
Abstract
The SEL1L-HRD1 complex represents the most conserved branch of endoplasmic reticulum (ER)-associated degradation (ERAD), a critical quality-control pathway that clears misfolded ER proteins. However, the molecular organization and pathogenic mechanisms of mammalian ERAD have remained elusive. Here, we report the cryo-EM structure of the core mammalian ERAD complex, comprising the ER lectin OS9, SEL1L, and the E3 ubiquitin ligase HRD1. The structure, validated by mutagenesis and crosslinking assays, reveals a dimeric assembly of the core complex in which SEL1L and OS9 form a claw-like configuration in the ER lumen that mediates substrate engagement, while HRD1 dimerizes within the membrane that may facilitate substrate translocation. Furthermore, pathogenic SEL1L mutations at the SEL1L-OS9 (Gly585Asp) and SEL1L-HRD1 (Ser658Pro) interfaces disrupt complex formation and impair ERAD activity. A newly identified disease-associated HRD1 variant (Ala91Asp), located in transmembrane helix 3, impairs HRD1 dimerization and substrate processing. These findings provide structural and functional insights for mammalian SEL1L-HRD1 ERAD and elucidate how mutations destabilizing this machinery contribute to human disease.
Similar content being viewed by others

Introduction
In eukaryotes, approximately 30% of newly synthesized proteins are directed to the endoplasmic reticulum (ER), where they fold and mature into their functional forms1,2. Proteins that fail to fold properly are eliminated via ER-associated degradation (ERAD), a pathway that retrotranslocates misfolded proteins from the ER lumen to the cytosol for proteasomal degradation3,4,5,6,7. Suppressor/Enhancer of Lin-12-like protein 1-like (SEL1L) and the E3 ubiquitin ligase HMG-CoA reductase degradation protein 1 (HRD1) complex, homologous to the yeast Hrd3-Hrd1 pair, forms the core of the mammalian ERAD machinery and represents its most extensively characterized module8,9,10,11,12. The discovery of pathogenic SEL1L and HRD1 variants in patients with ERAD-associated neurodevelopmental disorders with onset at infancy (ENDI syndrome)13,14,15, along with the severe phenotypes observed in global and cell type-specific Sel1L- or Hrd1-knockout (KO) mice12,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30, underscores the critical role of ERAD in health and disease. However, the molecular mechanism by which these variants impair ERAD remains poorly understood, primarily due to the lack of structural information on the human complex.
Hrd3/SEL1L and Hrd1/HRD1 form the ERAD core, with HRD1 acting as the E3 ubiquitin ligase31 and SEL1L stabilizing HRD119,32 and promoting complex assembly33,34. Additional cofactors include ER lectins Yos9/OS935,36,37 and XTP3B (also known as ERLEC1)38,39, Der1/Derlin40,41,42, and Usa1/HERP43, which assist in substrate recognition, retrotranslocation, and ubiquitination. Previous cryo-electron microscopy (cryo-EM) studies of yeast ERAD revealed both a homo-dimeric Hrd3-Hrd1 complex44 and monomeric Yos9-Hrd3-Hrd1 and Hrd3-Hrd1-Usa1-Derl subcomplexes45. While the dimeric structure suggested a protein-conducting channel formed by the Hrd1 dimer, the dimer interface was closed, and the aqueous channel interior was instead exposed to the surrounding lipid layer44. It was later proposed that this conformation is a detergent artifact with low ubiquitination activity45, favoring the monomeric model in yeast. In this monomeric model, Hrd1 and Der1 form complementary “half-channels” within a thinned ER membrane, while Hrd3 and Yos9 act as luminal substrate receptors45. Whether similar assemblies exist in mammals remains unknown.
Notably, both size-exclusion chromatography and chemical-mediated crosslinking assays of endogenous SEL1L-HRD1 complex from human cells support HRD1 oligomerization46 and dimerization47,48. These findings raise the possibility that unlike the yeast complex, SEL1L-HRD1 dimerization is functionally important in mammals. In this work, we characterize the architecture of the mammalian ERAD core complex, which is essential for understanding the molecular basis of ERAD-related diseases.
Results
The OS9-SEL1L-HRD1 complex forms the core of mammalian ERAD machinery
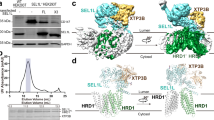
To define the composition of the SEL1L-HRD1 ERAD core complex, we independently analyzed SEL1L and HRD1 interactomes in HEK293T cells using immunoprecipitation (IP) followed by LC-MS/MS (Supplementary Fig. 1a)49. Each dataset was generated in triplicate using stringent filtering criteria, with SEL1L or HRD1 KO cells as negative controls. We identified 46 high-confidence SEL1L interactors and 24 HRD1 interactors (Supplementary Fig. 1b, c and Supplementary Tables 1-2). Cross-comparison revealed 10 shared interactors (Supplementary Fig. 1d and Supplementary Table 3), with OS9, XTP3B, SEL1L, HRD1, and FAM8A1 emerging as the most prominent components (Fig. 1a). OS9 and XTP3B are ER-resident lectins with partially redundant roles in HRD1-mediated ERAD39,50,51, with OS9 serving as the major partner34. Consistent with a previous study52, FAM8A1 deletion did not affect SEL1L-HRD1 protein levels or their function towards endogenous substrates such as IRE1α, CD147 and OS9 (Supplementary Figs. 1e–h).

a Heatmap showing the top 5 hits of the overlapping 10 hits of SEL1L- and HRD1- interacting proteins from immunoprecipitation-Mass Spec (IP-MS) experiments. Plotted values are peptide-spectrum matches (PSMs) normalized to their baits from three independent experiments. b Denaturing Strep-Tactin immunoprecipitation followed by Western blot analysis of proAVP(G57S) ubiquitination in HEK293F cells transfected with the ERAD substrate proAVP(G57S)*-Strep, OS9, SEL1L, and HRD1-WT-FLAG or -C2A (C291A/C294A)-FLAG, with 10 μM MG132 for 2 h. Input shown in Supplementary Fig. 2e and quantitation of ubiquitination levels shown in Supplementary Fig. 2f (three independent repeats). Source data are provided as a Source Data file. c Size exclusion chromatography (SEC) of OS9-SEL1L-HRD1 complex in transfected HEK293F cells (with FLAG-tagged HRD1, SEL1L and OS9) following FLAG-agarose purification (two independent repeats). The peak fractions (10-11 mL, shaded) were concentrated for cryo-EM sample preparation. The right panels show a Coomassie-stained SDS-PAGE gel of the concentrated OS9-SEL1L-HRD1 ERAD complex for cryo-EM sample preparation, and representative 2D class averages of picked particles. Source data of Coomassie-stained SDS-PAGE gel are provided as a Source Data file. d Cryo-EM map of the dimeric OS9-SEL1L-HRD1 complex. Inset showing the locally refined OS9-SEL1L map stitched with HRD1, as a monomer, for visualization. e Cartoon model of the OS9-SEL1L-HRD1 dimer rotated to show the side (left) and top (right) view of the structure.
To further define the core components of the complex, we co-expressed HRD1, SEL1L, OS9, HERP1, UBE2J1, and DERL2 in HEK293F cells, followed by HRD1 pull-downs. The OS9-SEL1L-HRD1 subcomplex co-purified robustly under multiple detergent conditions and maintained a stable stoichiometric ratio, except in Triton X-100 and NP-40 where complex integrity was lost (Supplementary Figs. 2a, b). In contrast, Derlin-2, HERP1 and UBE2J1 were not detected under any conditions tested (Supplementary Figs. 2a, b).
We next expressed a known ERAD substrate pro-arginine vasopressin (proAVP) carrying the Gly57Ser disease mutation53,54, tagged with an ER-retention signal KDEL to present secretion (proAVP G57S*, Supplementary Figs. 2c, d). Co-expression of OS9, SEL1L, and HRD1 in HEK293F cells resulted in ubiquitination of proAVP G57S* in an E3-dependent manner, as shown by the loss of ubiquitination with a ligase-dead HRD1 mutant (C291A/C294A; C2A)55 (Fig. 1b and Supplementary Figs. 2e, f). Notably, both Derlin-1 and Derlin-2 were required for efficient ERAD of proAVP G57S* (Supplementary Figs. 2g–i). Taken together, these data establish OS9-SEL1L-HRD1 as the core mammalian ERAD complex.
OS9-SEL1L-HRD1 ERAD core complex forms a dimer
To determine the molecular organization of the core complex, we co-expressed full length OS9, SEL1L and FLAG-tagged HRD1 in HEK293F cells. After membrane solubilization in lauryl maltose neopentyl glycol (LMNG), the complex was purified using FLAG-based immunoprecipitation followed by size-exclusion chromatography. This procedure yielded a single, well-defined peak containing all three components – OS9, SEL1L and HRD1 in near-stoichiometric amounts (Fig. 1c and Supplementary Fig. 2j). Peak fractions (#10-11, Fig. 1c) were collected, concentrated and immediately prepared for sample freezing and subsequent single-particle cryo-EM analysis (Supplementary Figs. 3a, b).
Initial 2D image analysis revealed the presence of a homogenous set of particles, most of which display visible C2 symmetry (Supplementary Fig. 3a). Subsequent 3D reconstruction with the selected particles revealed that the OS9-SEL1L-HRD1 complex adopts a dimeric architecture, with an overall resolution of 3.6 Å (Supplementary Figs. 3a, b and Supplementary Table 4). We then performed a focused 3D refinement using a mask around the visible regions of OS9 and SEL1L in the structure (Supplementary Fig. 3b). This process led to improvements in local resolutions to 3.3 Å for the OS9-SEL1L interface (Fig. 1d and Supplementary Fig. 3b). Model building into the EM density maps was guided using AlphaFold3-predicted structures56, followed by manual adjustments based on visible side chain densities for SEL1L-OS9 and the transmembrane helices of HRD1. This enabled accurate modeling of the three core components: OS9, SEL1L, and HRD1 (Fig. 1d, e and Supplementary Figs. 4a–e). The final 3D model captured all major domains of the core ERAD complex: the eight transmembrane helices of HRD1 (residues 1-265, blue), the luminal domain of SEL1L (residues 119-725, yellow), and two conserved domains of OS9 – the mannose-6-phosphate receptor homology (MRH) domain (residues 88-236) and a structured region comprising four β-sheets and an α-helix (termed “4β”, residues 28-87, 237-259, 633-652) (green) (Fig. 1d, e and Supplementary Figs. 4b–e). Regions not resolved in the map included the transmembrane domain of SEL1L, the cytosolic regions of both SEL1L and HRD1 (including the E3 ligase RING domain), and the remaining flexible portions of OS9’s luminal domain (dotted lines, Supplementary Fig. 4a).
SEL1L disease variants disrupt the OS9-SEL1L-HRD1 core complex
Our cryo-EM map of the OS9-SEL1L subcomplex revealed a crab claw-like configuration with a central pore (red asterisk, Fig. 2a), suggestive of a conduit for OS9-bound substrate delivery to HRD1. To probe this configuration, we mapped two disease-causing SEL1L variants, G585D and S658P13,49, to the structure. SEL1L-G585 resides at the SEL1L-OS9 interface (Fig. 2b, c and Supplementary Fig. 5a), while SEL1L-S658 lies near the SEL1L-HRD1 interface (Fig. 2d, e and Supplementary Fig. 5a). In SEL1L-/- HEK293T cells, re-expression of SEL1L-G585D selectively reduced OS9 binding by ~90% (lane 3, Fig. 2f), while S658P disrupted HRD1 interaction by ~ 70% (lane 4). The double mutant (G585D/S658P) nearly abolished both interactions (lane 5, Fig. 2f). Notably, neither mutation impaired binding to the E2 conjugating enzyme UBE2J1 (lane 2-5, Fig. 2f). These results demonstrate that disease-associated SEL1L variants selectively disrupt OS9 and HRD1 engagement, underscoring the OS9-SEL1L-HRD1 assembly as the structural and functional core of the mammalian ERAD machinery.

a, b View from ER lumen (a) or the side (b) with space-filling model for OS9-SEL1L complex. Red asterisk indicates the opening formed by SEL1L-OS9 in substrate-binding crab “claw”-like configuration. The position of SEL1L-G585 residue (purple sphere) shown in (b). 4β, the four β-sheet domain of OS9. MRH, mannose-6-phosphate receptor homology domain of OS9. APH, amphipathic helix of SEL1L. c Interface of OS9-SEL1L interaction with SEL1L-G585 residue shown as a purple sphere, with the cryo-EM map shown as mesh. d Close-up view with space-filling model for SEL1L-HRD1 showing the SEL1L-HRD1 interaction interface, with SEL1L-S658 residue shown as a red sphere. APH, amphipathic helix of SEL1L. e Interface of SEL1L-HRD1 interaction with SEL1L-S658 residue shown as a red sphere, with the cryo-EM map shown as mesh. f Immunoprecipitation of FLAG-agarose in SEL1L-/- HEK293T cells transfected with indicated SEL1L variants to examine their interaction with HRD1, OS9 and E2 UBE2J1, with quantitation of interaction levels with SEL1L shown on the right (three independent repeats. p value in HRD1: G585D p = 0.0101, S658P p < 0.0001, G585D;S658P p < 0.0001; p value in OS9: G585D p < 0.0001, S658P p > 0.9999, G585D;S658P p < 0.0001; p value in UBE2J1, G585D p > 0.9999, S658P p = 0.9935, G585D;S658P p = 0.4808). Values, mean ± SEM. n.s., not significant; *p < 0.05 and ****p < 0.0001 by one-way ANOVA test with comparison to WT. Source data are provided as a Source Data file.
In vivo formation of OS9-SEL1L dimers
Given that the yeast Yos9-Hrd3 structure is monomeric45, we next tested whether the mammalian OS9-SEL1L protomer adopts a closed dimer interface (Fig. 3a). Within this interface, OS9 residue D135 lies in close proximity to SEL1L residues D439 and M440 from the opposing protomer (Fig. 3b, c). To test whether this dimer exists in vivo rather than an artifact of detergent-based purification, we performed native disulfide crosslinking assays using Cys substitution at these position, with the more distal N442C serving as a negative control57. In the oxidizing ER lumen58, such spatial proximity enables disulfide bond formation between engineered Cys57. To avoid artifactual crosslinking during lysis, cells were directly lysed in denaturing SDS buffer lacking reducing agents and containing a cysteine-alkylating reagent N-ethylmaleimide (NEM), instead of non-denaturing detergents such as Triton X-100 or NP-40. Under these conditions, disulfide-linked complexes formed between OS9-D135C and SEL1L-D439C or M440C, were markedly reduced with SEL1L-N442C, and were absent with WT controls (lanes 7-8 vs. 9-10, Fig. 3d, e). Coomassie staining after HRD1-FLAG IP further confirmed OS9-SEL1L complex formation under native conditions (lane 1 vs. 2, Fig. 3f). As expected, all disulfide-linked species were eliminated by reducing agents (lane 2-3 vs. 7-8, Fig. 3d; lane 1 vs. 3, Fig. 3f). Moreover, 3D variability analysis reveals coordinated motions within the OS9-SEL1L luminal “substrate claw,” which alternates between compact and open conformations that likely facilitate dynamic substrate capture and handover to HRD1 (Supplementary Figs. 5b, c, Supplementary Movie 1). Together, these data demonstrate in vivo disulfide-mediated dimerization of the OS9-SEL1L complex.

a, b OS9-SEL1L-HRD1 model shown as a transparent surface. Zoomed-in view of the dashed box shown in (b). Key residues (SEL1L-D439, M440, and OS9-D135) at the interface are shown, with N442 as a negative control. c Distance between OS9-D135 and SEL1L-D439, M440 and N442. d, e (Non-)Reducing and Western blot analysis of the native disulfide crosslinking assays in HEK293F cells transfected with HRD1-FLAG, SEL1L WT and mutants (D439C, M440C, N442C), and OS9-D135C mutations, with quantitation of the percentage of disulfide-linked OS9-SEL1L shown in (e) (three independent repeats. p value in OS9-SEL1L/OS9: D439C p < 0.0001, M440C p < 0.0001, N442C p = 0.8804; p value in OS9-SEL1L/SEL1L: D439C p < 0.0001, M440C p = 0.0007, N442C p = 0.958). Lysates were treated with 15 mM N-ethylmaleimide (NEM) to preserve native disulfide-linked species during sample preparation. f (Non-)Reducing SDS-PAGE analysis (Coomassie blue staining) of HRD1-FLAG IP in HEK293F cells expressing indicated OS9 and SEL1L mutations (two independent repeats). Three panels in (f) were from the same gel, with the irrelevant lanes in the middle cut off. Values, mean ± SEM. n.s., not significant, ***p < 0.001 and ****p < 0.0001 by One-way ANOVA analysis with comparison to WT. Source data of Fig. 3d–f are provided as a Source Data file.
In vivo formation of HRD1 dimers
Our cryo-EM structure revealed that HRD1 dimerizes through interactions centered on transmembrane helix 3 (TM3) (Fig. 4a). In this interface, residues T93, F95 and R96 are positioned in close proximity (red spheres), while V94 and D97 are oriented away from the interface (gray spheres) (Fig. 4b and Supplementary Fig. 6a). Several of these residues are evolutionarily conserved (Fig. 4c). To test their functional contribution, we introduced Cys substitutions as performed above for SEL1L-OS9. T93C, F95C, and R96C, but not V94C and D97C, efficiently formed disulfide-linked HRD1 dimers, reaching ~50% dimerization (lanes 3, 5, 6, Fig. 4d). To further assess endogenous HRD1 dimerization, we treated HEK293T cells with the membrane-permeable crosslinker dithiobis-succinimidyl propionate (DSP), previously used to detect HRD1 dimers47,48. HRD1 dimers were readily observed in WT cells but markedly reduced in SEL1L-deficient cells (Supplementary Figs. 6b, c). In both crosslinking assays, dimeric species were eliminated by reducing agents, confirming disulfide/DSP-dependent linkage (Fig. 4d and Supplementary Figs. 6b, c). Together, these findings indicate that the OS9-SEL1L-HRD1 complex can assemble into a relatively stable dimeric form in vivo.

a, b Overview of the HRD1 homodimer, with zoomed-in view of the interface shown in (b). The numbers below the model refer to the TMs of HRD1. Key residues (HRD1-T93, F95 and R96) in HRD1 TM3 or the loop between TM3 and 4 are highlighted. c Sequence alignment of TM3 of HRD1 across various model organisms with the conserved amino acids shaded for various residues. d (Non-)Reducing and Western blot analysis of the native disulfide crosslinking assays in HRD1-/- HEK293T cells transfected with the indicated HRD1 mutants, with quantitation of the percent of dimeric HRD1 in total HRD1 shown below (three independent repeats. p value: T93C p < 0.0001, V94C p > 0.9999, F95C p < 0.0001, R96C p < 0.0001, D97C p > 0.9999). Cell lysates were treated with treated with 15 mM N-ethylmaleimide (NEM) to preserve native disulfide-linked species during sample preparation. Values, mean ± SEM. n.s. not significant, ****p < 0.0001 by One-way ANOVA analysis with comparison to WT. Source data are provided as a Source Data file.
TM3-mediated HRD1 dimerization is required for function
Because yeast Hrd1 dimers are considered inactive45, we next investigated the functional relevance of HRD1 dimers in mammalian cells. Our cryo-EM structure indicates that HRD1 homodimerization is stabilized by a polar interaction at T93 and a π-stacking interaction involving Y83 between opposing TM3 helices (Fig. 5a and Supplementary Fig. 7a). Unlike Y83, T93 is highly conversed from yeast to humans and appears to be the dominant stabilizing residue (Fig. 4c and Supplementary Fig. 7a). To test this, we introduced T93A and T93F mutants to disrupt the polar interaction and assessed dimerization using crosslinking assays. Both mutations abolished R96C-mediated disulfide-linked dimers (lane 4-5 vs. 3, Fig. 5b) and reduced DSP-crosslinked dimers by ~10-fold (lanes 3-4 vs. 2, Supplementary Fig. 7b). Consistently, both T93A and T93F markedly reduced HRD1-HRD1 interaction in transfected HEK293T cells (Fig. 5c), demonstrating that T93 is essential for TM3-mediated HRD1 dimerization.

a Side view of HRD1 TM3 at the interface. The density maps showing the polar interaction of T93-T93 and Y83-Y83 are shown in Supplementary Fig. 7a. S-S indicates the disulfide bond formed by HRD1-R96C. b (Non-)Reducing and Western blot analysis of the native disulfide crosslinking assays in HRD1-/- HEK293T cells transfected with the indicated HRD1 mutants, with quantitation of the percentage of dimeric HRD1 in total HRD1 shown below (three independent repeats. p value: R96C p < 0.0001, T93A/R96C p = 0.9809, T93F/R96C p = 0.9998). c Immunoprecipitation of FLAG-agarose in HRD1-/- HEK293T cells transfected with indicated HRD1 mutants to examine HRD1-HRD1 interaction, with quantitation shown below (three independent repeats. p value: T93A p < 0.0001, T93F p < 0.0001). d Turnover assays of proAVP(G57S)*-His in HRD1-/- HEK293T cells transfected with proAVP(G57S)*-His and indicated HRD1 mutants, assessed by cycloheximide (CHX) chase, with quantitation shown below (three independent repeats. p value: 2 h Empty p = 0.9986, 2 h C2A p = 0.5902, 2 h T93A p = 0.156, 2 h T93F p = 0.7403; 4 h Empty p = 0.0043, 4 h C2A p = 0.0009, 4 h T93A p < 0.0001, 4 h T93F p < 0.0001). (e–f) Denaturing Strep-Tactin immunoprecipitation in HRD1-/- HEK293T cells transfected with proAVP(G57S)*-Strep and indicated HRD1 mutants, treated with 10 μM MG132 for 2 h, to measure proAVP(G57S) ubiquitination. Quantitation of ubiquitination levels shown in (f) (three independent repeats, p value: Empty p < 0.0001, T93A p < 0.0001, T93F p < 0.0001, C2A p < 0.0001). g, h Immunoprecipitation of FLAG-agarose in HRD1-/- HEK293T cells transfected with indicated HRD1 mutants to examine their interaction with SEL1L, OS9 and Derlin-2, with quantitation shown in (h) (three independent repeats). Values, mean ± SEM. n.s., not significant, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by one-way ANONA test with comparison to WT (b, c, f, h), and two-way ANOVA test with comparison to WT (d). Source data of Fig. 5b–g are provided as a Source Data file.
We next asked whether HRD1 dimerization is required for ERAD activity. Expression of either T93 mutant in HRD1-/- HEK293T cells significantly prolonged the half-life of proAVP G57S*, and impaired its ubiquitination, phenocopying the ligase-dead HRD1-C2A mutant (Fig. 5d–f and Supplementary. 7c). Similar defects were observed with another ER-retained model substrate, POMC C28F*59 (Supplementary Figs. 8a, b). These functional impairments were not due to disruption of the OS9-SEL1L-HRD1 complex, as both T93 mutants retained OS9 and SEL1L binding, as well as Derlin-2 (Fig. 5g, h). Thus, T93-dependent TM3-mediated HRD1 dimerization is essential for ERAD function.
A disease-associated HRD1 variant disrupts dimerization and impairs ERAD function
We recently learned of a novel HRD1 variant of uncertain significance, A91D, identified in a one-year-old boy with congenital heart defects and early-onset pulmonary dysfunction (Lifera Omics Database). A91 is conserved from humans to Arabidopsis but not in C. elegans and yeast (Fig. 4c). Our cryo-EM structure places this residue within TM3 of HRD1 at the dimer interface (Fig. 6a). Expression of HRD1-A91D in HRD1-/- HEK239T cells markedly prolonged the half-life and impaired ubiquitination of proAVP G57S* (Fig. 6b, c and Supplementary Fig. 9a). Similar results were obtained for POMC C28F* (Supplementary Figs. 8a and 9b).

a A91 at the HRD1 dimer interface. b Turnover assays of proAVP(G57S)*-His in HRD1-/- HEK293T cells transfected with proAVP(G57S)*-His and indicated HRD1 mutants, assessed by cycloheximide (CHX) chase, with quantitation shown below (three independent repeats. p value: 2 h Empty p < 0.0001, 2 h C2A p = 0.0398, 2 h A91D p < 0.0001; 4 h Empty p < 0.0001, 4 h C2A p < 0.0001, 4 h A91D p < 0.0001). c Denaturing Strep-Tactin immunoprecipitation in HRD1-/- HEK293T cells transfected with proAVP(G57S)*-Strep and indicated HRD1 mutants, with or without 10 μM MG132 for 2 hr. Quantitation of poly-ub proAVP shown below (three independent repeats. p value: A91D p < 0.0001, C2A p < 0.0001). d (Non-)Reducing and Western blot analysis of the native disulfide crosslinking assays in HRD1-/- HEK293T cells transfected with indicated HRD1 mutants, with quantitation of the percent of dimeric HRD1 in total HRD1 shown below (three independent repeats. p value: WT vs. R96C p < 0.0001, WT vs. A91D/R96C p = 0.0005, R96C vs. A91D/R96C p = 0.0011). e, f Immunoprecipitation of FLAG-agarose in HRD1-/- HEK293T cells transfected with HRD1-WT and A91D variant to examine their interaction with SEL1L, OS9 and Derlin-2, with quantitation shown (f) (three independent repeats). Values, mean ± SEM. n.s., not significant, *p < 0.05 and ****p < 0.0001 by two-way ANONA test with comparison to WT (b), one-way ANOVA test with comparison to WT (d). Source data of Fig. 6b–f are provided as a Source Data file.
Substituting a nonpolar residue (A) with a negatively charged residue (D) would be expected to introduce both steric hindrance and electrostatic repulsion, potentially destabilizing HRD1 dimers. To test this, we examined dimerization using the crosslinking assays described above. Both native disulfide crosslinking (R96C reporter) and DSP crosslinking showed ~50% reduction in dimer formation by A91D (Fig. 6d and Supplementary Fig. 9c). Importantly, A91D retained normal binding to OS9, SEL1L and Derlin-2 (Fig. 6e, f), indicating that the defect is specific to HRD1 dimerization rather than disruption of the core complex. These findings show that HRD1 A91D patient variant selectively impairs HRD1 dimerization and ERAD function, linking dimer-interface integrity to human disease pathogenesis.
Proposed substrate handling by the OS9-SEL1L-HRD1 core complex
To understand how the core complex engages misfolded substrates, we examined the spatial arrangement of OS9-SEL1L relative to the HRD1 channel. Our structure reveals that the distal end of the putative substrate-binding region of OS9-SEL1L lies ~58 Å from the entrance of the HRD1 channel (Fig. 7a). The ~36 Å OS9-SEL1L pocket aligns precisely with the 12 Å pore entrance and the 18 Å HRD1 dimer channel, positioning the luminal module for efficient substrate delivery. Electrostatic analysis shows that OS9-SEL1L contains a negatively charged substrate-binding pocket (Supplementary Fig. 10a). Similar to the inward-bent helices that generate the substrate cavity in the yeast Hrd1-Der1 complex45, the HRD1 dimer in our structure appears to induce local membrane thinning or bending at the dimer interface, producing a cavity evident in the micelle density (Fig. 7b and Supplementary Fig. 10b). Notably, this channel is enriched in methionine residues (Fig. 7c), reminiscent of the “methionine bristles” that line the pores of the EMC and YidC membrane insertases60,61 as well as the M-domain of the signal-recognition particle62,63 (Fig. 7d and Supplementary Fig. 10c). These observations reveal a previously unrecognized methionine-lined conduit within the HRD1 dimer that may facilitate substrate engagement or translocation during ERAD.

a Distance measurement within the core complex. b Side view of the HRD1 model. The cryo-EM map filtered to 4 Å is shown as a transparent surface. The distances of the HRD1 interface (TM3) and the peripheral region are shown. c, d Surface model of the membrane-embedded region of HRD1 dimer (c) and human EMC3-6 subcomplex (d). Methionine (Met) residues in the putative substrate retrotranslocation channel are highlighted in red. EMC model is generated by AlphaFold-3 as methionine-conducted bristles are invisible in the EMC structure. Asterisk in (d) indicate the putative substrate retrotranslocation channel. e–g Comparison of the human HRD1 dimer (hHRD1) (e), yeast Hrd1 dimer (yHrd1) (PDB code: 5V6P) (f), and yeast Hrd1-Der1 structures (PDB code: 6vjz) (g). Asterisks indicate the putative substrate retrotranslocation channels. h, i Comparisons of our structure with recently reported structures of the XTP3B-SEL1L-HRD1 complex without (PDB: 8KET) (h) or with Derlin-engagement (PDB: 9UAV) (i).
Comparison with existing ERAD core structures
Our cryo-EM structure establishes an HRD1 dimer architecture that differs substantially from previously reported yeast ERAD complexes in both yeast44,45 and mammals64 (Fig. 7e–i and Supplementary Figs. 10d–f). In our model, the putative translocation channel is formed between TM1-4 of one HRD1 protomer and TM8 of the other (asterisks, Fig. 7e and Supplementary Fig. 10d). This arrangement contrasts with the yeast Hrd1-Hrd3 dimer, which lacks such a channel (Fig. 7f and Supplementary Fig. 10e), and the Hrd1-Der1 complex, where half-channels are formed by TM2-4 of Hrd1 and TM1-2, TM5 of Der1 (asterisks, Fig. 7g and Supplementary Fig. 10f).
Our structure also differs from the recently reported mammalian XTP3B-SEL1L-HRD1 complex64, which captured an asymmetric dimer in which only one HRD1 protomer binds XTP3B-SEL1L (2:1:1 stoichiometry) (Fig. 7h). That study proposed that Derlin engagement may trigger HRD1 dimer dissociation, although Derlin itself was not visualized. Together, these observations suggest that the HRD1 dimer likely represents one of several coexisting assembly states, with transitions potentially regulated by substrate load, Derlin engagement, or broader ERAD pathway dynamics.
Discussion
Using an unbiased IP-MS screen coupled with single-particle cryo-EM, we define the molecular architecture of the human OS9-SEL1L-HRD1 ERAD core complex (Fig. 8a). These integrated proteomic and structural analyses establish OS9, SEL1L, and HRD1 as the minimal components of the mammalian ERAD machinery. Within this assembly, SEL1L serves as the organizing scaffold, linking luminal lectin OS9 to membrane-embedded HRD1 and positioning misfolded substrates for entry into the HRD1 channel. The structure further reveals that the complex adopts an HRD1-centered dimer. Structure-guided mutagenesis – including patient-derived SEL1L and HRD1 variants – shows that disrupting this interface impairs dimerization and compromises ERAD activity, underscoring the functional importance of this dimeric configuration (Fig. 8a–c).

a The OS9-SEL1L-HRD1 ERAD core complex forms a homodimer in vivo, with HRD1 dimerization dependent on polar interactions between two T93 residues. In the ER lumen, the MRH domain of OS9 and SEL1L together form a substrate-binding “claw”-like configuration. b The HRD1 disease variant A91D disrupts dimerization, while the SEL1L variant S658P impairs the SEL1L-HRD1 interaction - both compromising ERAD complex formation and function. c ER-lumen view of the OS9-SEL1L complex highlights the substrate-binding “crab claw”-like configuration. SEL1L disease variant G585D disrupts the SEL1L-OS9 interaction and attenuates ERAD function. Created in BioRender. Wang, H. (BioRender.com/1290qkk).
Although pathogenic SEL1L and HRD1 variants are typically associated with ENDI syndrome – characterized by profound neurodevelopmental impairment and immunologic abnormalities in infancy13,14,15 – the A91D variant identified in a one-year-old boy presents with a distinctly different clinical picture involving congenital heart defects and early-onset pulmonary dysfunction. Our functional studies show that A91D selectively impairs HRD1 dimerization and ERAD activity while preserving interactions with OS9 and SEL1L, but why this variant produces a cardiopulmonary phenotype rather than the primarily neurological features seen in ENDI syndrome remains unclear. These findings suggest that A91D expands the clinical spectrum of ENDI and highlight the need for further work to understand how different HRD1 mutations give rise to divergent phenotypes.
The importance of HRD1 dimerization in ERAD has long been debated. In yeast, Hrd1 dimers are considered inactive because their lateral gates block Der1 engagement, which is required for retrotranslocating luminal ERAD-L substrates44,45,57. In contrast, the mammalian HRD1 dimer visualized here adopts a fundamentally different arrangement: instead of two sealed protomers, it forms an open, methionine-lined groove that extends across the interface, creating a plausible conduit for substrate passage (Fig. 8a, b). Recent work by Guo et al.64 reported a Derlin-engaged XTP3B-SEL1L-HRD1 structure, but the HRD1 construct used lacked a substantial portion of the cytosolic C-terminal region required for ERAD activity and Derlin recruitment48,52. Consistent with this, Derlin is absent from our purified OS9-SEL1L-HRD1 core complex, even when Derlin and HERP1 are overexpressed, supporting a model in which Derlins function at or near the retrotranslocation step, potentially in parallel with HRD140,41,42,65. Although our data indicate that dimerization of the OS9-SEL1L-HRD1 core complex is critical for ERAD, incorporation of Derlin is likely to remodel its structural and functional organization. We therefore propose that the structure presented here represents one conformational state of the mammalian HRD1 ERAD complex. Future studies will be required to determine whether the functional translocation unit operates as a dimeric conduit or as a monomer in association with Derlin, as proposed by Wang et al.66
Based on these findings, we propose a model in which the OS9-SEL1L “crab-claw” captures misfolded substrates and directs them toward the HRD1 channel. Each HRD1 protomer contributes TM1-3 and TM8 to form two methionine-rich hydrophobic grooves flanking the dimer axis (Fig. 8a). Methionine’s flexible, low-energy side chains may provide an adaptable interface that accommodates the diverse biochemical properties of ERAD substrates during transit across the membrane. Together, our study elucidates the molecular organization of the mammalian ERAD core complex, identifies a functionally essential HRD1 dimeric state, and provides a structural basis for understanding substrate recognition and retrotranslocation – processes whose dysregulation underlies a broad spectrum of human diseases.
Methods
Cell lines
HEK293T cells (ATCC, CRL-3216) were cultured at 37 °C with 5% CO2 in DMEM (Gibco) with 10% fetal bovine serum (Fisher Scientific). HEK293F cells (Gibco, A14527) were cultured at 37 °C with 5% CO2 in Expi293™ Expression Medium (Gibco) with shaking.
Protein expression and purification
0.6 mg plasmid was transfected to 0.5 L HEK293F cells by 1.6 mg polyethylenimine “Max” (PEI Max) (Polysciences) for protein expression. The medium was collected at 72 h post-transfection for protein purification. The pellet from 500 mL cells was resuspended in 30 mL buffer A (20 mM HEPES, 150 mM NaCl, pH7.5) supplemented by protease inhibitor (Sigma) and phosphatase inhibitor cocktail (Sigma) and disrupted by sonication. After 3500 × g centrifugation for 5 min, the resulting supernatant was then incubated with 1% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) at 4 oC for 2 h. The insoluble fraction was removed by centrifugation (21,000 × g, 4 oC, 30 min), and the supernatant was incubated with 1 mL anti-FLAG M2 resin (Sigma) at 4 oC for 4 h. The beads were then washed with 20 column volume (CV) buffer B (20 mM HEPES, 150 mM NaCl, 0.01% LMNG, pH7.5) by gravity flow. Proteins were eluted in 5 CV buffer B supplemented by 0.1 mg/mL 3xFLAG peptide (APExBIO). The eluate was concentrated and further purified by size-exclusion chromatography (SEC) (Superose 6 Increase, 10/300 GL column, Cytiva) in buffer C (20 mM HEPES, 150 mM NaCl, 0.02% glyco-diosgenin/GDN, pH7.5) for cryo-EM.
Cryo-EM sample preparation and data acquisition
Protein samples were concentrated using 100 kDa filter (Amino) to 4 mg/mL for grid preparation. Aliquots of 3 μl protein samples in detergent were applied to Quantifoil R1.2/1.3 400 mesh Cu holy carbon grids (Quantifoil) glow discharged with amylamine, blotted for 8 s at 4 °C and 100%, then plunge-frozen in liquid ethane using a Vitrobot Mark IV (FEI). Data was collected on the ThermoFisher Titan Krios equipped with a K3 direct electron detector. The microscope was operated at 300 kV at a nominal magnification of 130,000X with an energy filter slit width of 10 eV. A defocus range of −0.8 µm to −1.8 µm at 0.2 µm step sizes was used for data collection. Movies were collected at a dose of 60 e/Å2 across 40 frames per movie. A total of 6,350 movies were collected, and the calibrated pixel size was 0.652 Å.
Cryo-EM data processing
Cryo-EM data was processed using CryoSPARC v4.6.067. Movies were corrected for beam-induced motion and subjected to patch CTF estimation. Blob picker was initially used to select for particles with minimum and maximum diameters of 120 Å and 250 Å, respectively. 2D classification of the picked particles yielded classes with clear membrane-embedded protein domain elements at an approximate diameter of ~130 Å. These classes were used to generate templates and improve particle picking. Template picker was used to select for particles with a 200 Å diameter using the generated templates from selected 2D class averages. From the motion-corrected micrographs, 1,101,567 particles were extracted at a box size of 488 × 488 pixels. An initial 2D classification was conducted to select a subset of well-aligned particles from 2D class averages showing clear protein domain and secondary structure elements resolved within and outside the micelle. An initial model for the ERAD complex was then generated using ab-initio reconstruction from 163,759 selected particles.
Subsequent 3D classifications were performed to recover additional ERAD particles for 3D homogenous refinements. Starting with the initial 1,101,567 extracted particles, iterative heterogenous refinements were conducted to sort for ERAD particles and remove junk or poorly aligning particles. A final set of 192,786 particles were reconstructed using non-uniform refinement applying C2 symmetry. The average resolution of the final map was 3.64 Å, as determined by the gold standard Fourier Shell Correlation (FSC) from two independently processed half-particle sets at an FSC threshold of 0.143. A mask was then created for the SEL1L and OS9 domains visible in the cryo-EM map. Symmetry expansion, followed by local 3D refinement focused on SEL1L-OS9 using the generated mask, was conducted to overcome symmetry-breaking features such as subunit flexibility between the individual components in the ERAD dimer and improve the map resolution in the OS9-SEL1L interaction interface to 3.30 Å. Local resolution of the focused and unfocused map was determined in cryoSPARC at an FSC threshold of 0.143.
Model building and refinement
The predicted Alphafold 356 model of OS9, SEL1L, and HRD1 was docked as a rigid body into the cryo-EM map using ChimeraX (v1.9). Regions that were not resolved in the EM maps were removed from fitted model. We then broke the model into 5 rigid body elements: 2OS9, 2SEL1L, and the HRD1 dimer that were then adjusted into the cryo-EM map with COOT (v1.1.02). The transmembrane domains of HRD1 and secondary structural elements of SEL1L and OS9 were further rotated and positioned in the density map based on visible side chains in COOT. The final model was iteratively real space refined in PHENIX68 (v1.20.1) with 3 macrocycles applying Ramachandran and secondary structure restraints, and non-crystallographic symmetry constraints. Molprobity was used to validate the model and resolve clashes. All model and map figures were made with ChimeraX.
CRISPR/Cas9-based knockout (KO) HEK293T cells
To generate SEL1L-, HRD1-, FAM8A1-, Derlin-1-, and Derlin-2- deficient HEK293T cells, sgRNA oligonucleotides designed for human SEL1L (5’-GGCTGAACAGGGCTATGAAG-3’), human HRD1 (5’-GGACAAAGGCCTGGATGTAC-3’), FAM8A1 (gRNA1: 5’-GCGCGGCGGCTCCAATTTGT-3’, gRNA2: 5’-TTCGGCCTGGGGGTCGTCGC-3’), Derlin-1 (5’-GCTGCTGATCAGAATGGCGG-3’), and Derlin-2 (5’-GAAGGCCGAAGAAGTTCATG-3’) was inserted into lentiCRISPR v2 (plasmid 52961; Addgene). HEK293T cells grown in 10 cm petri dishes were transfected with indicated plasmids using 5 μl 1 mg/ml PEI (Sigma) per 1 μg of plasmids for HEK293T cells. Cells were cultured 24 h after transfection in medium containing 2 µg/ml puromycin for 24 h and then in normal growth media.
Plasmids
The following plasmids were used in the study (h denotes human genes; m denotes mouse genes): h-HRD1 cDNA with a C-terminal FLAG or Strep tag was cloned from HEK293T cDNA and inserted into the pcDNA3 to generate pcDNA3-h-HRD1(WT)-FLAG and pcDNA3-h-HRD1(WT)-Strep. The HRD1 mutant C2A (C291A/C294A), T93C, V94C, F95C, R96C, D97C, T93A, T93F and A91D were generated using the plasmid pcDNA-h-HRD1(WT)-FLAG or pcDNA3-h-HRD1(WT)-Strep as template. HRD1 T93A/R96C, T93F/R96C, and A91D/R96C mutants were generated using the plasmid pcDNA-h-HRD1(R96C)-FLAG as template. h-OS9 isoform 2 (OS9) cDNA was cloned from HEK293T cDNA and inserted into the pcDNA3 to generate pcDNA3-h-OS9(WT). The OS9 mutant D135C was generated using plasmid pcDNA3-h-OS9(WT) as the template. m-Sel1L cDNA, with/without a C-terminal FLAG tag was cloned from mouse liver cDNA and inserted into the pcDNA3 to generate pcDNA3-m-SEL1L(WT)-FLAG and pcDNA3-m-SEL1L(WT). The SEL1L-FLAG mutants S658P and G585D were generated using the plasmid pcDNA-m-SEL1L(WT)-FLAG as the template. The double mutations of SEL1L S658P/G585D was generated using the plasmid pcDNA-m-SEL1L(S658P)-FLAG as the template. The SEL1L mutants D439C, M440C and N442C were generated using the plasmid pcDNA-m-SEL1L(WT) as the template. The OS9 mutant D135C was generated using plasmid pcDNA3-h-OS9(WT) as the template. h-proAVP G57S with C-terminal 1xStrep or 10xHis tag and KDEL was cloned from the plasmid pcDNA3-h-proAVP G57S-HA53, to generate pcDNA3-h-proAVP G57S*. mPOMC C28F with 10xHis tag and KDEL in its C-terminal was cloned from the plasmid mPOMC C28F-HA59 to generate pcDNA3-m-POMC C28F*. All plasmids were validated by DNA sequencing. Residue numbering of SEL1L in figures corresponds to human SEL1L.
Western blot and antibodies (reducing SDS-PAGE)
HEK293T or HEK293F cells were harvested and snap-frozen in liquid nitrogen. The proteins were extracted by sonication in Western blot lysis buffer (50 mM Tris-HCl at pH7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA) with protease inhibitor (Sigma) and phosphatase inhibitor cocktail (Sigma). Lysates were incubated on ice for 15 min and centrifuged at 16,000 g for 10 min. Supernatants were collected and analyzed for protein concentration using the Bio-Rad Protein Assay Dye (Bio-Rad). 20–50 μg of protein were denatured at 95 °C for 5 min or 37 °C for 1 hr in 5x SDS sample buffer (250 mM Tris-HCl pH 6.8, 10% sodium dodecyl sulfate, 0.05% Bromophenol blue, 50% glycerol, and 1.44 M β-mercaptoethanol). Protein was separated on SDS-PAGE, followed by electrophoretic transfer to PVDF (Fisher Scientific) membrane. The blots were incubated in 2% BSA/Tri-buffered saline tween-20 (TBST) with primary antibodies overnight at 4 °C: anti-HSP90 (Santa Cruz, #sc-13119, 1:5000), anti-GAPDH (Proteintech, #60004-1, 1:5000), anti-SEL1L (home-made, 1:10,000)69, anti-HRD1 (Proteintech, #13473-1, 1:2000), anti-OS9 (Abcam, #ab109510, 1:5000), anti-CD147 (Proteintech, #11989-1, 1:3,000), anti-IRE1α (Cell Signaling, #3294, 1:2,000), anti-UBE2J1 (Santa Cruz, #sc-377002, 1:3,000), anti-Derlin-2 (gift, 1:1000), anti-Derlin-1 (gift, 1:1000), anti-FLAG (Sigma, #F1804, 1:1000), anti-HERPUD1 (HERP1) (Abcam, #ab150424, 1:3000), anti-FAM8A1 (Proteintech, #24746-1-AP,1:3000), anti-ubiquitin (Santa Cruz, #sc-8017, 1:1000), anti-His (Genscript, # A00174, 1:2000), anti-Strep (Sigma, # SAB2702216, 1:1000). Membranes were washed with TBST and incubated with secondary antibodies, HRP conjugated (Bio-Rad, 1:10,000) at room temperature for 1 h for ECL chemiluminescence detection system (Bio-Rad) development. Band intensity was determined using Image lab (Bio-Rad, v6.1.0) software.
Mass spectrometry
SEL1L- and HRD1- IP-MS in HEK293T cells have been described previously49 and were performed with three independent biological replicates. Briefly, IP of endogenous SEL1L or HRD1 in WT, SEL1L-/- or HRD1-/- HEK293T cells were performed using 10 mg proteins from each sample lysed with the IP buffer (150 mM NaCl, 0.2% NP-40, 0.1% Triton X-100, 25 mM Tris-HCl pH7.5) supplemented with protease inhibitors, protein phosphatase inhibitors, and 10 mM N-ethylmaleimide. The cell lysates were first incubated anti-SEL1L (home-made) or anti-HRD1 (home-made or Cell Signaling #14773) at 4 °C overnight, followed by incubation with Protein A agarose (Invitrogen, #20333) at 4 °C for 2 h. An IP reaction with IgG using cell lysate of SEL1L-/- or HRD1-/- cells was included as a negative control for SEL1L- or HRD1-IP, respectively.
The mass spectrometry was conducted by the Proteomics Resource Facility at the University of Michigan Medical School. The beads were resuspended in 50 µl of 0.1 M ammonium bicarbonate buffer (pH~8). Cysteines were reduced by adding 50 µl of 10 mM DTT and incubating at 45 °C for 30 min. Samples were cooled to room temperature and alkylation of cysteines was achieved by incubating with 65 mM 2-Chloroacetamide, under darkness, for 30 min at room temperature. An overnight digestion with 1 µg sequencing-grade modified trypsin was carried out at 37 °C with constant shaking in a Thermomixer. Digestion was stopped by acidification and peptides were desalted using SepPak C18 cartridges using manufacturer’s protocol (Waters). Samples were completely dried using vacufuge. Resulting peptides were dissolved in 0.1% formic acid/2% acetonitrile solution and were resolved on a nano-capillary reverse phase column (Acclaim PepMap C18, 2 micron, 50 cm, ThermoScientific) using a 0.1% formic acid/2% acetonitrile (Buffer A) and 0.1% formic acid/95% acetonitrile (Buffer B) gradient at 300 nl/min over a period of 180 min (2–25% buffer B in 110 min, 25–40% in 20 min, 40–90% in 5 min followed by holding at 90% buffer B for 10 min and re-equilibration with Buffer A for 30 min). Eluent was directly introduced into Q exactive HF mass spectrometer (Thermo Scientific, San Jose CA) using an EasySpray source. MS1 scans were acquired at 60 K resolution (AGC target=3 × 106; max IT = 50 ms). Data-dependent collision induced dissociation MS/MS spectra were acquired using Top speed method (3 s) following each MS1 scan (NCE ~ 28%; 15 K resolution; AGC target 1 × 105; max IT 45 ms). Proteins were identified by searching the MS/MS data against UniProt entries using Proteome Discoverer (v2.4, Thermo Scientific). Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.2 Da; two missed cleavages were allowed; carbamidomethylation of cysteine was considered fixed modification and oxidation of methionine, deamidation of asparagine and glutamine were considered as potential modifications. False discovery rate (FDR) was determined using Percolator and proteins/peptides with an FDR of ≤1% were retained for further analysis.
SEL1L- or HRD1- interacting proteins were selected based on the peptide spectrum matches (PSMs) from the label-free IP-MS results. For each protein hit, the PSM value from the IgG must be 0; the PSMs ratio of the bait KO sample (negative control) to WT sample must be smaller than the ratio of the corresponding bait. Proteomic data has been deposited into a public database PRIDE (PXD043674 and PXD041882).
Native disulfide crosslinking (non-reducing SDS-PAGE)
HEK293T or HEK293F cells transfected with the indicated OS9, SEL1L, or HRD1 variants were harvested and snap-frozen in liquid nitrogen. Cell pellets were resuspended in 1x non-reducing SDS loading buffer (50 mM Tris-HCl pH6.8, 2% sodium dodecyl sulfate, 0.01% Bromophenol blue, 10% glycerol), supplemented with 15 mM N-ethylmaleimide (NEM) to alkylate free cysteines and preserve native disulfide-linked species, and proteins were extracted by sonication. Lysates were incubated at room temperature for 15 min, followed by centrifugation at 16,000 × g for 10 min. The resulting supernatants were collected for SDS-PAGE and immunoblot analysis.
DSP crosslinking (non-reducing SDS-PAGE)
HEK293T cells transfected with the indicated HRD1 variants were treated with 2 mM DSP crosslinker (Thermo Science) or DMSO at room temperature for 2 h. The crosslinking reaction was quenched by adding 20 mM Tris-HCl (pH7.5). Treated cells were then harvested and snap-frozen in liquid nitrogen. Cell pellets were resuspended in 1x non-reducing SDS loading buffer (50 mM Tris-HCl pH6.8, 2% sodium dodecyl sulfate, 0.01% Bromophenol blue, 10% glycerol), and proteins were extracted by sonication. Lysates were incubated at room temperature for 15 min, followed by centrifugation at 16,000 × g for 10 min. The resulting supernatants were collected for SDS-PAGE and immunoblot analysis.
Co-immunoprecipitation (Co-IP) in HEK293T cells
HEK293T were snap-frozen in liquid nitrogen and whole cell lysate was prepared in IP buffer (150 mM NaCl, 0.2% NP-40, 0.1% Triton X-100, 25 mM Tris-HCl pH 7.5) for anti-SEL1L-FLAG or anti-HRD1-FLAG IP supplemented with protease inhibitors, and protein phosphatase inhibitors. A total of ~5 mg protein lysates were incubated with 10 μl anti-FLAG agarose (Sigma, #A2220) antibody overnight at 4 °C with gentle rocking. The incubated agaroses were washed three times with IP buffer and eluted in 0.1 mg/ml 3xFLAG peptides. The resulting elution was incubated with 5x SDS sample buffer (250 mM Tris-HCl pH 6.8, 10% sodium dodecyl sulfate, 0.05% Bromophenol blue, 50% glycerol, and 1.44 M β-mercaptoethanol) at room temperature for 15 min, followed by SDS-PAGE and immunoblot analysis.
Denaturing immunoprecipitation for ubiquitination assay
HEK293T cells were transfected with proAVP(G57S)-Strep and the indicated HRD1 variants. HEK293F cells were transfected with proAVP(G57S)-Strep, and OS9-SEL1L-HRD1 WT/C2A proteins. The transfected cells were treated with DMSO or 10 μM MG132 for 2 h. Treated cells were snap-frozen in liquid nitrogen and whole cell lysates was prepared in the Triton X-100 lysis buffer (50 mM Tris-HCl at pH7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA) with 1% SDS and 5 mM DTT, and denatured at 95 °C for 10 min and centrifuged at 16,000×g for 10 min. Subsequently, supernatants were diluted 1:10 with NP-40 lysis buffer and incubated with 10 μl Strep-Tatin agarose (IBA, #6-6350) overnight at 4 °C with gentle rocking. The incubated agaroses were washed three times with the Triton X-100 lysis buffer and eluted in 5 mM Biotin. The resulting elution was incubated with 5x SDS sample buffer (250 mM Tris-HCl pH 6.8, 10% sodium dodecyl sulfate, 0.05% Bromophenol blue, 50% glycerol, and 1.44 M β-mercaptoethanol) at 95 oC for 5 min followed by SDS-PAGE and Immunoblot. The samples were loaded into a 6-15% gradient SDS-PAGE gel for separation.
SEL1L and HRD1 disease variants
SEL1L G585D and S658P have been reported previously13,49,70. We recently identified through Lifera Omics Database a novel variant of HRD1 A91D, with unknown significance in a one-year-old boy with congenital heart defects and early-onset pulmonary dysfunction. Human studies were approved by Health Sciences Research University of Virginia (HSR230351).
Statistics
The cryo-EM data collection, refinement and validation statistics are shown in Supplementary Table 4. All experiments have been repeated at least two to three times and/or performed with multiple independent biological samples from which representative data are shown.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The materials and reagents used are either commercially available or available upon the request. Proteomic data are available in a public database PRIDE (PXD043674 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD043674) and PXD041882). The cryo-EM map and the corresponding atomic model for the ERAD complex structure have been deposited under the accession codes EMD70448 (https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-70448) (consensus map), EMD-70452 (locally refined map) and 9OG0. All data related to this study are available in the main text or the supplementary material. Source data are provided with this paper.
References
Kanapin, A. et al. Mouse proteome analysis. Genome Res 13, 1335–1344 (2003).
Benham, A. M. Protein secretion and the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 4, a012872 (2012).
Lippincott-Schwartz, J., Bonifacino, J. S., Yuan, L. C. & Klausner, R. D. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell 54, 209–220 (1988).
Sommer, T. & Jentsch, S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 365, 176–179 (1993).
Jensen, T. J. et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 (1995).
Ward, C. L., Omura, S. & Kopito, R. R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 (1995).
McCracken, A. A. & Brodsky, J. L. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132, 291–298 (1996).
Hampton, R. Y., Gardner, R. G. & Rine, J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044 (1996).
Bordallo, J., Plemper, R. K., Finger, A. & Wolf, D. H. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9, 209–222 (1998).
Biunno, I. et al. Isolation of a pancreas-specific gene located on human chromosome 14q31: expression analysis in human pancreatic ductal carcinomas. Genomics 46, 284–286 (1997).
Kaneko, M., Ishiguro, M., Niinuma, Y., Uesugi, M. & Nomura, Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett. 532, 147–152 (2002).
Hwang, J. & Qi, L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci. 43, 593–605 (2018).
Wang, H. H. et al. Hypomorphic variants of SEL1L-HRD1 ER-associated degradation are associated with neurodevelopmental disorders. J. Clin. Invest. https://doi.org/10.1172/JCI170054 (2024).
Weis, D. et al. Biallelic Cys141Tyr variant of SEL1L is associated with neurodevelopmental disorders, agammaglobulinemia, and premature death. J. Clin. Invest. https://doi.org/10.1172/JCI170882 (2024).
Wang, H. H., Biunno, I., Sun, S. & Qi, L. SEL1L-HRD1-mediated ERAD in mammals. Nat. Cell Biol. https://doi.org/10.1038/s41556-025-01690-1 (2025).
Qi, L., Tsai, B. & Arvan, P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 27, 430–440 (2017).
Bhattacharya, A. & Qi, L. ER-associated degradation in health and disease - from substrate to organism. J. Cell Sci. 132, jcs232850 (2019).
Sha, H. et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 20, 458–470 (2014).
Sun, S. et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 111, E582–E591 (2014).
Francisco, A. B. et al. Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J. Biol. Chem. 285, 13694–13703 (2010).
Fujita, H. et al. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J. 34, 1042–1055 (2015).
Yagishita, N. et al. Essential role of synoviolin in embryogenesis. J. Biol. Chem. 280, 7909–7916 (2005).
Ji, Y. et al. The Sel1L-Hrd1 endoplasmic reticulum-associated degradation complex manages a key checkpoint in B cell development. Cell Rep. 16, 2630–2640 (2016).
Sun, S. et al. Epithelial Sel1L is required for the maintenance of intestinal homeostasis. Mol. Biol. Cell 27, 483–490 (2016).
Bhattacharya, A. et al. Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH. EMBO J. 37, e99277 (2018).
Shrestha, N. et al. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGFβ signaling. J. Clin. Invest 130, 3499–3510 (2020).
Yoshida, S. et al. Endoplasmic reticulum-associated degradation is required for nephrin maturation and kidney glomerular filtration function. J. Clin. Invest. https://doi.org/10.1172/JCI143988 (2021).
Abdon, B. et al. Muscle specific ER-associated degradation maintains postnatal muscle hypertrophy and systemic energy metabolism. JCI Insight 8, e170387 (2023).
Ji, Y. et al. SEL1L-HRD1 endoplasmic reticulum-associated degradation controls STING-mediated innate immunity by limiting the size of the activable STING pool. Nat. Cell Biol. https://doi.org/10.1038/s41556-023-01138-4 (2023).
Thepsuwan, P. et al. Hepatic SEL1L-HRD1 ER-associated degradation regulates systemic iron homeostasis via ceruloplasmin. Proc. Natl. Acad. Sci. USA 120, e2212644120 (2023).
Bays, N. W., Gardner, R. G., Seelig, L. P., Joazeiro, C. A. & Hampton, R. Y. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 (2001).
Gardner, R. G. et al. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control Hrd1p Hrd3p. J. Cell Biol. 151, 69–82 (2000).
Mueller, B., Lilley, B. N. & Ploegh, H. L. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 175, 261–270 (2006).
Mueller, B., Klemm, E. J., Spooner, E., Claessen, J. H. & Ploegh, H. L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 105, 12325–12330 (2008).
Bhamidipati, A., Denic, V., Quan, E. M. & Weissman, J. S. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol. Cell 19, 741–751 (2005).
Kim, W., Spear, E. D. & Ng, D. T. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol. Cell 19, 753–764 (2005).
Szathmary, R., Bielmann, R., Nita-Lazar, M., Burda, P. & Jakob, C. A. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol. Cell 19, 765–775 (2005).
Christianson, J. C., Shaler, T. A., Tyler, R. E. & Kopito, R. R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 10, 272–282 (2008).
Hosokawa, N. et al. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 283, 20914–20924 (2008).
Lilley, B. N. & Ploegh, H. L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 102, 14296–14301 (2005).
Ye, Y., Shibata, Y., Yun, C., Ron, D. & Rapoport, T. A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429, 841–847 (2004).
Ye, Y. et al. Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 102, 14132–14138 (2005).
Carvalho, P., Goder, V. & Rapoport, T. A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 (2006).
Schoebel, S. et al. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548, 352–355 (2017).
Wu, X. et al. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, https://doi.org/10.1126/science.aaz2449 (2020).
Hwang, J. et al. Characterization of protein complexes of the endoplasmic reticulum-associated degradation E3 ubiquitin ligase Hrd1. J. Biol. Chem. 292, 9104–9116 (2017).
Huang, C. H., Hsiao, H. T., Chu, Y. R., Ye, Y. & Chen, X. Derlin2 protein facilitates HRD1-mediated retro-translocation of sonic hedgehog at the endoplasmic reticulum. J. Biol. Chem. 288, 25330–25339 (2013).
Huang, C. H., Chu, Y. R., Ye, Y. & Chen, X. Role of HERP and a HERP-related protein in HRD1-dependent protein degradation at the endoplasmic reticulum. J. Biol. Chem. 289, 4444–4454 (2014).
Lin, L. L. et al. SEL1L-HRD1 interaction is required to form a functional HRD1 ERAD complex. Nat. Commun. 15, 1440 (2024).
van der Goot, A. T., Pearce, M. M. P., Leto, D. E., Shaler, T. A. & Kopito, R. R. Redundant and Antagonistic Roles of XTP3B and OS9 in Decoding Glycan and Non-glycan Degrons in ER-Associated Degradation. Mol. Cell 70, 516–530.e516 (2018).
Fujimori, T., Kamiya, Y., Nagata, K., Kato, K. & Hosokawa, N. Endoplasmic reticulum lectin XTP3-B inhibits endoplasmic reticulum-associated degradation of a misfolded alpha1-antitrypsin variant. FEBS J. 280, 1563–1575 (2013).
Schulz, J. et al. Conserved cytoplasmic domains promote Hrd1 ubiquitin ligase complex formation for ER-associated degradation (ERAD). J. Cell Sci. 130, 3322–3335 (2017).
Shi, G. et al. ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Invest 127, 3897–3912 (2017).
Friberg, M. A., Spiess, M. & Rutishauser, J. Degradation of wild-type vasopressin precursor and pathogenic mutants by the proteasome. J. Biol. Chem. 279, 19441–19447 (2004).
Kikkert, M. et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 279, 3525–3534 (2004).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Pisa, R. & Rapoport, T. A. Disulfide-crosslink analysis of the ubiquitin ligase Hrd1 complex during endoplasmic reticulum-associated protein degradation. J. Biol. Chem. 298, 102373 (2022).
Hwang, C., Sinskey, A. J. & Lodish, H. F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 (1992).
Kim, G. H. et al. Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J. Clin. Invest 128, 1125–1140 (2018).
Pleiner, T. et al. Structural basis for membrane insertion by the human ER membrane protein complex. Science 369, 433–436 (2020).
Bai, L., You, Q., Feng, X., Kovach, A. & Li, H. Structure of the ER membrane complex, a transmembrane-domain insertase. Nature 584, 475–478 (2020).
Jomaa, A. et al. Structure of the quaternary complex between SRP, SR, and translocon bound to the translating ribosome. Nat. Commun. 8, 15470 (2017).
Jomaa, A. et al. Molecular mechanism of cargo recognition and handover by the mammalian signal recognition particle. Cell Rep. 36, 109350 (2021).
Guo, L. et al. Structural insights into the human HRD1 ubiquitin ligase complex. Nat. Commun. 16, 6007 (2025).
Rao, B. et al. The cryo-EM structure of the human ERAD retrotranslocation complex. Sci. Adv. 9, eadi5656 (2023).
Wang, Q. et al. Cryo-EM structure of the human Derlin-1/p97 complex reveals a hexameric channel in ERAD. Commun. Biol. 8, 1481 (2025).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Zhou, Z. et al. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 368, 54–60 (2020).
Kyostila, K. et al. A SEL1L mutation links a canine progressive early-onset cerebellar ataxia to the endoplasmic reticulum-associated protein degradation (ERAD) machinery. PLoS Genet 8, e1002759 (2012).
Acknowledgements
We thank the Qi, Jomaa, Sun labs (UVA), Arvan lab (UM), Drs. David Castle (UVA), Qinli Hu (UT Southwestern Medical Center), Maciej Gluc (UVA), Ruoya Ho (UVA), and Huan Bao (UVA) for technical support and insightful discussions; Drs. Michael Purdy and David Cooper (MEMC core, UVA), Haoran Yuan (UVA) and Riley Loyd (UVA) for assisting with the cryo-EM data collection and computational support; Dr. Chih-Chi Andrew Hu (Houston Methodist Hospital) for the Derlin-1 and Derlin-2 antibodies; and Drs. Fowzan Alkuraya and Khadijah Bakur for providing the information on the HRD1 A91D variant from the Lifera Omics Database. Cryo-EM data collection was conducted at the Molecular Electron Microscopy Core Facility (MEMC, RRID:SCR_019031) at the University of Virginia (UVA) School of Medicine (NIH G20-RR31199). We thank the Proteomics Resource Facility at University of Michigan Medical School for assisting proteomic assays. This work was supported by The Owens Family Foundation, the Searle Scholars Program (SSP-2023-106), American Cancer Society grant (#134088-IRG-19-143-33-IRG), and NIH 1R35 GM160490 to A.J.; NIH R01DK120047, R01DK120330, R35GM130292 to L.Q. L.L.L. was supported in part by National Ataxia Foundation Post-doctoral Fellowships NAF 918037. E.M. is supported by the cellular and molecular biology training program at UVA through NIH T32GM139787-3. L.E.Z. is supported by the American Heart Association Pre-doctoral Fellowship 25PRE1375196.
Author information
Authors and Affiliations
Contributions
This study was conceived by L.L.L., A.J. and L.Q. L.L.L. purified the ERAD core complex and collected cryo-EM data. E.M. processed cryo-EM data and built the atomic model. L.L.L. designed and performed all the biochemical experiments. L.E.Z. conducted the site directed point mutagenesis and plasmid preps. L.L.L. and L.Q. wrote the manuscript. All authors contributed to data analysis and final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Peer review
Peer review information
Nature Communications thanks Kenji Inaba and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, L.L., Maldosevic, E., Zhou, L.E. et al. Structural basis and pathological implications of the dimeric OS9-SEL1L-HRD1 ERAD Core Complex. Nat Commun 17, 2064 (2026). https://doi.org/10.1038/s41467-026-68777-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68777-7
