECSA = Cdl/Cs, assuming a specific capacitance of Cs = 60 μF cm−2. The turnover frequency (TOF) using the formula: TOF = (j × A)/(4 × F × n), where j is the anodic current density from polarization curves, A is the electrode area, F is the Faraday constant, and n is the number of active sites. The n value was determined by integrating the charge from CV curves in 1 M PBS (pH = 7 ± 0.1) at 50 mV s−1. Faradaic efficiency (ηF) was evaluated by chronoamperometry at a constant current density of 16.5 mA cm−2 in 1 M KOH for 30 h, with H2 and O2 products quantified by gas chromatography. The Faradaic efficiency of gaseous products is calculated according to the following equation: ηF = α × n × F/Q × 100%, where α is the electron transfer number, n represents the mole number of the product, F is the Faraday constant (96485 C mol−1), and Q denotes the total accumulated charge (C) during the testing process. TOF and Faradaic efficiency measurements were only performed once.
Fabrication and testing of AEMWE and PV-AEMWE systems
The anion exchange membrane water electrolysis (AEMWE) system is mainly composed of bipolar plate current collectors, catalytic electrodes, and an anion exchange membrane assembly. Specifically, the anion exchange membrane (FAA-3-30, 6 × 6 cm2) is immersed in a 1 M KOH solution for 24 h to activate the functional groups. This process enables the quaternary ammonium groups within the membrane to form stable coordination with hydroxide ions, significantly enhancing its ion-conduction efficiency. For the catalytic electrodes, NiFe LDH/FeMoO grown on an NF substrate and Y-NiMo/MoO2−x electrocatalysts are used as the cathode and anode, respectively. Without the need for a hot-pressing assembly process, the activated membrane material and the catalytic electrodes are physically stacked in a sandwich structure to construct a NiFe LDH/FeMoO||Y-NiMo/MoO2−x composite electrolysis system. For comparison, an IrO2||Pt/C electrolytic cell is also assembled using noble-metal catalysts. The system stabilization treatment is carried out in a constant-current mode, with a direct current of 10 mA cm−2 applied for a certain period. The electrochemical performance test is conducted in a 3 M KOH electrolyte system maintained at 60 ± 0.5 °C. The polarization curves were recorded using a potentiostat (Keysight E36154A) without iR compensation.
The fabrication of monolithic perovskite/silicon tandem solar cell is referenced to our group’s previous work63. Then, the perovskite/silicon tandem solar cell was connected as a power source to the NiFe LDH anode and the Y-NiMo/MoO2−x cathode to construct a photovoltaic-electrochemical (PV-EC) electrolysis system. The active area of the tandem solar cell was 4.0 cm2, which matched the effective electrode area of the AEMWE (~4.0 cm2). The performance of the PV-EC system was measured under an irradiance of 100 mW cm−2 on the surface of the solar cell, which was provided by a xenon light source solar simulator (Oriel, 9119) equipped with an AM 1.5G filter. The solar-to-hydrogen (STH) conversion efficiency of the photovoltaic-AEMWE system is determined by the following equation: \({{{\rm{S}}}}{{{\rm{TH}}}}=\frac{1.23\times {j}_{{{{\rm{op}}}}}\times {\eta }_{{{{\rm{F}}}}}}{{P}_{{{{\rm{in}}}}}\times {A}_{{{{\rm{sc}}}}}}\), where jop is the operating current, ηF is the Faradaic efficiency, Asc is the solar cell area, and Pin is the incident irradiation power.
Operando DEMS measurement
The DEMS experiment was conducted using an isotope labeling method in a typical three-electrode electrochemical cell, utilizing a QAS 100 system (Shanghai, Linglu Instruments). The electrolyte and the vacuum system were isolated from each other by a porous polytetrafluoroethylene (PTFE) membrane featuring a maximum pore size of 20 nm. Firstly, the Ni(OH)2, NiFe LDH, and NiFe LD/FeMoO catalysts were labeled with the 18O isotope. Specifically, CV was performed in a 1 M KOH electrolyte prepared with H218O, within the voltage range of 1.1–1.7 V vs. RHE at a scan rate of 5 mV s−1. The mass signals of gas products with different molecular weights were recorded in real time. Then, the electrode was washed with H216O to completely remove any residual H218O. Finally, four consecutive CV cycles were performed in a 1 M KOH electrolyte prepared with H216O, within the voltage range of 1.1–1.8 V vs. RHE at a scan rate of 5 mV s−1, and the mass signals of the gas products were recorded again.
In-situ ATR-SEIRAS measurement
All spectral measurements were acquired using a Fourier-transform infrared (FTIR) spectrometer (Nicolet iS50R, Thermo Fisher Scientific) equipped with a liquid-nitrogen-cooled MCT detector, with a spectral resolution of 4 cm−1. Gold-coated silicon prisms were prepared via a wet-chemical method: 0.045 g of HAuCl4 was dissolved in 10.5 mL of Milli-Q water, followed by the addition of 0.015 g of NaOH. Separately, 0.02 g of NH4Cl, 0.141 g of Na2SO3, and 0.093 g of Na2S2O3·5H2O were dissolved in 4.5 mL of deionized water. The two solutions were then mixed to form the gold-plating solution. Cleaned silicon prisms were immersed in a 40% ammonium fluoride solution for 90 s, followed by immersion in a mixture of 4 mL gold-plating solution and 0.034 mL hydrofluoric acid at 60 °C for 5 min to yield gold-coated prisms. The catalyst inks were prepared by dispersing 1 mg of catalyst in a mixture of 800 µL of deionized water, 190 µL of isopropanol, and 10 µL of Nafion. The mixture was sonicated for 30 min to obtain a uniformly dispersed ink solution. The ink was then dropped onto gold-coated prisms, dried under an infrared lamp, and the catalyst loading was determined to be 200 μg. The gold-coated prisms were mounted as working electrodes in a three-electrode electrochemical cell with an Ag/AgCl electrode serving as the reference electrode and a platinum wire serving as the counter electrode. The electrochemical cell was operated in chronoamperometric mode using a workstation, and Fourier-transform infrared (FTIR) spectra were recorded in real time at open-circuit potential, as well as under applied potentials ranging from 0.8 to 2.0 V versus RHE. Spectra were collected at 0.1 V intervals with an acquisition time of 45 s for each measurement.
In-situ Raman spectroscopy measurement
In-situ Raman spectroscopy for monitoring dynamic surface reconstruction was performed using a confocal Raman spectrometer (Horiba iHR320) with a 532 nm laser wavelength. The laser power was set at 25 mW, and the acquisition range covered 200–1000 cm−1. The as-prepared catalyst served as the working electrode, a platinum wire as the counter electrode, and an Ag/AgCl electrode as the reference electrode, with 1 M KOH solution used as the electrolyte. Raman signals were recorded at open-circuit potential (OCP) and applied potentials ranging from 1.02 to 1.41 V vs. RHE. All spectral data were subjected to baseline correction and background subtraction.
DFT calculation
All density functional theory (DFT) calculations were performed using the Vienna Ab Initio Package (VASP) within the generalized gradient approximation (GGA) with the PBE functional64,65,66. The projected augmented wave (PAW) method was employed to describe the ionic cores, and valence electrons were treated using a plane wave basis set with a kinetic energy cutoff of 500 eV67. Partial occupancies of the Kohn-Sham orbitals were allowed using the Gaussian smearing method and a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. A geometry optimization was considered convergent when the force change was smaller than 0.05 eV Å−1. Grimme’s DFT-D3 methodology was used to describe the dispersion interactions68. The Hubbard U had been considered in structures. And An on-site Hubbard term U-J was added to address the open-shell d-electrons (4.7 eV, 5 eV, and 4.6 eV) for Fe, Ni, and Mo. Ab initio molecular dynamics (AIMD) simulation was carried out to study disorder structures. During geometry optimization, the cut-off energy was set as 450 eV for structures, respectively. The PBE-D3 dispersion term was introduced to correct the van der Waals interactions69. AIMD simulations were run for 10 ps as equilibration with time steps of 1 fs, performing a constant temperature of 400 K in the Nosé-Hoover isokinetic ensemble. Surface slabs for NiFe LDH and the NiFe LDH/FeMoO heterostructure were built with a 20 Å vacuum. The NiFe LDH slab was cleaved along the (001) plane, and Fe2(MoO4)3 was cleaved along its (013) plane. The heterostructure was formed by placing the NiFe LDH slab atop the Fe2(MoO4)3 surface, aligning lattice vectors and relaxing the interfacial spacing to 2.5 Å. Full atomic relaxation was applied to the top NiFe LDH layers and the bottom Fe2(MoO4)3 layers. The Brillouin zone was sampled by a Monkhorst-Pack (MP) k-point grid of 3 × 3 × 1 for structure optimizations. The VESTA 3 software was used to visualize optimized structures70. The free energy of a gas-phase molecule or an adsorbate on the surface was calculated by the equation G = E + ZPE − TS, where E is the total energy, ZPE is the zero-point energy, T is the temperature in kelvin (298.15 K is set here), and S is the entropy. The OER via the AEM pathway involves four fundamental steps: formation of *OH, transformation to *O, subsequent oxidation to *OOH, and finally desorption of O271. Free energy of each step can be calculated by considering the standard states of H2O and H2, in order to describe the transfer of OH− and e−. The theoretical overpotential (η) can be determined by the potential-limiting step, which is identified as the maximum free energy increment among the four intermediate transitions. Alternatively, the LOM pathway considers the direct participation of lattice oxygen (Ol) in the oxygen evolution process. This five-stage mechanism involves the oxidation of lattice sites to *OlH, *Ol, *OlOH, and *OlO, leading to the subsequent coupling and evolution of O271. The energy barrier for these transitions can be evaluated by comparing the total energy of the respective surface state, in which the overpotential is defined by the step exhibiting the highest thermodynamic barrier.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. All other data are available from the corresponding author upon request. The source data generated in this study have been deposited in the figshare database under accession code https://doi.org/10.6084/m9.figshare.3051875972. Source data are provided with this paper.
References
Shih, A. J. et al. Water electrolysis. Nat. Rev. Method. Prim. 2, 84 (2022).
Li, Z. et al. Seed-assisted formation of NiFe anode catalysts for anion exchange membrane water electrolysis at industrial-scale current density. Nat. Catal. 7, 944–952 (2024).
Zhao, J. et al. Out-of-plane coordination of iridium single atoms with organic molecules and cobalt–iron hydroxides to boost oxygen evolution reaction. Nat. Nanotechnol. 20, 57–66 (2025).
Mei, Y. et al. MoZn-based high entropy alloy catalysts enabled dual activation and stabilization in alkaline oxygen evolution. Sci. Adv. 10, eadq6758 (2024).
Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011).
Wu, Y. et al. Triggering lattice oxygen activation of single-atomic Mo sites anchored on Ni–Fe oxyhydroxides nanoarrays for electrochemical water oxidation. Adv. Mater. 34, 2202523 (2022).
Wang, X., Zhong, H., Xi, S., Lee, W. S. V. & Xue, J. Understanding oxygen redox in the oxygen evolution reaction. Adv. Mater. 34, 2107956 (2022).
Li, X. et al. Unlocking the transition of electrochemical water oxidation mechanism induced by heteroatom doping. Angew. Chem. Int. Ed. 62, e202309732 (2023).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Zhu, Y. et al. Ru single atoms and sulfur anions dual-doped NiFe layered double hydroxides for high-current-density alkaline oxygen evolution reaction. Adv. Energy Mater. 15, 2500554 (2025).
Shen, S. et al. Negative-valent platinum stabilized by Pt─Ni electron bridges on oxygen-deficient NiFe-LDH for enhanced electrocatalytic hydrogen evolution. Adv. Mater. 37, 2500595 (2025).
He, Z. et al. Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat. Commun. 13, 2191 (2022).
Zhang, Q. et al. Unraveling the mechanism of self-repair of NiFe-based electrocatalysts by dynamic exchange of iron during the oxygen evolution reaction. ACS Catal. 13, 14975–14986 (2023).
Wang, S. et al. Achieving asymmetric redox chemistry for oxygen evolution reaction through strong metal-support interactions. J. Energy Chem. 96, 526–535 (2024).
Zhai, P. et al. Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density. Nat. Commun. 14, 1873 (2023).
Kim, E. et al. Activation of hidden catalytic sites in 2D basal plane via p–n heterojunction interface engineering toward efficient oxygen evolution reaction. Adv. Energy Mater. 15, 2403722 (2025).
Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction. Nat. Energy 5, 222–230 (2020).
Li, Z. et al. High-density cationic defects coupling with local alkaline-enriched environment for efficient and stable water oxidation. Angew. Chem. Int. Ed. 62, e202217815 (2023).
Zhai, Y. et al. Synergistic effect of multiple vacancies to induce lattice oxygen redox in NiFe-layered double hydroxide OER catalysts. Appl. Catal. B Environ. 323, 122091 (2023).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Lei, X. et al. Unraveling the oxygen vacancy site mechanism of a self-assembly hybrid catalyst for efficient alkaline water oxidation. ACS Catal. 14, 4523–4535 (2024).
Wu, F. et al. Engineering lattice oxygen regeneration of NiFe layered double hydroxide enhances oxygen evolution catalysis durability. Angew. Chem. Int. Ed. 64, e202413250 (2025).
Feng, C. et al. A self-healing catalyst for electrocatalytic and photoelectrochemical oxygen evolution in highly alkaline conditions. Nat. Commun. 12, 5980 (2021).
Tyndall, D. et al. Demonstrating the source of inherent instability in NiFe LDH-based OER electrocatalysts. J. Mater. Chem. A 11, 4067–4077 (2023).
Hu, Y. et al. Coordination stabilization of Fe by porphyrin-intercalated NiFe-LDH under industrial-level alkaline conditions for long-term electrocatalytic water oxidation. Adv. Funct. Mater. 35, 2413533 (2025).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704–1731 (2021).
Wang, Y.-H. et al. Oxygen defect engineering promotes synergy between adsorbate evolution and single lattice oxygen mechanisms of OER in transition metal-based (oxy)hydroxide. Adv. Sci. 10, 2303321 (2023).
Zhu, C. et al. Fe-Ni-Mo nitride porous nanotubes for full water splitting and Zn-air batteries. Adv. Energy Mater. 8, 1802327 (2018).
Xing, M. et al. Zipper-like interlocked heterostructure of NiFe layered double hydroxide-WN for super-stable oxygen evolution over 4500 h. Adv. Funct. Mater. 34, 2409559 (2024).
Luo, X. et al. Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation. Nat. Commun. 15, 8293 (2024).
Liu, S., Ren, S., Gao, R.-T., Liu, X. & Wang, L. Atomically embedded Ag on transition metal hydroxides triggers the lattice oxygen towards sustained seawater electrolysis. Nano Energy 98, 107212 (2022).
Luo, Y. et al. Morphology and surface chemistry engineering toward pH-universal catalysts for hydrogen evolution at high current density. Nat. Commun. 10, 269 (2019).
Li, X. et al. Adaptive bifunctional electrocatalyst of amorphous CoFe oxide @ 2D black phosphorus for overall water splitting. Angew. Chem. Int. Ed. 59, 21106–21113 (2020).
Ju, M. et al. Fe(III) docking-activated sites in layered birnessite for efficient water oxidation. J. Am. Chem. Soc. 145, 11215–11226 (2023).
Hu, Y. et al. Understanding the sulphur-oxygen exchange process of metal sulphides prior to oxygen evolution reaction. Nat. Commun. 14, 1949 (2023).
Wang, C. et al. Identification of the origin for reconstructed active sites on oxyhydroxide for oxygen evolution reaction. Adv. Mater. 35, 2209307 (2023).
Sun, K. et al. Interfacial water engineering boosts neutral water reduction. Nat. Commun. 13, 6260 (2022).
Thakur, N. et al. Identifying active sites of IrOx catalysts for OER: a combined operando XAS, SEIRAS, and theoretical study. J. Am. Chem. Soc. 147, 30613–30625 (2025).
Li, J. et al. Phase engineering boosting heterogeneous interface effect in RuO2/MnO2 catalysts for acidic oxygen evolution reaction. Chem. Eng. J. 496, 153921 (2024).
Lv, X. et al. Cobalt and yttrium doping to activate dual-site mechanism of amorphous NiFeOOH for large-current water electrooxidation. Adv. Sci. 12, e12638 (2025).
Song, H. et al. RuO2–CeO2 lattice matching strategy enables robust water oxidation electrocatalysis in acidic media via two distinct oxygen evolution mechanisms. ACS Catal. 14, 3298–3307 (2024).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659–23669 (2023).
Jia, C. et al. Shifting oxygen evolution reaction pathway via activating lattice oxygen in layered perovskite oxide. Adv. Funct. Mater. 33, 2301981 (2023).
Assat, G. & Tarascon, J.-M. Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat. Energy 3, 373–386 (2018).
Ren, X., Zhai, Y., Yang, N., Wang, B. & Liu, S. Lattice oxygen redox mechanisms in the alkaline oxygen evolution reaction. Adv. Funct. Mater. 34, 2401610 (2024).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 71, 035105 (2005).
Li, N. et al. Influence of iron doping on tetravalent nickel content in catalytic oxygen evolving films. Proc. Natl. Acad. Sci. USA 114, 1486–1491 (2017).
Xie, X. et al. Oxygen evolution reaction in alkaline environment: material challenges and solutions. Adv. Funct. Mater. 32, 2110036 (2022).
Wang, X. et al. Electrocatalytic selective oxygen evolution of FeOOH-modified perovskite for alkaline seawater electrolysis. J. Power Sources 614, 235017 (2024).
Jiang, C. et al. Transfer learning guided discovery of efficient perovskite oxide for alkaline water oxidation. Nat. Commun. 15, 6301 (2024).
Zhao, M. et al. Enriched edge sites of ultrathin Ni3S2/NiO nanomeshes promote surface reconstruction for robust electrochemical water splitting. Nano Energy 129, 110020 (2024).
Huang, Z., Liao, X., Zhang, W., Hu, J. & Gao, Q. Ceria-promoted reconstruction of Ni-based electrocatalysts toward efficient oxygen evolution. ACS Catal. 12, 13951–13960 (2022).
Lee, S., Bai, L. & Hu, X. Deciphering iron-dependent activity in oxygen evolution catalyzed by nickel–iron layered double hydroxide. Angew. Chem. Int. Ed. 59, 8072–8077 (2020).
Menezes, P. W. et al. Facile access to an active γ-NiOOH electrocatalyst for durable water oxidation derived from an intermetallic nickel germanide precursor. Angew. Chem. Int. Ed. 60, 4640–4647 (2021).
Deng, P.-J. et al. Layered double hydroxides with carbonate intercalation as ultra-stable anodes for seawater splitting at ampere-level current density. Adv. Energy Mater. 14, 2400053 (2024).
Tian, H., Wachs, I. E. & Briand, L. E. Comparison of UV and visible Raman spectroscopy of bulk metal molybdate and metal vanadate catalysts. J. Phys. Chem. B 109, 23491–23499 (2005).
Liu, M., Min, K.-A., Han, B. & Lee, L. Y. S. Interfacing or doping? Role of Ce in highly promoted water oxidation of NiFe-layered double hydroxide. Adv. Energy Mater. 11, 2101281 (2021).
Sha, Q. et al. Lattice oxygen mechanism induced on nickel sites by Cl– adsorption for efficient seawater oxidation reaction. J. Am. Chem. Soc. 147, 20716–20724 (2025).
Huang, L. F., Hutchison, M. J., Santucci, R. J., Scully, J. R. & Rondinelli, J. M. Improved electrochemical phase diagrams from theory and experiment: the Ni–water system and its complex compounds. J. Phys. Chem. C 121, 9782–9789 (2017).
Feng, C., She, X., Xiao, Y. & Li, Y. Direct detection of FeVI water oxidation intermediates in an aqueous solution. Angew. Chem. Int. Ed. 62, e202218738 (2023).
Cheung, P. C. W., Williams, D. R., Barrett, J., Barker, J. & Kirk, D. W. On the origins of some spectroscopic properties of “purple iron” (the tetraoxoferrate(VI) ion) and its Pourbaix safe-space. Molecules 26, 5266 (2021).
Liu, S. et al. Yttrium-doped NiMo-MoO2 heterostructure electrocatalysts for hydrogen production from alkaline seawater. Nat. Commun. 16, 773 (2025).
Dai, L. et al. Three-terminal monolithic perovskite/silicon tandem solar cell exceeding 29% power conversion efficiency. ACS Energy Lett. 8, 3839–3842 (2023).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Zhang, Y. et al. Restricting the over-oxidation of active sites in high-entropy electrocatalysts towards ultra-stabilized oxygen evolution in alkaline water electrolysis. J. Mater. Chem. A 12, 26909–26919 (2024).
Liu, S. et al. Defect-interface coupling for stable lattice-oxygen-driven oxygen evolution at industrial current densities. figshare https://doi.org/10.6084/m9.figshare.30518759 (2025).
Acknowledgements
The authors acknowledge support from the National Key Research and Development program of China (2022YFB3803600, Y.S.), National Natural Science Foundation of China (22372065, Y.S.), Hubei Provincial Key Research and Development Program (2023BAB113, Y.S.), and the Australian Research Council (FL250100099, CE230100017, IC200100023, DP220103294, C.Z.). The authors also thank the support from 2025 The University of New South Wales - Huazhong University of Science and Technology Strategic Partnership Research Seed Fund (5003187130, Y.S.). The authors thank engineer Jun Su in the Center of Optoelectronic Micro&Nano Fabrication and Characterizing Facility, WNLO of HUST, for the support in the SEM and TEM tests. The authors also thank the Analytical and Testing Center of HUST for assistance with the measurements.
Author information
Authors and Affiliations
Contributions
M.W., C.Z., and Y.S. directed the research project and supervised the experimental design. S.L. conceived the idea and performed most of the experiments. M.S., S.W., Z.Y., and Y.W. contributed to the first-principles calculation. S.L., N.L., and F.C. assisted in analyzing the data. L.D. fabricated the tandem solar cells. S.L., S.W., Y.S., C.Z., and M.W. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Junke Wang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Sun, M., Dai, L. et al. Defect-interface coupling for stable lattice-oxygen-driven oxygen evolution at industrial current densities. Nat Commun 17, 2135 (2026). https://doi.org/10.1038/s41467-026-68730-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68730-8
