
Abstract
In single-atom catalysts, the coordination microenvironment surrounding transition-metal centers is widely recognized as a key factor shaping their electronic structures and geometries, thereby governing catalytic behaviors. However, the synergetic catalysis between metal center and ligating atoms remains underexplored in single-atom catalysis. Herein, we construct the Pd(II)-N4 sites on carbon nitride to achieve photocatalytic semi-hydrogenation of alkynes. Under light irradiation, the Pd-N sites could transform to H-Pd···N-H moieties. The Pd-H center enhances alkyne insertion while the adjacent N-H group facilitates intramolecular proton transfer. This metal-ligand cooperativity of H-Pd···N-H sites, together with the steric-hindrance imposed by carbon nitride scaffold, lowers the energy barrier for Z-alkene formation and ensures exclusive stereoselectivity. Simultaneously, the H-Pd···N-H sites exhibit stronger adsorption toward alkynes than alkenes, effectively suppressing over-hydrogenation to alkanes. Across all evaluated substrates, Z-alkenes are the only isomers detected, excluding E-isomer and alkane. Notably, in a 5:95 mixture of diphenylacetylene and Z-stilbene, this method selectively converts only the alkyne to the Z-isomer without over-hydrogenation, highlighting its potential for product purification.
Similar content being viewed by others

Introduction
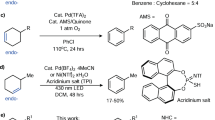
Z-olefinic structures are widespread in drugs and bioactive molecules1. Catalytic semi-hydrogenation of alkynes is among the most straightforward approaches to synthesize Z-alkenes2,3,4,5. However, formation of the corresponding E-isomer and over-hydrogenation to alkane is hard to avoid. Since Z- and E-isomers frequently exhibit distinct chemical or biological activities, but have nearly identical polarity and comparable molecular size, their separation is highly challenging3. Thus, achieving both high chemo- and stereo-selectivity in the alkyne semi-hydrogenation is of utmost significance. The Lindlar catalyst is a classical heterogeneous system for generating Z-alkenes and has been widely applied in the chemical industry6. However, the presence of adjacent metal-hydride (M-H) species or subsurface hydrogen species can lead to over-hydrogenation of the Z-alkene to alkane or formation of E-isomer via further β-H elimination (Fig. 1a). In single-atom catalysts (SACs), isolated transition-metal active sites could be built, eliminating interference from adjacent M-H species (Fig. 1b)7,8,9,10,11. The uniform chemical environment around metal active sites could deliver excellent chemoselectivity to prevent over-hydrogenation in alkyne semi-hydrogenation12,13,14,15,16,17,18,19. Nevertheless, trace amounts of E-isomers often remain in most of these catalytic systems (Table S1), highlighting the need for more sophisticated catalyst site architectures to replace mononuclear M-H species and improve the stereoselectivity.

a Lindlar catalytic system with complex M-H active sites. b Single-atom catalytic system featuring specific mononuclear M-H active sites. c Metal-ligand cooperativity in a homogeneous pincer catalytic system. d This work: metal-ligand cooperativity of H-Pd···N-H sites enable highly selective semi-hydrogenation under light irradiation. e Proposed mechanism. n.d., not detected.
In homogeneous catalysis, ligand engineering has enabled Ir-20, Ru-21, Mo-22, Co-23,24, and Mn-based25,26 pincer catalysts to achieve high stereoselectivity in semi-hydrogenation (Fig. 1c). Two primary catalytic mechanisms, inner-sphere and outer-sphere pathways, have been proposed to explain the origin of high stereoselectivity3. In these systems, either the unique H-N-M-H structure (outer-sphere) or steric hindrance on the ligand (inner-sphere) governs the attack spatial position of the alkyne, underscoring the crucial influence of the coordination microenvironment around the transition-metal center for stereoselectivity. Inspired by homogeneous pincer complexes, constructing a similar H-N-M-H microenvironment around single-atom sites on a bulky support offers the potential for precise regulation of stereoselectivity in heterogeneous catalysis (Fig. 1d). Herein, we build Pd(II)-N4 sites on graphitic carbon nitride (g-C3N4) through a photoinduced ligand exchange (PILE) method27. Meanwhile, the g-C3N4, a typical semiconductor with visible light absorption28,29,30, enables coupling of photocatalytic water splitting with the hydrogenation process for semi-hydrogenation of internal alkynes. Mechanistic studies (Fig. 1e) suggest that upon light irradiation, Pd(II) was initially reduced to Pd(0), which reacted with protons to form Pd(II)-H active intermediate. Simultaneously, N-H sites were formed adjacent to Pd(II)-H, forming H-Pd···N-H dual-atom sites. After alkyne insertion, the Pd···N-H sites, assisted by the steric-hindrance effect of the g-C3N4 scaffold, enabled intramolecular protonation along a lower energy pathway, facilitating the Z-alkene formation. Chemoselectivity was further controlled by the preferential adsorption of alkynes over alkenes on the H-Pd···N-H sites. Notably, using this method, the diphenylacetylene (1) could be converted to Z-stilbene (2) with > 99% chemoselectivity and > 99% stereoselectivity. In a 5:95 mixture of 1 and 2, selective reduction of alkyne impurities to Z-stilbene was achieved without over-hydrogenation. Remarkably, across all tested substrates, only Z-alkenes were detected, with no formation of E-isomer or alkane.
Results and discussion
Synthesis and characterization of Pd-N4/CN catalysts
The Pd(acac)2 was deposited onto g-C3N4 nanosheets by thoroughly mixing the two in dichloromethane (DCM), which was then followed by solvent evaporation. The resulting solid was dispersed in water and exposed to Xe lamp irradiation, during which ligand exchange occurred, enabling Pd to coordinate with N atoms on g-C3N4—a process referred to as the PILE strategy (Fig. 2a). After washing and drying, atomically dispersed Pd sites were successfully anchored on g-C3N4 framework. Inductively coupled plasma-emission spectrometry (ICP-OES) confirmed a Pd of 2.07 wt% (Table S2). Energy-dispersive X-ray spectroscopy (EDX) elemental mappings and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) imaging verified the uniform dispersion of Pd atoms without detectable aggregation (Figs. 2b and 2c). The atomic force microscopy (AFM) images revealed that the Pd-N4/CN maintained a two-dimensional structure with a thickness of ~2.2 nm (Fig. 2d). Compative X-ray photoelectron spectroscopy (XPS) analysis g-C3N4 and Pd-N4/CN revealed a shift of the C = N-C peak toward a higher binding energy, attributable to Pd-N bond formations (Fig. 2e). The Pd 3 d peaks at 342.26 eV and 337.00 eV further confirmed the presence of Pd(II) species (Fig. S5). Synchrotron X-ray absorption spectroscopy (XAS) provided complementary electronic and structural insights. In the Pd K-edge X-ray absorption near-edge structure (XANES) spectra, the absorption edge of Pd-N4/CN closely resembled that of Pd(acac)2, correlating with a + 2 oxidation state (Fig. 2f). In the R-space, Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra, a distinct Pd-N coordination peak ( ~ 1.47 Å) appeared with no evidence of Pd-Pd scattering, excluding nanoparticle formation (Fig. 2g). Furthermore, EXAFS fitting results indicated that four N atoms coordinated with Pd to form the initial coordination shell, forming a Pd-N4 structure (Fig. S6). As shown in Fig. S8, the diffraction peaks (2θ = 13.5, 28.4) of Pd-N4/CN correlated with g-C3N4 in X-ray diffraction (XRD) analysis, indicating that the Pd nanoparticle did not exist in the catalyst. Subsequently, the carrier separation and migration were studied via the transient photocurrent, steady photoluminescence (PL) spectra, and electrochemical impedance spectroscopy (EIS). Under light irradiation, the Pd-N4/CN exhibited a higher photocurrent density (~ 0.2 μA•cm-2) compared with g-C3N4 (Fig. S9). A smaller series resistance of Pd-N4/CN (121 Ω) was detected through EIS (Fig. S10). The reduced PL intensity demonstrated that the photoexcited electron was hard to recombine in Pd-N4/CN (Fig. S11). These experimental results consistently indicated that the incorporation of Pd enhanced charge separation and carrier migration efficiency.

a PILE strategy for developing Pd-N4/CN. b TEM image and EDX elemental mappings of Pd-N4/CN. c AC HAADF-STEM image of Pd-N4/CN. d AFM image of Pd-N4/CN. e N 1 s XPS spectra of g-C3N4 and Pd-N4/CN. f The XANES and g FT-EXAFS spectra at Pd K edge involving Pd foil, Pd(acac)2, Pd-N4/CN.
Photochemical semi-hydrogenation reactions
The photocatalytic semi-hydrogenation of 1 with Pd-N4/CN was selected as the model. A mixture of Pd-N4/CN (10 mg, 2 wt%), TEOA (0.5 mL), substrate 1 (0.1 mmol) in H2O/DCE (2/2 mL) was irradiated with 420 nm LEDs at room temperature for 4 h. After the reaction, only product 2 was detected in 99% yield, while E-1,2-diphenylethene (3) and 1,2-diphenylethane (4) were not detected by gas chromatography (GC) analysis. Control experiment results indicated that the Pd-N4/CN, light irradiation, and TEOA were all essential for the reaction. In the absence of the co-solvent DCE, the yield decreased to 77%, although selectivity remained >99% (Fig. 3a). As shown in Fig. 3b, g-C3N4 alone exhibited no catalytic activity. A physical mixture of Pd(acac)2 and g-C3N4 produced 2 in only 20% yield after 9 h. Notably, throughout the entire reaction, the selectivity for product 2 remained >99%, even after complete substrate conversion (Fig. 3b, pink and green lines), demonstrating that this method effectively suppressed over-hydrogenation.

Standard conditions: 1 (1.0 equiv., 0.1 mmol), Pd-N4/CN (2 wt%, 10 mg), TEOA (0.5 mL), H2O (2 mL), DCE (2 mL), rt, Ar, 4 h and 420 nm LEDs. The yields of 2, 3 and 4 were determined by GC analysis using biphenyl as the internal standard. a Control experiments. TEOA = nitrilotriethanol. DCE = 1,2-dichloroethane. n.d. = not detected. w/o = without. w/ = with. b Variation of yield and selectivity during the reaction process.
Investigations of the reaction mechanism
Light-on/off experiments confirmed that continuous irradiation is required for catalysis (Fig. 4a). Subsequently, in-situ XPS revealed a shift of Pd 3 d 5/2 and Pd 3 d 3/2 to lower binding energies after 90 min of irradiation on Pd-N4/CN (Fig. 4b), indicating photo-induced reduction of Pd(II). Stern–Volmer quenching experiments demonstrated that both Pd(II) and TEOA could quench the excited-state of g-C3N4 (Fig. 4c). On the other hand, the excited-state of Pd-N4/CN could be quenched by water, but cannot be quenched by substrate 1 (Fig. 4d). This quenching behavior supported an electron transfer pathway in which TEOA donated electrons to photoexcited g-C3N4, and the resulting photogenerated electrons subsequently reduced the Pd(II)-N4 sites before being transferred to H2O. After four cycles of Pd-N4/CN, the yield decreased to 78% (Table S4). Furthermore, AC HAADF-STEM revealed partial aggregation of Pd single atoms into nanoparticles (Fig. S13), providing an indirect evidence that Pd(II) can be reduced to Pd(0) during the reaction. Hydrogen evolution measurements further supported this conclusion: in the absence of alkyne, Pd-N4/CN could realize the hydrogen evolution reaction under light irradiation, whereas g-C3N4 alone exhibited no activity (Fig. 4e). It demonstrates that the single-atom Pd sites are the catalytic center of HER. In the presence of alkyne, no H2 was detected, suggesting either the hydrogenation of alkyne was faster compared with HER or that the generated H2 was rapidly consumed. Cyclic voltammetry (CV) curves of Pd-N4/CN exhibited a distinct oxidation peak with half-peak potential of −0.35 V (vs. Ag/AgCl), which decreased significantly upon alkyne addition (Fig. 4f)31,32. In contrast, g-C3N4 exhibited no such peak during CV test (Fig. S16). Based on literature reports33,34,35 and our experimental results, we propose that the proton reacted with Pd(0) to generate Pd-H species and subsequently reacted with alkyne.

a Time profiles of semi-hydrogenation with and without light. b In-situ Pd 3 d XPS spectra of Pd-N4/CN under Xe lamp irradiation. c Emission quenching of CN by TEOA or Pd(acac)2. I0 and I are intensities of the emission with or without the quencher. d Emission quenching of Pd-N4/CN by 1 or H2O. e Time-dependent photocatalytic H2 production performance. f CV curves of Pd-N4/CN with increasing concentrations of 1. w/o = without. w/ = with.
To elucidate the characteristics of active hydrogen species, a series of H sources was examined instead of H2O (Table 1). Upon utilizing g-C3N4 as the catalyst, the hydrogenation product was not detected under the H2 atmosphere (Table 1, Entries 1, 2). When using Pd-N4/CN as the catalyst and H2 as the H source, only the Z-isomer was detected and the yield of 2 could be significantly improved under light irradiation (Table 1, Entries 3, 4). It was proposed that H2 was activated by Pd(II)-N4 sites to form an H-Pd···N-H active center, which was accelerated under irradiation. Density functional theory (DFT) results supported this proposal, showing a spontaneous, low-energy barrier pathway (ΔG‡ = 4.5 kcal/mol, ΔG = −22.5 kcal/mol) (Fig. S17). Triethoxysilane, a typical hydride donor capable of generating M-H species, also yielded 2 as the sole hydrogenation product when used in place of H2O. These results collectively indicated that Pd-H was a key reactive intermediate. When D2O was utilized instead of H2O, deuterated Z-alkene was obtained in 95% yield with 59% deuterium incorporation rate. It was observed that the oxidized TEOA released protons and decreased the deuterated incorporation ratio. Thus, reducing the amount of TEOA led to an increase of 77% of the deuterium incorporation rate, but the yield decreased to 52% (Fig. S18). These data confirmed that the H source in the system was proton (from water or other proton donors).
Furthermore, the hydrogenation reactions were not completely inhibited by the addition of radical scavengers such as 2,2,6,6-tetramethyl-1-piperidinyloxy (22% yield) and 2,6-di-tert-butyl-4-methylphenol (89% yield), suggesting that a radical pathway was unlikely (Fig. S19). A Fourier transform infrared spectrometer (FTIR) was employed to monitor, structural changes of the catalyst under the photoexcitation state. Under a H2/Ar (5:95) atmosphere without light, the FTIR spectrum of Pd-N4/CN was collected as the background. Upon continuous light irradiation, a new vibrational band appeared at ~3480 cm-1, which was attributable to N-H stretching (Fig. 5a)36,37. In the control experiments, using g-C3N4 instead of Pd-N4/CN, no corresponding band appeared (Fig. 5b). These observations indicated that formation of the NH group required both illumination and the presence of Pd single-atom sites.

a In-situ FTIR spectra of Pd-N4/CN under H2/Ar (5:95) atmosphere with 390 nm LEDs irradiation. b In-situ FTIR spectra of g-C3N4 under H2/Ar (5:95) atmosphere with 390 nm LEDs irradiation. c Alkyne-TPD profiles of g-C3N4, Pd-N4/CN. The alkyne is prop-1-yn-1-ylbenzene. d Alkene-TPD profiles of g-C3N4, Pd-N4/CN. The alkene is Z-prop-1-en-1-ylbenzen. e DFT calculation results of adsorption strength. f Selective semi-hydrogenation of a 5:95 mixture of 1 and 2. w/ = with.
A series of kinetic experiments are used to reveal the rate-determining step. Via GC analysis, the yields were obtained at different times (0, 15, 30, 45, 60 min), which exhibited a linear relationship with reaction times. The slope stands for the initial reaction rate. Through experimental data, the initial rate vs. [1] and initial rate vs. light intensity both showed zero-order kinetics (Figs. S21 and S22). And the initial rates were positively correlated with [Pd] at low concentrations, while independent of [Pd] at higher concentrations (Fig. S23). Both the initial rate vs. [H2O] and initial rate vs. [TEOA] showed first-order kinetics (Figs. S24 and S25). Subsequently, we use D2O instead of H2O and stop the reaction at the initial stage (yield <30%). A significant reduction of yield was observed (Fig. S26). All hydrogen atoms of the H-Pd···N-H active species originate from H₂O or TEOA. Hence, according to kinetic experiment results, the rate-determining step might be either the generation of Pd-H intermediate between the Pd(0) and proton, or the intramolecular proton transfer from the N-H moiety to the Pd-alkenyl intermediate.
To compare the interactions of substrates with the catalyst, the prop-1-yn-1-ylbenzene and Z-prop-1-en-1-ylbenzene, which have a similar boiling point (185 oC and 175 oC), were selected as model molecules. Temperature programmed desorption (TPD) measurements revealed that Pd single-atom sites interacted with both alkyne and alkene, improving the desorption temperatures. However, the increase in desorption temperature was much more pronounced for the alkyne than the alkene (Figs. 5c, 5d). Theoretical calculations indicated that the ligand exchange with alkyne was exergonic for both unhydrogenated (Int-3a) and hydrogenated (Int-5) single-atom Pd(II)-N4 sites (Fig. 5e). Hence, alkyne exhibited stronger adsorption strength compared with the alkene on Pd-N4/CN. The difference in adsorption capacity resulted in high chemoselectivity and the suppression of over-hydrogenation. Consistently, when pure substrate 2 was introduced into the catalytic system, no further conversion was observed under standard conditions (Fig. S27). In industrial steam cracking, trace amounts of alkyne impurities were often generated alongside valuable alkenes. It is important to develop efficient methods to selectively remove these alkynes without over-hydrogenating the alkenes21. Upon treating a 5:95 mixture of 1 and 2 under standard conditions resulted in complete conversion of the alkyne to Z-alkene, with no detectable by-product (Fig. 5f).
DFT calculations
To elucidate the reaction mechanism and the selectivity control of hydrogenation, DFT calculations were conducted. The experimental studies demonstrated that single-atom Pd(II)-N4 sites activated H2 and adsorbed diphenylacetylene. Therefore, Int-1 served as the zero point of the free energy profile (Fig. 6). Int-1 underwent a rapid alkyne migratory insertion into the Pd-H bond via TS-1 to generate Int-2a with an activation free energy of 2.9 kcal/mol. The intramolecular hydrogenation of the alkenyl palladium Int-2a occurred through TS-2a to form the Z-alkene. The activation free energy was 23.3 kcal/mol. The E-alkene formed through H transfer from the isomeric intermediate Int-2a. However, the activation free energy of TS-2b was 8.7 kcal/mol higher compared with that of TS-2a. As shown in the three-dimensional structure of TS-2b, the steric clash between the ligand and the phenyl group of the substrate rendered this transition state kinetically less favored. Conversely, TS-2a suppressed such steric clashes, elucidating the exclusive formation of the Z-isomer. Moreover, the DFT calculations also identify the intramolecular protonolysis transition state (TS-2a) had the highest barrier in the catalytic cycle, implying the intramolecular proton transfer process might be the rate-determining step. However, the possibility, which the formation of the Pd-H species was involved in the rate-determining step, also cannot be excluded.

All energies were calculated at the ωB97XD/6-311 + G(d,p)-SDD/SMD(Water)//B3LYP-D3(BJ)/6-31 G(d)-LANL2DZ level of theory. The catalyst employed in DFT calculations is a single-atom Pd(II)-N4 site. See SI for computational details and disfavored pathways.
Substrate scope
The substrate scope of semi-hydrogenation of alkynes was investigated to affirm the mechanism. Initially, the scale-up reaction (1 mmol) was conducted, and the high chemo- and stereo-selectivity was maintained (Fig. 7, entry 1). The TON value is 101. The introduction of either alkyl groups (5) or methoxy groups (6) did not affect the conversion or selectivity. Other electron-rich aryl groups decreased the yield to 50% (7) and 75% (8). When using alkyl-1-yn-1-ylbenzene derivatives, good yields with single Z-isomer selectivity were obtained (9, 10). For alkyl internal alkyne, such as dodec-6-yne (11) and oct-3-yne (12), good yields with >99% stereoselectivity could be gained. And then, we tried to explore the substrates with sensitive functional groups. The -F (13), -Br (15), amino (16), amide (17), carbonyl (18), and ester groups (19) could be compatible and the corresponding products were obtained with moderate to excellent yields. The 1-Chloro-4-(phenylethynyl)benzene (14) could only get a 38% yield. The norethindrone derivative with alkenyl and carbonyl was compatible and got a 65% yield (20). For substrates that are difficult to synthesize, the ‘Intermolecular Reaction Screening’ method was used to evaluate functional group tolerance38,39. We repeated the semi-hydrogenation reaction of 1 with adding different additives. The conversion of 1, the yield of 2 and the recovery amount of the additive after reaction were determined by GC analysis. In this way, alkyl chloride (Entry 18), alkyl ketone (Entry 19) and alkyl cyan groups (Entry 20) could be compatible. The alkyl aldehyde, which is easily reduced, could be retain with a 49% recovery rate (Entry 21). The alkyl alcohol (Entry 22) and alkyl amine (Entry 23), which are easily oxidized, could not be retained. The PhNH2 (Entry 24) could be compatible, but the -SO2NH2 group (Entry 25) could not be remained.

Standard conditions: Alkyne (1.0 equiv., 0.1 mmol), Pd-N4/CN (2 wt%, 10 mg), TEOA (0.5 mL), H2O (2 mL), DCE (2 mL), rt, Ar, 4 h, 420 nm LEDs, isolated yield. The by-products were detected by GC or HPLC and 1H NMR analysis. aThe scale-up reaction: 1 (1.0 equiv., 1 mmol), Pd-N4/CN (2 wt%, 50 mg), TEOA (5 mL), H2O (20 mL), DCE (20 mL), rt, Ar, 12 h, 420 nm LEDs. bYields were determined by GC analysis using biphenyl as the internal standard. n.d. = not detected. Intermolecular reaction screening: 1 (1.0 equiv., 0.1 mmol), additive (1.0 equiv., 0.1 mmol), Pd-N4/CN (2 wt%, 10 mg), TEOA (0.5 mL), H2O (2 mL), DCE (2 mL), rt, Ar, 4 h, 420 nm LEDs. The yields and recoveries were determined by GC analysis using biphenyl as the internal standard. Green ‘√’ ( > 66%), yellow ‘−’ (34-66%), red ‘×’ ( < 34%).
Based on the above substrate scope results, only the Z-isomer was detected in all cases, without neither E-isomers or alkanes. Compared with previous single-atom catalytic semi-hydrogenation reactions (Table S1), the metal-ligand cooperative catalytic site (H-N···Pd-H) replaces the isolated metal‑hydride active site (M–H). And the intramolecular N-H proton transfer process results in a sufficiently large activation free energy difference between the Z- and E-isomers, demonstrating exclusive stereoselectivity. This novel synergistical catalysis highlights the strengths and distinguishing features of Pd-N4/CN catalytic system.
Conclusion
In summary, this study successfully established Pd(II)-N4 sites on g-C3N4 through the PILE strategy. The unique H-Pd···N-H microenvironment and steric constrants of g-C3N4 induce photocatalytic semi-hydrogenation of alkynes with exclusive stereoselectivity to Z-isomers. Additionally, the stronger adsorption capacity of alkynes on Pd-N4/CN helps suppress over-hydrogenation. This work highlighted the critical role of ligating atoms in single-atom catalysis and demonstrated that constructing coordination environments analogous to those in homogeneous metal complexes led to similar structure-activity relationships. These findings offered a novel perspective for designing single-atom catalysts and catalytic systems for organic transformations.
Methods
Synthesis of Pd-N4/CN photocatalyst
40 g of urea were placed into 50 mL crucibles, and transferred to a Muffle furnace. The temperature was increased to 550 °C at a rate of 0.5 °C/min, maintained for 5 h, and then allowed to cool naturally to room temperature, yielding a light-yellow powder (g-C3N4). First, 200 mg g-C3N4 and an appropriate amount of Pd(acac)2 were each dispersed in 10 mL dichloromethane (DCM), and ultrasonicated at room temperature for 2 h. Second, the homogeneous dispersions were then combined and vigorously stirred for 12 h to obtain precursor solution A. This precursor solution A was transferred to a 500 mL beaker and left in a fume hood for several hours to completely evaporate the DCM. Subsequently, 100 mL of deionized water was added, and evenly mixed to obtain dispersion solution B. As shown in Fig. S2, dispersion B (100 mL) was then placed into photo-reduction reactor. Under Xe lamp irradiation for 5 h, photo-reduction and ligand exchange of Pd(acac)2 adsorbed on the g-C3N4 framework occurred, resulting in the formation of isolated Pd atoms coordinated to the g-C3N4 matrix.
Photocatalytic semi-hydrogenation of alkynes
The semi-hydrogenation of diphenylacetylene was conducted in a 10 mL Schlenk tube. Photocatalyst (10 mg) and ultrapure water (2 mL) were initially added and sonicated for 5 min. Subsequently, dichloroethane (DCE, 2 mL), triethanolamine (TEOA, 0.5 mL), and alkyne (0.1 mmol) were introduced. The reactor was frozen using liquid nitrogen, evacuated, and purged with argon. This cycle was repeated three times. The reaction mixture was subsequently irradiated with a 410-420 nm blue LED at room temperature for 4 h under continuous stirring.
Data availability
The authors declare that the data generated in this study are provided in the Supplementary Information file. Data are available from the corresponding authors upon request.
References
Siau, W.-Y., Zhang, Y. & Zhao, Y. Stereoselective Synthesis of Z-Alkenes. Top Curr. Chem. 327, 33–58 (2012).
Decker, D., Drexler, H.-J., Heller, D. & Beweries, T. Homogeneous catalytic transfer semihydrogenation of alkynes – an overview of hydrogen sources, catalysts and reaction mechanisms. Catal. Sci. Technol. 10, 6449–6463 (2020).
Kusy, R. & Grela, K. Renaissance in Alkyne Semihydrogenation: Mechanism, Selectivity, Functional Group Tolerance, and Applications in Organic Synthesis. Chem. Rev. 125, 4397–4527 (2025).
Liu, K., Qin, R. & Zheng, N. Insights into the Interfacial Effects in Heterogeneous Metal Nanocatalysts toward Selective Hydrogenation. J. Am. Chem. Soc. 143, 4483–4499 (2021).
Zhang, L., Zhou, M., Wang, A. & Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 120, 683–733 (2019).
Lindlar, H. Ein neuer Katalysator für selektive Hydrierungen. Helv. Chim. Acta 35, 446–450 (1952).
Cui, X., Li, W., Ryabchuk, P., Junge, K. & Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 1, 385–397 (2018).
Gao, Y. & Wang, D. Atomically Dispersed Catalysts: Precise Synthesis, Structural Regulation, and Structure–Activity Relationship. CCS Chem. 6, 833–855 (2024).
Liang, X., Yao, S., Li, Z. & Li, Y. Challenge and Chance of Single Atom Catalysis: The Development and Application of the Single Atom Site Catalysts Toolbox. Acc. Chem. Res. 58, 1607–1619 (2025).
Liu, L. & Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Yang, X.-F. et al. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Guo, X. et al. Local Symmetry-Broken Single Pd Atoms Induced by Doping Ag Sites for Selective Electrocatalytic Semihydrogenation of Alkynes. ACS Nano 19, 2788–2798 (2025).
Jia, T. et al. Single-Atom Nickel on Carbon Nitride Photocatalyst Achieves Semihydrogenation of Alkynes with Water Protons via Monovalent Nickel. Angew. Chem. Int. Ed. 62, e202216511 (2023).
Liu, H. et al. Encapsulation of Pd Single-Atom Sites in Zeolite for Highly Efficient Semihydrogenation of Alkynes. J. Am. Chem. Soc. 146, 24033–24041 (2024).
Yang, B. et al. Incorporation of Pd Single-Atom Sites in Perovskite with an Excellent Selectivity toward Photocatalytic Semihydrogenation of Alkynes. Angew. Chem. Int. Ed. 63, e202410394 (2024).
Yang, F., Ding, S., Song, H. & Yan, N. Single-atom Pd dispersed on nanoscale anatase TiO2 for the selective hydrogenation of phenylacetylene. Sci. Chin. Mater. 63, 982–992 (2020).
Zhou, S. et al. Pd Single-Atom Catalysts on Nitrogen-Doped Graphene for the Highly Selective Photothermal Hydrogenation of Acetylene to Ethylene. Adv. Mater. 31, 1900509 (2019).
Jie, Y. et al. Acetylene Semihydrogenation over the Flexible Heptazine-Based g‑C3N4 Monolayer with Embedded Single Atoms (SA = Cu, Ag, Au, Ni, Pd, Pt). J. Phys. Chem. C 129, 13219–13231 (2025).
Wang, J. et al. Isolated Palladium Atoms Dispersed on Silicoaluminophosphate-31(SAPO-31) for the Semihydrogenation of Alkynes. ACS Appl. Nano Mater. 4, 861–868 (2021).
Wang, Y., Huang, Z. & Huang, Z. Catalyst as colour indicator for endpoint detection to enable selective alkyne trans-hydrogenation with ethanol. Nat. Catal. 2, 529–536 (2019).
Luo, J. et al. Controlled Selectivity through Reversible Inhibition of the Catalyst: Stereodivergent Semihydrogenation of Alkynes. J. Am. Chem. Soc. 144, 13266–13275 (2022).
Both, N. F., Spannenberg, A., Junge, K. & Beller, M. Low-Valent Molybdenum PNP Pincer Complexes as Catalysts for the Semihydrogenation of Alkynes. Organometallics 41, 1797–1805 (2022).
Fu, S. et al. Ligand-Controlled Cobalt-Catalyzed Transfer Hydrogenation of Alkynes: Stereodivergent Synthesis of Z- and E-Alkenes. J. Am. Chem. Soc. 138, 8588–8594 (2016).
Sheikh Mohammad, T., Sakharov, P., Raje, S. & de Ruiter, G. Z-Selective Semihydrogenation of Alkynes Catalyzed by a Co(I)PCNHCP Pincer Complex: A Simple, Fast, and Practical Methodology. ACS Catal. 15, 5370–5377 (2025).
Garbe, M. et al. Chemoselective semihydrogenation of alkynes catalyzed by manganese(i)-PNP pincer complexes. Catal. Sci. Technol. 10, 3994–4001 (2020).
Zubar, V., Sklyaruk, J., Brzozowska, A. & Rueping, M. Chemoselective Hydrogenation of Alkynes to (Z)-Alkenes Using an Air-Stable Base Metal Catalyst. Org. Lett. 22, 5423–5428 (2020).
Wang, G. et al. Mechanistic Investigation into Single-Electron Oxidative Addition of Single-Atom Cu(I)-N4 Site: Revealing the Cu(I)–Cu(II)–Cu(I) Catalytic Cycle in Photochemical Hydrophosphinylation. J. Am. Chem. Soc. 146, 8668–8676 (2024).
Ong, W.-J., Tan, L.-L., Ng, Y. H., Yong, S.-T. & Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 116, 7159–7329 (2016).
Rocha, G. F. S. R. et al. Carbon nitride based materials: more than just a support for single-atom catalysis. Chem. Soc. Rev. 52, 4878–4932 (2023).
Wang, J., Liu, Y., Zong, X., Lei, A. & Sun, Z. Recent advances in the heterogeneous photochemical synthesis of C–N bonds. Green Chem. 25, 5010–5023 (2023).
Ji, K. et al. Electrocatalytic Hydrogenation of 5-Hydroxymethylfurfural Promoted by a Ru1Cu Single-Atom Alloy Catalyst. Angew. Chem. Int. Ed. 61, e202209849 (2022).
Wu, Y., Liu, C., Wang, C., Lu, S. & Zhang, B. Selective Transfer Semihydrogenation of Alkynes with H2O (D2O) as the H (D) Source over a Pd-P Cathode. Angew. Chem. Int. Ed. 59, 21170–21175 (2020).
Guan, J. & Zhang, Q. Recent Progress in Single-Atom Catalysts for Photocatalytic Water Splitting. Solar RRL 4, 2000283 (2020).
Wang, P. et al. Single Pd atoms synergistically manipulating charge polarization and active sites for simultaneously photocatalytic hydrogen production and oxidation of benzylamine. Nano Energy 95, 107045 (2022).
Wang, N. et al. Design of Palladium-Doped g‑C3N4 for Enhanced Photocatalytic Activity toward Hydrogen Evolution Reaction. ACS Appl. Energy Mater. 1, 2866–2873 (2018).
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Guan, M.-H. et al. Cathode–Anode Synergy Electrosynthesis of Propanamide via a Bipolar C–N Coupling Reaction. J. Am. Chem. Soc. 147, 16301–16308 (2025).
Collins, K. D. & Glorius, F. A robustness screen for the rapid assessment of chemical reactions. Nat. Chem. 5, 597–601 (2013).
Collins, K. D. & Glorius, F. Intermolecular Reaction Screening as a Tool for Reaction Evaluation. Acc. Chem. Res. 48, 619–627 (2015).
Acknowledgements
We would like to thank Prof. Aiwen Lei (WHU), Prof. Qiang Liu (THU), Prof. Xiaotian Qi (WHU), Prof. Shan Tang (SJTU), Dr. Yan Li (WHU) for helpful discussions, Shanghai Synchrotron Radiation Facility (SSRF) BL11B beam-line and Beijing Synchrotron Radiation Facility (BSRF) 1W1B beamline for XAS testing, Large-scale Instruments and Equipment Sharing Platform (BJUT) for GC-MS, NMR testing. L. W. acknowledges the supercomputing system in the Super-computing Center of Wuhan University. National Natural Science Foundation of China 22272003 (ZS), 22301013 (YL), Beijing Natural Science Foundation (Z250018) (YL), The Fundamental Research Funds for Beijing Municipal Universities (055000546325510) (YL), National Key R&D Program of China 2023YFB3810800 (ZS), Beijing Outstanding Young Scientists Program BJJWZYJH01201910005017 (XW), R&D Program of Beijing Municipal Education Commission KZ20231000506 (ZS), The Project of Construction of Innovative Teams and Teacher Career Development for Universities and Colleges Under Beijing Municipality 11000024T000003219982 (DQ), Spark Program of Beijing University of Technology (XH-2025-09-05) (YY).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: YL, HM, ZS, Performed the experiments: HM, LW, YY, MM, GW, ZR, DC, PL, JW, PM, LA, DQ, YL, Analyzed the data: YL, HM, JW, MM, ZS, Contributed materials/analysis tools: KZ, XW, WH, Wrote the paper: YL, HM, JW, ZS.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Graham Ruiter, Jinshui Zhang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, H., Wang, L., Wang, J. et al. The H-Pd···N-H metal-ligand dual-atom sites synergistically catalyzed alkyne semi-hydrogenation with complete Z-selectivity. Nat Commun 17, 1972 (2026). https://doi.org/10.1038/s41467-026-68755-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68755-z
