
Abstract
Securinega alkaloids possess a distinctive tetracyclic scaffold and are pharmaceutically relevant, yet their biosynthesis in Flueggea suffruticosa remains unresolved. Here, we combine chemical logic with stable-isotope-labeled feeding experiments and single-cell transcriptomics to elucidate the core biosynthetic pathway. We identify a reductase, FsMS, that reduces premenisdaurilide to menisdaurilide, which then conjugates with 1-piperideine to generate the [2.2.2]-bicyclic neosecurinanes, (–)-virosine A and (–)-virosine B. We also identify two sulfotransferases, FsNSST1 and FsNSST2, that O-sulfate these neosecurinanes; the resulting sulfate esters undergo a spontaneous 1,2-amine shift to yield the [3.2.1]-bicyclic securinanes, allosecurinine and securinine. This transformation reveals an unexpected biosynthetic role of sulfotransferases, not as conventional tailoring enzymes, but as key mediators of scaffold remodeling. The newly identified biosynthetic genes are co-expressed within a specific vasculature-associated cell type, enabling prioritization of additional pathway candidates. These findings highlight the power of chemically guided single-cell transcriptomics in unraveling complex biosynthetic pathways.
Similar content being viewed by others

Introduction
Biosynthetic alkaloid scaffold remodeling is a common feature across alkaloid families and a major driver of their structural diversification. Alkaloid scaffold formation and remodeling are often mediated by enzymes that have acquired specialized catalytic activities from conventional metabolic roles. For example, scaffold formation in Lycopodium alkaloids involves neofunctionalized carbonic anhydrases1. Catharanthine is formed from dehydrosecodine via an α/β hydrolase-mediated [4 + 2]-cycloaddition2, and scaffold remodeling in colchicine biosynthesis is catalyzed by specialized cytochrome P4503. In contrast to other specialized metabolites, each alkaloids have their own unique precursor, making it difficult to predict the sequence of reactions leading to the core scaffold. This biochemical ambiguity further complicates the identification of biosynthetic genes using conventional approaches based on known enzyme functions.
To identify biosynthetic genes for plant specialized metabolites, tissue-specific RNA sequencing has been widely used. Co-expression analyses using known biosynthetic genes as baits are effective in uncovering biosynthetic pathways3,4,5,6,7,8,9,10,11. To enhance the resolution of co-expression analysis, researchers have adopted sampling strategies that consider tissue developmental stages1,12, inducible conditions13, and fine-scale tissue dissection. While these approaches improve the resolution of biosynthetic gene discovery, a gene expression analysis at the single-cell level would offer an even higher resolution. This advantage stems from the substantially increased number of samples (individual cells) and the ability to identify cell types in which biosynthetic pathway genes are co-expressed. Although some specialized metabolic pathways are organized in a non-cell autonomous manner, with sequential steps partitioned across different cell types and intermediates transported between them, single-cell RNA sequencing (scRNA-seq) based co-expression analysis has nonetheless been successfully applied to identify biosynthetic genes for benzyl acetone14, vinblastine15,16, hyperforin17, and taxol18,19.
In parallel, chemical insights into biosynthetic intermediates would aid in the elucidation of corresponding biosynthetic genes. Historically, biomimicry has played a pivotal role in guiding the synthesis of complex natural products20,21,22. Conversely, access to presumed biosynthetic precursors and insights into their inherent chemical reactivity have significantly advanced the understanding of biosynthetic pathways. Such information enables the prediction of biosynthetic transformations and candidate gene functions. In addition, isotopically labeled intermediates have proven critical for validating the involvement of candidate molecules in biosynthetic pathways. Hence, synthetic access to (multiply) isotope-labeled biosynthetic candidate intermediates can expedite the elucidation of biosynthetic pathway. Thus, the integration of chemical knowledge is essential to fully harness the potential of high-resolution transcriptomics, especially when most biosynthetic intermediates remain unidentified.
The biosynthesis of securinega alkaloids (SeAs) offers an ideal case study for applying an integrative, chemically guided single-cell transcriptomics approach. SeAs have captivated the scientific community for over six decades owing to their structural complexity and biological activities23,24,25. Recently, SeAs have emerged as promising medicinal compounds for the treatment of cancer26,27 and neurological diseases28,29,30. Despite their pharmaceutical significance, the biosynthetic origin of SeAs in plants remains largely unresolved. Beyond their known precursors, L-tyrosine (1) and the L-lysine-derived 1-piperideine (11), no intermediates bridging these precursors to the SeA scaffold have been identified31,32,33,34,35. Two recent papers have begun to define peripheral steps in SeA biosynthesis. In 2020, Xiao et al. identified FsBBE, which catalyzes the dehydrogenation of allosecurinine (8), a key step in the oxidative post-modification of the SeA scaffold36. More recently, Lichman et al. discovered FsPS (OLADO), which produces 1-piperideine (11) from lysine37. However, the mechanism underlying SeA scaffold formation and rearrangements has remained elusive. Nevertheless, extensive efforts toward the total synthesis of the securinane scaffold (8, 9) have proposed various candidate intermediates (Supplementary Fig. 1), offering critical insights into potential intermediates and core chemical transformations.
Here, we elucidate the biosynthetic pathway of monomeric SeAs in Flueggea suffruticosa by integrating chemical synthesis with single-cell transcriptomics. Putative intermediates labeled with stable isotopes were synthesized (Supplementary Fig. 27), and their intrinsic biochemical reactivities were evaluated (Fig. 1a). Single-cell transcriptomic analysis identified a specific cell type that exhibits highly enriched expression of the two previously known SeA-associated genes36,37 (Fig. 1b). This multidisciplinary approach led to the identification of key biosynthetic intermediates and the discovery of enzymes responsible for their conversion into SeA (Fig. 1c).

a To elucidate L-tyrosine-derived biosynthetic intermediates of SeAs, hypothetical intermediates doubly labeled with 13C were synthesized. b Single-cell transcriptomics and coexpression analysis were performed on F. suffruticosa leaves. Intermediates doubly labeled with 13C were biochemically assessed, and candidate biosynthetic genes were functionally validated both in vitro and in planta. c We discovered the scaffold formation and remodeling of SeAs in F. suffruticosa (Gray box). FsMS menisdaurilide synthase, FsPS piperideine synthase, FsNSST1/2 neosecurinane sulfotransferase 1/2.
Results
Single-cell RNA sequencing proposed the cell cluster responsible for SeA biosynthesis
To address the absence of a reference genome, de novo genome assembly and gene annotation of F. suffruticosa were performed. Using PacBio HiFi sequencing, over 65 Gbps of reads with an average length of 15.9 kb and a Phred quality score of Q27 were generated from genomic DNA extracted from F. suffruticosa leaves. These reads were assembled into a high-quality genome, from which 34,960 protein-coding genes were predicted. The ploidy of F. suffruticosa is 2n = 26, and the estimated genome size is 450 Mbps. A contig N50 of 30.93 Mbps and a BUSCO gene completeness ratio of 95.8% demonstrated the reliability of the genome assembly and gene annotation (Fig. 2a).

a Statistics of F. suffruticosa genome assembly and gene annotation. b Uniform manifold approximation and projection (UMAP) plot of cells after processing showed clusters classified into five major cell types. Cluster numbers were assigned in descending order based on the number of cells in each cluster. c Dot plot for genes involved in sulfate assimilation, L-Lys biosynthesis, L-Tyr/L-Phe biosynthesis, and SeA biosynthesis across cell clusters; they were highly enriched in clusters 7. FsAPSK2, APS kinase 2; FsATPS2, ATP sulfurylase 2; FsStr, sulfite transferase; FsTauE/SafE, sulfite transporter; FsDAPEpi, diaminopimelate epimerase; FsDAPAT, diaminopimelate aminotransferase; FsCS, chorismate synthase; FsCM, chorismate mutase; FsPS, piperideine synthase; FsBBE2, berberine bridge-like enzyme 2. d Co-expression analysis using FsPS as a bait gene. Genes are ranked based on their Spearman correlation to FsPS, up to rank 80. FsPS, piperideine synthase; FsNSST1 and 2, neosecurinane sulfotransferase 1 and 2; FsMS, menisdaurilide synthase. Genes related to the following biosynthetic pathways were color-coded: sulfate assimilation (red), L-Lys biosynthesis (green), L-Tyr/L-Phe biosynthesis (purple), and SeA biosynthesis (blue).
With a reference genome in hand, tissue-specific expression patterns of two known SeA biosynthetic genes, FsBBE236 and FsPS37, were examined. FsBBE2, which catalyzes the conversion of allosecurinine (8) to 2,3-dehydroallosecurinine, showed higher expression in leaves compared to other tissues. In contrast, FsPS, an early-stage biosynthetic gene, showed no tissue-specific expression pattern (Supplementary Fig. 2). The D2O labeling assay, which provides qualitative information on biosynthetic reactivity in the tissue of interest by measuring the incorporation of deuterium into the target metabolite, showed that the leaf of F. suffruticosa is capable of producing securinine (9) (Supplementary Fig. 3)38,39. Therefore, scRNA-seq was performed on F. suffruticosa leaves to identify candidate genes in the SeA biosynthetic pathway through co-expression analysis with higher resolution.
For scRNA-seq experiments, protoplasts were isolated from 5-week-old F. suffruticosa leaves and used to generate 10X Genomics libraries in two biological replicates. Microscopic observation and viability staining confirmed that both replicates yielded high-quality protoplasts suitable for downstream analyses (Supplementary Fig. 4). The libraries were sequenced, aligned, and demultiplexed by using the assembled F. suffruticosa genome and annotation. Replicates 1 and 2 captured 6313 and 7146 cells, with median genes per cell of 1425 and 1782, and median reads per cell of 19,482 and 17,590, respectively. Mapping showed 34.9% and 45.6% of reads confidently aligned to the genome, with 32.8% and 42.9% mapped to exonic regions, respectively (Supplementary Fig. 5a). The validated cell-by-gene matrices were used for downstream analyses.
The single cells were grouped into 12 distinct cell clusters based on their gene expression levels (Fig. 2b and Supplementary Fig. 5b). By comparing known cell-type markers of leaf tissues with cluster-specific marker genes, the clusters were predicted as mesophyll, epidermis, vasculature, and guard cells15,40,41,42,43,44. The marker genes used for cell type annotation and their expression level across clusters were shown in a dot plot (Supplementary Fig. 6). Interestingly, two known SeA biosynthetic genes, FsPS and FsBBE2, showed enriched expression in cluster 7 (Fig. 2c and Supplementary Fig. 5c). In addition, genes enriched in cluster 7 were associated with processes such as L-lysine (10) biosynthesis via the diaminopimelate pathway (GO:009089), aromatic amino acid family biosynthesis (GO:0009073), and sulfate assimilation (GO:0000103) (Fig. 2c and Supplementary Fig. 7). In a hdWGCNA analysis, cluster 7 module 39 included the FsPS and genes involved in the biosynthesis of lysine and tyrosine, and this module showed a cluster 7-specific expression pattern (Supplementary Fig. 8). Therefore, we hypothesized that cluster 7 is the cell cluster responsible for SeA production in a F. suffruticosa leaf.
Identification and biosynthesis of menisdaurilide, a precursor to neosecurinane alkaloids
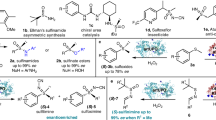
L-Tyrosine (1) and L-lysine-derived 1-piperideine (11) have been identified as biosynthetic intermediates of SeA by radioactive isotope feeding assays31,32,33,34,35 (Fig. 1c). However, the specific molecule derived from L-tyrosine (1) that forms the scaffold with 1-piperideine (11) has remained elusive33. Inspired by previous biomimetic syntheses of SeAs (Fig. 3a), we postulated menisdaurilide (5) as a potential biosynthetic intermediate. In 2008, de March et al. reported the synthesis of allosecurinine (8) employing vinylogous Mannich reaction as a key step45 (Fig. 3a). The silyl enol ether derivative of the O-TBDPS menisdaurilide (13) was allowed to react with iminium ion intermediate 14 via Diels–Alder-like transition state to furnish 15, which was further converted to allosecurinine (8) through a four-step transformation. In 2017, the Gademann group reported that compound 15 can be converted into (–)-virosine A (6) via an intramolecular aza-Michael reaction as a key step46. Recently, our group reported that lithium enolate 17 from O-TBDPS menisdaurilide (12) reacts with enone 18 to furnish vinylogous Michael adduct 19, which was further transformed into compound 21 with neosecurinane core47 (Fig. 3a).

a Chemical reactivity of menisdaurilide (5) and 1-piperideine (11) (TBDPS tert-butyldiphenylsilyl, TIPS triisopropylsilyl, Boc tert-Butyloxycarbonyl). b Possible reaction pathways regarding the formation of neosecurinane scaffold from menisdaurilide (5) and 1-piperideine (11) (TS transition state). c Reaction between menisdaurilide (5) and 1-piperideine (11) in aqueous buffer with varying pH levels (N = 4, mean ± SEM, two-sided Student’s t test, ***p < 0.001; ****p < 0.0001). d [13C2]-menisdaurilide (5) was supplied with or without 1-piperideine (11) to native (left) or boiled (right) leaf lysate (pH 8). Produced [13C2]-virosine A ([13C2]−6) and [13C2]-virosine B ([13C2]−7) were measured (N = 4, mean ± SEM. One-way ANOVA, post hoc Tukey’s HSD). Letters indicate statistical differences among treatments. Source data are provided as a Source Data file.
These synthetic precedents allowed us to envision that menisdaurilide (5) would react with 1-piperideine (11) to generate neosecurinane scaffold (Fig. 3b). We hypothesized that menisdaurilide (5) and 1-piperideine (11) would react to form four different transition states (Fig. 3b, TS-A to D) and produce (–)-virosine A (6), (–)-virosine B (7), (+)-episecurinol A (23), and (+)-securinol A (24), respectively. Although previous works employed highly activated derivatives of menisdaurilide (13, 17) and 1-piperideine (14, 18), we exposed a mixture of these biosynthetically relevant fragments in a buffer at physiologically reasonable pH levels. Surprisingly, the formation of (–)-virosine A (6), (–)-virosine B (7), and (+)-securinol A (24) was observed when the mixture of menisdaurilide (5) and 1-piperideine (11) was treated with phosphate buffer at pH 7 or 8 (Fig. 3c). Importantly, the production of these monomeric SeAs was more efficient under a more basic pH. Intrigued by these observations, we conducted a feeding experiment of [13C2]-menisdaurilide ([13C2]-5) to the leaf lysate of F. suffruticosa (pH 8) to further verify its biosynthetic plausibility (Fig. 3d). Consistent with the observed chemical reactivity, [13C2]-menisdaurilide ([13C2]-5) was incorporated into (–)-virosine A (6) and (–)-virosine B (7), regardless of the prior thermal denaturation of the leaf lysate, suggesting that this vinylogous Mannich reaction can occur in the absence of enzymes. Hence, we presumed that menisdaurilide (5) is the L-tyrosine (1)-derived metabolite that can react with 1-piperideine (11) to generate the neosecurinane scaffold. However, this does not, by any means, undermine the possibility of the existence of enzymes that mediate this biosynthetic event.
We further analyzed the distribution of menisdaurilide (5), 1-piperideine (11), and SeAs across the leaf, stem, and root of 5-week-old F. suffruticosa. Menisdaurilide (5), 1-piperideine (11), (–)-virosine A (6), (–)-virosine B (7), allosecurinine (8), and securinine (9) were detected in all three tissue types (Supplementary Fig. 9). Additionally, (+)-securinol (24) was also identified in all tissues at levels comparable to those of (–)-virosine A/B (6, 7), whereas (+)-episecurinol (23) was not detected within our detection limit. These observations are consistent with the results of the in vitro assay, in which mixing menisdaurilide (5) and 1-piperideine (11) in an aqueous buffer did not yield detectable levels of (+)-episecurinol (23). Furthermore, the existence of menisdaurilide (5) in other SeA-producing plants belonging to the Flueggea and Phyllanthus genera48,49, further suggests its role as a common biosynthetic precursor derived from L-tyrosine (1). Notably, a wide range of neonorsecurinane alkaloids containing a pyrrolidine A ring have been isolated from Phyllanthus genera. By analogy with our proposed mechanism for neosecurinane formation, we speculate that a reaction employing 1-pyrroline instead of 1-piperideine (11) could, in principle, give rise to neonorsecurinane scaffolds.
Encouraged by the successful coupling of menisdaurilide (5) with 1-piperideine (11), we focused on identifying the penultimate biosynthetic precursor of menisdaurilide and the enzyme responsible for its formation. Menisdaurilide (5) is a bicyclic molecule composed of a cyclohexenol (C ring) and a butenolide (D ring) (Fig. 4a). From a retrobiosynthetic perspective, the C ring of menisdaurilide (5) likely originates from the phenol moiety of L-tyrosine (1), and the D ring is presumed to be formed via an intramolecular oxa-Michael reaction50. This hypothesis led us to propose that the allylic alcohol moiety on the C ring is formed by reducing the enone group after the D-ring is generated. Accordingly, we came up with two plausible penultimate precursors of menisdaurilide (5): a ketone candidate (3) requiring reduction and a diol candidate (4) requiring dehydration to be converted into the target molecule, menisdaurilide (5) (Fig. 4a).

a Retrobiosynthetic perspective of the menisdaurilide (5) biosynthesis. Ketone intermediate (3) and diol intermediate (4) are shown. b Lysate feeding assay with cofactor supplements. [13C2]-4 was supplemented with ATP or PAPS, while [13C2]-3 was supplemented with reductants. PAPS 3’-phosphoadenosine-5’-phosphosulfate. c [13C2]-premenisdaurilide ([13C2]-3) was converted to [13C2]-menisdaurilide ([13C2]-5) in crude lysate reductant dependently. [13C2]-menisdaurilide ([13C2]-5) peak area was measured (N = 4, mean ± SEM. One-way ANOVA, post hoc Tukey’s HSD. Letters indicate statistical difference among treatments). d Functional characterization of menisdaurilide synthase (FsMS) with other ketoreductase candidates in N. benthamiana. The HPLC-MS/MS chromatograms of menisdaurilide (5) are shown, and the peak area was measured 24 h post-premenisdaurilide (3) supplementation (N = 6, except for the g21280 where N = 5; mean ± SEM. One-way ANOVA, post hoc Tukey’s HSD). Letters indicate statistical differences among agroinfiltrated genes. FsMS Flueggea suffruticosa menisdaurilide synthase, EV empty vector (negative control). Source data are provided as a Source Data file.
[13C2]-labeled putative precursors were synthesized (Supplementary Fig. 29) and incubated with crude leaf lysates of F. suffruticosa in the presence of appropriate cofactors for each reaction. ATP and PAPS were selected as initial candidates because phosphorylation and sulfation are major biochemical transformations that can convert a hydroxyl group into a better leaving group. Notably, incubation of the [13C2]-ketone candidate ([13C2]-3) in the presence of NAD(P)H resulted in the formation of [13C2]-menisdaurilide ([13C2]-5) (Fig. 4b, c). In contrast, no [13C2]-menisdaurilide ([13C2]-5) was observed from the [13C2]-diol ([13C2]-4) candidate. Thus, we identified the ketone candidate, named premenisdaurilide (3), as the biosynthetic precursor of menisdaurilide (5) and proposed that a NAD(P)H-dependent ketoreductase is responsible for this biotransformation. It is noteworthy that premenisdaurilide (3) is unstable in water, which likely accounts for its absence in plant extracts (Supplementary Fig. 10). Markedly, the premenisdaurilide (3) has been frequently employed as a penultimate precursor to menisdaurilide in chemical syntheses51,52,53 (Supplementary Fig. 11).
With the precursor identified and the enzyme’s functional ontology defined, we focused on searching for the candidate ketoreductase on cluster 7, which exhibits enriched expression of genes associated with monomeric SeA biosynthesis (Fig. 2d and Supplementary Fig. 5d). Ten ketoreductases that showed enriched expression in cluster 7 were selected as candidate genes (Supplementary Fig. 12). Upon transient expression of these candidates in Nicotiana benthamiana leaves followed by feeding with premenisdaurilide (3), a significant increase in the menisdaurilide (5, 3.02 min) production was observed in N. benthamiana expressing g07207 (Fig. 4d). By contrast, transient expression of g16283 in N. benthamiana led to a modest increase in menisdaurilide (5) accumulation, significantly lower than that observed for g07207 (Supplementary Fig. 12a). Furthermore, g16283 showed weaker co-expression with FsPS and FsBBE2 than g07207 and was not assigned to the cluster 7-specific module (cluster 7 module 39), whereas g07207 was a member of this module (Supplementary Figs. 8d and 12b). Taken together, these findings indicate that g07207 plays the major role in menisdaurilide biosynthesis in F. suffruticosa. We thus annotated g07207 as a menisdaurilide synthase (FsMS).
After identifying the gene, we purified the corresponding protein expressed in E. coli, and performed an in vitro assay. We found that NADPH is markedly superior to NADH as a cofactor, regardless of pH, suggesting that NADPH is indeed the true cofactor for FsMS (Supplementary Fig. 13). We believe that NADH in the crude lysate facilitated the reaction indirectly, possibly through the reduction of NADP+ to NADPH. An additional peak with the same fragmentation pattern at 3.35 min is postulated to correspond to aquilegiolide (22), which is known to rapidly establish an equilibrium with menisdaurilide (5) in various solvents54.
Neosecurinanes are converted to securinanes via sulfotransferase-mediated 1,2-amine shift
In 2017, Gademann et al. reported a chemical transformation converting the neosecurinane scaffold into the securinane scaffold46 (Fig. 5a). This transformation involves chemical activation (mesylation) of the anti-periplanar hydroxyl group on the neosecurinane scaffold, which results in the formation of an aziridinium ion intermediate (27) via an intramolecular SN2 reaction, followed by an elimination reaction to yield the securinane scaffold. Based on this observed chemical reactivity, they proposed that neosecurinane alkaloids are biosynthetic precursors of securinane alkaloids. Shortly afterward, Peixoto et al. reported a general 1,2-amine shift from neo(nor)securinane scaffold into (nor)securinane scaffold using Mitsunobu’s alcohol-activating conditions55.

a Chemical activation of the hydroxyl group on the neosecurinane scaffold triggers its remodeling into the securinane scaffold. b Chemical mimicry of acyltransferase and sulfotransferase activity. c [13C2]-(–)-virosine B ([13C2]−7) was supplemented with either PAPS or ATP in pH 8.0 F. suffruticosa seedling lysates (N = 3, mean ± SEM. One-way ANOVA, post hoc Tukey’s HSD). Leaf disc feeding assay of (–)-virosine A (6) (d) and (–)-virosine B (7) (e) (N = 3, mean ± SEM. One-way ANOVA, post hoc Tukey’s HSD). f–k Functional validation of FsNSST1/2. HPLC-MS/MS chromatograms of in vitro enzyme assay products (f, i). Allosecurinine (8) or securinine (9) was observed only when PAPS was supplemented (g, j). Candidate sulfotransferases were transiently expressed in N. benthamiana, and either (–)-virosine A (6) (h) or (–)-virosine B (7) (k) was supplemented (N = 6, mean ± SEM. One-way ANOVA, post hoc Tukey’s HSD). Letters indicate statistical differences among treatments. Empty vector (EV) or eGFP was used as a negative control. Source data are provided as a Source Data file.
While these chemical syntheses provided insights regarding the scaffold remodeling, there was no direct evidence that this conversion occurs in SeA-producing tissues of F. suffruticosa. Moreover, the mode of hydroxyl group activation in neosecurinane alkaloids had remained unclear, as the leaving group is eliminated from the molecule during the transformation. To solve this enigma, we first chemically converted the hydroxyl group of (–)-virosine B (7) into plausible biological leaving groups: the acetyl (28) and the sulfate (29) groups (Fig. 5b). O-Acetylvirosine B (28) was isolated from the reaction mixture in 92% yield, implying that a stronger activating group might be required to facilitate the desired transformation. However, treatment of the hydrochloride salt of (–)-virosine B (7) with SO3•pyridine, followed by incubation in phosphate buffer (pH 8), resulted in the formation of securinine (9) in 64% and 90% yield after 10 min and 6 h, respectively (Fig. 5b and Supplementary Information, Section 3.5.2). The reaction between the hydrochloride salt of (–)-virosine B (7•HCl) and SO3•pyridine would furnish the O-sulfated intermediate 29 and pyridinium hydrochloride, maintaining an acidic reaction environment. The ammonium moiety of 29 would be deprotonated only upon exposure to pH 8 buffer, enabling a 1,2-amine shift. This observation suggests that the 1,2-amine shift may proceed spontaneously, without enzymatic assistance.
Building on these results, we conducted a feeding experiment using [13C2]-(–)-virosine B ([13C2]-7) to the crude F. suffruticosa seedling lysate supplemented with either ATP or PAPS (3’-phosphoadenosine-5’-phosphosulfate). This experiment compared sulfation and phosphorylation in planta, due to the difficulty associated with chemical phosphorylation in mild conditions (Fig. 5c). We observed a significantly higher conversion of [13C2]-(–)-virosine B ([13C2]-7) into [13C2]-securinine ([13C2]-9) with the addition of PAPS to the lysate, compared to ATP-supplemented lysate and control groups. In addition, we supplemented [13C2]-(–)-virosine A ([13C2]−6) and virosine B ([13C2]−7) into F. suffruticosa leaf discs and confirmed that allosecurinine (8) and securinine (9) were specifically derived from (–)-virosine A (6) and (–)-virosine B (7), respectively, in F. suffruticosa leaves (Fig. 5d, e). These observations confirmed that neosecurinane alkaloids are biosynthetic precursors of securinane alkaloids and suggested that this biosynthetic transformation requires sulfation.
To identify the sulfotransferase responsible for securinane biosynthesis in F. suffruticosa, we selected candidate genes annotated as sulfotransferases and enriched in cluster 7. Three genes were used as promising baits for co-expression analysis: FsPS, FsMS, and FsBBE2. Two sulfotransferases originally annotated as flavonol-4-sulfotransferases strongly correlated with FsPS and showed high expression levels in cluster 7 at the single cell level (Fig. 2c, d). Three additional sulfotransferases highly expressed in leaves, g00640, g21405, and g01123, were also included in the candidates. In vitro enzyme assay revealed that two cluster 7-specific sulfotransferases produce allosecurinine (8) and securinine (9) from (–)-virosine A (6) and (–)-virosine B (7), respectively (Fig. 5f, g, i, j and Supplementary Fig. 14). The catalytic activity was PAPS-dependent in vitro (Fig. 5g, j and Supplementary Fig. 15a, b). The in planta enzyme assays in N. benthamiana reproduced the in vitro product profiles (Fig. 5h, k and Supplementary Fig. 15c, d). A difference in enzymatic activity between in vitro and in planta assays was observed, which likely reflects variation in transient expression among infiltrated leaves, differential availability of endogenous PAPS in expressing cells, distinct biochemical environments between the two systems, and possible downstream metabolism of the products in N. benthamiana. Thus, we named the two genes having a PAPS-dependent securinane biosynthetic activity FsNSST1 and FsNSST2 (Neosecurinane sulfotransferase 1 and 2). Other candidate sulfotransferases did not show catalytic activity except g21405, which showed weak activity on (–)-virosine B (7).
Interestingly, FsNSST1/2 can convert menisdaurilide (5) into its O-sulfated derivative (Supplementary Fig. 16), supporting the sulfating reactivity of FsNSST1/2 and the involvement of O-sulfated neosecurinane alkaloids as intermediates in the 1,2-amine shift. Attempts to detect the sulfated intermediate 29 were unsuccessful, presumably due to its chemical instability. We also found that higher pH conditions lead to a more rapid 1,2-amine shift, whereas the presence of FsNSST1/2 does not facilitate the production of securinine (9) from the sulfated intermediate 29 in vitro (Supplementary Information, Section 3.5.3). These results suggest that FsNSST1/2 would function as canonical sulfotransferases and are unlikely to further accelerate the 1,2-amine shift of sulfated intermediates.
FsNSST1 and FsNSST2 are homologous sulfotransferases encoded within a sulfotransferase-rich locus of approximately 200 kb on Contig_00010 (Supplementary Fig. 17). This locus contains 21 genes in total, 19 of which are annotated as sulfotransferases, and FsNSST1 and FsNSST2 are separated by about 35 kb within this array. FsNSST1 and FsNSST2 share 53% amino acid sequence identity, and g21405 shares 51% and 60% identity with FsNSST1 and FsNSST2, respectively, placing these three enzymes in the same sulfotransferase clade (Supplementary Fig. 18a, b). AlphaFold-based structural models show that FsNSST1, FsNSST2, and g21405 share a similar overall fold and conserved PAPS-binding motifs but minor differences at residues predicted to contact the substrate (Supplementary Fig. 18c, d). Notably, these differences include a phenylalanine-to-methionine substitution in g21405 at a predicted substrate-contacting position, which may contribute to its markedly reduced activity.
Discussion
SeAs and their precursor were isolated across leaves, stems, and roots of F. suffruticosa (Supplementary Fig. 9). Such tissue-wide distribution complicates the discovery of candidate biosynthetic genes via traditional bulk transcriptomics and does not, on its own, distinguish sites of biosynthesis from potential transport and sequestration. However, scRNA-seq only requires any active biosynthetic tissues for proposing biosynthetic candidate genes, enabling the identification of distinct cell types within the tissue. Through scRNA-seq analysis of the leaf, we discovered a specialized cell cluster that showed enriched expression of two known SeA biosynthetic genes. Cluster 7 was predicted to correspond to a vasculature-associated cell type based on known marker genes (Fig. 2c and Supplementary Figs. 5c, 6). Within these cells, we found that genes involved in producing L-tyrosine (1) and L-lysine (10), the two starting molecules of SeA biosynthesis, were actively expressed. We combined a simple Spearman correlation-based ranking using FsPS as bait with a higher-order co-expression analysis using hdWGCNA. This approach revealed a cluster 7-specific module (module 39) comprising 73 genes (0.2% of total protein-coding genes) that showed a distinct cluster 7 expression pattern and encompassed all SeA biosynthetic genes characterized in this study, while excluding negatively tested ketoreductase candidates (Supplementary Fig. 8). Together, these analyses indicate that integrating simple correlation with hdWGCNA provides a robust and biologically coherent framework for delineating the core SeA biosynthetic pathway and its potential regulators within the vasculature-associated SeA-producing cell population.
Batch effects between replicates were evaluated by performing Seurat-based integration after initial replicate-wise analyses. In the integrated dataset, the SeA-associated cluster (cluster 9) was preserved and showed robust, consistent signatures across replicates (Supplementary Fig. 19). All core SeA biosynthetic genes characterized in this study (FsPS and FsBBE2) remained specifically enriched in cluster 9 and retained strong co-expression relationships, indicating that their coordinated expression is not driven by replicate-specific technical variation (Supplementary Fig. 19). Notably, SUC2, a canonical vasculature marker, emerged as a significant cluster 9 marker only after integration, further reinforcing the assignment of the SeA-producing population to vasculature-associated cells. In addition, the integrated UMAP topology, co-expression patterns, and dot plots for genes of interest and cell-type markers were highly concordant between replicate 1 and replicate 2 when examined with a focus on the SeA biosynthesis cluster.
Deciphering the transcriptome at the cell type level provides valuable insights into alkaloid biosynthesis in plants (Fig. 6). In qPCR analysis, monomeric SeA biosynthetic genes FsPS, FsMS, FsNSST1, and FsNSST2 showed no significant differences in expression across tissues (Supplementary Figs. 1, 20), suggesting that tissue-specific bulk RNA-seq would provide limited insight for identifying these genes. This limitation was successfully overcome by scRNA-seq analysis. ATP sulfurylase (ATPS) and adenylyl-sulfate kinase (APSK) are enzymes producing PAPS, which is a cofactor of sulfotransferase, from ATP. While two ATPSs and two APSKs are present in the F. suffruticosa genome, only FsATPS2 and FsAPSK2 were specifically expressed in cluster 7, suggesting their potential specialized roles in SeA biosynthesis. In addition, further modification of allosecurinine (8) may occur in cluster 7 and cluster 4 (Fig. 2c and Supplementary Fig. 5c), as the key downstream enzyme FsBBE236, which is the first enzyme to direct the flux from allosecurinine (8) to various SeAs with elevated oxidation levels, was also enriched in these clusters. This suggests the existence of additional SeA biosynthesis-associated cell clusters and potential mechanisms that mediate the intercellular movement of SeAs or their intermediates. One plausible scenario is that SeAs or reactive intermediates produced in vasculature-associated cells are redistributed via vascular transport and subsequently sequestered in sink tissues. Transporters implicated in specialized metabolite movement remain plausible components, although their expression may be distributed across multiple cell types and thus not emerge as cluster-specific markers. Notably, the enrichment of catalases and peroxidases in this cluster implies that detoxification of H2O2 produced during 1-piperideine (11) biosynthesis and sulfur assimilation may be activated (Fig. 6). Interestingly, genes related to jasmonate (JA) biosynthesis and signaling were also enriched in cluster 7 (Supplementary Fig. 7), consistent with previous reports that FsBBE2 was induced by JA36. This suggests that the SeA biosynthesis might be regulated by JA signaling in cluster 7. This single-cell transcriptome of F. suffruticosa could facilitate the identification of additional SeA-associated genes, including transporters, downstream biosynthetic enzymes, and regulatory transcription factors involved in enriched primary metabolism or in mechanisms that support recovery from amino acid depletion, as well as additional SeA-associated cell clusters.

Single-cell transcriptomic analysis identified cluster 7 as the SeA biosynthetic cell type. This cluster showed enrichment not only of core SeA biosynthetic genes (green boxes and arrows) but also of auxiliary genes (purple boxes and arrows) expected to facilitate SeA biosynthesis.
Although the overall and exonic alignment rates in our 10x Genomics scRNA-seq datasets are lower than ideal for plant single-cell studies, multiple lines of evidence indicate they remain sufficient for the objectives of this work. Mapping of published bulk RNA-seq data to the same genome and annotation yielded 90% of alignment rate, and BUSCO and gene model statistics support annotation adequacy (Fig. 2a). Reprocessing with STARsolo following a recent plant single-cell study led to a moderate increase in mapping rates (Supplementary Table 5). As 94% of genome-aligned reads are exonic, the main limitation lies in genome alignment rather than exon assignment. Guided by recent plant sc/snRNA-seq QC recommendations56, we therefore quantified rRNA/organelle contributions and found substantial contamination of ~18–21% across the two 10x replicates (Supplementary Table 6), indicating that rRNA/plastid carryover resulted in reduced alignment. Here, we established F. suffruticosa as a molecular and single-cell system, although protoplast isolation protocols are still being optimized. Protoplast isolation was challenging due to the hydrophobic outer leaf layer, cell viability tended to decline, and FACS enrichment was not performed. These factors likely increased organelle leakage and ambient RNA. Nevertheless, our alignment metrics are comparable to those in a previous plant scRNA-seq study that successfully identified benzyl acetone biosynthetic genes (Supplementary Table 4). The SeA-producing cell population (cluster 7) was consistent across the replicates and enriched for vasculature markers and SeA pathway genes, and protein-coding gene coverage after filtering retains 60%. Independent PIP-seq data from the same developmental stage revealed an improved alignment rate of 77%. Analysis of the PIP-seq data reproduced the clustering and gene expression patterns observed in 10x (Supplementary Fig. 21). This suggests that the impact of low alignment on our primary objective of identifying the SeA-associated cell population and prioritizing pathway genes is likely to be limited.
Menisdaurilide (5) has been isolated from multiple plant genera48,49,54,57,58,59,60,61, and its glucoside (phyllanthurinolactone) acts as the leaf-closing factor of Phyllanthus urinaria62,63. Despite its abundance and ecological significance, its biosynthetic pathway remains completely unknown. In this study, we found that menisdaurilide (5) is produced from premenisdaurilide (3), and we identified a reductase, FsMS, responsible for this conversion. FsMS showed a strong co-expression with FsPS and FsBBE2, further supporting the role of menisdaurilide (5) as a precursor of SeA. We observed spontaneous formation of the neosecurinane scaffold from menisdaurilide (5) and 1-piperideine (11) under conditions of pH greater than 7. This suggests two possible scenarios: either the reaction occurs within subcellular compartments with a basic pH (e.g., mitochondrial matrix, peroxisome, and chloroplast stroma), or a specialized scaffold-forming protein provides a suitable microenvironment that facilitates the reaction. Further investigation is required to elucidate the subcellular localizations of the biosynthetic enzymes and to identify potential scaffold-forming proteins.
The intermediates linking L-tyrosine (1) to premenisdaurilide (3) remain unidentified. Following Parry33 and Spenser’s hypothesis35, which proposed 4-hydroxyphenylpyruvic acid (4HPP) (2) as a putative precursor from L-tyrosine (1), we chemically synthesized [13C2]-4HPP ([13C2]-2, Supplementary Fig. 30) and fed it to the leaf of F. suffruticosa, but we were unable to detect any significant increase of [13C2]-allosecurinine ([13C2]-8), securinine ([13C2]-9), and menisdaurilide ([13C2]-5) compared to the control (Supplementary Fig. 22). Furthermore, orthologs of the L-tyrosine aminotransferase, which converts L-tyrosine (1) into 4HPP (2), did not show meaningful expression correlation with SeA biosynthetic genes in our single-cell transcriptomics (Fig. 2 and Supplementary Fig. 23). These results imply the involvement of an alternative biosynthetic scenario that converts L-tyrosine (1) to premenisdaurilide (3) and, eventually, menisdaurilide (5). We propose that the elucidation of the biosynthetic pathway from L-tyrosine (1) to premenisdaurilide (3) be pursued through an integrated approach combining comparative genomics and chemically guided single-cell transcriptomics.
Sulfotransferase is a ubiquitous enzyme class that transfers sulfate groups to the hydroxyl groups of various substrates, using PAPS as a biological sulfate donor64,65. While numerous sulfotransferases have been discovered across different organisms, plant sulfotransferases are primarily known for enhancing solubility or facilitating the catabolism of metabolites65,66,67. Although sulfotransferases have been occasionally reported as tailoring enzymes in the biosynthesis of plant specialized metabolites68,69,70, their involvement in biosynthetic pathways associated with skeletal arrangement remains largely unexplored71. Scaffold formation or remodeling via alcohol activation is a well-documented mechanism in complex natural product biosynthesis. However, compared to phosphorylation72,73, acetylation6, and malonylation11, which are commonly observed in biosynthetic modifications, the role of sulfation in scaffold remodeling has not been previously described. In this study, we identified two sulfotransferases that catalyze the sulfation of the [2.2.2]-bicyclic neosecurinane scaffold, thereby initiating its conversion into the [3.2.1]-bicyclic securinane scaffold. In our in vitro assays, the sulfation-dependent 1,2-amine shift proceeds more efficiently under basic conditions, suggesting that scaffold remodeling in planta may be facilitated by locally alkaline subcellular compartments. These findings expand the known roles of sulfotransferases by linking canonical sulfation to a subsequent 1,2-amine shift-driven alkaloid scaffold remodeling pathway.
The scaffold remodeling step from neo(nor)securinane to (nor)securinane represents a crucial point of structural diversification in SeA biosynthesis. Although belonging to the same genus, Flueggea virosa is rich in norsecurinine and its oligomers, which are absent in F. suffruticosa74,75,76. In addition, F. virosa contains virosecurinine and viroallosecurinine, the enantiomers of securinine (9) and allosecurinine (8), respectively77. A wide range of securinane-type alkaloids has been discovered in other genera of the Phyllanthaceae family, Phyllanthus78, Margaritaria79, and Breynia80 species. These observations suggest that the emergence of a sulfotransferase capable of converting neosecurinane to securinane played a pivotal role in driving the chemical diversification of securinega alkaloids. In particular, understanding why F. suffruticosa maintains two NSST paralogs within a sulfotransferase-rich genomic array (Supplementary Fig. 17), and how this array has evolved will be an important topic for future investigation. One possibility is that partial functional redundancy between FsNSST1 and FsNSST2 increases the robustness of SeA production under variable developmental or environmental conditions. Exploring the evolutionary trajectory of these sulfotransferases across Phyllanthaceae species and correlating their presence with metabolomic profiles offers promising avenues for future research. Such studies may illuminate the evolutionary adaptation of sulfotransferases in alkaloid biosynthesis and their contribution to the metabolic diversity observed within this plant family.
The biosynthetic pathway of monomeric SeA in F. suffruticosa was revealed by combining biomimetic synthesis and scRNA-seq. Guided by a biosynthetic hypothesis, putative biosynthetic intermediates labeled with stable isotopes were synthesized, and their innate biochemical reactivity was examined. Single-cell transcriptomic analysis revealed a specific cell type with highly enriched expression of the only two previously known SeA biosynthetic genes. Based on these findings, premenisdaurilide and menisdaurilide were identified as L-tyrosine-derived SeA intermediates, and the enzyme responsible for menisdaurilide biosynthesis (FsMS) was characterized. In addition, two sulfotransferases (FsNSST1 and FsNSST2) were found to catalyze the sulfation of the neosecurinane ([2.2.2]-bicyclic) scaffold, after which the resulting intermediates undergo a sulfation-dependent 1,2-amine shift to yield the securinane ([3.2.1]-bicyclic) scaffold. This highlights a specialized function of sulfotransferases, a universally distributed enzyme class across all domains of life, in scaffold remodeling of plant alkaloids.
Methods
Analysis of securinega alkaloids (SeAs), menisdaurilide, and 1-piperideine
Securinanes, neosecurinanes, and their intermediate menisdaurilide (5) were analyzed using an HPLC-MS/MS (LCMS-8050, Shimadzu, Japan). Leaf, stem, and root tissue from 5-week-old F. suffruticosa seedlings were ground in liquid nitrogen, and 100 mg of ground materials were extracted with 1 mL of extraction solution (0.1% formic acid aqueous solution). Extracts were filtered and diluted 100-fold in methanol. Samples were injected into the Acquity UPLC® BEH C18 column (1.7 µm, 2.1 × 100 mm; Waters, Milford, MA, USA). The solvents were (A) 20 mM ammonium acetate (aq.) and (B) methanol. The LC-time program was as follows ((B) concentration in %): 0–3 min: 5–20%, 3–15 min: 20–40%, 15–17 min: 40–85%, 17–17.5 min: 85–95%, 17.5–20.5 min: 95%, 20.5–21 min: 5%, 21–24 min: 5%. The flow rate was 0.3 mL/min, the injection volume was 1 µL, and the column oven was set at 40 °C. Retention times, precursor ions, and product ions of target metabolites (including 13C-labeled and unlabeled) were compared with synthesized authentic compounds. Subsequent mass spectrometry was performed in the positive-ion mode via ESI (interface voltage, 3 kV; interface temperature, 300 °C; DL temperature, 250 °C; heat block temperature, 400 °C; nebulizing gas, 3 L/min; drying gas, 10 L/min; heating gas, 10 L/min). Multiple reaction monitoring (MRM) was used to detect allosecurinine (8), securinine (9), (–)-virosine A (6), (–)-virosine B (7), menisdaurilide (5), and menisdaurilide O-sulfate (S37) are listed in Supporting Information (Supplementary Table 1).
To analyze 1-piperideine (11), samples were injected into the XBridgeⓇ Amide Column (3.5 µm, 2.1 × 150 mm) (Waters). The mobile phase and LC-time program were used as the ref. 81. For the relative quantification, the 1-piperideine dimer peak was used for the peak integration.
To assess the chemical stability of premenisdaurilide (3) in aqueous solution, 1 mg of premenisdaurilide (3) was dissolved in 1 mL of water and diluted at different time points. Diluted samples were immediately injected into the XBridgeⓇ Amide Column (3.5 µm, 2.1 × 150 mm) (Waters). The solvents were (A) 10 mM ammonium hydroxide (aq.) and (B) acetonitrile. The LC-time program was as follows ((B) concentration in %): 0–5 min: 98−60%, 5–6 min: 60–10%, 6−7 min: 10–5%, 7–10 min: 5%, 10–11 min: 5–98%, 11–14 min: 98%. The flow rate was 0.3 mL/min. Injection volume and column oven temperature were identical to those above. ESI negative-ion mode was used. MRM transitions and MS parameters are listed in Supplementary Table 1.
Plant materials
F. suffruticosa leaves were collected from a natural population at Bulgok-san Mountain, Gyeonggi-do, Republic of Korea (37°21'20.5“N 127°07'40.7“E) for genomic DNA isolation and sequencing. For other biochemical assays, commercially available F. suffruticosa seeds were obtained from a local vendor (Cheonnyangssiat, Hwacheon-gun, Republic of Korea) and germinated in the laboratory. The seeds were sterilized using 2% (w/v) sodium dichloroisocyanurate dihydrate (Sigma-Aldrich) and 0.1% (v/v) Tween 20 (Glentham), and sown in moist soil. The seeds were incubated under a 16 h dim light /8 h dark cycle. After germination, the plants were grown under 16 h light and 8 h dark conditions in a plant growth room. The growth condition was 26 ± 2 °C with 60% relative humidity (RH). Nicotiana benthamiana plants were also grown in the plant growth room under the same conditions: 16 h–8 h long day cycle, 26 °C, and 60% RH. Three to four-week-old plants were used for Agrobacterium infiltration in transient expression experiments.
Genomic DNA and total RNA sequencing of F. suffruticosa
Genomic DNA from F. suffruticosa leaves (2n = 26)82 was extracted following the cetyltrimethyl ammonium bromide (CTAB) method by Macrogen (Seoul, Republic of Korea)83. To estimate the genome size of F. suffruticosa, the extracted DNA was sequenced using the TruSeq Nano DNA (350) library kit and the HiSeqXten platform. Additionally, the genomic DNA was sequenced for genome assembly using the Revio platform from Pacific Biosciences (PacBio, Menlo Park, CA, USA) with the PacBio HiFi Library Kit 3.0. For gene annotation, total RNA was extracted from seven leaves, three stems, and five roots of F. suffruticosa using the RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) in combination with the RNAse-free DNase I (Qiagen). Each extracted RNA was prepared using the TruSeq Stranded Total RNA with Ribo-Zero Plant Kit and sequenced on the NovaSeq6000 platform.
Genome assembly and annotation of F. suffruticosa
Python (v3.9.10), Anaconda3 (v22.9.0), and R (v4.3.3) were used for the subsequent analysis. Genome assembly and annotation generally followed the pipeline outlined in our previous reports84,85. The de novo genome assembly of F. suffruticosa was generated using the Hifiasm genome assembler (v0.18.5-r499) with default options86. The quality of the genome assembly was evaluated by using the benchmarking universal single-copy ortholog (BUSCO, v5.2.2)87 analysis on the eudicots_odb10 database and assembly-stats (v1.0.1). The primary genome assembly was used for further analysis based on the quality assessment.
The repetitive elements of the genome assembly were annotated by using RepeatModeler (v2.0.3) with NINJA (NINJA-0.95-cluster_only) database and RepeatMasker (v4.1.2)88,89. The specific repeat library for F. suffruticosa and the predefined eudicot repeat library were used to annotate repetitive elements with default options. The plant canonical telomeric repeat sequence “TTTAGGG” and its synonymous forms were identified using a “grep” command in the masked repetitive elements88,89.
To annotate protein-coding genes, 15 bulk RNA sequencing reads were aligned to the soft-masked genome assembly with hisat2 (v2.2.1) with –no-unal –dta --max-introlen 5000 and other default options90. The protein-coding genes of the F. suffruticosa genome were annotated by using the BRAKER pipeline (v2.1.6 using Genemark-ES/ET/EP v.4.61_lic). The Viridiplantae protein database of OrthoDB (v11)91,92,93,94,95,96,97,98,99,100 and the 11 bulk RNA-seq alignment BAM files were used in the BRAKER pipeline to generate hints to train Augustus. The annotation of protein-coding genes was evaluated by using BUSCO (v5.2.2) analysis on the eudicots_odb10 database and assembly-stats (v1.0.1)87. The protein-coding genes were functionally annotated by using MMseqs2 (v13.4511) against the UniProt Knowledgebase (release 2023.01) and the NCBI NR database101,102. The Gene Ontology (GO) terms and Enzyme Commission (EC) numbers were assigned through UniProt API based on the functional annotation results.
D₂O labeling assay
For the D₂O labeling assay, leaves from 5-week-old F. suffruticosa plants were carefully excised and placed in individual 10 mL vials containing either 2 mL of H₂O (control) or 99% D₂O, following the protocol38,39. The vials were incubated for 6 days in a growth room under a 16 h light/8 h dark photoperiod. After the incubation period, 8 mm (diameter) leaf discs were collected from each leaf using a leaf borer and immediately frozen in liquid nitrogen. The frozen samples were ground using a TissueLyser II (Qiagen) at 27 Hz for 2 min. The ground tissue was extracted with 400 µL of 0.1% formic acid by vortexing for 1 min, followed by sonication at 50% amplitude for 5 min. The extracts were centrifuged at 18,000 × g for 5 min, filtered through a 0.22 µm PTFE filter, and diluted 20-fold in methanol before being subjected to LC-MS analysis.
The LC-MS analysis was conducted using the same LC time program previously optimized for the separation of SeAs, with the mass spectra scanned to identify isotope ratios. The isotope ratios were statistically analyzed to assess the incorporation of D₂O into allosecurinine (8) and securinine (9).
Protoplast isolation in F. suffruticosa leaves
The protoplasts were isolated from leaves of 5-week-old F. suffruticosa plants. Young leaves (the 10th to 12th from the base of the main stem) were used for each replicate in scRNA-seq. Seven leaves per plant were diagonally incised across the primary vein using a scalpel. The cut leaves were treated under vacuum with 25 mL of enzyme solution containing 1% (v/v) Viscozyme L, 0.5% (v/v) Celluclast, 0.5% (v/v) Pectinex (all from Novozymes, Bagsværd, Denmark), 9% (w/v) mannitol, 5 mM 2-(N-morpholino) ethanesulfonic acid (MES), 1 mM KNO3, 1 mM MgSO4, 0.2 mM KH2PO4, 10 μM CaCl2, 0.1 μM KI, and 0.1 μM CuSO4 (all from Duchefa Biochemie, Haarlem, the Netherlands). The vacuum treatment and subsequent release were performed for 5 and 15 min, respectively. Leaves were then incubated in the dark for 1 h at 25 °C on an orbital shaker (25 rpm). The resulting protoplast solution was filtered through a 40 μm cell strainer (Corning Inc., Corning, NY, USA) to remove debris. The protoplasts were gently collected by centrifugation at 100 × g for 5 min with low brake in pre-chilled 9% (w/v) mannitol solution. The resulting protoplasts were then filtered again using a 40 μm tip strainer (Bel-Art SP Scientificware, Wayne, NJ, USA), right before loading into nanoliter-scale Gel Beads-in-Emulsion (GEMs).
To validate the viability and integrity of enzymatically isolated protoplasts, fluorescein diacetate (FDA; Thermo Fisher Scientific, MA, USA) staining was performed. FDA was dissolved in acetone at a concentration of 5 mg/mL to prepare the stock solution. For the FDA working solution, 4 μL of the stock solution was added to 1 mL of 9% (w/v) mannitol solution. A total of 100 μL FDA working solution was added to 1 mL of the protoplast suspension and gently mixed. The stained protoplasts were immediately observed under a fluorescent microscope (M205FA, Leica microsystems, Wetzlar, Germany) with the optical filter set Leica 10447408 (Excitation, 450–490 nm; emission, 500–550 nm).
Generation and analysis of single-cell transcriptome data from F. suffruticosa leaves
To generate single-cell transcriptome data from F. suffruticosa leaves, protoplasts were isolated and validated as described above. Two biological replicates of the protoplast suspensions were used for scRNA-seq library preparation. Libraries were prepared by using the 10X Genomics Chromium single-cell microfluidics device and the Chromium single-cell 3’ RNA library v4 kit (10X Genomics, Pleasanton, CA, USA) according to the manufacturer’s protocol. The prepared libraries were sequenced on a NovaSeq 6000 platform (Illumina, San Diego, CA, USA) at Macrogen to generate paired-end NGS reads. Raw sequencing data were processed using the Cell Ranger pipeline (v8.0.1, 10X Genomics) for demultiplexing, alignment to the F. suffruticosa reference genome, and generation of gene expression matrices with mkref and count mode with default options. These matrices were subsequently used for downstream analyses, including cell clustering, differential gene expression, and functional annotation.
The cell-by-gene matrices were further analyzed and visualized using the R package Seurat (v5.1.0)103. Ambient RNA contamination in the raw matrices was removed using SoupX (v1.6.2) with the roundToInt=TRUE option in the adjustCounts function and other default parameters104. The data were then normalized using the SCTransform method105. Cells with less than 300 or more than 3000 detected features (nFeature_RNA) were filtered out based on the distribution observed in the violin plots of nFeature_RNA and nCount_RNA.
The pre-processed matrices were subjected to dimensionality reduction, and 20 principal components (PCs) were selected for clustering and UMAP analysis based on the elbow plot of the PCA. DoubletFinder106 was applied to identify and remove doublets in a Seurat object using 20 PCs. The optimal pK value was determined using the sweep.stats function and corresponding plots. The estimated doublet formation ratio, neighborhood size, and ratio of artificial doublet were set to 0.08, 0.08, and 0.25, respectively. The identified doublets were subsequently removed from further analysis. PCA, UMAP dimensionality reduction, and clustering were re-performed using only the singlet cells as described above.
Marker genes for each cluster were identified using the FindAllMarkers function with the parameters only.pos=TRUE, min.pct=0.1, and logfc.threshold=0.2. Cell-type marker genes were compiled from previously published leaf single-cell transcriptome studies (Supplementary Data 1), experimental analyses with spatial validation such as in situ hybridization, and from genes whose known functions are consistent with the characteristic features of the corresponding cell types15,40,41,42,43,44. For each candidate, we identified putative orthologs in the F. suffruticosa genome and annotation and retained only those that satisfied three criteria for cross-species transfer: (1) sequence similarity thresholds of ≥80% query coverage and ≥40% identity (with most orthologs showing ≥90% coverage and ≥60% identity); (2) best BLAST hit in the F. suffruticosa genome, defined by the lowest e-value (and, when the e-value was 0, the highest query coverage and identity); and (3) strong cluster specificity in our dataset, operationalized as ranking within the top 200 cluster markers with p < 0.05. Only transferred marker genes that also overlapped with cluster marker genes under these criteria were ultimately used for cell-type annotation. The specificity of the selected markers was further evaluated by visual inspection of feature plots generated from the integrated Seurat object (Supplementary Fig. 24)15,40,41,42,43,44. From the 124 cell type marker genes, 35 experimentally validated genes were primarily used as markers to assign cell types to each cluster. Among the marker genes of each cluster, those that ranked within the top 200, expressed at levels greater than 10, and identified as orthologs by a BLAST search against the proteome of F. suffruticosa were selected as cell type indicators107. The expression patterns of the marker genes used to predict the cell type of each cluster were depicted as a DotPlot function in the Seurat package (Supplementary Fig. 25).
To identify specialized biological processes associated with each cluster, Gene Ontology (GO) enrichment analysis was performed on the marker gene sets of each cluster. Marker genes were annotated with GO terms derived from functional annotations as described above. GO annotations were processed to extract biological process terms using the GO.db (v3.18.0) and GOSemSim (v2.28.1) R packages108,109. Enrichment analysis was performed using the enricher function of the clusterProfiler (v4.10.1) package, with a p value cutoff of 0.1. GO terms were filtered, and descriptions of enriched terms were retrieved from the GOTERM database110. Dot plots and bar plots were generated to display the most significantly enriched GO terms, labeled with their descriptions.
Co-expression analysis was performed on the processed cell-by-gene matrices. Spearman’s correlation coefficient matrix was calculated based on gene expression levels across all cells. FsBBE2 and FsPS (g19768) were used as bait genes for the co-expression analysis36,37. The expression levels and patterns of FsBBE2, FsPS, and candidate genes involved in SeA biosynthesis were visualized using the VlnPlot and FeaturePlot functions in the Seurat package111.
Co-expression analysis was performed using the hdWGCNA package112. After Seurat-based processing, we constructed metacells using the hdWGCNA metacell workflow with fraction = 0.01. Co-expression modules of the cluster 7 were then inferred with default hdWGCNA parameters. The resulting modules were visualized using the hdWGCNA dendrogram, module eigengene feature plots, and dot plots summarizing the expression of genes within selected modules.
PIP-seq analysis
PIP-seq scRNA-seq libraries were generated from 5-week-old F. suffruticosa young leaves using the PIP-seq T2 workflow113. Protoplasts were isolated following an optimized VCP-enzyme protocol: 0.3 g of the 3rd young leaves from the apex were gently surface-cleaned, thinly sliced without crushing, immediately floated on thawed VCP enzyme solution, vacuum-infiltrated for 5 min (300–400 mmHg), and incubated in the dark on an orbital shaker at 25 rpm for 2 h at 25 °C. The digestion mixture was filtered through a 70 µm cell strainer, diluted in pre-equilibrated 9% (w/v) mannitol, and protoplasts were gently pelleted by low-speed centrifugation (80 × g, 20 °C, 6 min). After careful resuspension in 9% mannitol, intact protoplasts were enriched by sucrose sedimentation on 30% (w/v) sucrose solution (80 × g, 20 °C, 5 min), and the protoplast band was collected with wide-bore tips immediately prior to loading into nanoliter-scale Gel Beads-in-Emulsion (GEMs). The prepared libraries were sequenced on a NovaSeq 6000 platform (Illumina, San Diego, CA, USA) at Macrogen to generate paired-end NGS reads. Raw PIP-seq reads were processed with PIPseeker (v3.3.0) for alignment. Downstream analyses (filtering, normalization, clustering, and marker identification) followed the Seurat-based pipeline described above.
Bulk RNA-seq alignment analysis
Open-source bulk RNA-seq data from F. suffruticosa (PRJCA007529) were used to assess the compatibility of our genome assembly and annotation for read alignment. Raw paired-end reads were quality-trimmed with Trimmomatic (v0.39)114, and the trimmed reads were aligned to the same soft-masked genome and GTF annotation used for the scRNA-seq analyses. A STAR genome index was generated with splice-junction guidance (STAR --runMode genomeGenerate; --sjdbGTFfile; --sjdbOverhang 100; --genomeSAindexNbases 13) using STAR (v2.7.11b)115. Each sample was then mapped in paired-end mode with STAR (v2.7.11b), producing coordinate-sorted BAM files (--outSAMtype BAM SortedByCoordinate) and transcriptome-aligned BAMs for quantification (--quantMode TranscriptomeSAM).
Quantification of rRNA and organelle-derived RNA contamination in scRNA-seq libraries
To quantify rRNA and organelle-derived RNA contamination in scRNA-seq libraries, we applied a reference-based QC procedure following plant sc/snRNA-seq quality-control recommendations56. Nuclear rRNA loci were first predicted directly from the F. suffruticosa genome using Barrnap (kingdom = euk), extracted to FASTA, and de-redundified. Chloroplast and mitochondrial references were obtained from the closest available relatives (Flueggea virosa chloroplast genome, NCBI BK059210.1; Ricinus communis mitochondrial genome, NCBI NC_015141.1) and used solely for contamination estimation. rRNA regions in these organellar genomes were identified with Barrnap (v0.9, kingdom = bac for chloroplast; kingdom = mito for mitochondria) and extracted (https://github.com/tseemann/barrnap). Raw paired-end reads (10x Genomics or PIP-seq) were mapped to (1) nuclear rRNA, (2) chloroplast rRNA and the full chloroplast genome, and (3) mitochondrial rRNA and the full mitochondrial genome using Bowtie2116 (v2.5.4) in very-sensitive mode, and contamination fractions were estimated from mate-level overall alignment rates reported by SAMtools flagstat (v1.22.1)95. To obtain a non-redundant estimate of total contamination, nuclear rRNA, chloroplast genome, and mitochondrial genome references were concatenated into a single “union” reference and reads were remapped; the resulting overall alignment rate was taken as the redundancy-removed rRNA/organelle contamination fraction.
Identification of nonenzymatic reaction to produce neosecurinane scaffold from menisdaurilide and 1-piperideine
A standard nonenzymatic reaction contained 100 mM 1-piperideine (11) and 100 mM menisdaurilide (5) dissolved in buffer solutions having various pH (50 mM phosphate buffer (pH 6, 7, and 8)). Reaction mixtures were incubated for 48 h at 30 °C in a 500 rpm mixing block (Bioer, Hangzhou, China). After the incubation, reactants were extracted with the same volume of 0.1% formic acid water and 20-fold diluted with methanol, filtered, and neosecurinanes were measured as described above.
F. suffruticosa crude lysate feeding assay
To confirm whether hypothetical metabolites are the biosynthetic intermediates of allosecurinine (8) or securinine (9), 13C-labeled potential intermediates were added to the crude tissue lysate with various cofactors. Five-week-old F. suffruticosa seedlings were harvested from the soil and rinsed with deionized water before sampling. Sampled seedlings were snap-frozen in liquid nitrogen and ground using a mortar and pestle with liquid nitrogen. A total of 50 mg of ground seedlings were transferred into 2 mL microcentrifuge tubes and were redissolved in 1 mL of pre-chilled lysis buffer to obtain crude seedling lysates. To assess the enzymatic transition from premenisdaurilide (3) to menisdaurilide (5), 50 mM HEPES buffer (pH 7.0) was used as the lysis buffer, while 100 mM Tris-HCl buffer (pH 8.0) was used to validate the enzymatic transition from neosecurinane to the securinane scaffold. Crude leaf lysates were also prepared using the same method. For the negative control, lysates boiled at 98 °C for 10 min were used. Prepared lysates were aliquoted into 0.2 mL clear PCR tubes, placed on ice, and used for the assay within 30 min.
To evaluate premenisdaurilide reductase activity assay in crude tissue lysate, [13C2]-premenisdaurilide ([13C2]-3) was added (final concentration 1 mM) to crude leaf lysate and boiled leaf lysate. NADH or NADPH was added (final concentration 500 µM) as a cofactor. Substrate-fed lysates were incubated at 30 °C for 20 h with mild agitation. After the incubation, reacted lysates were diluted 100-fold in 50% methanol and filtered for [13C2]-menisdaurilide quantification using the HPLC-MS/MS system as described above. Lysates prepared from four independent plants were used as biological replicates.
To assess neosecurinane sulfotransferase activity in crude tissue lysate, [13C2]-(–)-virosine A ([13C2]−6) and [13C2]-(–)-virosine B ([13C2]−7) were supplied to crude lysate from whole seedlings (final concentration 500 µM). PAPS and ATP were tested as cofactors. Substrate-fed lysates were incubated at 30 °C for 20 h with mild agitation. Reacted lysates were diluted 10-fold in DW and serially diluted 10-fold in methanol. Diluted samples were filtered and analyzed using the HPLC-MS/MS system.
Expression and purification of proteins for in vitro enzyme assay
Coding sequences of putative neosecurinane sulfotransferases were amplified from cDNA produced from RNA from F. suffruticosa leaves. The pET50 plasmids for the expression of candidate sulfotransferases with C-terminal 8×His tag were transformed into Escherichia coli RosettaTM (DE3) strain. Transformed cells were cultured in 250 mL of LB broth media containing kanamycin (50 mg/L) and chloramphenicol (30 mg/L) in a shaking incubator (200 RPM, 37 °C) until the optical density measured at 600 nm (OD600) reached 0.4–0.6. At the OD600, protein induction was started by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. The cells were then incubated at 18 °C for 20 h and harvested by centrifugation.
Cell pellets were resuspended in 10 mL of lysis buffer solution (50 mM Tris-Cl (pH 8.0), 300 mM NaCl, 20 mM imidazole, 10 mM phenylmethylsulfonyl fluoride, 1 tablet of protease inhibitor cocktail (cOmplete mini EDTA-Free) (Roche), 10% (v/v) glycerol, 0.05% (v/v) tween-20) and subsequently lysed by sonication on ice using the Q125 sonicator (10 min, 30% amplitude, 4 s:2 s / on:off) (Qsonica, Newtown, CT, USA). Whole-cell lysates were centrifuged at 16,500 × g, 4 °C for 40 min, and soluble fractions were loaded onto Ni-NTA agarose resin (Thermo Fisher Scientific). After washing with washing buffer (lysis buffer with 50 mM imidazole), the resin was eluted with elution buffer (lysis buffer with 250 mM imidazole). The purification result was confirmed by SDS-PAGE. The purified proteins were then desalted using the PD-10 column (Cytiva, Marlborough, MA, USA) following the manufacturer’s instructions. Protein concentrations of desalted proteins were measured using the PierceTM BCA assay kits (Thermo Fisher Scientific) and were immediately used for the in vitro enzyme assay.
In vitro sulfotransferase activity assay
Purified, desalted proteins (10 µg) were added into 200 µL of the reaction mixture (final concentration of 100 mM Tris-HCl pH 8.0, 10 mM MgCl2, 2 mM 3’-phosphoadenosine-5’-phosphosulfate (PAPS) (Merck KGaA, Darmstadt, Germany), and 0.5 mM (–)-virosine A (6) or (–)-virosine B (7)) and incubated for 22 h at 30 °C. The reaction mixtures were diluted 100-fold with methanol and filtered using syringe filters (0.22 µm) for metabolite analysis. Produced allosecurinine (8) or securinine (9) was measured using HPLC-MS/MS as described above.
Functional evaluation of biosynthetic genes in N. benthamiana
A pCAMBIA-derived binary vector harboring the CDS of target genes downstream of the 35S promoter was transformed into the Agrobacteria tumefaciens AGL1 strain. Transformed strains were cultured overnight in LB medium with antibiotics (50 µg/mL kanamycin, 10 µg/mL rifampicin) at 28 °C with shaking at 220 rpm, followed by centrifugation at 2150 × g for 8 min. The bacterial pellets were resuspended in infiltration buffer containing 10 mM MES (pH 5.60), 10 mM MgCl2, and 150 µM acetosyringone to reach a final OD600 of 0.5 and subsequently incubated in the dark at 25 °C for 2 h. Infiltration was performed on the abaxial surface of rosette leaves from 5-week-old N. benthamiana plants using a sterile 1 mL needleless syringe. To minimize biological variation, two leaves per plant were infiltrated across three independent plants, totaling six biological replicates per strain. After 72 h of infiltration, substrates were fed by injecting 1 mM intermediate solutions (premenisdaurilide (3), (–)-virosine A (6), and (–)-virosine B (7)) into the AGL1-infiltrated leaf. All substrate solutions were prepared and used immediately before use. After 24 h of substrate feeding, the leaf was collected, and products were extracted as described above.
F. suffruticosa leaf disc assay
Leaf discs (8 mm diameter) were excised from 5-week-old F. suffruticosa plants and submerged in a feeding solution containing 1 mM of 13C-labeled compounds ((–)-virosine A (6), (–)-virosine B (7), and 4HPP (2)) or in control buffer (50 mM HEPES, pH 7.0) using 24 well plates. The plates were incubated in a plant growth chamber for 72 h under controlled temperature and light conditions (26 °C, 16 h light / 8 h dark). After incubation, leaf discs were harvested and snap-frozen for extraction as described above. Ground leaf discs were extracted with 1 mL of methanol, filtered, and diluted ten-fold for HPLC-MS/MS analysis.
Sequence and structural analysis of FsNSST1, FsNSST2, and g21405
A maximum-likelihood phylogenetic tree was constructed using amino acid sequences of Arabidopsis sulfotransferases (AtSOT1–AtSOT18) together with all predicted F. suffruticosa sulfotransferases (FsSOTs). Sequences were aligned with MAFFT (v7.522)117 using the L-INS-i algorithm (mafft-linsi --thread 80 --maxiterate 1000), and the resulting alignment was used for tree inference with RAxML-NG v1.2.0 under the JTT + G4 + F substitution model118. Branch support was assessed with 1000 bootstrap replicates, and the final tree was visualized and annotated in iTOL119. Three-dimensional structures were predicted from the full-length protein sequences using AlphaFold3 with default parameters120. Predicted structures were aligned with the reported crystal structure of Arabidopsis sulfotransferase (AtSOT16, PDB: 8K9Y) and visualized in PyMol to compare overall folds, PAPS-binding motifs, and residues surrounding the putative substrate-binding pocket. The crystal structure of (–)-virosine B (29) reported by Kim et al.121 (XYZ file) was imported into AutoDockTools (v1.5.7) and placed into the putative substrate-binding pocket defined from the structural alignment; the pose was manually adjusted with minor refinements before residue–substrate contacts were evaluated. Candidate substrate-contacting residues were inspected by measuring distances between side chains and the docked substrate in the aligned models.
Statistical analysis
To determine statistical significance, all data in this study were analyzed using GraphPad Prism (v.10.4.1) with one of the following statistical tests: one-way ANOVA followed by Tukey’s HSD post hoc test, two-sided Student’s t test, or Welch’s t test. All analyses used independent biological samples (not technical repeats). Sample sizes were not predetermined by statistical methods; unless otherwise stated, experiments were conducted with three or more biological replicates. Exact p values and complete test statistics for each comparison (including one-way ANOVA and post hoc results) are provided in Supplementary Data 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The F. suffruticosa genome assembly, the corresponding HiFi genome sequencing data, and the bulk RNA-seq data used for gene annotation are available from NCBI under BioProject accession PRJNA1284022. Single-cell RNA-seq data from F. suffruticosa leaf tissue (10x Genomics; replicates 1 and 2) and the PiP-seq T2 dataset are publicly available on Zenodo (https://doi.org/10.5281/zenodo.18040926). Source data are publicly available on Figshare (https://doi.org/10.6084/m9.figshare.28979417). All data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Nett, R. S. et al. Plant carbonic anhydrase-like enzymes in neuroactive alkaloid biosynthesis. Nature 624, 182–191 (2023).
Caputi, L. et al. Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 360, 1235–1239 (2018).
Nett, R. S., Lau, W. & Sattely, E. S. Discovery and engineering of colchicine alkaloid biosynthesis. Nature 584, 148–153 (2020).
Boachon, B. et al. A promiscuous CYP706A3 reduces terpene volatile emission from arabidopsis flowers, affecting florivores and the floral microbiome. Plant Cell 31, 2947–2972 (2019).
Wisecaver, J. H. et al. A global coexpression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 29, 944–959 (2017).
De La Peña, R. et al. Complex scaffold remodeling in plant triterpene biosynthesis. Science 379, 361–368 (2023).
Nett, R. S. & Sattely, E. S. Total biosynthesis of the tubulin-binding alkaloid colchicine. J. Am. Chem. Soc. 143, 19454–19465 (2021).
Reed, J. et al. Elucidation of the pathway for biosynthesis of saponin adjuvants from the soapbark tree. Science 379, 1252–1264 (2023).
De La Peña, R. & Sattely, E. S. Rerouting plant terpene biosynthesis enables momilactone pathway elucidation. Nat. Chem. Biol. 17, 205–212 (2021).
Chavez, B. G. et al. Elucidation of tropane alkaloid biosynthesis in Erythroxylum coca using a microbial pathway discovery platform. Proc. Natl. Acad. Sci. USA 119, e2215372119 (2022).
Hong, B. et al. Biosynthesis of strychnine. Nature 607, 617–622 (2022).
Mehta, N., Meng, Y., Zare, R., Kamenetsky-Goldstein, R. & Sattely, E. A developmental gradient reveals biosynthetic pathways to eukaryotic toxins in monocot geophytes. Cell 187, 5620–5637.e5610 (2024).
Sohrabi, R. et al. In planta variation of volatile biosynthesis: an alternative biosynthetic route to the formation of the pathogen-induced volatile homoterpene DMNT via triterpene degradation in arabidopsis roots. Plant Cell 27, 874–890 (2015).
Kang, M., Choi, Y., Kim, H. & Kim, S. G. Single-cell RNA-sequencing of Nicotiana attenuata corolla cells reveals the biosynthetic pathway of a floral scent. N. Phytol. 234, 527–544 (2022).
Li, C. et al. Single-cell multi-omics in the medicinal plant Catharanthus roseus. Nat. Chem. Biol. 19, 1031–1041 (2023).
Sun, S. et al. Single-cell RNA sequencing provides a high-resolution roadmap for understanding the multicellular compartmentation of specialized metabolism. Nat. Plants 9, 179–190 (2023).
Wu, S., Morotti, A. L. M., Yang, J., Wang, E. & Tatsis, E. C. Single-cell RNA sequencing facilitates the elucidation of the complete biosynthesis of the antidepressant hyperforin in St. John’s wort. Mol. Plant 17, 1439–1457 (2024).
McClune, C. J. et al. Multiplexed perturbation of yew reveals cryptic proteins that enable a total biosynthesis of baccatin III and Taxol precursors. bioRxiv https://doi.org/10.1101/2024.11.06.622305 (2024).
Zhan, X. et al. Mass spectrometry imaging and single-cell transcriptional profiling reveal the tissue-specific regulation of bioactive ingredient biosynthesis in Taxus leaves. Plant Commun. 4, 100630 (2023).
Novak, A. J. E., Grigglestone, C. E. & Trauner, D. A biomimetic synthesis elucidates the origin of preuisolactone A. J. Am. Chem. Soc. 141, 15515–15518 (2019).
Piettre, S. & Heathcock, C. H. Biomimetic total synthesis of Proto-daphniphylline. Science 248, 1532–1534 (1990).
Godfrey, R. C., Green, N. J., Nichol, G. S. & Lawrence, A. L. Total synthesis of brevianamide A. Nat. Chem. 12, 615–619 (2020).
Chirkin, E., Atkatlian, W. & Porée, F-H. Chapter one—The Securinega alkaloids. in The Alkaloids: Chemistry and Biology (ed. Knölker, H.-J.) (Academic Press, 2015).
Weinreb, S. M. Total synthesis of the Securinega alkaloids. Nat. Prod. Rep. 26, 758–775 (2009).
Wehlauch, R. & Gademann, K. Securinega alkaloids: complex structures, potent bioactivities, and efficient total syntheses. Asian J. Org. Chem. 6, 1146–1159 (2017).
Perez, M. et al. Synthesis and antiproliferative and metabolic evaluations of novel securinine derivatives. ACS Med. Chem. Lett. 7, 403–407 (2016).
Hou, W. et al. Novel virosecurinine bivalent mimetics as potent reversal agents against P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 183, 111726 (2019).
Hou, W., Huang, H., Wu, X.-Q. & Lan, J.-X. Bioactivities and mechanism of action of securinega alkaloids derivatives reported prior to 2022. Biomed. Pharmacother. 158, 114190 (2023).
Xiao, H. et al. Securinine promotes neuronal development and exhibits antidepressant-like effects via mTOR activation. ACS Chem. Neurosci. 12, 3650–3661 (2021).
Lin, X. & Jun-tian, Z. Neuroprotection by D-securinine against neurotoxicity induced by beta-amyloid (25–35). Neurol. Res. 26, 792–796 (2004).
Parry, R. J. Biosynthesis of the securinega alkaloids-I. Specific incorporation of tyrosine. Tetrahedron Lett. 15, 307–310 (1974).
Parry, R. J. Biosynthesis of the Securinega alkaloids. Stereospecificity of hydrogen loss from C-3 of tyrosine. J. Chem. Soc. Chem. Commun. 5, 144–145 (1975).
Parry, R. J. Biosynthesis of the Securinega alkaloids. Investigations of the early and late stages of the pathway. Bioorg. Chem. 7, 277–288 (1978).
Sankawa, U., Ebizuka, Y. & Yamasaki, K. Biosynthesis of securinine, the main alkaloid of Securinega suffruticosa. Phytochemistry 16, 561–563 (1977).
Golebiewski, W. M., Horsewood, P. & Spenser, I. D. Biosynthesis of securinine: the mode of incorporation of lysine. J. Chem. Soc. Chem. Commun. 6, 217–218 (1976).
Fan, Z. et al. Discovery and biosynthesis of ascorbylated Securinega alkaloids. ACS Catal. 11, 8818–8828 (2021).
Wood, C. X. et al. Nonsymmetric formation of Δ1-piperideine from lysine in plants via a bacterial-like PLP-dependent enzyme. bioRxiv https://doi.org/10.1101/2024.06.04.597157 (2024).
Nett, R. S., Dickschat, J. S. & Peters, R. J. Labeling studies clarify the committed step in bacterial gibberellin biosynthesis. Org. Lett. 18, 5974–5977 (2016).
Nett, R. S. et al. D2O Labeling to measure active biosynthesis of natural products in medicinal plants. AIChE J. 64, 4319–4330 (2018).
Kim, J.-Y. et al. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell 33, 511–530 (2021).
Liu, Z. et al. Global dynamic molecular profiling of stomatal lineage cell development by single-cell RNA sequencing. Mol. Plant 13, 1178–1193 (2020).
Lopez-Anido, C. B. et al. Single-cell resolution of lineage trajectories in the Arabidopsis stomatal lineage and developing leaf. Dev. Cell 56, 1043–1055.e1044 (2021).
Procko, C. et al. Leaf cell-specific and single-cell transcriptional profiling reveals a role for the palisade layer in UV light protection. Plant Cell 34, 3261–3279 (2022).
Xia, K. et al. The single-cell stereo-seq reveals region-specific cell subtypes and transcriptome profiling in Arabidopsis leaves. Dev. Cell 57, 1299–1310.e1294 (2022).
Bardají, G. G. et al. Diastereoselective synthesis of allosecurinine and viroallosecurinine from menisdaurilide. J. Org. Chem. 73, 7657–7662 (2008).
Wehlauch, R. et al. Investigating biogenetic hypotheses of the Securinega alkaloids: enantioselective total syntheses of Secu’amamine E/ent-Virosine A and Bubbialine. Org. Lett. 19, 548–551 (2017).
Park, S., Kang, G., Kim, C., Kim, D. & Han, S. Collective total synthesis of C4-oxygenated securinine-type alkaloids via stereocontrolled diversifications on the piperidine core. Nat. Commun. 13, 5149 (2022).
Park, K. J. et al. Securinega alkaloids from the twigs of Securinega suffruticosa and their biological activities. J. Nat. Prod. 82, 1345–1353 (2019).
Bachmann, T. L., Ghia, F. & Torssell, K. B. G. Lignans and lactones from Phyllanthus anisolobus. Phytochemistry 33, 189–191 (1993).
Brown, P. D., Willis, A. C., Sherburn, M. S. & Lawrence, A. L. Total synthesis of incarviditone and incarvilleatone. Org. Lett. 14, 4537–4539 (2012).
Audran, G. & Mori, K. Synthesis of phyllanthurinolactone, the leaf-closing factor of Phyllanthus urinaria L., and its three stereoisomers. Eur. J. Org. Chem. 1998, 57–62 (1998).
Honzumi, M. & Ogasawara, K. A synthesis of cyclohexanoid butenolides isolated from Sinomenium acutum. Tetrahedron Lett. 43, 1047–1049 (2002).
Cantó, M. et al. First synthesis of (+)-rengyolone and (+)- and (−)-menisdaurilide. Tetrahedron Asymmetry 13, 455–459 (2002).
McNulty, J. et al. Discovery of the apoptosis-inducing activity and high accumulation of the butenolides, menisdaurilide and aquilegiolide in Dicentra spectabilis. Planta Med. 73, 1543–1547 (2007).
Antien, K. et al. Bio-inspired total synthesis of twelve Securinega alkaloids: structural reassignments of (+)-virosine B and (−)-episecurinol A. Chem. Eur. J. 25, 11574–11580 (2019).
Grones, C. et al. Best practices for the execution, analysis, and data storage of plant single-cell/nucleus transcriptomics. Plant Cell 36, 812–828 (2024).
Ren, W. et al. Phytochemical and chemotaxonomic study on the dried rhizome of Menispermum dauricum DC. Biochem. Syst. Ecol. 94, 104189 (2021).
Otsuka, H. et al. Butenolides from Sinomenium acutum. Phytochemistry 33, 389–392 (1993).
Elo Manga, S. S., Messanga, B. B. & Sondengam, B. L. 7,8-Dihydrobenzofuranones from Ouratea reticulata. Fitoterapia 72, 706–708 (2001).
Yi, X.-X. et al. Four new cyclohexylideneacetonitrile derivatives from the hypocotyl of Mangrove (Bruguiera gymnorrhiza). Molecules 20, 14565–14575 (2015).
Wang, L. et al. Chemical constituents of Pteris excelsa and their antioxidant activity. Biochem. Syst. Ecol. 106, 104564 (2023).
Ueda, M., Shigemori-Suzuki, T. & Yamamura, S. Phyllanthurinolactone, a leaf-closing factor of nyctinastic plant, Phyllanthus urinaria L. Tetrahedron Lett. 36, 6267–6270 (1995).
Ueda, M., Asano, M., Sawai, Y. & Yamamura, S. Leaf-movement factors of nyctinastic plant, Phyllanthus urinaria L.; the universal mechanism for the regulation of nyctinastic leaf-movement. Tetrahedron 55, 5781–5792 (1999).
Koprivova, A. & Kopriva, S. Sulfation pathways in plants. Chem. Biol. Interact. 259, 23–30 (2016).
Günal, S., Hardman, R., Kopriva, S. & Mueller, J. W. Sulfation pathways from red to green. J. Biol. Chem. 294, 12293–12312 (2019).
Gidda, S. K. et al. Biochemical and molecular characterization of a hydroxyjasmonate sulfotransferase from Arabidopsis thaliana*. J. Biol. Chem. 278, 17895–17900 (2003).
Marsolais, F. et al. Molecular and biochemical characterization of two brassinosteroid sulfotransferases from Arabidopsis, AtST4a (At2g14920) and AtST1 (At2g03760). Planta 225, 1233–1244 (2007).
Piotrowski, M. et al. Desulfoglucosinolate sulfotransferases from Arabidopsis thaliana catalyze the final step in the biosynthesis of the glucosinolate core structure*. J. Biol. Chem. 279, 50717–50725 (2004).
Klein, M., Reichelt, M., Gershenzon, J. & Papenbrock, J. The three desulfoglucosinolate sulfotransferase proteins in Arabidopsis have different substrate specificities and are differentially expressed. FEBS J. 273, 122–136 (2006).
Hashiguchi, T. et al. Identification of a novel flavonoid glycoside sulfotransferase in Arabidopsis thaliana. J. Biochem. 155, 91–97 (2013).
Reynolds, E. et al. Elucidation of gene clusters underlying withanolide biosynthesis in ashwagandha through yeast metabolic engineering. bioRxiv https://doi.org/10.1101/2024.12.24.630284 (2024).
Yamaguchi, S., Sun, T. -P., Kawaide, H. & Kamiya, Y. The GA2 locus of Arabidopsis thaliana encodes ent-kaurene synthase of gibberellin biosynthesis. Plant Physiol. 116, 1271–1278 (1998).
Henry, L. K. et al. Contribution of isopentenyl phosphate to plant terpenoid metabolism. Nat. Plants 4, 721–729 (2018).
Iketubosin, G. O. & Mathieson, D. W. The isolation of hordenine and norsecurinine from Securinega Virosa (Baill.). The structure of Norsecurinine. J. Pharm. Pharmacol. 15, 810–815 (1963).
Zhang, H., Han, Y.-S., Wainberg, M. A. & Yue, J.-M. Anti-HIV Securinega alkaloid oligomers from Flueggea virosa. Tetrahedron 71, 3671–3679 (2015).
Zhang, H., Wei, W. & Yue, J.-M. From monomer to tetramer and beyond: the intriguing chemistry of Securinega alkaloids from Flueggea virosa. Tetrahedron 69, 3942–3946 (2013).
Saito, S. et al. Determination of securinine and its stereoisomers in plants of the Securinega species, and isolation of viroallosecurinine and virosine. Yakugaku Zasshi 84, 1126–1133 (1964).
Joshi, B. S., Gawad, D. H., Pelletier, S. W., Kartha, G. & Bhandary, K. Isolation and structure (X-ray analysis) of Ent-norsecurinine, an alkaloid from Phyllanthus niruri. J. Nat. Prod. 49, 614–620 (1986).
Arbain, D. et al. The alkaloids of Margaritaria indica. Part 2. The structures of 4-epiphyllanthine, margaritarine and the structural revision of securinol A. J. Chem. Soc. Perkin Trans. 1, 1863–1869 (1991).
Lajis, N., Guan, O., Sargent, M., Skelton, B. & White, A. Viroallosecurinine and ent-Phyllanthidine from the leaves of Breynia coronata (Euphorbiaceae). Aust. J. Chem. 45, 1893–1897 (1992).
Nett, R. S., Dho, Y., Low, Y.-Y. & Sattely, E. S. A metabolic regulon reveals early and late acting enzymes in neuroactive Lycopodium alkaloid biosynthesis. Proc. Natl. Acad. Sci. USA 118, e2102949118 (2021).
Andriyanova, E. A., Lomonosova, M. N. & Berkutenko, A. N. IAPT/IOPB chromosome data 17. Taxon 63, 1148–1155 (2014).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15 (1997).
Kim, T. et al. Genome assembly of Hibiscus sabdariffa L. provides insights into metabolisms of medicinal natural products. G3 Genes Genomes Genetics 14, jkae134 (2024).
Kim, J. & Kim, C. A beginner’s guide to assembling a draft genome and analyzing structural variants with long-read sequencing technologies. STAR Protoc. 3, 101506 (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Saha, S., Bridges, S., Magbanua, Z. V. & Peterson, D. G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294 (2008).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res. 51, D445–D451 (2022).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, lqaa108 (2021).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinforma. 7, 62 (2006).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119–e119 (2014).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2015).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2, lqaa026 (2020).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
TheUniProtConsortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2022).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Young, M. D. & Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience 9, giaa151 (2020).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e324 (2019).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
GeneOntologyConsortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Yu, G. et al. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978 (2010).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287 (2012).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Morabito, S., Reese, F., Rahimzadeh, N., Miyoshi, E. & Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 3, 100498 (2023).
Clark, I. C. et al. Microfluidics-free single-cell genomics with templated emulsification. Nat. Biotechnol. 41, 1557–1566 (2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Kim, T., Kim, S., Chung, G., Park, K. & Han, S. Re-examining the stereochemistry of polycyclic suffruticosine via TDDFT calculations, ECD spectroscopy, and chemical synthesis. Org. Chem. Front. 10, 5123–5129 (2023).
Acknowledgements
We thank Dr. Moonyoung Kang at the Max Planck Institute for Chemical Ecology for valuable advice on scRNA-seq experiments. This work was supported by grants from the National Research Foundation of Korea (NRF) grant fund (NRF-2021R1A2C2011203, NRF-2024-00400556, RS-2024-00352749), Rural Development Administration (RDA) grants (RS-2024-00322407, RS-2024-00322354), KAIST Cross-Generation Collaborative Lab Project, and KAIST Ecological Research Program.
Author information
Authors and Affiliations
Contributions
S.H. and S.-G.K. conceived and supervised the project. S.C., G.K., T.K., S.K., S.H., and S.-G.K. wrote the manuscript. S.C., G.K., T.K., H.Y., and Y.H. performed biochemical experiments. G.K. and S.K. conducted chemical synthesis. S.C., G.K., T.K., H.Y., and H.L. performed metabolite analysis. T.K., H.Y., H.K., and K.M. conducted single-cell transcriptomic analysis. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ryan Nett, COLIN KIM, Dijun Chen, Zhihua Liao, and the other anonymous reviewers for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Choung, S., Kang, G., Kim, T. et al. Chemically guided single-cell transcriptomics reveals sulfotransferase-mediated scaffold remodeling in securinine biosynthesis. Nat Commun 17, 1954 (2026). https://doi.org/10.1038/s41467-026-68816-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68816-3
